

Abstract
Herpes simplex virus type 1 (HSV-1) packages its microns-long double-stranded (ds) DNA genome into a nanometer-scale protein shell, termed the capsid. Upon confinement within the capsid, neighboring DNA strands experience repulsive electrostatic and hydration forces as well as bending stress associated with the tight curvature required of packaged DNA. By osmotically suppressing DNA release from HSV-1 capsids, we provide the first experimental evidence of a high internal pressure of tens of atmospheres within a eukaryotic human virus, resulting from the confined genome. Furthermore, the ejection is progressively suppressed by increasing external osmotic pressures, which reveals that internal pressure is capable of powering ejection of the entire genome from the viral capsid. Despite billions of years of evolution separating eukaryotic viruses and bacteriophages, pressure-driven DNA ejection has been conserved. This suggests it is a key mechanism for viral infection and thus presents a new target for antiviral therapies.
Introduction
Herpes simplex virus type 1 (HSV-1) is one of 8 human pathogenic herpesviruses known today, including Epstein-Barr virus (EBV), cytomegalovirus (CMV), and Kaposi’s sarcoma-associated herpesvirus (KSHV)1. Herpesviruses consist of a double-stranded DNA (dsDNA) molecule contained within a rigid protein shell, termed the capsid. HSV-1 is a prototypical model system to study the general infection mechanisms of herpesviruses and other viruses that release their genome into the cell nucleus without capsid disassembly2. During viral replication and infection, DNA enters and presumably exits the HSV-1 capsid through a specialized opening formed by the portal vertex3. Additional proteins and a lipid membrane surround the DNA-filled capsid, which facilitate viral entry into host cells4. Figure 1 illustrates the HSV-1 infection process as observed by transmission electron microscopy. After binding at the outer membrane (Figure 1a), viruses enter the cell cytoplasm and are transported toward the nucleus (Figure 1b). The viral capsid ejects its genome upon docking to a nuclear pore complex5, which forms a passageway for molecular traffic into the nucleus (Figure 1c).
Figure 1.
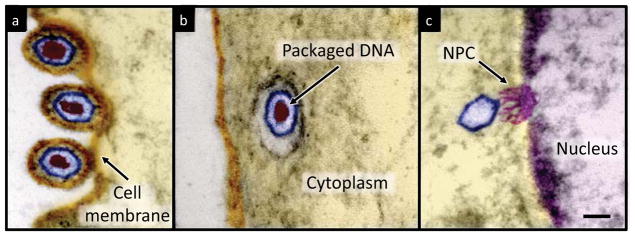
Visualization of HSV-1 infection process. Artificially colored electron micrographs of HSV-1 at the cell membrane (a) in transport to the nucleus (b) and bound at a nuclear pore complex (NPC) embedded within the nuclear envelope (c). The dsDNA genome appears as an electron-dense region within the capsid, which is visible in (a) & (b) but absent in (c) due to DNA ejection upon NPC binding. Scale bar, 50 nm.
Current drug therapies targeting specific viral proteins6 have poor long-term success due to the development of drug resistance that often results from high mutation rates during viral genome replication. Resistance to many antiviral drugs can occur from single amino acid substitutions in the targeted viral protein7. This highlights the importance of understanding the less specific physical properties of the virus particle regulating replication and infectivity8. Recent investigations include the increasing mechanical stability of HSV-1 during capsid maturation9 as well as correlation between human immunodeficiency virus (HIV) stiffness and its ability to enter a host cell10.
The HSV-1 DNA packaging process resembles that of the more extensively studied dsDNA bacteriophages11,12 (viruses that infect bacteria), which package their microns-long genome into a nanometer-scale capsid. This confinement requires DNA to bend along radii that are energetically unfavorable given its 50 nm persistence length*, creating bending stress on the packaged genome13–15. DNA can fill as much as 60% of the internal capsid volume with the remaining volume fraction occupied by water molecules and small ions that freely diffuse through the capsid wall. At these high packaging densities, DNA inter-axial spacings as small as 25 to 30 Å (corresponding to 5 – 10 Å surface separations) create repulsive electrostatic forces between the negatively charged strands of DNA. Furthermore, removal of water molecules from their hydration layers between neighboring DNA helices causes additional repulsive hydration forces16 on the tightly packaged genome. This hydration force scales directly with the inter-axial spacings and dominates over the electrostatic and van der Waals interactions as DNA surfaces approach to within 10 Å of each other16.
To overcome these forces viral DNA packaging motors generate over 50 pico-Newtons, making them the strongest known molecular machine17,18. The work done by the motor during genome packaging is stored as the repulsive and bending energies described above, which create an internal pressure within the capsid that is responsible for DNA ejection13–15,19. The energetics and mechanical properties associated with tight genome confinement have been probed using microcalorimetry20 and atomic force microscopy (AFM)8,21,22. We have investigated this internal pressure for HSV-1 using solutions containing an osmotic stress agent, polyethylene glycol with molecular weight 8000 g/mol (PEG 8000)23. The capsid wall is permeable to water and small ions but not to PEG 8000, thus creating an osmotic pressure gradient between the bulk solution and DNA within the capsid. By determining the PEG concentration required to completely suppress genome ejection, an internal capsid pressure of tens of atmospheres was first confirmed for bacteriophage λ19. Although measured for several bacteriophages, an internal genome pressure has not been measured in a eukaryotic virus. A similar packaging process11,12 and DNA packaging density24,25 for HSV-1 compared to dsDNA bacteriophages has led to years of speculation for the existence of internal pressure within HSV-1 capsids26,27. By osmotically suppressing DNA ejection from HSV-1, this work provides the first experimental evidence of a high internal pressure within a eukaryotic human virus.
Results and Discussion
Mild trypsin treatment induces DNA ejection from HSV-1 capsids in vitro by cleaving the portal protein (UL63) without degrading the major capsid protein (VP5) or causing morphological damage to capsids28. Approximately 40% of capsids eject their DNA in response to trypsin treatment, as determined by UV absorbance spectroscopy (Supporting Information Figure S1). Although this proportion increases with higher trypsin concentrations, a lower concentration reduces the risk of morphological damage or degradation of the major capsid protein, which was not observed to occur for the trypsin concentrations used here28. Southern blot analysis confirms that trypsin-induced DNA ejection follows the expected directionality, with ejection beginning at the S end of the HSV-1 genome29 (Supporting Information Figure S2). This directional ejection supports the notion of DNA release occurring through the portal vertex, as expected for genome ejection in vivo.
After triggering ejection with trypsin, DNA released into solution is degraded by addition of DNase. The non-ejected DNA within capsids was then extracted by SDS and protease K treatment for analysis by pulse field gel electrophoresis (PFGE). Extraction was done in the presence of excess EDTA, which chelates Mg ions, to inactivate DNase from further DNA degradation; Figure 2a illustrates the experimental assay. In standard buffer conditions (Figure 2b, lane 2) only full-length genomes (151 kbp, GenBank accession number JQ780693) are observed, representing the proportion of capsids not opened by trypsin. Absence of DNA molecules smaller than the full genome length confirms that the ~40% of capsids opened by trypsin release their entire genome. We then investigated internal pressure within HSV-1 by inducing DNA ejection in the presence of varying concentrations of PEG. After ejection equilibrates, and the ejected DNA portion is digested by DNase, the non-ejected DNA was extracted from capsids for PFGE analysis as described above. In this way, we could monitor DNA ejection from HSV-1 as a function of the external solution osmotic pressure set by the PEG concentration23. When HSV-1 capsids are opened by trypsin treatment in the presence of PEG, DNA exits the capsid until the decreasing ejection force equals the resisting force imposed by the external osmotic pressure. Higher external osmotic pressures (increasing PEG concentrations) impose larger resisting forces, which suppress longer DNA lengths within the capsid. This results in shorter than full-length genomes retained within capsids, observed for PEG concentrations between 5% and 25% (w/w) (Figure 2b lanes 3–7). Since not all capsids are opened by trypsin treatment, partial-genomes (opened capsids) and full-length genomes (unopened capsids) are present for PEG concentrations of 25% (w/w) and below. In the presence of 30% (w/w) PEG, complete suppression of DNA ejection occurs, yielding only full-length genomes observed by PFGE (Figure 2b, lane 8). This corresponds to an internal pressure of ~18 atmospheres within HSV-1 capsids balanced by an external osmotic pressure of equal magnitude. These results demonstrate the existence of genome pressure within HSV-1 and reveal that this pressure is responsible for ejection of the entire packaged DNA molecule out of the viral capsid.
Figure 2.

Osmotic suppression of HSV-1 genome ejection, (a) Schematic of experimental assay. DNA ejection from HSV-1 capsids is initiated by trypsin digestion in the presence (or absence) of PEG and DNase I at 37°C. Non-ejected DNA was extracted from capsids by sodium dodecyl sulfate (SDS) and protease K treatment and analyzed by pulse field gel electrophoresis (PFGE). (b) PFGE of osmotically suppressed DNA remaining inside viral capsids in the presence of varying concentrations of PEG (lanes 2–8). For all experiments, DNA within unopened capsids resulted in an additional band of approximately 147 kbp. This band was also present for DNA isolated from mature virions as well as phenol/chloroform extracted capsid DNA, independent of PEG or trypsin (not shown). The source of this additional band is not clear, but may relate to variations in a-repeats49 or an alternative genetic discrepancy involved in viral replication or recombination present in a subpopulation of viruses. The faint smears of lower molecular weight DNA present in lanes 6–8 represent sheared DNA fragments that occur from manipulation of large DNA molecules. (c) The fraction of viral genome ejected from HSV-1 capsids (circles) as a function of the external osmotic pressure (conversion of PEG concentration to osmotic pressure is described in the Materials and Methods section), compared to our earlier results for a bacteriophage λ mutant containing 78% of the wild type DNA length (triangles). Error bars represent the standard deviation of the gel band intensity profile. The dashed line is shown as a visual guide for the fraction ejected from HSV-1.
The fraction of HSV-1 DNA ejected as a function of the external osmotic pressure follows a similar dependence as our earlier results19,30 for a bacteriophage λ mutant containing 78% of the wild type DNA length (Figure 2c). As in the case for HSV-1, full suppression of DNA ejection from the λ mutant occurs at ~18 atmospheres, compared to approximately 25 atmospheres pressure required to suppress ejection from wild type DNA length λ30. Cryo-electron microscopy (cryo-EM) reconstructions of HSV-1 and the 78% DNA length λ mutant show the packaged genome as multiple layers along the inner capsid wall with 26 Å inter-layer spacings for both viruses25,31. This suggests that internal capsid pressure directly correlates with the measured DNA inter-layer spacings of the packaged genome. At these high packaging densities electrostatic and hydration forces attempt to maximize spacings to reduce the repulsive interactions between neighboring DNA strands. Yet, the packaging density (volume of DNA divided by the internal capsid volume) is approximately 20% lower for HSV-1 than the λ mutant9, revealing that packaging density alone does not determine DNA-DNA spacings and the resulting internal capsid pressure. X-ray scattering measurements of DNA in solution condensed by PEG demonstrate that DNA adopts a hexagonal packaging structure at the DNA-DNA spacings found in viral capsids16. If the 26 Å inter-layer distance is applied to the entire hexagonally packaged HSV-1 genome, then a significant volume unoccupied by DNA would exist toward the center of the capsid. However, as observed by cryo-EM reconstructions25, DNA is in fact dispersed throughout the HSV-1 capsid with densely packaged layers at the periphery of the capsid volume. This suggests that a balance between repulsive interactions and bending stress results in a lower DNA packaging density toward the center of the capsid, and the consequently decreased spacings along the capsid wall.
Our results suggest that DNA ejection during HSV-1 infection is initially a passive process powered by the internal pressure of the tightly packaged genome. Accordingly, a significant fraction of HSV-1 DNA is released into isolated cell nuclei independent of cellular metabolic energy32. It may be that after a portion of DNA is ejected, which reduces the internal capsid pressure and the corresponding ejection force, cellular or enzymatic processes could facilitate release of the remaining viral DNA32. A similar enzymatic mechanism has been proposed for delivering the final portion of phage genomes during infection33,34. In addition to enzymatic processes, there are alternative suggestions focusing on the osmotic pressure differential across the lipid membrane to promote genome ejection35. Furthermore, to directly measure internal capsid pressure, we use DNase to digest the ejected DNA so that the resulting osmotic pressure balance is determined only by the DNA remaining within the capsid. However, during viral infection in vivo the ejected DNA is not digested and additionally contributes to the pressure balance. As previously shown for bacteriophage λ, in the absence of DNase the external solutions osmotic pressure causes ejected DNA to condense, resulting in a force that pulls additional DNA out of the capsid36. Thus, the osmotic pressure within the cell nucleus created by the high concentration of macromolecules could similarly promote condensation37 of the ejected HSV-1 DNA, thereby pulling out of the capsid any non-ejected portion of the genome. Additionally, this process may also be facilitated by DNA condensing proteins binding to incoming HSV-1 DNA38.
The similarities between osmotically suppressed DNA ejection for a eukaryotic human virus and an E. coli-infecting bacteriophage demonstrates the universality of internal capsid pressure resulting from tight genome confinement. This protease-based assay permits investigation of viral genome pressure independent of a receptor protein to initiate DNA release, a requirement that has previously limited osmotic suppression experiments to a few bacteriophages for which purified receptors were available19,34,39. Determining the presence of internal pressure for other eukaryotic viruses with high genome packaging densities, such as cytomegalovirus40, would be of particular interest. The dependence of internal capsid pressure on genome bending stress and non-specific electrostatic and hydration forces suggests that pressure may also play a role in replication of the double-stranded RNA viruses that utilize an energy-dependent packaging motor41 and have RNA inter-axial spacings comparable to those of pressurized dsDNA viruses42–44. Potential viral replication pathways utilizing internal capsid pressure are summarized in Figure 3.
Figure 3.
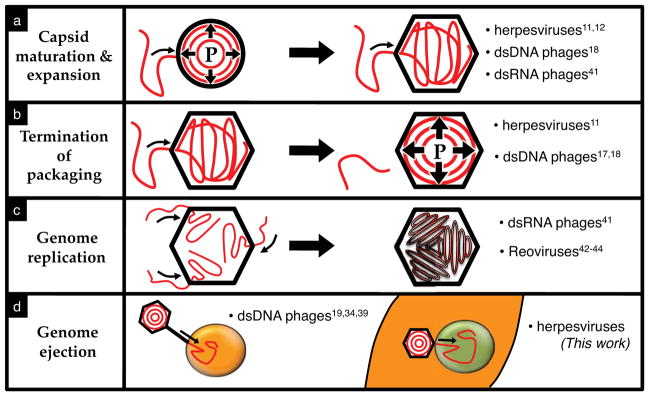
Potential roles of internal capsid pressure during viral replication, (a) An increasing internal pressure (“P”) during genome packaging triggers expansion and maturation of the preassembled immature procapsid. (b) As the genome packaging density increases, the mounting internal pressure demands larger forces of the packaging motor, which may serve as the signal to terminate the packaging reaction. (c) During genome packaging eukaryotic reoviruses replicate single-stranded RNA to double-stranded RNA inside the capsid analogous to dsRNA bacteriophage φ6, which results in genome packaging densities similar to dsDNA viruses. Such intra-capsid replication could be regulated, at least in part, by the generation of an internal pressure resulting from the increasing genome packaging density as newly synthesized dsRNA continues to fill the internal capsid volume. Furthermore, as packaged single-stranded RNA is replicated into double-stranded RNA within the capsid, pressure-induced genome ordering may help organize the tightly packaged genome in a manner that facilitates efficient intra-capsid transcription. (d) Internal capsid pressure powering genome ejection into the host cell (dsDNA bacteriophages) or nucleus (herpesviruses).
As we have recently shown for bacteriophage λ, a one percent increase in the length of the packaged genome above the wild type length leads to a 10-fold decrease in the viral titer. This occurs from an imbalance between the packaging force of the terminase motor and the internal capsid pressure45. Bending energy and repulsive forces acting on the densely packaged viral genome yield a stressed state distinct from condensed cellular DNA. Variations in the degree of DNA condensation can alter the binding of small molecules and proteins to DNA46,47. Additionally, the associated thermodynamics of these interactions are influenced by the number of co-ions released upon binding DNA, which can vary depending on its condensation state48. Developing ways to alter this stressed state of the packaged viral genome could potentially serve as a future antiviral drug target to interfere with genome packaging or ejection during viral replication. The unique physical properties of the tightly confined viral genome, as opposed to a specific nucleotide sequence or encoded protein, represent an evolutionarily static target for antiviral therapies. This limits the potential for development of drug resistance that can occur due to rapid adaptive mutations of viral genomes.
Conclusions
Genome ejection from HSV-1 was progressively suppressed by solutions of varying external osmotic pressure. This result provides the first experimental demonstration of internal genome pressure within a human virus, which is of the same order of magnitude as in dsDNA bacteriophages. The evolutionary conservation of pressure-driven DNA ejection for bacteriophages and eukaryotic viruses suggests that it is a key mechanism for viral infection.
Materials and Methods
HSV-1 capsid isolation
African green monkey kidney cells (Vero) grown in Dulbeccos Modified Eagle’s Medium (Cellgro) with 5% fetal calf serum (GeneMate) and 5% penicillin/streptomycin (Cellgro) were infected with HSV-1 KOS strain at a multiplicity of infection (MOI) of 5 PFU/cell for 20 hours at 37 °C. Cells were scraped into solution and centrifuged at 3,500 RPM for 10 minutes in a JLA-16.250 rotor. The cell pellet was resuspended in 20 mM Tris buffer (pH 7.5) on ice for 20 minutes and lysed by addition of 1.25% (v/v) Triton X – 100 (Alfa Aesar) for 30 minutes on ice. Samples were centrifuged at 2,000 RPM for 10 minutes and the nuclei pellet resuspended in TNE (10 mM Tris, 0.5 M NaCl, 1 mM EDTA) buffer with protease inhibitor cocktail (Complete; Roche). Nuclei were disrupted by sonication for 30 seconds. Large debris were cleared by brief centrifugation and the supernatant spun in a 20–50 % (w/w) TNE sucrose gradient at 24,000 rpm in a Beckman SW41 rotor for 1 hour. The C-capsid band was isolated by side puncture, diluted in TNE buffer and centrifuged at 24,000 rpm for an additional 1 hour. Capsids were resuspended in TNE and stored at 4 °C.
Trypsin-induced HSV-1 genome ejection
HSV-1 C-capsids were diluted into TM buffer (50 mM Tris, 10 mM MgSO4, pH 7.4). To determine the total amount of protected viral DNA present, capsids were thermally ruptured by incubation at 90 °C for 15 minutes followed by slow cooling. Heated capsids, as well as intact capsids with and without trypsin (MP Biomedicals) were incubated at 37 °C for 1.5 hours in the presence of 0.1 mg/mL DNase I (Akron) to digest DNA released from capsids, followed by filtration (Pall, 10 kDa MWCO) to remove capsids and their non-ejected DNA. The digested nucleotide concentration of the filtrate was determined by the 260 nm absorbance peak (Supporting Information Figure S1) using an Agilent 8453 Spectrophotometer with a 1 cm path length quartz cuvette. Samples containing trypsin and DNase (but without HSV-1 capsids) were prepared to account for the absorbance contribution due to the presence of trypsin and DNase. The amount of HSV-1 DNA ejected in response to trypsin treatment was determined by comparing the UV absorbance intensity to that of the heated and untreated capsids (Supporting Information Figure S1). Trypsin concentrations were adjusted so that ~40% of DNA is ejected, which corresponded to approximately 2 μg/mL.
Osmotic suppression of DNA ejection
HSV-1 C-capsids along with varying concentrations of 8000 MW polyethylene glycol (PEG) (Fisher) were incubated at 37 °C for 1.5 hours with trypsin and DNase as described above. The corresponding osmotic pressure (Π) as a function of the PEG weight-weight percentage (w) was determined by the empirical relation19 Π(atm) = −1.29G2T + 140G2 + 4G, where G=w/(100 – w) and T is the temperature (°C). Horizontal error bars of Figure 2c represent the standard error in the osmotic pressure resulting from pipetting viscous PEG solutions. Non-ejected DNA was extracted from capsids by addition of 10 mM ethylenediaminetetraacetic (EDTA) (Duchefa), 0.5% (w/v) SDS (Sigma), and 50 μg/mL protease K (Amresco) followed by a 1 hour incubation at 65 °C. The length of osmotically suppressed DNA within capsids was determined by pulse field gel electrophoresis using a Bio-Rad CHEF II DR at 6 V/cm with initial and final switch times of 4 and 13 seconds respectively. Gels were stained with SybrGold and size estimations performed with UVP VisionWorksLS software using the Mid-range molecular weight standard from New England BioLabs as a reference.
Southern blot hybridization was used to identify the genome end osmotically suppressed within capsids in the presence of 5% (w/w) PEG. Samples were prepared as described above, except that gels were stained with ethidium bromide. Blotting was then performed with probes specific to the L and S segments of the HSV-1 genome (GenBank accession number JQ780693).
Electron microscopy
Vero cells were infected at an MOI of 300 PFU/cell and fixed with 2% gluteraldehyde in phosphate buffered saline (PBS) 1 to 3 hours post infection, followed by 1 hour incubation with 1% osmium tetroxide in PBS. Samples were dehydrated by a graded ethanol series and propylene oxide, embedded in Epon, and further contrasted with lead citrate and uranyl acetate.
Supplementary Material
Acknowledgments
We gratefully acknowledge David Marino for helping in the early development of the experimental assay, William Newcomb for providing essential advice with designing the trypsin-induced ejection assay, Joseph Suhan for assistance in obtaining EM images, Abhishek Mandal for technical assistance, James Conway and Nathan Urban for helpful advice during preparation of the manuscript. This work was supported by the Swedish Research Council, VR grant 622-2008-726 (A.E) and NSF grant CHE-1152770 (A.E.). Support was also provided by Public Health Service grant AI060836 from the National Institutes of Health (F.L.H). D.W.B. has received support from the Mellon College of Science Dean’s Fund.
Footnotes
Persistence length defines the stiffness of a polymer, describing the minimum radius of curvature it can adopt by the available thermal energy. Bending it to a smaller radius requires additional work.
Supporting Information Available
Supporting Information Figures S1 – S3. This information is available free of charge via the internet at http://pubs.acs.org.
References
- 1.Davison AJ, Eberle R, Ehlers B, Hayward GS, McGeoch DJ, Minson AC, Pellett PE, Roizman B, Studdert MJ, Thiry E. Arch Virol. 2009;154:171–177. doi: 10.1007/s00705-008-0278-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Whittaker GR, Kann M, Helenius A. Annu Rev Cell Dev Biol. 2000;16:627–651. doi: 10.1146/annurev.cellbio.16.1.627. [DOI] [PubMed] [Google Scholar]
- 3.Newcomb WW, Juhas RM, Thomsen DR, Homa FL, Burch AD, Weller SK, Brown JC. J Virol. 2001;75:10923–10932. doi: 10.1128/JVI.75.22.10923-10932.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Grünewald K, Desai P, Winkler DC, Heymann JB, Belnap DM, Baumeister W, Steven AC. Science. 2003;302:1396–1398. doi: 10.1126/science.1090284. [DOI] [PubMed] [Google Scholar]
- 5.Sodeik B, Ebersold MW, Helenius A. J Cell Biol. 1997;136:1007–1021. doi: 10.1083/jcb.136.5.1007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Baines JD. Trends Microbiol. 2011;19:606–613. doi: 10.1016/j.tim.2011.09.001. [DOI] [PubMed] [Google Scholar]
- 7.Thomsen DR, Oien NL, Hopkins TA, Knechtel ML, Brideau RJ, Wathen MW, Homa FL. J Virol. 2003;77:1868–1876. doi: 10.1128/JVI.77.3.1868-1876.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Roos WH, Bruinsma R, Wuite GJL. Nat Phys. 2010;6:733–743. [Google Scholar]
- 9.Roos WH, Radtke K, Kniesmeijer E, Geertsema H, Sodeik B, Wuite GJL. Proc Natl Acad Sci USA. 2009;106:9673–9678. doi: 10.1073/pnas.0901514106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kol N, Shi Y, Tsvitov M, Barlam D, Shneck RZ, Kay MS, Rousso I. Biophy J. 2007;92:1777–1783. doi: 10.1529/biophysj.106.093914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Baines JD, Weller SK. In: Viral Genome Packaging Machines: Genetic, Structure, and Mechanism. Catalano CE, editor. Landes; Georgetown: 2005. pp. 135–150. [Google Scholar]
- 12.Conway JF, Homa FL. In: Alphaherpesviruses. Weller SK, editor. Caister; Norwich: 2011. pp. 175–193. [Google Scholar]
- 13.Kindt J, Tzlil S, Ben-Shaul A, Gelbart WM. Proc Natl Acad Sci USA. 2001;98:13671–13674. doi: 10.1073/pnas.241486298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Purohit PK, Kondev J, Phillips R. Proc Natl Acad Sci USA. 2003;100:3173–3178. doi: 10.1073/pnas.0737893100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Tzlil S, Kindt JT, Gelbart WM, Ben-Shaul A. Biophys J. 2003;84:1616–1627. doi: 10.1016/S0006-3495(03)74971-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Rau DC, Lee B, Parsegian VA. Proc Natl Acad Sci USA. 1984;81:2621–2625. doi: 10.1073/pnas.81.9.2621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Smith DE, Tans SJ, Smith SB, Grimes S, Anderson DL, Bustamante C. Nature. 2001;413:748–752. doi: 10.1038/35099581. [DOI] [PubMed] [Google Scholar]
- 18.Fuller DN, Raymer DM, Rickgauer JP, Robertson RM, Catalano CE, Anderson DL, Grimes S, Smith DE. J Mol Biol. 2007;373:1113–1122. doi: 10.1016/j.jmb.2007.09.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Evilevitch A, Lavelle L, Knobler CM, Raspaud E, Gelbart WM. Proc Natl Acad Sci USA. 2003;100:9292–9295. doi: 10.1073/pnas.1233721100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Jeembaeva M, Jönsson B, Castelnovo M, Evilevitch A. J Mol Biol. 2010;395:1079–1087. doi: 10.1016/j.jmb.2009.11.069. [DOI] [PubMed] [Google Scholar]
- 21.Ivanovska I, Wuite G, Jönsson B, Evilevitch A. Proc Natl Acad Sci USA. 2007;104:9603–9608. doi: 10.1073/pnas.0703166104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hernando-Pérez M, Miranda R, Aznar M, Carrascosa JL, Schaap IAT, Reguera D, de Pablo PJ. Small. 2012;8:2366–2370. doi: 10.1002/smll.201200664. [DOI] [PubMed] [Google Scholar]
- 23.Parsegian VA, Rand RP, Fuller NL, Rau DC. Meth Enzymol. 1986;127:400–416. doi: 10.1016/0076-6879(86)27032-9. [DOI] [PubMed] [Google Scholar]
- 24.Booy FP, Newcomb WW, Trus BL, Brown JC, Baker TS, Steven AC. Cell. 1991;64:1007–1015. doi: 10.1016/0092-8674(91)90324-r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zhou ZH, Chen DH, Jakana J, Rixon FJ, Chiu W. J Virol. 1999;73:3210–3218. doi: 10.1128/jvi.73.4.3210-3218.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gelbart WM, Knobler CM. Science. 2009;323:1682–1683. doi: 10.1126/science.1170645. [DOI] [PubMed] [Google Scholar]
- 27.Liashkovich I, Hafezi W, Kühn JM, Oberleithner H, Shahin V. J Mol Recognit. 2011;24:414–421. doi: 10.1002/jmr.1120. [DOI] [PubMed] [Google Scholar]
- 28.Newcomb WW, Booy FP, Brown JC. J Mol Biol. 2007;370:633–642. doi: 10.1016/j.jmb.2007.05.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Newcomb WW, Cockrell SK, Homa FL, Brown JC. J Mol Biol. 2009;392:885–894. doi: 10.1016/j.jmb.2009.07.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Grayson P, Evilevitch A, Inamdar M, Purohit P, Gelbart W, Knobler C, Phillips R. Virology. 2006;348:430–436. doi: 10.1016/j.virol.2006.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lander GC, Johnson JE, Rau DC, Potter CS, Carragher B, Evilevitch A. Nucleic Acids Res. 2013;41:4518–4524. doi: 10.1093/nar/gkt137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ojala PM, Sodeik B, Ebersold MW, Kutay U, Helenius A. Mol Cell Biol. 2000;20:4922–4931. doi: 10.1128/mcb.20.13.4922-4931.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.González-Huici V, Salas M, Hermoso JM. Mol Microbiol. 2004;52:529–540. doi: 10.1111/j.1365-2958.2004.03993.x. [DOI] [PubMed] [Google Scholar]
- 34.São-José C, de Frutos M, Raspaud E, Santos MA, Tavares P. J Mol Biol. 2007;374:346–355. doi: 10.1016/j.jmb.2007.09.045. [DOI] [PubMed] [Google Scholar]
- 35.Molineux IJ. Virology. 2006;344:221–229. doi: 10.1016/j.virol.2005.09.014. [DOI] [PubMed] [Google Scholar]
- 36.Jeembaeva M, Castelnovo M, Larsson F, Evilevitch A. J Mol Biol. 2008;381:310–323. doi: 10.1016/j.jmb.2008.05.081. [DOI] [PubMed] [Google Scholar]
- 37.Teif VB, Bohinc K. Prog Biophys Mol Biol. 2011;105:208–222. doi: 10.1016/j.pbiomolbio.2010.07.002. [DOI] [PubMed] [Google Scholar]
- 38.Oh J, Fraser NW. J Virol. 2008;82:3530–3537. doi: 10.1128/JVI.00586-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Leforestier A, Brasilès S, de Frutos M, Raspaud E, Letellier L, Tavares P, Livolant F. J Mol Biol. 2008;384:730–739. doi: 10.1016/j.jmb.2008.09.035. [DOI] [PubMed] [Google Scholar]
- 40.Bhella D, Rixon FJ, Dargan DJ. J Mol Biol. 2000;295:155–161. doi: 10.1006/jmbi.1999.3344. [DOI] [PubMed] [Google Scholar]
- 41.Poranen MM, Pirttimaa MJ, Bamford DH. In: Viral Genome Packaging Machines: Genetic, Structure, and Mechanism. Catalano CE, editor. Landes; Georgetown: 2005. pp. 117–134. [Google Scholar]
- 42.Gouet P, Diprose JM, Grimes JM, Malby R, Burroughs JN, Zientara S, Stuart DI, Mertens PP. Cell. 1999;97:481–490. doi: 10.1016/s0092-8674(00)80758-8. [DOI] [PubMed] [Google Scholar]
- 43.McClain B, Settembre E, Temple BRS, Bellamy AR, Harrison SC. J Mol Biol. 2010;397:587–599. doi: 10.1016/j.jmb.2010.01.055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Prasad BV, Rothnagel R, Zeng CQ, Jakana J, Lawton JA, Chiu W, Estes MK. Nature. 1996;382:471–473. doi: 10.1038/382471a0. [DOI] [PubMed] [Google Scholar]
- 45.Nurmemmedov E, Castelnovo M, Medina E, Catalano CE, Evilevitch A. J Mol Biol. 2012;415:263–273. doi: 10.1016/j.jmb.2011.11.015. [DOI] [PubMed] [Google Scholar]
- 46.Teif VB. Biophys J. 2005;89:2574–2587. doi: 10.1529/biophysj.105.063909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Porschke D. J Mol Biol. 1991;222:423–433. doi: 10.1016/0022-2836(91)90220-z. [DOI] [PubMed] [Google Scholar]
- 48.Burak Y, Ariel G, Andelman D. Biophys J. 2003;85:2100–2110. doi: 10.1016/s0006-3495(03)74638-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Umene K, Oohashi S, Yoshida M, Fukumaki Y. J Gen Virol. 2008;89:841–852. doi: 10.1099/vir.0.83467-0. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.