

Abstract
A series of inhibitors of the soluble epoxide hydrolase (sEH) containing two urea groups has been developed. Inhibition potency of the described compounds ranges from 2.0 μM to 0.4 nM. 1,6-(hexamethylene)bis[(adamant-1-yl)urea] (3b) was found to be a potent slow tight binding inhibitor (IC50 = 0.5 nM) with a strong binding to sEH (Ki = 3.1 nM) and a moderately long residence time on the enzyme (koff = 1.05×10−3 s−1; t1/2 = 11 min).
Keywords: soluble epoxide hydrolase, inhibitor, adamantane, isocyanate, diurea
In mammals, the soluble epoxide hydrolase (sEH, E.C. 3.3.2.10) is involved in the metabolism of epoxy-fatty acids to vicinal diols through a catalytic addition of a water molecule.1,2 Endogenous substrates for the sEH include cytochrome P450 metabolites of arachidonic acid, such as epoxyeicosatrienoic acids (EETs), and of docosahexaenoic acid, known as EpDPEs.3,4 EETs exert vasodilatory effects through the activation of the Ca2+-activated K+ channels in endothelial cells, which are beneficial in many renal and cardiovascular diseases.5,6 Furthermore, the EETs have some anti-inflammatory and analgesic properties.7 Their conversion to dihydroxyeicosatrienoic acids (DHETs) by sEH produces a molecular that is readily conjugated and removed from the site of action. The inhibition of sEH in vivo by highly selective inhibitors results in an increase of the concentration of EETs and is accompanied by a reduction in angiotensin driven blood pressure in rodent models, but also reduction of inflammatory and painful states, thereby suggesting that sEH is a target for the treatment of hypertension, inflammatory diseases and pain.8–10
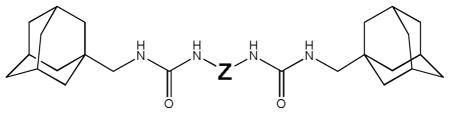
Small N,N′-disubstituted symmetric ureas, such as 1,3-dicyclohexyl urea, were found to be very potent inhibitors of sEH.11–15 However, because of their strong crystalline lattice, these kinds of compounds have poor solubility in many solvents. To improve solubility, asymmetric ureas with a flexible side chain, such as AUDA (12-(3-adamantylureido)-dodecanoic acid), were tested and found to be potent sEH inhibitors. While this class of sEH inhibitor shows biological effects when tested in vivo, they are rapidly metabolized, limiting their utility. In contrast, compounds that lack flexible side chain usually have poor physical properties so they show limited biological effects in vivo without careful formulation.16,17 Therefore, to improve the metabolic stability, a third class of conformationally restricted inhibitors, such as AEPU (1-adamantyl-3-(1-acetylpiperidin-4-yl)-urea) or t-AUCB (trans-4-((4-(3-adamantylureido)-cyclohexyl)oxy)-benzoic acid), were designed. This latest series includes very potent and more metabolically stable sEH inhibitors that permit in vivo studies. However, these compounds have in general poor solubility, and are quite expensive to synthesize since several steps (3 to 5) are required. Here, we report the testing of symmetric di-ureas that are simpler to obtain as sEH inhibitors. As shown on Figure 1, a flexible chain was incorporated at the center of the molecules to improve physical properties, while adamantane and urea groups were placed at both ends of the molecules to protect the central flexible chain from metabolism, and to provide the additional possibility of hydrogen bonding to improve potency and solubility.
Figure 1.
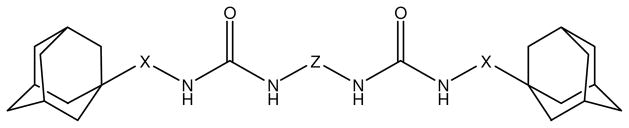
General structure of synthesized diureas
As described on scheme 1, two simple (one step) and complementary approaches were used to obtain the desired compounds in high yield (> 95%). Commercially available 1-isocyanatemethyl adamantane or various adamantyl containing isocyanates18 were reacted with various amines containing 2, 4, 6 or 8 carbons that are usually used in supramolecular chemistry as guest-monomers.19–21. To vary the X–parameter, several commercially available hydrochlorides of amines were reacted with alkyl di-isocyanates. Compounds containing phenyl and piperidine rings between the urea groups were synthesized as well because those groups commonly confer properties found to be valuable in medicinal chemistry.22–24 Structures of the obtained chemicals were assessed by NMR, while purity was assessed by mass spectrometry and elemental analysis (see supplemental materials for details).
Scheme 1.

Reagents and conditions: (a) adamant-2-ylmethyl isocyanate (1.9 equiv), DMF, rt, 12 h; (b) triethylamine (2 equiv), DMF, 0–25 °C, 12h.
The inhibitor potency of the synthesized compounds was measured using recombinant purified human sEH and CMNPC (cyano(6-methoxynaphthalen-2-yl)methyl ((3-phenyloxiran-2-yl)methyl) carbonate) as a substrate as described.25 For the di-adamantyl urea-based compounds (1a–1f), increasing the length of the flexible chain between the urea groups from 2 to 6 carbons in the compounds 1a–c lead to a > 400-fold increase in potency (lower IC50). Further increase of chain length to 8 carbons resulted in a 15-fold decrease of inhibition potency for compound 1d, suggesting an optimal length for interaction with the enzyme. 1,4-Diaminobenzene (1e) and piperidine (1f) based disubstituted diureas also showed poor potency, presumably because the significant reduction of flexibility between the urea groups did not permit an optimal positioning of the compounds inside the enzyme active site.
In the 2, 3 and 4 series, not only the length and nature of the chain between the urea groups (Z) but also the spacer connecting the urea groups with adamantane (X) were altered as well (Table 2). As found with the first series (Table 1), the presence of an alkyl chain in the middle of the molecule (series 2 and 3) yielded globally more potent inhibitors than the presence of a phenyl group (series 4). While, as observed for series 1, the length of the middle chain influenced potency (globally, series 2 (with 4 carbon) yielded more potent compounds than series 3 (8 carbon)), the IC50s were markedly influenced by the spacer between the adamantanes and ureas (X), especially in the 3 series. This provides evidence for the orientation of the inhibitor in the active site of human sEH, and raises the possibility that the second urea makes strong polar interactions with the enzyme. Interestingly, changing the bond from the ureas to the adamantane from a 1- (2a and 3a) to a 2- (3a to 3d) position does not alter the potency of the compounds with short central alkyl chains (2a and 2d), but dramatically (≈500-fold) decreases the potency of a compound with a longer central alkyl chain (3d).
Table 2.
IC50 values for diadamantyl-based sEH inhibitors 2a–d, 3a–d and 4a–b*
| ||||
|---|---|---|---|---|
| # | X | Z | IC50 (nM)a | mp (°C) |
| 2a | - |
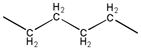
|
7.2 | 251.1–253.4 |
| 2b |
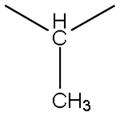
|
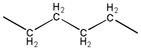
|
1.2 | 257.1–258.9 |
| 2c |
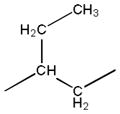
|
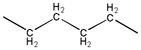
|
2.3 | 210.9–213.7 |
| 3a | - |
|
997 | 256.3–257.8 |
| 3b |
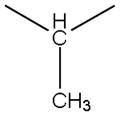
|
|
0.5 | 191.8–192.9 |
| 3c |
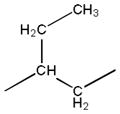
|
|
1440 | 130.1–131.4 |
| 4a | - |
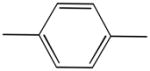
|
19.5 | 362.4–364.1 |
| 4b |
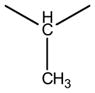
|
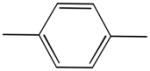
|
161 | 285.2–286.1 |
|
| ||||
| 2d | - |
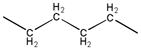
|
10.4 | 274.2–276.0 |
| 3d | - |
|
2.6 | 254.6–255.4 |
As determined via a kinetic fluorescent assay.25
Table 1.
IC50 values for diadamantyl urea-based sEH inhibitors 1a–f*.
| |||
|---|---|---|---|
| Z | n | # | IC50 (nM)a |
| -(CH2)n- | 2 | 1a | 179.2 |
| 4 | 1b | 26.3 | |
| 6 | 1c | 0.4 | |
| 8 | 1d | 7.3 | |
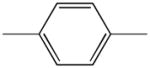
|
1e | 779.6 | |
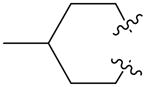
|
1f | 39.7 | |
As determined via a kinetic fluorescent assay.25
For the more potent diadamantyl diureas, we evaluated the inhibitor binding constant (Ki), the inhibitor off rate (koff), and the inhibitor residence time in the enzyme (t1/2) using a Förster resonance energy transfer (FRET) competitive displacement assay developed for the human sEH (Table 3).26 Compounds with the same molecular weight (two adamantane groups, two urea groups and eight carbons comprising various spacers between those groups) were selected for further testing to ensure that any difference arises from specific structural feature and not due to a general effect of the molecules.
Table 3.
IC50, Ki and koff for a selection of the best compounds
In general, the trends in potency measured by a kinetic IC50 assay correlated with the equilibrium Ki determination. While the IC50 of compounds 1c, 2b, 3b and 3d was distributed over a 5-fold difference range, the Ki for these four diureas only ranged between 2.4 and 3.4 nM. These values are very similar and are almost equal with the variation of the assay over a narrow range of Kis. Because the urea inhibitors are slow-tight binding ligands of sEH, the IC50s measured after only 5 minutes incubation, as shown here, probably underestimated the true potency of some compounds, explaining the apparent divergence between IC50 and Ki values.26 The Ki is a ratio of kinetic off and on rates which are independently altered by structure. Ki reflects the strength of the interaction between the inhibitor and the sEH. The inhibitor is only effective in blocking the hydrolysis of endogenous bioactive fatty acid epoxides while it is bound to the target enzyme. Because of this, determination of residence time, the period when the drug is bound to enzyme, is very important for predicting biological activity. 26 Interestingly, among the 4 compounds tested, the koff were distributed over a 5-fold difference range and parallel the IC50s. These data indicate that spacers between the urea group and the the adamantane lead to a slower koff and have an optimal length. These results suggest that the molecular flexibility given by the alkyl spacers allows a better fit to the enzyme’s active site, including formation of a new hydrogen bond by the second urea group. For the diureas without such flexible spacers, formation of new hydrogen bonds is hampered by the rigidity of the inhibitor, which does not allow optimal positioning.
To predict bioavailability of disubstituted biureas, the solubility of four the most potent inhibitors were measured in phosphate buffered water and their cLogP calculated (Table 4). Diadamantyl diureas with a flexible chain between urea groups are more potent and more soluble that the simple symmetric ureas (DCU and DAU), the first potent sEH inhibitors.11,27 Interestingly, the cLogP values suggest that compounds 1c and 2b have drug-like solubility, and thus should have good bioavailability.28 While the four compounds tested have the same mass and atomic composition, 1c is markedly more soluble than the other three. Because the cLogP values suggest that the compounds are quite hydrophobic, it is possible that the observed inhibition is due to the aggregation of the chemicals on the enzyme.29 Across the compound tested, increasing the BSA concentration in the buffer by 10-fold resultedin an increase in inhibition potencies (IC50s) by around 2-fold (Table 4), suggesting that the observed inhibition is neither due to non-specific interaction nor to aggregation.29 To furtherassess the properties of the compounds, we measured their stability in human liver microsomes (Table 4). While the compounds were stable in the absence of NADPH, they were metabolized in its presence, probably by P450s. Compounds 2b and 3d were the most stable, suggesting that secondary adamantyl are more stable than tertiary. Compared to 1b, in presence of NADPH, 2b and 3b are more stable suggesting that a methyl on the carbon linker between the urea and adamantyl is preferable.
Table 4.
IC50 and water solubility of the selected compounds
| # | IC50 nMa
|
Solubilityb (μM) | cLogPd | Microsomal stability (%)e
|
||
|---|---|---|---|---|---|---|
| BSA 0.1 mg/mL | BSA 1.0 mg/mL | NADPH
|
||||
| − | + | |||||
| 1c | 0.4 | 0.7 | 50 < S < 75 | 4.54 | 102 | 3 |
| 2b | 1.2 | 2.5 | 10 < S < 25 | 4.34 | 92 | 24 |
| 3b | 0.5 | 0.8 | 10 < S < 25 | 6.01 | 100 | 11 |
| 3d | 2.6 | 4.1 | 5 < S < 10 | 4.96 | 100 | 36 |
|
| ||||||
| DCUc | 3.0 | 5.1 | 5 < S < 10 | 2.28 | 100 | 22 |
|
| ||||||
| DAUc | 1.2 | 1.3 | 5 < S < 10 | 2.84 | 93 | 19 |
As determined via a kinetic fluorescent assay.25
Solubilities were measured in sodium phosphate buffer (pH 7.4, 0.1 M) containing 1% of DMSO.
DCU: 1,3-dicyclohexyl urea; DAU: 1,3-diadamantyl urea.
Calculated using ChemBioDraw Uultra v12.0 (PerkinElmer, Waltham, MA).
Percent of compound (1 μM) remaining after 30 minutes incubation with human liver microsomes (1 mg/mL) at 37°C with or without NADPH generating system. Results are triplicate average. 5 to 10% errors were obtained.
To understand the increased potency of the diureas, compound 3b was manually docked into the active site of the human sEH using the “bioMedCAChe 5.0” software (Fujitsu computer Systems Corporation). For this, the published X-ray crystal structure of the human sEH complexed with a urea-based ligand (PDB accession number 1ZD3) was used. After docking, the ligand and the amino acid residues within 8.0 Å from the ligand were minimized on MM geometry (MM3) as described.17 As expected, one of the urea groups of 3b forms strong hydrogen bonds with the catalytic residues (Asp335, Tyr383 and Tyr466) of the sEH. Surprisingly, the other urea group of 3b forms a hydrogen bond with Ser374 side chain. This is a novel hydrogen bond for this family of sEH ligands, and probably explains the unexpected high potency of compound 3b compared to 3a and 3c. For these later compounds, the steric bulky groups (adamantane for 3a and 1-(2-methylbutyl)adamantane for 3c) next to the urea function should impede the formation of this extra bond with the enzyme.
We described the synthesis and structure-activity relationships of a series of di-adamantyl diureas containing various spacers between the two urea groups. The data show that ureas with small flexible linkers between the adamantane and the urea group show excellent inhibition potency along with good binding parameters. Diureas with phenyl spacers between urea groups showed poor activity due to the small size of molecule and the lack of flexibility. We showed that compounds with flexible adamantanes have significantly better binding to the enzyme than those without, as described by the Ki and koff values. This can be explained by the fact that the flexible chain allows second urea group to form additional hydrogen bonds with Ser374. This is the first time that this residue is shown to be involved in sEH ligand binding. Lower mp of the flexible diureas could have a positive indirect effect on their ease of formulation as well.
Fig. 1.

Compound 3b (blue) docked into the active site of epoxide hydrolase domain of human sEH (cyan). Hydrogen bonds are indicated by dashed lines. However, hydrogen bonds between Tyr466, Tyr383 and the carbonyl of the urea group are not indicated for the sake of clarity.
Acknowledgments
The reported study was partially funded by National Institute of Environmental Health Sciences (NIEHS) grant R01 ES002710, NIEHS Superfund Research Program grant P42 ES004699 and Russian Foundation for Basic Research (RFBR) grant No. 12-03-33044.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Arand M, Grant DF, Beetham JK, Friedberg T, Oesch F, Hammock BD. FEBS Lett. 1994;338:251. doi: 10.1016/0014-5793(94)80278-5. [DOI] [PubMed] [Google Scholar]
- 2.Oesch F. Xenobiotica. 1973;3:305. doi: 10.3109/00498257309151525. [DOI] [PubMed] [Google Scholar]
- 3.Zeldin DC, Kobayashi J, Falck JR, Winder BS, Hammock BD, Snapper JR, Capdevilla JH. J Biol Chem. 1993;268:6402. [PubMed] [Google Scholar]
- 4.Spector AA, Fang X, Snyder GD, Weintraub NL. Prog Lipid Res. 2004;43:55. doi: 10.1016/s0163-7827(03)00049-3. [DOI] [PubMed] [Google Scholar]
- 5.Fleming I, Rueben A, Popp R, Fisslthaler B, Schrodt S, Sander A, Haendeler J, Falck JR, Morisseau C, Hammock BD, Busse R. Arterioscler Thromb Vasc Biol. 2007;27:2612. doi: 10.1161/ATVBAHA.107.152074. [DOI] [PubMed] [Google Scholar]
- 6.Imig JD. Expert Opin Drug Metab Toxicol. 2008;4:165. doi: 10.1517/17425255.4.2.165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Yu Z, Xu F, Huse LM, Morisseau C, Draper AJ, Newman JW, Parker C, Graham L, Engler MM, Hammock BD, Zeldin DC, Kroetz DL. Circ Res. 2000;87:992. doi: 10.1161/01.res.87.11.992. [DOI] [PubMed] [Google Scholar]
- 8.Spiecker M, Liao JK. Arch Biochem Biophys. 2005;433:420. doi: 10.1016/j.abb.2004.10.009. [DOI] [PubMed] [Google Scholar]
- 9.Imig JD, Zhao X, Capdevilla JH, Morisseau C, Hammock BD. Hypertension. 2002;39:690. doi: 10.1161/hy0202.103788. [DOI] [PubMed] [Google Scholar]
- 10.Imig JD, Zhao X, Zaharis CZ, Olearczyk JJ, Pollock DM, Newman JW, Kim IH, Watanabe T, Hammock BD. Hypertension. 2005;46:975. doi: 10.1161/01.HYP.0000176237.74820.75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Morisseau C, Goodrow MH, Dowdy D, Zheng J, Greene JF, Sanborn JR, Hammock BD. Proc Natl Acad Sci USA. 1999;96:8849. doi: 10.1073/pnas.96.16.8849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.McElroy NR, Jurs PC, Morisseau C, Hammock BD. J Med Chem. 2003;46:1066. doi: 10.1021/jm020269o. [DOI] [PubMed] [Google Scholar]
- 13.Kim IH, Morisseau C, Watanabe T, Hammock BD. J Med Chem. 2004;47:2110. doi: 10.1021/jm030514j. [DOI] [PubMed] [Google Scholar]
- 14.Kim IH, Heirtzler FR, Morisseau C, Nishi K, Tsai HJ, Hammock BD. J Med Chem. 2005;48:3621. doi: 10.1021/jm0500929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Schmelzer KR, Kubala L, Newman JW, Kim IH, Eiserich JP, Hammock BD. Proc Natl Acad Sci USA. 2005;102:9772. doi: 10.1073/pnas.0503279102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hwang SH, Wecksler AT, Zhang G, Morisseau C, Nguyen LV, Fu SH, Hammock BD. Bioorg Med Chem Lett. 2013;23:3732. doi: 10.1016/j.bmcl.2013.05.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hwang SH, Tsai HJ, Liu JY, Morisseau C, Hammock BD. J Med Chem. 2007;50:3825. doi: 10.1021/jm070270t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Butov GM, Pershin VV, Burmistrov VV. Russian J of Org Chem. 2011;47:606. [Google Scholar]
- 19.Butov GM, Burmistrov VV, Saad KR. J Chem Chem Eng. 2012;6:774. [Google Scholar]
- 20.Raymo FM, Stoddart JF. Chem Rev. 1999;99:1643. doi: 10.1021/cr970081q. [DOI] [PubMed] [Google Scholar]
- 21.Zubovich EA, Burmistrov VV, Butov GM. Butlerov Comm. 2013;33:65. [Google Scholar]
- 22.Kowalski JA, Swinamer AD, Muegge I, Eldrup AB, Kukulka A, Cywin CL, De Lombaert S. Bioorg Med Chem Lett. 2010;20:3703. doi: 10.1016/j.bmcl.2010.04.078. [DOI] [PubMed] [Google Scholar]
- 23.Kato Y, Fuchi N, Saburi H, Nishimura Y, Watanabe A, Yagi M, Nakadera Y, Higashi E, Yamada M, Aoki T. Bioorg Med Chem Lett. 2013;23:5975. doi: 10.1016/j.bmcl.2013.08.054. [DOI] [PubMed] [Google Scholar]
- 24.Pecic S, Pakhomova S, Newcomer ME, Morisseau C, Hammock BD, Zhu Z, Deng SX. Bioorg Med Chem Lett. 2013;23:417. doi: 10.1016/j.bmcl.2012.11.084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Jones PD, Wolf NM, Morisseau C, Whetstone P, Hock B, Hammock BD. Anal Biochem. 2005;343:66. doi: 10.1016/j.ab.2005.03.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lee KSS, Morisseau C, Yang J, Wang P, Hwang SH, Hammock BD. Anal Biochem. 2013;434:259. doi: 10.1016/j.ab.2012.11.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Morisseau C, Goodrow MH, Newman JW, Wheelock CE, Dowdy DL, Hammock BD. Biochem Pharmacol. 2002;63:1599. doi: 10.1016/s0006-2952(02)00952-8. [DOI] [PubMed] [Google Scholar]
- 28.Lipinski CA, Lombardo F, Dominy BW, Feeney PJ. Adv Drug Deliv Rev. 2001;46:3. doi: 10.1016/s0169-409x(00)00129-0. [DOI] [PubMed] [Google Scholar]
- 29.McGovern SL, Caselli E, Grigorieff N, Shoichet BK. J Med Chem. 2002;45:1712. doi: 10.1021/jm010533y. [DOI] [PubMed] [Google Scholar]