

Abstract
Metastatic melanoma is highly resistant to drug treatment, and the underlying mechanisms of this resistance remain unclear. Increased tissue stiffness is correlated with tumor progression, but whether increased tissue stiffness contributes to treatment resistance in melanoma is not known. To investigate the effect of substrate stiffness on melanoma cell treatment responsiveness, PEG hydrogels were utilized as a cell culture system to precisely vary matrix elasticity and investigate melanoma cell responses to a commercially available pharmacological inhibitor (PLX4032). The tensile moduli were varied between 0.6 and 13.1 kPa (E) and the effects of PLX4032 on metabolic activity, apoptosis, and proliferation were evaluated on human cell lines derived from radial growth phase (WM35) and metastatic melanoma (A375). The A375 cells were found to be stiffness-independent; matrix elasticity did not alter cell morphology or apoptosis with PLX4032 treatment. The WM35 cells, however, were more dependent on substrate modulus, displaying increased apoptosis and smaller focal adhesions on compliant substrates. Culturing melanoma cells on PEG hydrogels revealed stage-dependent responses to PLX4032 that would have otherwise been masked if cultured strictly on TCPS. These findings demonstrate the utility of PEG hydrogels as a versatile in vitro culture platform with which to investigate the molecular mechanisms of melanoma biology and treatment responsiveness.
Keywords: BRAF, Cell adhesion, Cell viability, ECM (extracellular matrix), Elasticity, Hydrogel, Photopolymerization
Introduction
Melanoma is an aggressive form of skin cancer that is difficult to treat in later stages by conventional chemotherapeutics. Early stage radial growth phase (RGP) melanoma is easily treated by surgical excision [1], while vertical growth phase (VGP) has a propensity to metastasize [2]. Metastatic melanoma, however, is notoriously drug resistant [3], characterized by a median survival time of 6–10 months [4]. One promising new drug, PLX4032 (Vemurafenib), was approved by the FDA in 2011 for metastatic melanoma treatment. PLX4032 is a small molecule inhibitor that has high specificity towards mutated BRAF proteins and has been shown to cause both cell cycle arrest and induce apoptosis in melanoma [5–7] and potent growth arrest in thyroid carcinoma cells [8,9]. More than 60% of all melanomas have been found to contain a BRAF mutation (V600E) that renders this protein constitutively active [10]. Clinical trials showed marked patient responses to PLX4032 [11,12] and follow up studies reported 16-month overall survival [13]. Yet while PLX4032 is among the most promising melanoma treatments, patients eventually relapse and the mechanisms and contributing factors of melanoma drug resistance remain elusive.
As one approach to aid in preclinical compound screening and better understanding of the molecular mechanisms that might contribute to melanoma drug resistance, improved in vitro culture systems are being explored. Traditional tissue culture-treated polystyrene (TCPS) is often the initial culture platform used for drug screening, but it is orders of magnitude stiffer than most soft tissues in the body and may lead to physiologically irrelevant cellular morphologies or responses [14–16]. Matrix elasticity has been shown to regulate cell function in a number of different cell types, such as mesenchymal stem cells [17] and smooth muscle cells [18], and clinically, tumors are often found to be stiffer than the surrounding or healthy tissues [19,20]. In vitro, increased substrate elasticity has been shown to induce malignant morphology in healthy mammary epithelial cells and lead to increased invasion of breast cancer cells [21,22]. The underlying substrate can also alter intracellular signaling [21,23], which ultimately may change the efficacy of drug treatments. In fact, Weigelt et. al. showed that when breast cancer cells were cultured on TCPS or Matrigel, the reduction of proliferation to clinically available drugs was altered [24].
Many studies have shown the importance of matrix elasticity on breast cancer cells, but the same is not yet known for melanoma. Unlike epithelial-derived breast cancer cells, melanoma is derived from melanocytes which arise from the neural crest [25], and so it is difficult to assume melanocytes and epithelial cells will respond similarly to a microenvironmental change like substrate elasticity. We hypothesized that matrix elasticity is important for assessing melanoma responses to drug treatment and that softer materials may provide better insight into physiologically relevant cellular responses. To investigate melanoma's dependence on substrate modulus, we utilized peptide functionalized poly(ethylene glycol) (PEG) hydrogels as a highly tunable, hydrated, and chemically defined cell culture substrate that can be designed to recapitulate important aspects of the extracellular matrix (ECM) [26,27]. In particular, the thiol-ene “click” chemistry was exploited to form crosslinked networks via step-growth kinetics involving the reaction of an –ene functionalized multi-arm PEG with cysteine-containing peptides (-thiol) [28]. Cell-matrix interactions can be altered by the concentration of ECM molecule peptide mimics, such as the fibronectin-derived peptide RGDS [26]. Matrix remodeling can be controlled by inclusion of matrix metalloproteinase (MMP) degradable peptide sequences, allowing cell-mediated degradation [29]; alternatively, the hydrogel can also be rendered nondegradable by the inclusion of crosslinkers such as PEG-dithiols [30]. Finally, bulk biophysical properties, such as modulus or equilibrium water content, can be controlled by changing the network crosslinking density, which may be tuned by changing the concentration, molecular weight, or number of arms of the PEG [28,31]. This innate tunability of this biomaterial provides an attractive cell culture platform to answer fundamental questions about cellular responses to microenvironmental changes.
Here, we sought to answer whether matrix stiffness would alter melanoma cell morphology and responses to PLX4032 treatment using this synthetic ECM mimic. Formulations based on a 4-arm norbornene-functionalized PEG and bifunctional cysteine-containing MMP- degradable peptides were crosslinked using the thiol-ene photopolymerization approach. The matrix elasticity was varied from 0.6 to 13.1 kPa (E, Young's modulus) with the aim of spanning a range of mechanical properties reported for healthy and pathologic tissue, and the resulting gels were then seeded with either RGP or metastatic melanoma cells. Cell morphology and cell-matrix interactions were assessed via immunostaining and focal adhesion size then viability was challenged with PLX4032 treatment. To test cell responsiveness to this inhibitor as a function of the microenvironment, metabolic activity, apoptosis, and proliferation were quantified and correlated to substrate elasticity.
Materials and Methods
Reagents
All chemicals were purchased from Sigma-Aldrich unless otherwise noted. Cell culture reagents were purchased from Life Technologies unless otherwise noted. Antibodies used: monoclonal paxillin (Y113; Millipore) and goat anti-rabbit Alexa-Fluor 488. TRITC-phalloidin (Sigma-Aldrich) stocks were prepared at 0.06 mg/mL and DAPI stocks were 0.1 mg/mL. PLX4032 (ChemieTek) stocks were dissolved in DMSO (Sigma-Aldrich) at 100 mM.
Synthesis and characterization of macromers and peptides
Four-arm PEG-norbornene (MW: 20,000) (Figure 1) was synthesized as previously described [28]. Briefly, norbornene acid was coupled to form norbornene anhydride in dichloromethane (DCM) via N,N'- diisopropylcarbodiimide (DIC) coupling. PEG hydroxyl (JenKem Technology USA) was dissolved in DCM and reacted with the norbornene anhydride in the presence of pyridine and 4-(dimethylamino)pyridine (DMAP) overnight. The product was precipitated in cold diethyl ether 3 times, dried, and characterized by proton NMR for degree of functionalization. In all studies, PEG with >95% functionalization was used.
Figure 1.
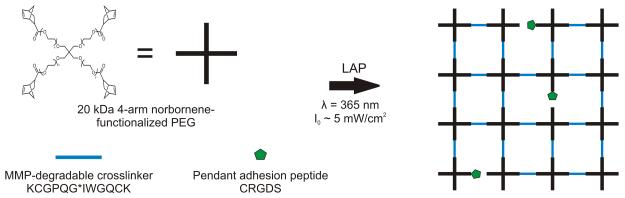
Schematic of network formation utilizing cysteine-containing peptides and norbornene-functionalized PEG.
The photoinitiator lithium phenyl-2,4,6-trimethylbenzoylphosphinate (LAP) was synthesized as previously described [32]. Briefly, 2,4,6-trimethylbenzoyl chloride was added to dimethyl phenylphosphonite (Acros Organics) and allowed to stir overnight. Lithium bromide and 2-butanone was then added to the reaction, heated to 50°C for 10 minutes, and then cooled. The product was filtered, washed and filtered 3 times with 2-butanone, and allowed to dry. The product was verified by proton NMR.
All peptides were purchased from American Peptide Company, Inc. An MMP-degradable crosslinker (KCGPQG*IWGQCK) and pedant adhesion peptide RGD (CRGDS) were used.
Formation and characterization of PEG-norbornene hydrogels
Gels were formed between a glass slide that was dipped in Sigmacote and a thiolated coverslip to create flat hydrogels for cell seeding. Coverslips were passed through a flame to remove contaminates, and then reacted in 95% ethanol (pH ~5.5) with 0.55% (v/v) 3-mercaptotrimethoxy silane for 3 minutes. Coverslips were then rinsed with 95% ethanol and allowed to dry in an 80°C oven for 15 minutes.
Macromer solutions were prepared with the concentrations of reagents in Table 1 with 1.7 mM LAP. In a sterile cell culture hood, 30 μL drops of macromer solution were placed on sterile Sigmacote-coated slides and then covered with a thiolated 18 mm circle coverslip (Fisher Scientific). The solutions were placed under a UV lamp centered around 365 nm light at ~5 mW/cm2 for 3 minutes. The formed gels were then allowed to swell in PBS for at least 2 hours, then sterilized with 5% IPA in PBS at room temperature for 1 hour. Before cell seeding, the gels were rinsed twice with PBS. Unless otherwise noted, 3 gels were formed per condition for each of 3 independent experiments.
Table 1.
Macromolecular monomer compositions for gel formulations.
| [PEG] (mM) | [Crosslinker] (mM) | [Pendant RGD] (mM) | |
|---|---|---|---|
| Stiff | 7.5 | 14.25 | 1.5 |
| Medium | 3 | 4.5 | 1.5 |
| Soft | 3 | 3.9 | 1.5 |
For characterization, 30 μL gels were formed in the cut-end of 1 mL syringe. The same gel formulations as those used for cell culture were made (Table 1). To determine the equilibrium swollen gel mass, the gels were allowed to swell overnight in PBS then weighed the next day. The mass swelling ratio q (Table 2) was calculated from the equilibrium swollen mass and the calculated theoretical dry mass. The water content (% Water) was calculated using the mass swelling ratio (1-q−1) and converted to a percent. The shear modulus (G) was measured on a DHR-3 shear rheometer (TA Instruments) and converted to Young's modulus, where E = 2G(1 + ν) assuming a Poisson's ratio (ν) of 0.5 [33]. Strain and frequency sweeps were performed to ensure measurements were within the linear viscoelastic regime. Averages represent 3 independent experiments.
Table 2.
Measured values of modulus and mass swelling ratio.
| Equilibrium Mass Swelling Ratio q | Young's Modulus E (kPa) | Crosslink Density1 ρXL (mM) | % Water | |
|---|---|---|---|---|
| Stiff | 21 ± 1 | 13.1 ± 0.4 | 5.17 ± 0.40 | 95.3 ± 0.2 |
| Medium | 45 ± 1 | 1.6 ± 0.1 | 0.83 ± 0.09 | 97.7 ± 0.1 |
| Soft | 62 ± 1 | 0.6 ± 0.1 | 0.36 ± 0.05 | 98.4 ± 0.1 |
Crosslink density was calculated from rubber elasticity theory: ρXL = GQ1/3(RT)−1
Cell Culture
Both cell lines were a generous gift from Professor Natalie Ahn, Department of Chemistry and Biochemistry, University of Colorado. All cell lines were cultured in 10% fetal bovine serum (FBS) in RPMI 1640 without phenol red. For experiments, cells were seeded at 1 × 105 cells/ cm2 in 1% FBS in RPMI 1640 and subsequently cultured in 1% FBS for the remainder of any experiment. Cells were seeded on gels or TCPS and allowed to adhere overnight. DMSO control (0.01% v/v) or 1 μM PLX4032 diluted in media was then added to the corresponding samples and incubated for 48 hours.
Metabolic Activity
At the end of the 48-hour drug incubation time, CellTiter-Glo (Promega) was added to samples per the manufacturer's instructions. Samples were placed on an orbital shaker for 2 minutes, and then 100 μL of lysate was transferred to a white 96-well plate and the luminescence was measured on a BioTek H1 Synergy plate reader (BioTek). To measure basal metabolic activity, standard curves of ATP and DNA were generated. After applying CellTiter-Glo to samples, the cell lysate was then diluted to quantify DNA content by Quant-it PicoGreen (Life Technologies).
Apoptosis
After 48 hours of PLX4032 treatment, the EnzChek Caspase-3 Assay Kit #2 (Life Technologies) was used to assess apoptosis. Briefly, floating cells were collected by centrifugation and attached cells were removed with TrypLE Select for 5–10 minutes at 37 °C and then agitated to remove as many cells possible. Cell pellets were then lysed with the provided lysis buffer and then the company protocol was followed. DNA content was measured via PicoGreen (Life Technologies), and caspase 3 activity was normalized to the DNA content in a given sample. For analysis, each PLX4032-treated sample was normalized to its corresponding DMSO control sample to show a fold change in caspase 3 activity with PLX4032 treatment.
Proliferation
During the last 16 hours of incubation with or without the inhibitor, the cells were pulsed with EdU to detect cells entering S-phase. After a total of 48 hours, samples were processed according to the protocol for the Click-iT EdU Alexa-Fluor 488 kit (Life Technologies). One exception was to invert the coverslips on 10 μL drops of the Alexa-Fluor 488 solution. For these experiments, 12 μL gels were formed on 12 mm circle coverslips (Fisher Scientific). Samples were then imaged on an inverted Nikon TE2000; three images per gel were taken and duplicate gels were prepared for each condition. Three independent experiments were performed. EdU quantification was performed using an ImageJ (NIH) macro to threshold images and count the number of nuclei positive for DAPI (blue) and EdU (green).
Immunostaining
For immunostaining experiments, cells were fixed after 24 hours of culture. Half the volume of media in the well was removed and replaced with 1:10 buffered formalin (Sigma-Aldrich) for 20 minutes at room temperature and rinsed twice with 0.05% Tween 20 (Sigma-Aldrich) in PBS. Samples were then permeabilized with 0.1% Triton-X (Fisher Scientific) for 5 minutes, washed twice, and then blocked for 30 minutes with 1% bovine serum albumin (BSA) in PBS. Samples were incubated with the primary antibody diluted in blocking buffer for 1 hour at room temperature, washed 3 times, and the secondary antibody was applied for 45 minutes at room temperature. Antibodies were used at: paxillin 1:400, goat anti-rabbit AlexaFluor 488 1:200, TRITC-Phalloidin 1:200, DAPI 1:1000. Samples were imaged on a Zeiss NLO LSM 710 (Zeiss) at 40X.
Focal adhesion quantification
ImageJ (NIH) was used to quantify the focal adhesion area on a per cell basis. For each cell that formed visible focal adhesions, the cell was cropped from the image and the RGB channels separated. If there was high cytoplasmic staining of paxillin in green, then the background was subtracted using a rolling ball radius of 15 pixels. The image was thresholded, made binary, and the area was measured using the Analyze Particles function. At least 10 different cells were analyzed for focal adhesion size from each of 3 independent experiments.
Statistical analysis
To compare metabolic activity and apoptosis samples, a one-way ANOVA was performed in Prism 5 (GraphPad Software, Inc). To compare treated and control EdU staining samples within each stiffness condition, a two-way ANOVA was performed with Bonferroni posttests in Prism. The data was then transposed and a two-way ANOVA with Bonferroni posttests was performed that compared the control samples for each stiffness to each other and then compared the treated samples for each stiffness to each other. For focal adhesion quantification, a Shapiro-Wilk test for normality was performed in R for each cell type/condition from each independent experiment. The distributions were found to be non-normal, and therefore a modified Levene's test was performed in R to determine if the variances among the experiments for each condition were different. Because the samples were deemed to have statistically similar variances, we pooled the data (>30 cells for each condition) from all three experiments and used a Kruskal-Wallis test in Prism to compare culture conditions within each cell type.
Results
Regulation of cell-matrix adhesion by substrate elasticity
To assess the effects of matrix elasticity on melanoma responses to drug treatment in a range of physiologically relevant moduli, PEG-based hydrogels were engineered such that the matrix moduli were systematically altered through changes in the network crosslinking density. Specifically, in these studies, 4-arm 20 kDa norbornene-functionalized PEG was crosslinked with a bifunctional cysteine-containing MMP-degradable peptide (Figure 1). Gels were formed via the thiol-ene polymerization reaction, which was photoinitiated with LAP under ~5 mW/cm2 of UV light centered around 365 nm. To promote cell adhesion, 1.5 mM of the pendant fibronectin-derived RGDS peptide was incorporated. Fibronectin is present in melanoma tumors and can promote invasion [34], and the integrin pair αvβ3, which readily binds to RGDS, is upregulated in tumorigenic melanoma cells [35]. An MMP degradable crosslinker was selected to better recapitulate cell-mediated degradation of the local microenvironment observed in vivo and to enable comparisons to 3D studies where degradability is necessary for cell spreading, survival, and proliferation. While the mechanical properties of the gels may change with cells present, due to matrix deposition and/or MMP degradation, changes in modulus were minimized by the use of PEG (to minimize protein adsorption), low cell density, and relatively short culture times. Additionally, by using a range of moduli over three orders of magnitude, the mechanical properties of the gels may change over the course of the experiment, but the large relative difference between the soft and rigid gels remains, as evidenced by the differences in cell spreading observed at the beginning and end of the experimental timeline.
The bulk modulus of the materials was varied by changing either the concentration of PEG (7.5 mM or 3 mM) or altering the concentration of the MMP crosslinker (Table 1). Using these formulations, we were able to achieve a range of moduli from approximately 13.1 kPa (E, Young's modulus) to 0.6 kPa. The crosslinking density was calculated from rubber elasticity theory [33](see Table 2 footnote), and the equilibrium mass swelling ratio was also measured to determine the water content of each gel (Table 2). The modulus of skin and melanoma tumors is not well established in the literature, so we chose a range of moduli: the softest gel formulation of 0.6 kPa (soft), 1.6 kPa (medium), and 13.1 kPa (stiff). This modulus range encompasses physiologically relevant tissues from breast tissue (0.1–0.5 kPa) to relaxed muscle (approximately 10 kPa) [17,36]. Because increased tissue stiffness is often correlated with tumor formation [37], several gel elasticities were used for subsequent studies. TCPS samples were included as controls in order to compare results to the established literature related to studying melanoma cell responses to BRAF inhibition.
To evaluate whether substrate modulus affected cell morphology and cell-matrix interactions, immunostaining for the actin cytoskeleton and the focal adhesion component, paxillin, was performed on all substrates under control conditions (Figure 2A). Increased focal adhesion formation has been linked to increased ERK signaling [21,22], an important survival pathway. Therefore, we reasoned that melanoma cell morphology and cell-matrix interactions could be regulated by matrix elasticity, where stiff substrates induce a more spread morphology and mature focal adhesion formation compared to soft substrates.
Figure 2.

Effect of substrate elasticity on overall cell morphology and focal adhesion formation. (A) Immunostaining for paxillin (green), f-actin (red) and nuclei (blue) on TCPS, stiff, medium, and soft gels of both cell types. Inset image is magnification of outlined area. (B) Quantification of focal adhesion area based on image analysis. n = 3 ± SEM. *p < 0.05. Scale bars, 20 μm.
The RGP WM35 cells were well spread on TCPS with distinct f-actin fibers around the peripheral edges and punctate paxillin staining (Figure 2A). In contrast, when cultured on softer PEG gels, the WM35 cells appeared more rounded and exhibited fewer f-actin stress fibers. Depending on the stiffness of the gel, approximately 50–75% of cells exhibited punctate paxillin staining. Of the WM35 cells that had distinct focal adhesion formation, cells formed much smaller adhesions as the gel elasticity was decreased. Focal adhesion area was quantified using image analysis, and there was a general downward trend of decreasing focal adhesion size with decreasing substrate modulus (0.70 μm2 on the stiffest to 0.44 μm2 on softest gels) (Figure 2B). Overall, results demonstrate that the WM35 cells are sensitive to the underlying substrate elasticity and consequently form stiffness-dependent focal adhesions.
In contrast to the WM35 cells, the metastatic A375 cells were much smaller and had more irregular morphologies, often exhibiting f-actin patterns that indicated lamellipodia formation on TCPS, stiff, and medium gels. Furthermore, when cultured on gels, the A375 cells appeared slightly rounded though still able to form focal adhesions, based on punctate paxillin staining. Again, focal adhesion size was quantified and found to be approximately the same (between 0.44 – 0.54 μm2), independent of the underlying substrate elasticity, while the size of the adhesions were generally smaller than that observed for the RGP WM35s (Figure 2B).
Effect of substrate elasticity on cell responsiveness to drug treatment
To determine if basic cell function, with no drug treatment, was altered due to substrate elasticity-induced changes in morphology, basal metabolic activity was assessed on TCPS and stiff, medium, and soft gels. Basal metabolic activity was measured by normalizing ATP to DNA content within a sample to provide a measure of metabolic activity on a “per cell” basis (Figure 3). For both cell types, the basal metabolic activity was not significantly different between the different culture conditions.
Figure 3.

Basal metabolic activity (ATP normalized to DNA content) measured under control conditions on all culture substrates. None of the samples were statically different. n = 3 ± SEM.
To evaluate how matrix elasticity regulates the response of melanoma cells to drug treatment, WM35 and A375 cells were cultured on TCPS and stiff, medium, and soft gels and treated with PLX4032. As an initial measure of drug-induced effects, metabolic activity was measured after 48 hours of 1 μM PLX4032 treatment (Figure 4). The metabolic activity in each treated sample was normalized to its respective control (i.e., the TCPS PLX4032-treated sample was normalized to a TCPS DMSO-containing sample) to yield relative metabolic activity present after drug treatment. The WM35 cells exhibited a significant response on all substrates as measured by a reduction in metabolic activity with drug treatment to approximately 0.55 of the control conditions. Similarly, the A375 cells exhibited a significant decrease in metabolic activity to no more than 0.43 of the control conditions on all culture substrates. Among the different substrates, however, the change in metabolic activity was not significant. Because the WM35 cells exhibited stiffness-dependent focal adhesion formation, we had hypothesized that there would be increased cell death on soft substrates. However, here we observed that PLX4032 drastically reduced metabolic activity on both TCPS and gels and there was no significant change with substrate stiffness. Metabolic activity measurements, however, cannot differentiate between the cytotoxic (cell death) and cytostatic (anti-proliferative) effects of PLX4032 and may not capture the specific effects of PLX4032 treatment. Therefore, we next measured cell death directly via apoptosis.
Figure 4.

Relative fraction of metabolic activity for PLX4032-treated cells compared to cells cultured on each respective substrate without inhibitor. The dashed line represents the control samples. None of the treated samples for either cell type were statistically different metabolic activity based on culture substrate. n = 3 ± SEM.
Decoupling cytostatic and cytotoxic effects of drug treatment
Apoptosis was measured via caspase 3 activity, and treated samples were normalized to their respective controls (i.e., the TCPS PLX4032-treated sample was normalized to a TCPS DMSO-containing sample) to show the fold change in caspase 3 activity (Figure 5). Interestingly, the WM35 cells exhibited increased sensitivity to PLX4032 as the substrate modulus decreased. On TCPS there was a 2-fold increase in caspase 3 activity with drug treatment, while on the soft gels, there was a nearly 3-fold increase. In contrast, the A375 cells did not exhibit major changes in the levels of apoptosis as a function of substrate properties. There was a smaller increase in apoptosis on TCPS (approximately 1.7-fold), but no significant increases were observed for cells cultured on gels. Based on this evidence, we concluded that the WM35 cells were more susceptible to PLX4032 treatment on gels as compared to the A375 cells. Because PLX4032 has been shown to both induce apoptosis and cause cell cycle arrest, we next investigated whether or not the decrease in A375 metabolic activity could be the result of PLX4032's antiproliferative effect rather than its cytotoxic effects.
Figure 5.

Apoptosis measured via caspase 3 activity on different culture substrates. Each bar represents the PLX4032-treated sample normalized to its respective control showing the fold increase in caspase 3 activity with drug treatment. None of the A375 changes in apoptosis were statistically different. n = 3 ± SEM. *p < 0.05
To measure proliferation, an EdU assay was used and the percentage of cells that were stained positive was quantified (Figure 6). In general, drug treatment led to less than 1% of cells undergoing DNA synthesis on any substrate. The WM35 cells cultured on stiff and soft gels, as well as the A375 cells on soft samples, that were treated with PLX4032 had no cells entering S phase. Interestingly, in both cell types, there was an increase in the percent of proliferating cells when cultured on top of gels without drug treatment. Specifically, we observed ~39% of A375 cells on TCPS undergoing proliferation, but that number increased to as high as 68% on the gel formulations. The WM35 cells exhibited a less dramatic increase from 46% on TCPS to a maximum of 64% on medium gels, but the increase was statistically significant.
Figure 6.

Percent of cells entering S phase as quantified by EdU staining. White bars (left y-axis) represent control conditions; grey bars (right y-axis) represent PLX4032-treated samples. n = 3 ± SEM. Three fluorescent images were taken for each gel or TCPS well, the percent of positive EdU cells quantified, and then averaged for each sample. *p < 0.01 compared to TCPS control; #p < 0.0001 for PLX4032 samples compared to corresponding control; **p < 0.01.
Discussion
Here, using PEG hydrogels as a 2D cell culture platform, we assessed how substrate elasticity affects melanoma cells' sensitivity to drugs and whether or not the treatment responsiveness was tumor stage-dependent. The modulus of healthy and tumorigenic skin tissue is not well established [37], and so a range of elasticities was chosen to capture both states. Previous studies have measured that a range in Young's modulus from 0.2 kPa to 1.2 kPa will elicit vastly different morphologies in mammary epithelial cells in vitro [21]. Breast tissue, however, is among the softest tissues, so here we used a broad range of elastic moduli in an effort to encompass both healthy and tumorigenic tissue stiffness (0.6 to 13.1 kPa).
To examine the effect of drug treatment on melanoma cell function, metabolic activity was chosen as an initial indication of drug sensitivity, however, it ultimately was not indicative of the phase-dependent responses to substrate modulus. The WM35 cells showed large decreases in metabolic activity in response to PLX4032, and further analysis revealed this decrease was due to increased apoptosis on compliant gels coupled with almost complete inhibition of proliferation. The increase in apoptosis on more compliant substrates appeared to counteract the increased proliferation that we observed, resulting in similar losses of metabolic activity with PLX4032 treatment.
In comparison, the A375 cells exhibited slightly larger decreases in metabolic activity than the WM35 cells. Similarly to the WM35 cells, the observed decrease in metabolic activity showed an incomplete picture of the cellular responses to drug treatment. We found that A375 levels of apoptosis did not change with matrix elasticity. When cultured on any of the gels, A375 cells proliferated more than when cultured on TCPS; more cells were entering S-phase based on EdU staining. PLX4032 treatment induced potent cell cycle arrest, where very few cells were undergoing DNA synthesis. Taken together, A375 metabolic activity might have been low on PLX4032-treated hydrogel substrates due in large part to inhibition of proliferation rather than increasing levels of apoptosis on compliant gels.
We note that many reports have demonstrated that 3D cellular architecture or culture platforms are more physiologically relevant (reviewed in [38] and [39]) and can elicit very different cellular responses to the same stimuli when compared to TCPS culture [40]. Cells cultured in 3D microenvironments using synthetic hydrogels [40] or naturally derived materials like collagen [41,42], exhibit resistance to drug treatment as compared to 2D TCPS culture. While naturally derived hydrogels, such as reconstituted basement membrane (Matrigel), can promote more physiologically relevant multicellular acini structures [41] and altered responses to clinically available drugs compared to TCPS samples [24], 3D cell culture is not without complexity. One difficulty relates to performing experiments on encapsulated samples; in order to perform certain assays, the gels must be degraded in order to collect the cells. A second drawback of 3D culture is that cells locally remodel their environment in 3D (e.g., degrading the local matrix to allow morphological changes), which can make it difficult to control and quantify the local material properties in the pericellular region. Thus, we decided to first study melanoma cells on the surfaces of hydrogels, as a simple alternative to TCPS or encapsulated cells and explore the mechanisms of melanoma responses to matrix elasticity and drug treatment in a highly controlled environment. Specifically, the substrate elasticity is tunable and constant, and the drug treatment is introduced in a relatively homogeneous manner. While the PEG hydrogel system is completely compatible with future 3D cell culture studies, by using a 2D system, assays such as western blotting and apoptosis can be performed similarly to TCPS experiments.
Although 2D cell culture has its benefits, there are many excellent studies that exploit the advantages of 3D culture to recapitulate important aspects of cell-cell adhesion or a 3D microenvironment to study melanoma drug responsiveness [43,44]. Three-dimensional melanoma spheroid studies have provided platforms with which to observe decreased efficacy of drugs in vitro that may prove crucial to elucidating drug resistant mechanisms or ineffective drugs in preclinical studies. For example, metastatic melanoma cells survive in 3D aggregates with the MEK inhibitor UO126 while radial and vertical growth phase cells are more susceptible [45]. Additionally, another MEK inhibitor AZD6244 was shown to be cytostatic on collagen-embedded spheroids, and wash out experiments showed the effects were reversible [46]. Unlike the MEK inhibitors, however, PLX4032 was found to be effective in 3D to induce apoptosis and prevent aggregate outgrowth in collagen [5,6]. While spheroid studies show V600E mutated melanoma susceptibility, clinical efficacy suffers from patient relapse [11,13]. Therefore, better methods are needed to study melanoma in vitro to provide new insight into treatment responsiveness and cellular mechanisms. While naturally derived materials such as collagen and reconstituted basement membrane provide physiologically relevant moduli, there is also inherent variability in the material, sequestration of growth factors, and ill-defined chemistry [47]. Alternatively, PEG hydrogels serve as a highly defined bioinert scaffold that allows precise tuning of properties such as modulus or cell-mediated degradation [47]. Using PEG hydrogels as a 2D substrate for cell culture, we observed stiffness-independent apoptotic responses in the metastatic A375 cells while the RGP WM35 cells exhibited increased sensitivity when cultured on more compliant substrates. These findings correlate with previous 3D culture studies, but using the 2D PEG hydrogel system, we were able to investigate how matrix stiffness specifically affects treatment responsiveness in melanoma cells.
To better understand the effect of substrate rigidity on apoptosis, cell morphology and focal adhesion size were examined for a possible correlation between cell-matrix interactions and response to PLX4032 treatment. Metastatic A375 cell morphology, focal adhesion size, and levels of apoptosis showed a weak dependence on matrix elasticity compared to the RGP WM35 cells. The WM35 cells formed smaller focal adhesions on softer substrates and appeared more rounded. Despite the different morphologies, this did not significantly alter basal metabolic activity. However, when cell survival was challenged with drug treatment, increased levels of apoptosis were observed in cells cultured on more compliant gels. Matrix stiffness revealed different responses to PLX4032 between the two cell lines that were not seen on TCPS. Taken together, the metastatic cells were able to adhere and survive equally well on soft (0.6 kPa) and very stiff (TCPS) substrates, exhibiting stiffness-independent survival while the WM35 cells were dependent on the underlying substrate modulus when cell survival was challenged with drug treatment.
Based on our findings, we hypothesize that the WM35 cells interact with their microenvironment by forming matrix stiffness-induced focal adhesions. As a result, the WM35 cells retain the ability to sense the rigidity of the underlying substrate, and on softer materials, they form fewer, smaller focal adhesions, decreasing survival signaling, and ultimately increasing PLX4032 sensitivity as indicated by increased apoptosis (Figure 7). Metastatic A375 cells, on the other hand, appear to have decreased mechanosensing abilities, and changes in substrate compliance did not alter focal adhesion formation or PLX4032 sensitivity. The A375 cells were able to maintain similar levels of cell-matrix interactions on all substrates and showed little differences in apoptosis.
Figure 7.

Schematic of the effect of substrate stiffness on WM35 and A375 cells. WM35 cells exhibit dependence on substrate modulus by forming stiffness-dependent focal adhesions (green dots) and increase f-actin stress fiber formation (red lines). We hypothesize that for the WM35 cells, increased signaling from focal adhesion formation, perhaps an indication of stronger cell-matrix interactions, can increase survival signaling to inhibit apoptosis induced by PLX4032. In comparison, A375 cells show little dependence on the underlying matrix elasticity, always forming similarly sized matrix interactions. The lack of large focal adhesions, however, does not increase levels of apoptosis. Instead, A375 cells may have very different intracellular signaling that compensates for the lack of stiffness dependence for survival.
Focal adhesion formation may regulate susceptibility to PLX4032 treatment based on survival signaling through BRAF-MEK-ERK and/or PI3K-AKT. Matrix elasticity can regulate focal adhesion formation in breast cancer and increase ERK signaling [21,22]; the same phenomenon may occur in melanoma. In addition, PI3K signaling via AKT has been correlated with increased survival of melanoma with MEK or BRAF inhibitor treatment [48]. Boisvert-Adamo and Aplin showed that AKT contributes to adhesion-dependent protection from anoikis in melanoma [49], and that increased AKT levels have been found in drug resistant melanoma cell lines [48]. Focal adhesion signaling can regulate both BRAF-MEK-ERK and PI3K-AKT signaling, and thereby affect treatment responsiveness. Perhaps PI3K-AKT signaling is increased in the A375 cells, which promotes an anti-apoptotic phenotype, while the WM35 cells are heavily oncogene addicted to BRAF-MEK-ERK. The ability to sense the local microenvironment, however, may be stage-dependent in melanoma. The interaction between pathways, involvement of focal adhesion formation and the subsequent signaling cascade may provide clues to melanoma drug responses. Ultimately, this interplay between pathways and the interaction of melanoma tumor cells with the microenvironment may alter the efficacy of BRAF and MEK inhibitors.
Conclusions
We investigated the use of PEG-based hydrogels for 2D cell culture to determine how extracellular matrix elasticity affects melanoma responses to PLX4032 treatment. Metastatic A375 cells can better mitigate the cytotoxic effects of PLX4032 on more compliant substrates, which led to decreased proliferation more than an increase in apoptosis. Conversely, early stage RGP melanoma cells exhibited increased susceptibility to PLX4032 on compliant substrates due to increased apoptosis. We hypothesize that these differences may be linked to the ability to form cell-matrix interactions via substrate rigidity-regulated focal adhesion formation. Overall, culturing melanoma cells on compliant hydrogel platforms may provide a more physiologically relevant culture system than TCPS and provide better insight into what microenvironmental factors contribute to the drug resistant nature of melanoma.
Supplementary Material
Acknowledgements
The authors thank Dr. Natalie Ahn (University of Colorado, Boulder) for kindly providing the melanoma cell lines. The authors also wish to thank Dr. Huan Wang and David Cate for helpful technical discussions, Dr. Chelsea Kirschner and Dr. William Wan for statistical analysis advice, and Caitlin Jones for technical assistance. This work was supported by the National Institutes of Health (# R01 CA132633) and the Howard Hughes Medical Institute.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- [1].Breslow A. The surgical treatment of stage I cutaneous melanoma. Cancer Treat Rev. 1978;5:195–8. doi: 10.1016/s0305-7372(78)80013-9. [DOI] [PubMed] [Google Scholar]
- [2].Bogenrieder T, Herlyn M. Cell-surface proteolysis, growth factor activation and intercellular communication in the progression of melanoma. Crit Rev Oncol Hematol. 2002;44:1–15. doi: 10.1016/S1040-8428(01)00196-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Soengas MS, Lowe SW. Apoptosis and melanoma chemoresistance. Oncogene. 2003;22:3138–51. doi: 10.1038/sj.onc.1206454. [DOI] [PubMed] [Google Scholar]
- [4].Manola J, Atkins M, Ibrahim J, Kirkwood J. Prognostic factors in metastatic melanoma: a pooled analysis of Eastern Cooperative Oncology Group trials. J Clin Oncol. 2000;18:3782–93. doi: 10.1200/JCO.2000.18.22.3782. [DOI] [PubMed] [Google Scholar]
- [5].Tsai J, Lee JT, Wang W, Zhang J, Cho H, Mamo S, et al. Discovery of a selective inhibitor of oncogenic B-Raf kinase with potent antimelanoma activity. Proc Natl Acad Sci USA. 2008;105:3041–6. doi: 10.1073/pnas.0711741105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Lee JT, Li L, Brafford PA, van den Eijnden M, Halloran MB, Sproesser K, et al. PLX4032, a potent inhibitor of the B-Raf V600E oncogene, selectively inhibits V600E-positive melanomas. Pigment Cell Melanoma Res. 2010;23:820–7. doi: 10.1111/j.1755-148X.2010.00763.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Bollag G, Hirth P, Tsai J, Zhang J, Ibrahim PN, Cho H, et al. Clinical efficacy of a RAF inhibitor needs broad target blockade in BRAF-mutant melanoma. Nature. 2010;467:596–9. doi: 10.1038/nature09454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Salerno P, De Falco V, Tamburrino A, Nappi TC, Vecchio G, Schweppe RE, et al. Cytostatic activity of adenosine triphosphate-competitive kinase inhibitors in BRAF mutant thyroid carcinoma cells. J Clin Endocrinol Metab. 2010;95:450–5. doi: 10.1210/jc.2009-0373. [DOI] [PubMed] [Google Scholar]
- [9].Sala E, Mologni L, Truffa S, Gaetano C, Bollag GE, Gambacorti-Passerini C. BRAF silencing by short hairpin RNA or chemical blockade by PLX4032 leads to different responses in melanoma and thyroid carcinoma cells. Mol Cancer Res. 2008;6:751–9. doi: 10.1158/1541-7786.MCR-07-2001. [DOI] [PubMed] [Google Scholar]
- [10].Davies H, Bignell G, Cox C, Stephens P, Edkins S, Clegg S, et al. Mutations of the BRAF gene in human cancer. Nature. 2002;417:949–54. doi: 10.1038/nature00766. [DOI] [PubMed] [Google Scholar]
- [11].Flaherty KT, Puzanov I, Kim KB, Ribas A, McArthur GA, Sosman JA, et al. Inhibition of mutated, activated BRAF in metastatic melanoma. N Engl J Med. 2010;363:809–19. doi: 10.1056/NEJMoa1002011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Chapman PB, Hauschild A, Robert C, Haanen JB, Ascierto P, Larkin J, et al. Improved survival with vemurafenib in melanoma with BRAF V600E mutation. N Engl J Med. 2011;364:2507–16. doi: 10.1056/NEJMoa1103782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Sosman JA, Kim KB, Schuchter L, Gonzalez R, Pavlick AC, Weber JS, et al. Survival in BRAF V600-mutant advanced melanoma treated with vemurafenib. N Engl J Med. 2012;366:707–14. doi: 10.1056/NEJMoa1112302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Cukierman E. Taking cell-matrix adhesions to the third dimension. Science. 2001;294:1708–12. doi: 10.1126/science.1064829. [DOI] [PubMed] [Google Scholar]
- [15].Debnath J, Brugge JS. Modelling glandular epithelial cancers in three-dimensional cultures. Nat Rev Cancer. 2005;5:675–88. doi: 10.1038/nrc1695. [DOI] [PubMed] [Google Scholar]
- [16].Yamada K, Cukierman E. Modeling tissue morphogenesis and cancer in 3D. Cell. 2007;130:601–10. doi: 10.1016/j.cell.2007.08.006. [DOI] [PubMed] [Google Scholar]
- [17].Engler AJ, Sen S, Sweeney HL, Discher DE. Matrix elasticity directs stem cell lineage specification. Cell. 2006;126:677–89. doi: 10.1016/j.cell.2006.06.044. [DOI] [PubMed] [Google Scholar]
- [18].Peyton S, Raub C, Keschrumrus V. The use of poly (ethylene glycol) hydrogels to investigate the impact of ECM chemistry and mechanics on smooth muscle cells. Biomaterials. 2006;27:4881–93. doi: 10.1016/j.biomaterials.2006.05.012. [DOI] [PubMed] [Google Scholar]
- [19].Levental I, Levental KR, Klein EA, Assoian R, Miller RT, Wells RG, et al. A simple indentation device for measuring micrometer-scale tissue stiffness. J Phys: Condens Matter. 2010;22:194120. doi: 10.1088/0953-8984/22/19/194120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Bhatia KS, Yuen EH, Cho C, Tong CS, Lee YY, Ahuja AT. A pilot study evaluating real-time shear wave ultrasound elastography of miscellaneous non-nodal neck masses in a routine head and neck ultrasound clinic. Ultrasound Med Biol. 2012;38:933–42. doi: 10.1016/j.ultrasmedbio.2012.02.034. [DOI] [PubMed] [Google Scholar]
- [21].Paszek MJ, Zahir N, Johnson KR, Lakins JN, Rozenberg GI, Gefen A, et al. Tensional homeostasis and the malignant phenotype. Cancer Cell. 2005;8:241–54. doi: 10.1016/j.ccr.2005.08.010. [DOI] [PubMed] [Google Scholar]
- [22].Provenzano PP, Inman DR, Eliceiri KW, Keely P. Matrix density-induced mechanoregulation of breast cell phenotype, signaling and gene expression through a FAK–ERK linkage. Oncogene. 2009;28:4326–43. doi: 10.1038/onc.2009.299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Levental KR, Yu H, Kass L, Lakins JN, Egeblad M, Erler JT, et al. Matrix crosslinking forces tumor progression by enhancing integrin signaling. Cell. 2009;139:891–906. doi: 10.1016/j.cell.2009.10.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Weigelt B, Lo AT, Park CC, Gray JW, Bissell MJ. HER2 signaling pathway activation and response of breast cancer cells to HER2-targeting agents is dependent strongly on the 3D microenvironment. Breast Cancer Res Treat. 2010;122:35–43. doi: 10.1007/s10549-009-0502-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Ernfors P. Cellular origin and developmental mechanisms during the formation of skin melanocytes. Exp Cell Res. 2010;316:1397–407. doi: 10.1016/j.yexcr.2010.02.042. [DOI] [PubMed] [Google Scholar]
- [26].Lutolf MP, Raeber GP, Zisch AH, Tirelli N, Hubbell JA. Cell- responsive synthetic hydrogels. Adv Mater. 2003;15:888–92. [Google Scholar]
- [27].Hoyle CE, Bowman CN. Thiol–ene click chemistry. Angew Chem Int Ed. 2010;49:1540–73. doi: 10.1002/anie.200903924. [DOI] [PubMed] [Google Scholar]
- [28].Fairbanks BD, Schwartz MP, Halevi AE, Nuttelman CR, Bowman CN, Anseth KS. A versatile synthetic extracellular matrix mimic via thiol-norbornene photopolymerization. Adv Mater. 2009;21:5005–10. doi: 10.1002/adma.200901808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Patterson J, Hubbell JA. Enhanced proteolytic degradation of molecularly engineered PEG hydrogels in response to MMP-1 and MMP-2. Biomaterials. 2010;31:7836–45. doi: 10.1016/j.biomaterials.2010.06.061. [DOI] [PubMed] [Google Scholar]
- [30].Anderson SB, Lin C-C, Kuntzler DV, Anseth KS. The performance of human mesenchymal stem cells encapsulated in cell-degradable polymer-peptide hydrogels. Biomaterials. 2011;32:3564–74. doi: 10.1016/j.biomaterials.2011.01.064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Gould ST, Darling NJ, Anseth KS. Small peptide functionalized thiol–ene hydrogels as culture substrates for understanding valvular interstitial cell activation and de novo tissue deposition. Acta Biomater. 2012;8:3201–9. doi: 10.1016/j.actbio.2012.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Fairbanks B, Schwartz M, Bowman C, Anseth K. Photoinitiated polymerization of PEG-diacrylate with lithium phenyl-2, 4, 6-trimethylbenzoylphosphinate: polymerization rate and cytocompatibility. Biomaterials. 2009;30:6702–7. doi: 10.1016/j.biomaterials.2009.08.055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Bryant SJ, Anseth KS. Photopolymerization of Hydrogel Scaffolds. In: Ma PX, Ellisseeff J, editors. Scaffolding in Tissue Engineering. CRC Press; Boca Raton, FL: 2006. pp. 1–20. [Google Scholar]
- [34].Gaggioli C, Robert G, Bertolotto C, Bailet O, Abbe P, Spadafora A, et al. Tumor-derived fibronectin is involved in melanoma cell invasion and regulated by V600E B-Raf signaling pathway. J Investig Dermatol. 2007;127:400–10. doi: 10.1038/sj.jid.5700524. [DOI] [PubMed] [Google Scholar]
- [35].Albelda SM, Mette SA, Elder DE, Stewart R, Danmjanovich L, Herlyn M, et al. Integrin distribution in malignant melanoma: association of the β3 subunit with tumor progression. Cancer Res. 1990;50:6757–64. [PubMed] [Google Scholar]
- [36].Butcher DT, Alliston T, Weaver VM. A tense situation: forcing tumour progression. Nat Rev Cancer. 2009;9:108–22. doi: 10.1038/nrc2544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Gambichler T, Moussa G, Sand M, Sand D, Altmeyer P, Hoffmann K. Applications of optical coherence tomography in dermatology. J Derm Sci. 2005;40:85–94. doi: 10.1016/j.jdermsci.2005.07.006. [DOI] [PubMed] [Google Scholar]
- [38].Tibbitt MW, Anseth KS. Hydrogels as extracellular matrix mimics for 3D cell culture. Biotechnol Bioeng. 2009;103:655–63. doi: 10.1002/bit.22361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Kim JB. Three-dimensional tissue culture models in cancer biology. Semin Cancer Biol. 2005;15:365–77. doi: 10.1016/j.semcancer.2005.05.002. [DOI] [PubMed] [Google Scholar]
- [40].Loessner D, Stok KS, Lutolf MP, Hutmacher DW, Clements JA, Rizzi SC. Bioengineered 3D platform to explore cell-ECM interactions and drug resistance of epithelial ovarian cancer cells. Biomaterials. 2010;31:8494–506. doi: 10.1016/j.biomaterials.2010.07.064. [DOI] [PubMed] [Google Scholar]
- [41].Szot CS, Buchanan CF, Freeman JW, Rylander MN. 3D in vitro bioengineered tumors based on collagen I hydrogels. Biomaterials. 2011;32:7905–12. doi: 10.1016/j.biomaterials.2011.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Chen L, Xiao Z, Meng Y, Zhao Y, Han J, Su G, et al. The enhancement of cancer stem cell properties of MCF-7 cells in 3D collagen scaffolds for modeling of cancer and anti-cancer drugs. Biomaterials. 2012;33:1437–44. doi: 10.1016/j.biomaterials.2011.10.056. [DOI] [PubMed] [Google Scholar]
- [43].Smalley K, Lioni M, Herlyn M. Life isn't flat: taking cancer biology to the next dimension. In Vitro Cell Dev Biol-Animal. 2006;42:242–7. doi: 10.1290/0604027.1. [DOI] [PubMed] [Google Scholar]
- [44].Halaban R, Zhang W, Bacchiocchi A, Cheng E, Parisi F, Ariyan S, et al. PLX4032, a selective BRAFV600E kinase inhibitor, activates the ERK pathway and enhances cell migration and proliferation of BRAFWT melanoma cells. Pigment Cell Melanoma Res. 2010;23:190–200. doi: 10.1111/j.1755-148X.2010.00685.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Smalley K, Haass N, Brafford P, Lioni M, Flaherty K, Herlyn M. Multiple signaling pathways must be targeted to overcome drug resistance in cell lines derived from melanoma metastases. Mol Cancer Ther. 2006;5:1136–44. doi: 10.1158/1535-7163.MCT-06-0084. [DOI] [PubMed] [Google Scholar]
- [46].Haass N, Sproesser K, Nguyen T, Contractor R, Medina C, Nathanson K, et al. The mitogen-activated protein/extracellular signal-regulated kinase kinase inhibitor AZD6244 (ARRY-142886) induces growth arrest in melanoma cells and tumor regression when combined with docetaxel. Clin Cancer Res. 2008;14:230–9. doi: 10.1158/1078-0432.CCR-07-1440. [DOI] [PubMed] [Google Scholar]
- [47].Cushing M, Anseth K. Hydrogel cell cultures. Science. 2007;316:1133–4. doi: 10.1126/science.1140171. [DOI] [PubMed] [Google Scholar]
- [48].Shao Y, Aplin AE. Akt3-mediated resistance to apoptosis in B-RAF-targeted melanoma cells. Cancer Res. 2010;70:6670–81. doi: 10.1158/0008-5472.CAN-09-4471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Boisvert-Adamo K, Aplin AE. B-RAF and PI-3 kinase signaling protect melanoma cells from anoikis. Oncogene. 2006;25:4848–56. doi: 10.1038/sj.onc.1209493. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.