

Abstract
Stimulation of the CD95/Fas/Apo-1 receptor leads to apoptosis through activation of the caspase family of cysteine proteases and disruption of the mitochondrial transmembrane potential (Δψm). We show that, in Jurkat human T cells and peripheral blood lymphocytes, Fas-induced apoptosis is preceded by 1) an increase in reactive oxygen intermediates (ROI) and 2) an elevation of Δψm. These events are followed by externalization of phosphatidylserine (PS), disruption of Δβm, and cell death. The caspase inhibitor peptides, DEVD-CHO, Z-VAD.fmk, and Boc-Asp.fmk, blocked Fas-induced PS externalization, disruption of Δψm, and cell death, suggesting that these events are sequelae of caspase activation. By contrast, in the presence of caspase inhibitors, ROI levels and Δψm of Fas-stimulated cells remained elevated. Because ROI levels and Δψm are regulated by the supply of reducing equivalents from the pentose phosphate pathway (PPP), we studied the impact of transaldolase (TAL), a key enzyme of the PPP, on Fas signaling. Overexpression of TAL accelerated Fas-induced mitochondrial ROI production, Δψm elevation, activation of caspase-8 and caspase-3, proteolysis of poly(A)DP-ribose polymerase, and PS externalization. Additionally, suppression of TAL diminished these activities. Therefore, by controlling the balance between mitochondrial ROI production and metabolic supply of reducing equivalents through the PPP, TAL regulates susceptibility to Fas-induced apoptosis. Early increases in ROI levels and Δψm as well as the dominant effect of TAL expression on activation of caspase-8/Fas-associated death domain-like IL-1β-converting enzyme, the most upstream member of the caspase cascade, suggest a pivotal role for redox signaling at the initiation of Fas-mediated apoptosis.
A poptosis, a form of programmed cell death (PCD),3 is indispensable for normal development and homeostasis within multicellular organisms (1). Defects in PCD may underlie the etiology of neurodegenerative diseases, cancer, autoimmune diseases, and AIDS (2, 3). Thus, the delivery of signals through the APO-1/Fas/CD95 Ag and the structurally related TNF family of cell surface death receptors has emerged as a major pathway in the elimination of unwanted cells under physiological and disease conditions (4). Fas and the type I TNF receptor may mediate cell death by a similar mechanism via cytoplasmic death domains shared by both receptors (5, 6). Signaling through the receptors involves the assembly of a death-inducing signaling complex (DISC) with IL-1β converting enzyme (ICE)/caspase-1-like activity (7–11). The process of death by Fas stimulation starts out with the activation of caspase-8 (FLICE/MACHα1/Mch5) recruited via its N-terminal death effector domain to DISC (11, 12). Sequential activation of ICE/caspase-1, caspase-3, and related cysteine proteases results in the proteolysis of several cellular substrates, which, in turn, leads to the characteristic morphologic and biochemical changes of apoptosis (4, 10). Nevertheless, cross-linking of the Fas receptor on different cell types may lead to different outcomes. For example, Fas transduces an activation signal and stimulates proliferation in freshly isolated PBL (13, 14) or in certain tumor cell lines (15). The mechanisms of processing biologically opposing signals through Fas stimulation have not yet been determined.
Reactive oxygen intermediates (ROIs) have long been considered toxic by-products of aerobic existence; however, evidence is now accumulating that controlled levels of ROIs modulate various aspects of cellular function and are necessary for signal transduction pathways, including those mediating apoptosis (16–22). Because apoptosis and Bcl-2 protection were demonstrated in very low oxygen pressure, ROI may not be absolutely required for PCD (23). Nevertheless, increased production of ROIs was demonstrated in TNF (24–26) and Fas-mediated cell death (27–32). A cell may normally generate 1011 ROI molecules/day (33). ROI production during apoptosis may be controllable by increased synthesis of reducing equivalents (34). A normal reducing atmosphere, required for cellular integrity, is maintained by GSH, which protects the cell from damage by excess ROIs (35). Synthesis of GSH from its oxidized form, glutathione disulfide, depends on NADPH produced by the pentose phosphate pathway (PPP) (35). In fact, a fundamental function of PPP is to maintain glutathione in a reduced state and thereby protect sulfhydryl groups and cellular integrity from emerging oxygen radicals.
The PPP comprises two separate, oxidative and nonoxidative, phases. Reactions in the oxidative phase are irreversible, whereas all reactions in the nonoxidative phase are fully reversible. The two phases are functionally connected. The nonoxidative phase converts ribose 5-phosphate to glucose 6-phosphate for utilization by the oxidative phase and thus indirectly contributes to generation of NADPH. Different enzymes are rate limiting in the two phases. The oxidative phase primarily depends on glucose 6-phosphate dehydrogenase (G6PD) (36), whereas transaldolase (TAL) is the rate-limiting enzyme for the nonoxidative phase (27, 37). TAL catalyzes the transfer of a 3-carbon fragment, corresponding to dihydroxyacetone, to d-glyceraldehyde 3-phosphate, d-erythrose 4-phosphate, and a variety of other acceptor aldehydes (38). TAL expression and enzymatic activity are regulated in a tissue-specific (37, 39, 40) and development-specific manner (41). TAL overexpression lowers G6PD and 6-phosphogluconate dehydrogenase (6PGD) activities and NADPH and GSH levels and renders the cell highly susceptible to apoptosis induced by serum deprivation, hydrogen peroxide, nitric oxide, TNF-α, and anti-Fas mAb (27). When TAL levels are reduced, G6PD and 6PGD activities and GSH levels are increased, and apoptosis is inhibited. TAL activity profoundly impacts the balance between the two branches of PPP and the ultimate output of NADPH and GSH (27). These findings are consistent with an overwhelming influence of TAL-catalyzed dihydroxyacetone transfer reactions on the distribution of flux between the PPP and nucleotide metabolism, which determines the overall propagation of biochemical signals in a metabolic network (42).
The present study shows that Fas-induced disruption of Δψm, phosphatidylserine (PS) externalization, and cell death are preceded by elevation of mitochondrial and cytosolic ROI levels and hyperpolarization of Δψm. The degree of TAL expression can determine the amount of Fas-induced ROI production, PS externalization, and cell death. Moreover, TAL is shown to influence 1) activation of caspases, including caspase-8, and 2) changes in Δψm. Overexpression of TAL accelerated Fas-induced ROI production, activation of caspases, PS externalization, and cell death in Jurkat human T cells. In contrast, suppression of TAL activity abrogated these effects and inhibited Fas-induced PCD. A key finding is that increased ROI production serves as an early and defining step in Fas-induced apoptosis.
Materials and Methods
Cell culture and apoptosis assays
PBMC were isolated from heparinized venous blood on a Ficoll-Hypaque gradient and resuspended in RPMI 1640 medium supplemented with 10% FCS, 2 mM L-glutamine, 100 IU/ml penicillin, and 100 μg/ml gentamicin. Cells (106/ml) were prestimulated with 5 μg/ml Con A for 5–7 days; subsequently, monocytes/macrophages were removed by adherence (43), and apoptosis was induced with 1 μg/ml Fas Ab CH-11 (MBL, Watertown, MA) (44–46). To measure Con A-induced cell proliferation, 105 PBLs were incubated in each well of a microtiter plate using six parallel samples. The plates were incubated at 37°C in a humidified atmosphere with 5% CO2 for 72 h. The cultures were pulsed with 0.4 μCi of [3H]TdR for 8 h, and [3H]TdR incorporation was measured as previously described (43). Jurkat cell lines, producing increased (L26-3/1 and L26-3/2D1) and suppressed (L18-3/1 and L18-3/1D9) levels of TAL, were previously described (27). Twenty-four hours before assays, Jurkat cells were fed fresh medium and seeded at a density of 2 × 105 cells/ml, and cell death was induced with 50 or 100 ng/ml anti-Fas mAb CH-11. Apoptosis was monitored by observing cell shrinkage and nuclear fragmentation and was quantified by trypan blue exclusion (46). DNA fragmentation during apoptosis was monitored by agarose gel electrophoresis (27). Apoptosis was also measured by flow cytometry after concurrent staining with fluorescein-conjugated annexin V (annexin V-FITC; R&D Systems, Minneapolis, MN; FL-1) and propidium iodide (FL-2) as previously described (47, 48). Staining with phycoerythrin-conjugated annexin V (annexin V-PE; R&D Systems) was used to monitor PS externalization (FL-2) in parallel with measurement of ROI levels and Δψm, using DHR, DCF, or DiOC6 fluorescence, respectively (FL1, see below). Staining with annexin V alone or in combination with DHR or DiOC6 was conducted in 10 mM HEPES (pH 7.4), 140 mM NaCl, and 2.5 mM CaCl2. Using three-color fluorescence, Fas-induced changes in mitochondrial ROI levels, Δψm, and PS externalization within CD4 and CD8 T cells were concurrently analyzed by parallel staining with DHR or DiOC6 (FL-1), annexin V-PE (FL-2), and Quantum
Red-conjugated CD3, CD4, and CD8 mAbs (Sigma, St. Louis, MO; FL-3). Quantum Red contains two covalently linked fluorochromes, PE and Cy5. PE absorbs light energy at 488 nm and emits at 670 nm, in the excitation range of Cy5, which acts as an acceptor dye.
Flow cytometric analysis of ROI production, Δψm, and membrane integrity
The production of ROIs was estimated fluorometrically using oxidation-sensitive fluorescent probes 5,6-carboxy-2′,7′-dichlorofluoresceindiacetate (DCFH-DA), DHR, and hydroethidine (HE; Molecular Probes, Eugene, OR) as previously described (27, 49, 50). Following apoptosis assay, cells were washed three times in 5 mM HEPES-buffered saline, pH 7.4, incubated in HEPES-buffered saline with 0.1 μM DHR for 2 min, 1 μM DCFH-DA for 15 min, or 1 μM HE for 15 min, and samples were analyzed using a Becton Dickinson FACStar Plus flow cytometer (Mountain View, CA) equipped with an argon ion laser delivering 200 mW of power at 488 nm. Fluorescence emission from DCF (green) or DHR (green) was detected at a wavelength of 530 ± 30 nm. Fluorescence emission from oxidized HE, ethidium (red), was detected at a wavelength of 605 nm. Dead cells and debris were excluded from the analysis by electronic gating of forward and side scatter measurements. ROI levels, as determined by fluorescence of control cells, served as a baseline for assessment of increased ROI levels in response to Fas stimulation. While R123, the fluorescent product of DHR oxidation, binds selectively to the inner mitochondrial membrane, ethidium and DCF remain in the cytosol of living cells (50). The Δψm was estimated by staining with 40 nm DiOC6 (Molecular Probes), a cationic lipophilic dye (28, 51, 52), for 15 min at 37°C in the dark before flow cytometry (excitation, 488 nm; emission, 525 nm recorded in FL-1). The fluorescence of DiOC6 is oxidation independent and correlates with Δψm (52). DiOC6 staining was complete after a 15-min incubation. DiOC6 fluorescence was diminished 3- to 4-fold by 5 μM carbonyl cyanide m-chlorophenylhydrazone (mClCCP) and 10-fold or more by 50 μM mClCCP as previously described (53, 54). Δψm was also quantitated using a potential-dependent J-aggregate-forming lipophilic cation, 5,5′,6,6′-tetrachloro-1,1′,3,3′-tetraethylbenzimidazolocarbocyanine iodide (JC-1) (55). JC-1 selectively incorporates into mitochondria, where it forms monomers (fluorescence in green, 527 nm) or aggregates, at high transmembrane potentials (fluorescence in red, 590 nm) (55, 56). Cells were incubated with 1 μM JC-1 for 15 min at 37°C before flow cytometry. Cotreatment with a protonophore, 5 μM mClCCP (Sigma), for 15 min at 37°C resulted in decreased DHR, DiOC6, and JC-1 fluorescence and served as a positive control for disruption of mitochondrial transmembrane potential (52). Staining with 150 nM bis-(1–3-dibutylbarbituric acid)trimethixine oxonol (DiBAC4; excitation, 488 nm; emission, 525 nm) for 10 min at room temperature was used to assess cell membrane potential (57).
TAL and G6PD activities and glutathione levels
TAL enzyme activity was tested in the presence of 3.2 mM d-fructose 6-phosphate, 0.2 mM erythrose 4-phosphate, 0.1 mM NADH, and 10 μg of α-glycerophosphate dehydrogenase/triosephosphate isomerase at a 1:6 ratio at room temperature by continuous absorbance reading at 340 nm for 6 min (58). The enzyme assays were conducted in the activity range of 0.001–0.01 U/ml. G6PD was measured in the presence of 120 mM Tris (pH 7.7), 10 mM MgCl2, 2 mM glucose 6-phosphate, 0.9 mM NADP, and 0.1 U/ml 6PGD (59). TAL activity levels correlated with changes in TAL expression as determined by densitometry (model GS-700, Bio-Rad, Hercules, CA). The total glutathione content was determined by the enzymatic recycling procedure essentially as described by Tietze (60). Cells (106) were resuspended in 50 μl of 4.5% 5-sulfosalicylic acid. The acid-precipitated protein was pelleted by centrifugation at 4°C for 10 min at 15,000 × g. The total protein content of each sample was determined using the Lowry assay (61). The GSH content of the aliquot assayed was determined in comparison to reference curves generated with known amounts of GSH (27).
Caspase-3/CPP32 enzyme assay and protease inhibitors
CPP32 activity was measured by incubating cell extracts in 50 μl of 80 μM DEVD-7-amino-4-trifluoromethyl-coumarin (DEVD-AFC; Calbiochem, La Jolla, CA), 250 mM sucrose, 20 mM HEPES-KOH (pH 7.5), 50 mM KCl, 2.5 mM MgCl2, and 1 mM DTT for 15 min at 37°C (62, 63). The protein content of cell extracts was determined by the Lowry assay (61). Fluorescence (400–505 nm) after addition of 1 ml of ice-cold water was compared with a standard curve of AFC (Sigma). The specificity of the enzymatic reaction was tested by using caspase-3/CPP32 inhibitor DEVD-CHO and caspase-1/ICE inhibitor YVAD-CMK (Bachem, King of Prussia, PA) at a concentration range of 50–300 μM (62). Caspase inhibitors Z-Val-Ala-Asp(Ome).fmk (Z-VAD) and Boc-Asp.fmk (Boc-Asp) as well as noncaspase cysteine protease inhibitor, Z-Phe-Ala.fmk (Z-FA; Enzyme Systems Products (Livermore, CA) were tested at concentrations of 20, 50, and 300 μM (64).
Western blot analysis
Forty micrograms of total cell lysate in 10 μl/well was separated by SDS-PAGE and electroblotted to nitrocellulose (65). For visualization of poly-(ADP-ribose) polymerase (PARP), cells were lysed in 62.5 mM Tris-HCl (pH 6.8), 6 M urea, 10% glycerol, 2% SDS, 0.00125% bromophenol blue, and 5% 2-ME; sonicated for 15 s; and boiled for 5 min (66). Nitrocellulose strips were incubated in 100 mM Tris (pH 7.5), 0.9% NaCl, 0.1% Tween-20, and 5% skim milk with the primary Abs, anti-PARP mAb C-2-10 (66), anti-FLICE/Mch5/caspase-8 (mAb 5F7 directed to C-terminal amino acids 176–460 of human FLICE; Panvera, Madison, WI), anti-transaldolase Ab 170 (38), and actin mAb C4 (Boehringer Mannheim, Indianapolis, IN) at a 1000-fold dilution at room temperature overnight. After washing, the blots were incubated with biotinylated secondary Abs and subsequently with horseradish peroxidase-conjugated avidin (Jackson ImmunoResearch Laboratories, West Grove, PA). Between incubations, the strips were vigorously washed in 0.1% Tween-20, 100 mM Tris (pH 7.5), and 0.9% NaCl. The blots were developed with a substrate comprised of 1 mg/ml 4-chloronaphthol and 0.003% hydrogen peroxide.
Statistics
Alterations in cell survival, ROI levels, caspase-3 and PPP enzyme activities, and GSH levels were analyzed by Student's t test. Changes were considered significant at p < 0.05.
Results
Elevation of ROI levels and Δψm precede PS externalization during Fas-induced apoptosis
In accordance with previous observations (27, 28), Fas-induced apoptosis was associated with increased ROI production (Fig. 1A). To assess changes in intracellular ROI levels we used oxidation-sensitive fluorescent probes DHR, DCFH-DA (27, 50, 67), and HE (53). DHR is nonfluorescent, uncharged, and readily taken up by cells, whereas R123, the product of DHR oxidation, is fluorescent, is positively charged, and binds selectively to the inner mitochondrial membrane of living cells (50). The fluorescence of this dye is an indicator of mitochondrial ROI production and membrane integrity. DCFH-DA is also readily taken up by cells and, after deacetylation to DCFH, is oxidized to its fluorescent derivative, DCF, and remains in the cytosol (50). HE is oxidized into ethidium by ROIs and remains in the cytosol (53). Using all three oxidation-dependent fluorescent probes, significant increases in ROI levels were observed in Jurkat cells at 4°C as early as 20 min after addition of Fas Ab CH-11 (Fig. 1A).
FIGURE 1.

Flow cytometric analysis of mitochondrial ROI production and transmembrane potential (ΔΨm) in Fas-stimulated Jurkat cells. Cells were analyzed after exposure to 50 ng/ml Fas Ab for 20 min (on ice) and 1 h (at 37°C). Dead cells and debris were gated out by forward (FSC) and side (SSC) scatter measurements. A, PS externalization and ROI production were concurrently monitored by staining with annexin V-PE (FL-2) and DHR (FL-1), respectively (dot plot, left column). The Fas-induced increase in ROI levels is shown by overlay of DHR fluorescence of annexin V-negative populations (histograms, right column). Open curves correspond to control cells, while shaded curves represent Fas-treated cells. The x-axis shows the log FL-1 fluorescence intensity; the y-axis indicates the cell number. Values over curves indicate the mean channel of DHR fluorescence of control (0 min) and Fas-treated cells (20 min and 1 h). B, Left column, DiOC6 fluorescence (FL-1) of annexin V-PE-negative cells. The mean channel of histograms and the percentage of cells with increased ΔΨm (in parentheses) are shown for control (open curve) and Fas-treated (shaded curve) cells. B, Right column, JC-1 fluorescence (FL-2) of Fas-stimulated (shaded histogram) and control (open histogram) cells. As a control, reduced ΔΨm was measured in the presence of mClCCP, an uncoupling agent that reduces ΔΨm. C, Time course of ROI production and changes in Δ Ψm in response to stimulation of Jurkat cells with 50 ng/ml CH-11. Survival was assessed by flow cytometric determination of the percent of annexin V-PE-negative cells at the time points indicated. ROI production was measured in log fluorescence intensity after labeling with DHR. ΔΨm was assessed by DiOC6 (FL-1) and JC-1 fluorescence (FL-2). The fluorescence of control cells served as a baseline for each experiment. Data represent the mean ± SE of four or more independent experiments. Based on six independent experiments, the mean channel of DHR fluorescence was increased after 20-min stimulation with Fas Ab from a baseline of 7.8 ± 0.5 to 11.5 ± 0.7 by 3.7 ± 0.7 (p < 0.01). D, Flow cytometric analysis of intracellular ROI levels in Fas-stimulated Jurkat cells using DCFH-DA, HE, and DHR. PS externalization and ROI production were concurrently monitored by annexin V-PE (FL-2) and DFC (FL-1) or annexin V-FITC (FL-1) and HE (FL-2) staining, respectively (dot plots, columns 1 and 3). The Fas-induced increase in ROI levels is shown by an overlay of DCF or HE fluorescence of annexin V-negative populations (histograms, columns 2 and 4). DHR fluorescence was measured in parallel (column 5). Values over curves indicate the mean channel of DCF, HE, or DHR fluorescence of control (0 min) and Fas-treated cells (20 min and 1 h). Open curves correspond to control cells, while shaded curves represent Fas-treated cells. Data are representative of four independent experiments.
PS, which is normally confined to the inner leaflet of the plasma membrane, is exported to the outer plasma membrane leaflet during apoptosis. PS externalization is an early event of PCD that may serve as a flag allowing phagocytes to recognize and engulf these apoptotic cells before they become leaky and rupture (47, 48). To assess the timing of PS externalization with respect to mitochondrial ROI production, cells undergoing Fas-induced apoptosis were analyzed by concurrent staining with annexin V-PE (FL-2) and DHR (FL-1). As shown in Fig. 1A, DHR fluorescence increased in annexin V-negative cells, suggesting that Fas-induced elevation of ROI levels in mitochondria occurred before PS externalization. ROI production continued to increase twofold or more over the baseline on a logarithmic scale (Fig. 1C). DCF and ethidium fluorescence were also increased in annexin V-negative cells, indicating elevated levels of ROI in the cytosol upon Fas stimulation (Fig. 1D). ROI levels remained elevated in annexin V-positive cells until they underwent apoptotic shrinking as determined by forward angle light scattering as a direct measure of particle size (data not shown). With precipitous decline of cell viability and size, 12–24 h after Fas stimulation DHR fluorescence returned toward baseline levels, in correlation with a decrease in Δψm (see below), possibly reflecting leakage of ROI secondary to damage of mitochondrial and cellular membranes (50–52).
Previous studies suggested that a decline of Δψm may be an early event in apoptosis, including Fas-dependent signaling (68). Since an early increase in DHR fluorescence is dependent on the integrity of a negatively charged inner mitochondrial membrane, we also examined changes in Δψm using the potentiometric dyes, DiOC6 (51, 52) and JC-1 (55, 56). As shown in Fig. 1B, DiOC6 fluorescence was increased in annexin V-negative cells as early as 20 min after stimulation with Fas Ab. Red fluorescence of JC-1 (FL-2) was also increased in Fas-treated cells (Fig. 1B). Δψm of annexin V-negative cells remained elevated for several hours after Fas stimulation (Fig. 1C). Similarly, increased DiOC6 staining in annexin V-negative cells in response to Fas stimulation was noted by fluorescence microscopy (Fig. 2). DiOC6 staining was diminished in shrunken annexin V-positive cells. Control cells stained with JC-1 showed green fluorescence, while Fas-treated cells gained red JC-1 fluorescence (Figs. 1B and 2), consistent with a higher Δψm (55, 56).
FIGURE 2.

Fluorescence microscopy of control (A, C, and E) and Fas-treated Jurkat cells (B, D, and F) stained with potentiometric dyes DiOC6 and JC-1. Jurkat cells were cultured for 1 h in the absence (control) or the presence of 50 ng/ml Fas Ab CH-11. A and B were stained with annexin V-PE. In A, in the absence of fluorescence, cells illuminated with visible light are shown. C and D were stained with annexin V-PE (red) and DiOC6 (green). C shows green fluorescence only. D shows increased green (DiOC6) fluorescence of annexin V-negative cells; bright yellow cells show concurrent DiOC6 and annexin V-PE staining; shrunken cells with red annexin V-PE staining displayed diminished green (DiOC6) fluorescence. E and F were stained with JC-1. Green fluorescence was detected in control cells (E), while green and red fluorescence was noted in Fas-stimulated cell populations (F). Magnifications, ×400.
DiBAC4 fluorescence significantly increased in annexin V-positive, but not in annexin V-negative, cells (data not shown), indicating that changes in external cell membrane potential occurred at a later stage of PCD.
Fas-induced cell death is associated with increased ROI production in peripheral blood T lymphocytes
Stimulation of freshly isolated PBL for up to 3 days with 50 ng/ml, 100 ng/ml, or 1 μg/ml CH-11 Fas Ab had no toxic effect but, rather, increased cell survival (data not shown). These results are consistent with data from other laboratories suggesting that stimulation of the Fas receptor alone transduces activation rather than death signals (13, 14). Incubation of PBL for 20 min on ice with 1 μg/ml Fas Ab increased ΔΨm, as detected by DiOC6 and JC-1 fluorescence (Fig. 3A). Previous studies suggested that prestimulation of T lymphocytes with mitogenic lectins increases susceptibility to apoptotic signaling through the Fas receptor (13). We used the lectin Con A to trigger polyclonal activation of T lymphocytes. Stimulation with Con A dramatically elevated ΔΨm. After incubation with Fas Ab, ΔΨm of Con A-prestimulated PBL was further increased (Fig. 3A). The activation signals associated with short term Fas or Con A stimulation did not significantly change mitochondrial ROI levels as measured by DHR fluorescence (Fig. 3A). In contrast, prestimulation with Con A for 5–7 days sensitized PBL to Fas-induced apoptosis (Fig. 3, B and C). Fas stimulation of PBL that had been preincubated with Con A for at least 5 days triggered a rapid increase in mitochondrial ROI production and ΔΨm. These changes occurred in annexin V-negative cells (Fig. 3, B and C). Three-color fluorescence staining with DHR or DiOC6 (FL-1), annexin V-PE (FL-2), and Quantum Red-conjugated CD3, CD4, and CD8 mAbs (FL-3) revealed that Fas-induced ROI production and elevation of ΔΨm occurred in both CD4+ and CD8+ T cells (Fig. 3, B and C). With decline of cell viability and size, 12–24 h after Fas stimulation DHR fluorescence returned toward baseline levels in parallel with a decrease in DiOC6 and JC-1 fluorescence (data not shown), possibly reflecting leakage of ROI secondary to damage of mitochondrial and cellular membranes (50–52). Thus, Fas-induced oxidative stress and ΔΨm elevation preceded PS externalization in both Jurkat lymphoblastoid T cells and PBL (Figs. 1–3).
FIGURE 3.
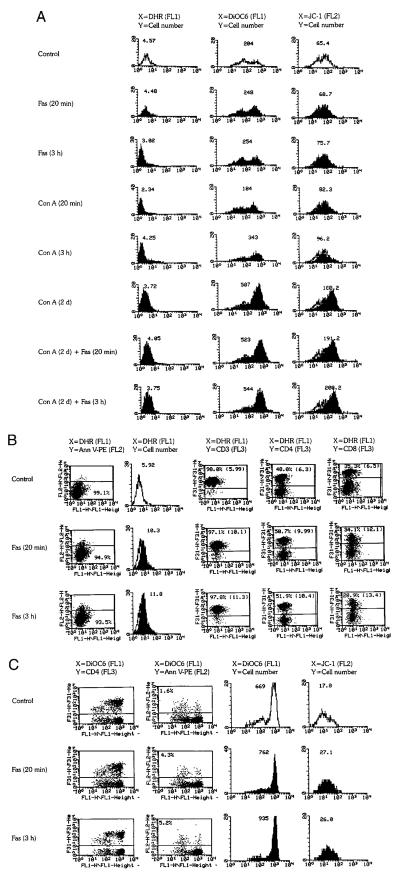
Flow cytometric analysis of ROI production and mitochondrial transmembrane potential (ΔΨm) in PBL. A, PBL, freshly purified or prestimulated with Con A for 2 days (Con A, 2d), were incubated with 1 μg/ml Fas mAb CH-11 for 20 min on ice or 3 h at 37°C. As controls, freshly isolated PBL stimulated with Con A for 20 min on ice or 3 h at 37°C were also examined. Dead cells and debris were gated out by FSC/SSC measurements. ROI production was assessed by DHR fluorescence (mean channel, FL-1) of annexin V-PE (FL-2)-negative cells. Open curves correspond to control cells, while shaded curves represent Con A- and/or Fas-treated cells, as indicated for each histogram. The x-axis shows the log FL-1 fluorescence intensity; the y-axis indicates the cell number. Δψm was measured by DiOC6 fluorescence (FL-1) of annexin V-PE-negative cells or JC-1 fluorescence (FL-2) of live cells based on forward/side scatter (FSC/SSC) gating. Values over curves indicate the mean channels of DHR, DiOC6, and JC-1 fluorescence. B, Fas-induced mitochondrial ROI production in PBL prestimulated with 5 μg/ml Con A for 5 days. After prestimulation with Con A, PBL were incubated with 1 μg/ml Fas Ab CH-11. ROI production was assessed by DHR fluorescence (FL-1). PS externalization was determined by annexin V-PE staining (FL-2). Column 1 shows the percentage of annexin V-PE-negative cells and their right shift on the FL-1 axis. Column 2 shows histogram and mean channel number of DHR fluorescence of annexin V-PE-negative cells. T cells and their CD4 and CD8 subsets were identified by staining with Quantum Red-conjugated CD3, CD4, and CD8 Abs, respectively (FL-3). Columns 3–5 indicate the percentages of CD3-, CD4-, and CD8-stained cells and their mean DHR fluorescence (in parentheses) gated on annexin V-PE-negative cells. C, Fas-induced ΔΨm changes in PBL prestimulated with Con A for 5 days. Column 1 shows increased DiOC6 (FL-1) fluorescence of both CD4− and CD4+ compartments of PBL (FL-3) gated on annexin V-PE-negative cells in response to Fas stimulation. Column 2 shows the percentage of annexin V-PE-positive cells and increased DiOC6 fluorescence (right shift) of annexin V-negative cells. Column 3 indicates the histogram and mean channel of DiOC6 fluorescence gated on annexin V-PE-negative cells. Column 4 shows the histogram and mean channel of JC-1 fluorescence (FL-2) of live cells based on FSC/SSC gating. Data are representative of five independent experiments.
Effects of caspase inhibitors on ROI production, ΔΨm, and PS externalization during Fas-induced apoptosis
To assess the timing of ROI production with respect to caspase activation, the effects of caspase inhibitors DEVD, Z-VAD, and Boc-Asp (62) on Fas-induced cell death were investigated. Jurkat cells were pretreated for 3 h with caspase inhibitors (62) before addition of Fas Ab CH-11. Following pretreatment with 300 μM DEVD-CHO, PS externalization did not occur, while ROI levels in Fas-stimulated cells remained significantly elevated (up to twofold on a logarithmic scale) compared with those in control cells (p < 0.001; Fig. 4, A and B). An increase in ΔΨm was detectable in annexin V-negative cells as early as 20 min after stimulation on ice with Fas Ab (Fig. 4C). ΔΨm diminished in annexin V-positive cells (Fig. 4C). DEVD pretreatment did not influence the Fas-induced elevation in ΔΨm (Fig. 4C). However, DEVD blocked PS externalization and the drop in ΔΨm was confined to annexin V-positive Jurkat cells. In agreement with earlier results (62, 69), DEVD completely blocked Fas-induced cell death of Jurkat cells. Similar to DEVD-CHO, ZVAD.fmk (50 μM) and Boc-Asp.fmk (50 μM) completely blocked PS externalization without affecting Fas-induced elevation in ΔΨm (Fig. 4D) or increases in ROI levels assessed by DHR, DCF, and ethidium fluorescence (not shown). Pretreatment with the caspase-1 inhibitor YVAD-CMK or the cysteine protease inhibitor Z-FA.fmk (up to 300 μM for 3 h) did not significantly inhibit cell death (data not shown), in accordance with a dominant role of caspase-8 and caspase-3 in Fas-dependent apoptosis (11).
FIGURE 4.


Effects of caspase-3 inhibitor DEVD-CHO on Fas-induced mitochondrial ROI production, Δψm, and cell death. Jurkat cells were preincubated in the presence or the absence of DEVD (300 μM) for 3 h. A, Survival and ROI levels were evaluated by trypan blue exclusion and DHR fluorescence, respectively, 24 h after stimulation with Fas Ab. The fluorescence of control cells served as the baseline for each experiment. The data show the mean ± SE of four experiments. In comparison to untreated control cells, DHR fluorescence was elevated in Fas-treated or Fas- plus DEVD-treated cells (p < 0.001). B, Time course of Fas-induced mitochondrial ROI production in DEVD-pretreated cells (300 μM, 3 h). ROI levels were assessed by DHR fluorescence (FL-1) in annexin V-PE (FL-2)-negative cells after stimulation with 50 ng/ml CH11 Fas Ab for 20 min on ice or 1 h at 37°C. C, Effect of Fas stimulation on Δψm of DEVD-pretreated cells. Δψm was assessed by DiOC6 fluorescence (FL-1) in annexin V-PE (FL-2)-negative and -positive cells. Left panel, Increased DiOC6 fluorescence in annexin V-PE (FL-2) negative and decreased DiOC6 fluorescence in annexin V-PE-positive cells in response to Fas stimulation. The percentage of annexin V-positive cells is shown in the upper left corner. Right panel, The histogram and mean channel number of DiOC6 fluorescence in annexin V-PE (FL-2)-negative cells. D, Effect of Fas stimulation on Δψm of Jurkat cells pretreated for 3 h with 300 μM DEVD, 50 μM Z-VAD, or 50 μM Boc-Asp. The Δψm was assessed by DiOC6 fluorescence (FL-1) in annexin V-PE (FL-2)-negative and -positive cells (left panels). The percentage of annexin V-positive cells is shown in the upper left corner. Right panels, The histogram and mean channel number of DiOC6 fluorescence in annexin V-PE (FL-2)-negative cells. Data are representative of three independent experiments.
Fas-induced ROI production, ΔΨm elevation, caspase activation, and cell death are regulated by transaldolase
Disruption of the mitochondrial membrane potential has been proposed as the point of no return in apoptotic signaling (28, 54, 68). Mitochondrial membrane permeability is subject to regulation by an oxidation-reduction equilibrium of ROIs, pyridine nucleotides (NADH/NAD and NADPH/NADP), and GSH levels (70). Since the overall NADPH output of PPP, GSH levels, and sensitivity to apoptosis are regulated by TAL, we investigated the effect of changes in TAL activity at various checkpoints of Fas-induced cell death. Stimulation of Jurkat L26-3/4 and L26-3/2D1 cells with increased TAL activity and decreased NADPH, NADH, and GSH levels (27) resulted in accelerated PS externalization and cell death (Figs. 5A) as well as increased mitochondrial ROI production compared with those of control Jurkat cells (Fig. 5B). Increases in DiOC6 fluorescence within annexin V-negative compartments and the drop in DiOC6 fluorescence within annexin V-positive compartments were accelerated in Fas-treated L26-3/4 and L26-3/2D1 cells (Fig. 6A). Along the same line, JC-1 fluorescence was enhanced in Fas-treated L26-3/4 and L26-3/2D1 cells (Fig. 6B). Thus, increases in ΔΨm were larger and occurred earlier in cells with increased TAL expression (Fig. 6, A and B). By contrast, mitochondrial ROI production (Fig. 5C), changes in ΔΨm (Fig. 6), and cell death were inhibited in Jurkat L18-3/1 and L18-3/1D9 cells with decreased TAL activity and increased GSH content (Fig. 5A).
FIGURE 5.

Fas-induced mitochondrial ROI production and cell death in the presence and the absence of DEVD-CHO in Jurkat cells stably transfected with TAL-H expression vectors. L26-3/4 and L26-3/2D1 cells were transfected with the sense construct. L18-3/1 and L18-3/1D9 cells were transfected with the antisense construct. After pretreatment with or without 300 μM DEVD-CHO for 3 h, cells were stimulated with Fas Ab CH-11. A, The rate of cell death was determined by staining with annexin V-FITC. Data represent the mean ± SE of five experiments. Survival was diminished in L26-3/4 (p < 0.001) and L26-3/2D1 cells (p < 0.005). Cell death was inhibited in L18-3/1 and L18-3/1D9 cells (p < 0.001). B, Changes in ROI levels in response to stimulation with 50 ng/ml Fas Ab for 1 h were monitored by DHR fluorescence (FL-1) in annexin V-PE-negative cells. The histogram and mean channel number of DHR fluorescence, with and without DEVD-CHO pretreatment, in L26-3/4, L26-3/2D1, L18-3/1, L18-3/1D9, and control Jurkat cells are indicated.
FIGURE 6.

Fas-induced changes in Δψm following 1-h stimulation with 50 ng/ml CH-11 Ab of Jurkat cells stably transfected with TAL-H expression vectors. A, Δψm was assessed by DiOC6 fluorescence (FL-1) in annexin V-PE (FL-2)-positive and -negative cells. The mean channel of DiOC6 fluorescence of annexin V-PE-negative and -positive cells (percent distribution in parentheses) is indicated in each dot plot. B, Δψm was assessed by measurement of JC-1 fluorescence (FL-2) in live (R1) and dead (R2) cell populations based on FSC/SSC measurements. Values over histograms indicate the mean channel of JC-1 fluorescence.
DEVD pretreatment for 3 h completely abrogated Fas-induced PS externalization and cell death in all cell lines (Figs. 4 and 5). A drop in ΔΨm, occurring in annexin V-positive cells (Figs. 4C and 6A), was also eliminated by DEVD (Fig. 4C), suggesting that activation of caspases was required for disruption of ΔΨm. In contrast, accelerated mitochondrial ROI production (Fig. 5C) and early increases in ΔΨm (Fig. 4C) were not affected by DEVD pretreatment. These results indicated that stimulation of mitochondrial ROI production and increases in ΔΨm preceded or occurred independently from activation of caspases in Fas-induced apoptosis.
Activation of cysteine proteases have been described as both a result and a cause of oxidative stress. To test the effect of TAL activity on activation of proteases through Fas, we quantitated cleavage of DEVD-AFC, a substrate of caspase-3 (62, 63) and caspase-8 (11, 71), and monitored proteolysis of caspase-8/FLICE and PARP, a signature substrate of caspase-3 (72) in Jurkat cells with altered levels of TAL expression. In control Jurkat cells, significant increases in DEVD-AFC cleavage activity were noted 2 h after stimulation with 50 ng/ml Fas Ab at 37°C (p < 0.01; Fig. 7). Proteolysis of FLICE, isoforms caspase-8/a and caspase-8/b (73), required at least 1-h stimulation with 50 ng/ml Fas Ab at 37°C. PARP cleavage was detectable after 4-h Fas stimulation. Cleavage of DEVD-AFC (Fig. 7), PARP (Fig. 8), and FLICE (Fig. 9) was accelerated in L26-3/4 and L26-3/2D1 cells with increased TAL expression compared with that in control Jurkat cells. By contrast, cleavage of DEVD-AFC, PARP, and FLICE was inhibited in L18-3/1 and L18-3/1D9 cells with suppressed TAL expression (Figs. 7–9).
FIGURE 7.

Rate of caspase-3 activity in Jurkat cells stably transfected with TAL-H expression vectors during Fas-induced apoptosis. Protease activity was measured by cleavage of DEVD-AFC in cell extracts prepared at the time points indicated. Data show the mean ± SE of four experiments.
FIGURE 8.

A, Cleavage of PARP (116 kDa) to 85-kDa fragment during Fas-induced apoptosis of Jurkat cells stably transfected with TAL-H expression vectors. Six hours after stimulation with 50 ng/ml Fas Ab, PARP cleavage was accelerated in L26-3/4 and L26-3/2D1 cells and was abrogated in L18-3/1 and L18-3/1D9 cells compared with that in control Jurkat cells (left panel). PARP was not cleaved in lysates of cells unstimulated with Fas Ab (right panel). Cell lysates containing 40 μg of total protein/lane were prepared and analyzed by Western blot using anti-PARP mAb C-2-10. B, Monitoring of TAL expression using Ab 170. Levels of TAL expression were reduced in L18-3/1D9 (−29%) and L18-3/1 (−48%) cells and were increased in L26-3/4 (2.5-fold) and L26-3/2D1 (+31%) cells compared with those in control Jurkat cells as previously described (27). C, Actin was detected with mAb C4.
FIGURE 9.
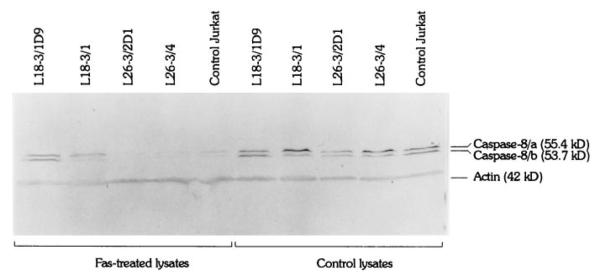
Proteolysis of FLICE/caspase-8 isoforms caspase-8/a and caspase-8/b during Fas-induced apoptosis of Jurkat cells stably transfected with TAL-H expression vectors. Four hours after stimulation with 50 ng/ml Fas Ab, cleavage of caspase-8 was accelerated in L26-3/4 and L26-3/2D1 cells and was abrogated in L18-3/1 and L18-3/1D9 cells compared with that in control Jurkat cells (left panel). Caspase-8 was not cleaved in lysates of cells unstimulated with Fas Ab (right panel). Cell lysates containing 40 μg of total protein/lane were prepared and analyzed by Western blot using anti-caspase-8 mAb 5F7. Actin was detected with mAb C4.
Discussion
Controlled levels of ROIs are necessary for the operation of signal transduction pathways, including those mediating apoptosis (16–22). However, the place of ROIs in the Fas signaling pathway has not been clearly defined (2). It is currently believed that apoptotic signaling transduced through the Fas receptor involves sequential activation of initiator and executioner caspases, such as caspase-8 and caspase-3, respectively (74). The sequential activation of cysteine proteases is followed by the disruption of ΔΨm. The latter event appears to be the point of no return in the effector phase of PCD, as it leads to the mitochondrial release of apoptogenic proteins, which, in turn, activate downstream caspases and endonucleases (4, 10, 68, 75–78). It has been proposed that lymphocytes treated with dexamethasone or stimulated with Fas first reduce their ΔΨm by an unknown mechanism and then hyperproduce ROI that serve as PCD effector molecules (53, 68). While a disruption of ΔΨm with the uncoupling of electron transport can indeed lead to respiratory burst, a reverse scenario, i.e., uncontrolled production of ROI initiating the decline of ΔΨm, is more consistent with the generation of mitochondrial permeability transition (79). Mitochondrial function depends on the integrity of its inner lipid bilayer. Agents that cause permeability transition or pore formation are either pro-oxidants or direct cross-linkers of SH groups (79).
We previously showed that Fas-mediated cell death was associated with ROI production, and its rate correlated with intracellular GSH levels as regulated by TAL expression (27). Similar findings were reported at the same time by other laboratories (28–32). In the present studies, ROI production was assessed using oxidation-sensitive fluorescent probes. In living cells, R123, the fluorescent product of DHR oxidation, binds selectively to the inner mitochondrial membrane, whereas DCF, the fluorescent product of DCFH oxidation, and ethidium, the fluorescent product of HE oxidation, remain in the cytosol (50). Thus, increased ROI levels in response to Fas stimulation were elevated both inside and outside the mitochondria. In both PBL and Jurkat cells, ROI production increased as early as 20 min after Fas stimulation before activation of caspases, PS externalization, or a precipitous decline of cell viability. The selective increase in DHR fluorescence reflected increased mitochondrial ROI production at a time when mitochondrial transmembrane potential and membrane integrity were maintained (50). DiOC6 and JC-1 fluorescence were increased as early as 20 min after stimulation with Fas Ab, suggesting that an early increase in ROI production was accompanied by ΔΨm elevation, that is, a hyperpolarization of the inner mitochondrial membrane. The dependence of elevated ROI levels on the maintenance of ΔΨm was underlined by the parallel inhibition of DHR, DiOC6, and JC-1 fluorescence in the presence of the uncoupling agent mClCCP. DiOC6 and JC-1 fluorescence decreased in annexin V-positive cells with diminished size, possibly reflecting a disruption of ΔΨm (68). In correlation with Salvioli et al.
(80), the staining profile of DiOC6 was the most variable among the potentiometric dyes. However, all three dyes, DiOC6, rhodamine 123, and JC-1, consistently demonstrated an early elevation of Δψm/mean channel fluorescence in response to Fas stimulation. Previous studies suggested that a decline of Δψm may be an early event in apoptosis, including Fas-dependent signaling (68). Our data indicate that increased ROI production and Δψm elevation precede PS externalization and a disruption of Δψm. Similar findings of an early increase in DHR fluorescence were recently suggested to represent a key event in PCD that preceded cytochrome c release and disruption of Δψm (54). The release of cytochrome c, beginning 2 h or later following Fas stimulation, appears to be a consequence of mitochondrial membrane damage (81).
Activation of caspases has been considered a hallmark of PCD (4, 10, 74). An increased cleavage of DEVD-AFC, a substrate of caspase-3 (62, 63) and caspase-8 (11, 71), was demonstrated in Jurkat cells as early as 2 h after stimulation with Fas Ab, in agreement with previous findings (64). Pretreatment with the caspase inhibitors, DEVD, Z-VAD, and Boc-Asp, completely abrogated Fas-induced PS externalization, indicating that activation of caspase-3, caspase-8, and related cysteine proteases was absolutely required for cell death (7–9, 74). ROI levels were partially inhibited in DEVD-treated Jurkat cells, suggesting that caspase-3 activation, perhaps through damage of mitochondrial membrane integrity, contributes to ROI production and serves as a positive feedback loop at later stages of the apoptotic process. Nevertheless, ROI levels remained significantly elevated after pretreatment with caspase inhibitors (up to twofold on a logarithmic scale compared with control cells). This suggested that activation of caspase-3 or caspase-8 was not required for increased ROI production and Δψm hyperpolarization. By contrast, DEVD, Z-VAD, and Boc-Asp blocked PS externalization and the decline of Δψm in annexin V-positive Jurkat cells, suggesting that disruption of Δψm (1) was a relatively late event with respect to ROI production and Δψm hyperpolarization and (2) depended on activation of caspase-3 and related proteases. Disruption of Δψm was previously suggested as a point of no return in Fas-induced apoptosis of Jurkat cells, secondary to activation of caspases (68, 74). The present data indicate that increases in ROI levels and Δψm precede disruption of Δψm during Fas-induced apoptosis of Jurkat cells. Moreover, increased ROI production in addition to caspase activation may be required to cause death of Fas-stimulated PBL.
Prestimulation with mitogenic lectins for 5 days was required to sensitize peripheral blood T lymphocytes to apoptotic signaling through the Fas receptor, in accordance with observations by others (13, 14, 82). Stimulation of freshly isolated PBL for up to 3 days with Fas Ab was not toxic. However, incubation of PBL for as few as 20 min on ice with 1 μg/ml Fas Ab or Con A increased Δψm (detected by DiOC6 and JC-1 fluorescence) without increasing ROI levels. By contrast, prestimulation with Con A for 5–7 days sensitized PBL to Fas-induced apoptosis. Fas stimulation of PBL after at least 5 days of incubation with Con A triggered a rapid increase in ROI production and Δψm in annexin V-negative peripheral blood CD4+ and CD8+ T lymphocytes. Thus, in two different cell preparations (Jurkat lymphoblastoid T cells and PBL), Fas-induced apoptosis was associated with increased mitochondrial ROI levels and Δψm elevation before PS externalization.
The Fas receptor is expressed on a variety of freshly isolated normal cells, among them PBL (4, 13, 14). Direct stimulation of PBL through Fas does not induce apoptosis but, rather, leads to cellular activation. To convert Fas-mediated signals from activation to apoptosis, 5 days of prestimulation with Con A were required. The process was accompanied by an early increase in mitochondrial ROI levels. This signaling conversion may mimic a state of antigenic and/or mitogenic overstimulation and depict a physiological process that eliminates unwanted cells. Sensitization to Fas-induced ROI production and apoptosis may be related to changes in metabolism, i.e., GSH depletion and accelerated DNA synthesis in activated T lymphocytes (21, 83). Interestingly, the PPP is the sole source of the NADPH needed for GSH synthesis and the ribose 5-phosphate used for synthesis of the DNA and other nucleic acids (35).
DEVD-AFC cleavage activity and proteolysis of caspase-8 and PARP were accelerated in cells with increased TAL expression and were inhibited in cells with suppressed TAL expression. Proteolytic activation of caspase-8 (FLICE/MACH) appears to be the first step in the death cascade at the cytosolic face of the Fas receptor (12). Cleavage of caspase-8 is initiated by its recruitment to DISC (11, 12, 74). Alternatively, caspase-8 can be activated by dimerization (71, 84). The dominant effect of TAL expression on cleavage of caspase-8 is consistent with a redox control mechanisms at the pinnacle of the death cascade. The redox mechanisms influencing assembly of DISC, oligomerization, and/or cleavage of FLICE are critical questions to be investigated.
TAL regulates key checkpoints in Fas-mediated apoptosis: mitochondrial ROI formation, activation of caspase-8 and caspase-3, Δψm, PS externalization, and, ultimately, cell death, through regulation of the PPP, where the enzyme controls the levels of intra-cellular reducing equivalents. TAL overexpression in Jurkat or H9 human T cells down-regulates G6PD and 6PGD activities and decreases NADPH, NADH, and GSH levels. Alternatively, decreased TAL expression up-regulates G6PD and 6PGD activities and increases GSH (27, 85). TAL overexpression increases sensitivity while suppression of TAL diminishes sensitivity to Fas-induced cell death. In cells overexpressing TAL, glucose 6-phosphate is profoundly depleted (27) which may be directly responsible for the diminished G6PD activities and NADPH/GSH levels and increased sensitivity to apoptosis. Alternatively, increased G6PD activities and GSH levels of cells with suppressed TAL activity may result from increased availability of glucose 6-phosphate for generation of NADPH. Of note, glucose transport can be lost within minutes of the stimulation of Jurkat cells with Fas Ab (86), thereby blocking the cell's ability to combat oxidative stress and diminishing the survival of Fas-stimulated cells. The results indicate that TAL is a dominant regulator of glucose utilization and propagation of apoptotic signals in Fas-stimulated T cells. The impact of transaldolase on Fas-induced mitochondrial ROI production and changes in Δψm as reported here implies a central role for this enzyme in the PPP and propagation of biochemical signals dependent on NADPH and GSH production (27, 42). These findings are compatible with a pivotal role we describe for ROIs in Fas signaling.
Acknowledgments
We thank Drs. Anthony Martonosi and Sandy Livnat for helpful discussions and Dr. Paul Phillips for continued encouragement and support.
Footnotes
This work was supported in part by Grant RO1DK49221 from the National Institutes of Health, Grant RG2466A1/3 from the National Multiple Sclerosis Society, and the Central New York Community Foundation.
Abbreviations used in this paper: PCD, programmed cell death; DISC, death-inducing signaling complex; ICE, IL-1β-converting enzyme; FLICE, Fas-associated death domain-like ICE; ROI, reactive oxygen intermediate; GSH, reduced glutathione; PPP, pentose phosphate pathway; G6PD, glucose 6-phosphate dehydrogenase; TAL, transaldolase; 6PGD, 6-phosphogluconate dehydrogenase; Δψm, mitochondrial transmembrane potential; PS, phosphatidylserine; annexin V-FITC, fluorescein-conjugated annexin V; annexin V-PE, phycoerythrin-conjugated annexin V; DHR, dihydrorhodamine 123; DCF, 5,6-carboxy-2′,7′-dichlorofluorescein; DiOC6, 3,3′-dihexyloxacarbocyanine iodide; DCFH-DA; DCF-diacetate; HE, hydroethidine; mClCCP, carbonyl cyanide m-chlorophenylhydrazone; JC-1, 5,5′, 6,6′-tetrachloro-1,1′,3,3′-tetraethylbenzimidazolocarbocyanine iodide; DEVD, Asp-Glu-Val-Asp; AFC, 7-amino-4-trifluoromethyl-coumarin; YVAD, Tyr-Val-Ala-Asp; Z-VAD, Z-Val-Ala-Asp; Z-FA, Z-Phe-Ala; PARP, poly(ADP-ribose) polymerase.
References
- 1.Wyllie AH, Kerr JFR, Currie AE. Cell death: the significance of apoptosis. Int. Rev. Cytol. 1980;68:251. doi: 10.1016/s0074-7696(08)62312-8. [DOI] [PubMed] [Google Scholar]
- 2.Nagata S, Golstein P. The Fas death factor. Science. 1995;267:1449. doi: 10.1126/science.7533326. [DOI] [PubMed] [Google Scholar]
- 3.Thompson CB. Apoptosis in the pathogenesis and treatment of disease. Science. 1995;267:1456. doi: 10.1126/science.7878464. [DOI] [PubMed] [Google Scholar]
- 4.Nagata S. Apoptosis by death factor. Cell. 1997;88:355. doi: 10.1016/s0092-8674(00)81874-7. [DOI] [PubMed] [Google Scholar]
- 5.Itoh N, Nagata S. A novel protein domain required for apoptosis: mutational analysis of human Fas antigen. J. Biol. Chem. 1993;268:10932. [PubMed] [Google Scholar]
- 6.Tartaglia LA, Ayres TM, Wong GHW, Goeddel GV. A novel domain within the 55 kd TNF receptor signals cell death. Cell. 1993;74:845. doi: 10.1016/0092-8674(93)90464-2. [DOI] [PubMed] [Google Scholar]
- 7.Tewari M, Dixit VM. Fas- and tumor necrosis factor-induced apoptosis is inhibited by the Poxvirus crmA gene product. J. Biol. Chem. 1995;270:3255. doi: 10.1074/jbc.270.7.3255. [DOI] [PubMed] [Google Scholar]
- 8.Los M, Van de Craen M, Penning LS, Schenk H, Westendorp M, Baeuerle PA, Droge W, Krammer PH, Flers W, Schulze-Osthoff K. Requirement of an ICE/CED-3 protease for Fas/Apo-1-mediated apoptosis. Nature. 1995;375:81. doi: 10.1038/375081a0. [DOI] [PubMed] [Google Scholar]
- 9.Enari M, Hug H, Nagata S. Involvement of an ICE-like protease in Fas-mediated apoptosis. Nature. 1995;375:78. doi: 10.1038/375078a0. [DOI] [PubMed] [Google Scholar]
- 10.Martin SJ, Green DR. Protease activation during apoptosis: death by a thousand cuts. Cell. 1995;82:349. doi: 10.1016/0092-8674(95)90422-0. [DOI] [PubMed] [Google Scholar]
- 11.Boldin MP, Goncharov TM, Goltsev YV, Wallach D. Involvement of MACH, a novel MORT1/FADD-interacting protease, in Fas/Apo-1- and TNF receptor-induced cell death. Cell. 1996;85:803. doi: 10.1016/s0092-8674(00)81265-9. [DOI] [PubMed] [Google Scholar]
- 12.Muzio M, Chinnaiyan AM, Kischkel FC, O'Rourke K, Shevchenko A, Ni J, Scaffidi K, Bretz JD, Zhang M, Gentz R, et al. FLICE, a novel FADD-homologous ICE/CED-3-like protease, is recruited to the CD95 (Fas/Apo1) death-inducing signaling complex. Cell. 1996;85:817. doi: 10.1016/s0092-8674(00)81266-0. [DOI] [PubMed] [Google Scholar]
- 13.Miyawaki T, Uehara T, Nibu R, Tsuji T, Yachie A, Yonehara S, Taniguchi N. Differential expression of apoptosis related Fas antigen on lymphocyte subpopulations in human peripheral blood. J. Immunol. 1992;149:3753. [PubMed] [Google Scholar]
- 14.Alderson MK, Armitage RJ, Maraskovsky E, Tough TW, Roux E, Schooley K, Ramsdell F, Lynch DH. Fas transduces activation signals in normal human T lymphocytes. J. Exp. Med. 1993;178:2231. doi: 10.1084/jem.178.6.2231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Owen-Schaub LB, Radinsky R, Kruzel E, Berry K, Yonehara S. Anti-Fas on nonhemopoietic tumors: levels of Fas/Apo-1 and Bcl-2 are not predictive of biological responses. Cancer Res. 1992;54:1580. [PubMed] [Google Scholar]
- 16.Lipton SA, Choi Y, Pan Z, Lei SZ, Chen HV, Sucher NJ, Loscalzo J, Singel DJ, Stamler JS. A redox-based mechanism for the neuro-protective and neurodestructive effects of nitric oxide and related nitroso-compounds. Nature. 1993;364:626. doi: 10.1038/364626a0. [DOI] [PubMed] [Google Scholar]
- 17.Halliwell B, Gutteridge JM. Role of free radicals and catalytic metal ions in human disease: an overview. Methods Enzymol. 1990;186:1. doi: 10.1016/0076-6879(90)86093-b. [DOI] [PubMed] [Google Scholar]
- 18.Stamler JS. Redox signaling: nitrosylation and related target interactions of nitric oxide. Cell. 1994;78:931. doi: 10.1016/0092-8674(94)90269-0. [DOI] [PubMed] [Google Scholar]
- 19.Korsmeyer SJ. Regulators of cell death. Trends Genet. 1995;11:101. doi: 10.1016/S0168-9525(00)89010-1. [DOI] [PubMed] [Google Scholar]
- 20.Buttke TM, Sandstrom PA. Oxidative stress as a mediator or apoptosis. Immunol. Today. 1994;15:7. doi: 10.1016/0167-5699(94)90018-3. [DOI] [PubMed] [Google Scholar]
- 21.Hamilos DL, Wedner HJ. The role of glutathione in lymphocyte activation. I. Comparison of inhibitory effects of buthionine sulfoximine and 2-cyclohexene-1-one by nuclear size transformation. J. Immunol. 1985;135:2740. [PubMed] [Google Scholar]
- 22.Los M, Droge W, Stricker K, Bauerle PA, Schulze-Osthoff K. Hydrogen peroxide as a potent activator of T lymphocyte functions. Eur. J. Immunol. 1995;25:159. doi: 10.1002/eji.1830250127. [DOI] [PubMed] [Google Scholar]
- 23.Jacobson MD, Raff MC. Programmed cell death and bcl-2 protection in very low oxygen. Nature. 1995;374:814. doi: 10.1038/374814a0. [DOI] [PubMed] [Google Scholar]
- 24.Meier B, Radeke HH, Selle S, Younes M, Sies H, Resch K, Habermehl GG. Human fibroblasts release reactive oxygen species in response to interleukin-1 or tumor necrosis factor-α. Biochem. J. 1989;263:539. doi: 10.1042/bj2630539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hennet T, Richter C, Peterhans E. Tumor necrosis factor-α induces superoxide anion generation in mitochondria of L929 cells. Biochem. J. 1993;289:587. doi: 10.1042/bj2890587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Schulze-Osthoff K, Krammer PH, Droge W. Divergent signaling via APO-1/Fas and the TNF receptor, two homologous molecules involved in physiological cell death. EMBO J. 1994;13:4587. doi: 10.1002/j.1460-2075.1994.tb06780.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Banki K, Hutter E, Colombo E, Gonchoroff NJ, Perl A. Glutathione levels and sensitivity to apoptosis are regulated by changes in transaldolase expression. J. Biol. Chem. 1996;271:32994. doi: 10.1074/jbc.271.51.32994. [DOI] [PubMed] [Google Scholar]
- 28.Xiang J, Chao DT, Korsmeyer SJ. BAX-induced cell death may not require interleukin 1β-converting enzyme-like proteases. Proc. Natl. Acad. Sci. USA. 1996;93:14559. doi: 10.1073/pnas.93.25.14559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kasahara Y, Iwai K, Yachie A, Ohta K, Konno A, Seki H, Miyawaki T. Involvement of reactive oxygen intermediates in spontaneous and CD95 (Fas/APO-1)-mediated apoptosis of neutrophils. Blood. 1997;89:1748. [PubMed] [Google Scholar]
- 30.Williams MS, Henkart PA. Role of reactive oxygen intermediates in TCR-induced death of T cell blasts and hybridomas. J. Immunol. 1996;157:2395. [PubMed] [Google Scholar]
- 31.Gulbins E, Brenner B, Schlottmann K, Welsch J, Heinle H, Koppenhoefer UL, Coggeshall KM, Lang F. Fas-induced programmed cell death is mediated by a Ras-regulated O2- synthesis. Immunology. 1996;89:205. doi: 10.1046/j.1365-2567.1996.d01-743.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Um HD, Orenstein JM, Wahl SM. Fas mediates apoptosis in human monocytes by a reactive oxygen intermediate dependent pathway. J. Immunol. 1996;156:3469. [PubMed] [Google Scholar]
- 33.Ames BN. Endogenous oxidative DNA damage, aging, and cancer. Free Rad. Res. Commun. 1989;7:121. doi: 10.3109/10715768909087933. [DOI] [PubMed] [Google Scholar]
- 34.Sarafian TA, Bredesen DE. Is apoptosis mediated by reactive oxygen species? Free Rad. Res. 1994;21:1. doi: 10.3109/10715769409056549. [DOI] [PubMed] [Google Scholar]
- 35.Mayes P. The pentose phosphate pathway & other pathways of hexose metabolism. In: Murray R, Granner D, Mayes P, Rodwell V, editors. Harper's Biochemistry. 23th Ed. Appleton & Lange; Norwalk, CT: 1993. p. 201. [Google Scholar]
- 36.Wood T. The Pentose Phosphate Pathway. Academic Press; New York: 1985. [Google Scholar]
- 37.Heinrich PC, Morris HP, Weber G. Behavior of transaldolase (EC 2.2.1.2.) and transketolase (EC 2.2.1.1.) in normal neoplastic, differentiating, and regenerating liver. Cancer Res. 1976;36:3189. [PubMed] [Google Scholar]
- 38.Banki K, Halladay D, Perl A. Cloning and expression of the human gene for transaldolase: a novel highly repetitive element constitutes an integral part of the coding sequence. J. Biol. Chem. 1994;269:2847. [PubMed] [Google Scholar]
- 39.Novello F, McLean P. The pentose phosphate pathway of glucose metabolism: measurements of the nonoxidative reactions of the cycle. Biochem. J. 1968;107:775. doi: 10.1042/bj1070775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Banki K, Colombo E, Sia F, Halladay D, Mattson D, Tatum A, Massa P, Phillips PE, Perl A. Oligodendrocyte-specific expression and autoantigenicity of transaldolase in multiple sclerosis. J. Exp. Med. 1994;180:1649. doi: 10.1084/jem.180.5.1649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Baquer NZ, Hothersall JS, McLean P, Greenbaum AL. Aspects of carbohydrate metabolism in developing brain. Dev. Med. Child Neurol. 1977;19:81. doi: 10.1111/j.1469-8749.1977.tb08027.x. [DOI] [PubMed] [Google Scholar]
- 42.Ni T, Savageau MA. Application of biochemical systems theory to metabolism in human red blood cells: signal propagation and accuracy of representation. J. Biol. Chem. 1996;271:7927. doi: 10.1074/jbc.271.14.7927. [DOI] [PubMed] [Google Scholar]
- 43.Perl A, Gonzalez-Cabello R, Lang I, Gergely P. Effector activity of OKT4+ and OKT8+ T-cell subsets in lectin-dependent cell-mediated cytotoxicity against adherent HEp-2 cells. Cell. Immunol. 1984;84:185. doi: 10.1016/0008-8749(84)90089-3. [DOI] [PubMed] [Google Scholar]
- 44.Yonehara S, Ishii A, Yonehara M. A cell-killing monoclonal antibody (anti-Fas) to a cell surface antigen co-downregulated with the receptor of tumor necrosis factor. J. Exp. Med. 1989;169:1747. doi: 10.1084/jem.169.5.1747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Itoh N, Yonehara S, Ashii A, Yonehara M, Mizushima S, Sameshima M, Hase A, Seto Y, Nagata S. The polypeptide encoded by the cDNA for human cell surface antigen Fas can mediate apoptosis. Cell. 1991;66:233. doi: 10.1016/0092-8674(91)90614-5. [DOI] [PubMed] [Google Scholar]
- 46.Trauth BC, Klas C, Peters AMJ, Matzku S, Moller P, Falk W, Debatin K, Krammer PH. Monoclonal antibody-mediated tumor regression by induction of apoptosis. Science. 1989;245:301. doi: 10.1126/science.2787530. [DOI] [PubMed] [Google Scholar]
- 47.Vermes I, Haanen C, Steffens-Nakken H, Reutelingsperger C. A novel assay for apoptosis: flow cytometric detection of phosphatidylserine expression on early apoptotic cells using fluorescein labelled annexin V. J. Immunol. Methods. 1995;184:39. doi: 10.1016/0022-1759(95)00072-i. [DOI] [PubMed] [Google Scholar]
- 48.Martin SJ, Reutelingsperger CPM, McGahon AJ, Rader JA, van Schie CAA, LaFace DM, Green DR. Early redistribution of plasma membrane phosphatidylserine is a general feature of apoptosis regardless of the initiating stimulus: inhibition by overexpression of Bcl-2 and Abl. J. Exp. Med. 1995;182:1545. doi: 10.1084/jem.182.5.1545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Burow S, Valet G. Flow-cytometric characterization, free radical formation, peroxidase activity and phagocytosis of human granulocytes with 2,7-dichlorofluorescein (DCF) Eur. J. Cell. Biol. 1987;43:128. [PubMed] [Google Scholar]
- 50.Royall JA, Ischiropoulos H. Evaluation of 2′,7′-dichlorofluorescein and dihydrorhodamine 123 as fluorescent probes for intracellular H2O2 in cultured endothelial cells. Arch. Biochem. Biophys. 1993;302:348. doi: 10.1006/abbi.1993.1222. [DOI] [PubMed] [Google Scholar]
- 51.Petit PX, O'Connor JE, Grunwald D, Brown SC. Analysis of the membrane potential of rat- and mouse-liver mitochondria by flow cytometry and possible applications. Eur. J. Biochem. 1990;194:389. doi: 10.1111/j.1432-1033.1990.tb15632.x. [DOI] [PubMed] [Google Scholar]
- 52.Tanner MK, Wellhausen SR, Klein JB. Flow cytometric analysis of altered mononuclear cell transmembrane potential induced by cyclosporin. Cytometry. 1993;14:59. doi: 10.1002/cyto.990140111. [DOI] [PubMed] [Google Scholar]
- 53.Zamzami N, Marchetti P, Castedo M, Decaudin D, Macho A, Hirsch T, Susin SA, Petit PX, Mignotte B, Kroemer G. Sequential reduction of mitochondrial transmembrane potential and generation of reactive oxygen species in early programmed cell death. J. Exp. Med. 1995;182:367. doi: 10.1084/jem.182.2.367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Vander Heiden M, Chandel NS, Williamson EK, Schumaker PT, Thompson CB. Bcl-XL regulates the membrane potential and volume homeostasis of mitochondria. Cell. 1997;91:627. doi: 10.1016/s0092-8674(00)80450-x. [DOI] [PubMed] [Google Scholar]
- 55.Smiley ST, Reers M, Mottola-Hartshorn C, Lin M, Chen A, Smith TW, Steele GD, Jr., Bo Chen L. Intracellular heterogeneity in mitochondrial membrane potentials revealed by a J-aggregate-forming cation JC-1. Proc. Natl. Acad. Sci. USA. 1991;88:3671. doi: 10.1073/pnas.88.9.3671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Cossarizza A, Franceschi C, Monti D, Salvioli S, Bellesia E, Rivabene R, Biondo L, Rainaldi G, Tinari A, Malorni W. Protective effect of N-acetylcysteine in tumor necrosis factor-α-induced apoptosis in U937 cells: the role of mitochondria. Exp. Eye Res. 1995;220:232. doi: 10.1006/excr.1995.1311. [DOI] [PubMed] [Google Scholar]
- 57.Brauner T, Hulser DF, Strasser RJ. Comparative measurements of membrane potentials with microelectrodes and voltage-sensitive dyes. Biochim. Biophys. Acta. 1984;771:208. doi: 10.1016/0005-2736(84)90535-2. [DOI] [PubMed] [Google Scholar]
- 58.Pontremoli S, Prandini DB, Bonsignore A, Horecker BL. The preparation of crystalline transaldolase from Candida utilis. Proc. Natl. Acad. Sci. USA. 1961;47:1942. doi: 10.1073/pnas.47.12.1942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Rudack D, Chisholm EM, Holten D. Rat liver glucose 6-phosphate dehydrogenase. J. Biol. Chem. 1971;246:1249. [PubMed] [Google Scholar]
- 60.Tietze F. Enzymic method for quantitative determination of nanogram amounts of total and oxidized glutathione: applications to mammalian blood and other tissues. Anal. Biochem. 1969;27:502. doi: 10.1016/0003-2697(69)90064-5. [DOI] [PubMed] [Google Scholar]
- 61.Lowry OH, Rosebrough NJ, Farr AL, Randall RJ. Protein measurement with the Folin phenol reagent. J. Biol. Chem. 1951;193:265. [PubMed] [Google Scholar]
- 62.Nicholson DW, Ali A, Thornberry NA, Vaillancourt JP, Ding CK, Gallant M, Gareau Y, Griffin PR, Labelle M, Lazebnik YA, et al. Identification and inhibition of ICE/CED-3 protease necessary for mammalian apoptosis. Nature. 1995;376:37. doi: 10.1038/376037a0. [DOI] [PubMed] [Google Scholar]
- 63.Kluck RM, Bossy-Wetzel E, Green DR, Newmeyer DD. The release of cytochrome c from mitochondria: a primary site for Bcl-2 regulation of apoptosis. Science. 1997;275:1132. doi: 10.1126/science.275.5303.1132. [DOI] [PubMed] [Google Scholar]
- 64.Armstrong RC, Aja T, Xiang J, Gaur S, Krebs JF, Hoang K, Bai X, Korsmeyer SJ, Karanevsky DS, Fritz LC, et al. Fas-induced activation of the cell death-related protease CPP32 is inhibited by Bcl-2 and by ICE family protease inhibitors. J. Biol. Chem. 1996;271:16850. doi: 10.1074/jbc.271.28.16850. [DOI] [PubMed] [Google Scholar]
- 65.Towbin HH, Staehelin T, Gordon J. Electrophoretic transfer of proteins from polyacrylamide gels to nitrocellulose sheets: procedure and some applications. Proc. Natl. Acad. Sci. USA. 1979;76:4350. doi: 10.1073/pnas.76.9.4350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Lamarre D, Talbot B, De Murcia G, LaPlante C, LeDuc Y, Mazen A, Poirier GG. Structural and functional analysis of poly(ADP-ribose) polymerase: an immunological study. Biochim. Biophys. Acta. 1988;950:147. doi: 10.1016/0167-4781(88)90007-3. [DOI] [PubMed] [Google Scholar]
- 67.Packham G, Ashmun RA, Cleveland JL. Cytokines suppress apoptosis independent of increases in reactive oxygen levels. J. Immunol. 1996;156:2792. [PubMed] [Google Scholar]
- 68.Susin SA, Zamzami N, Castedo M, Daugas E, Wang H, Geley S, Fassy F, Reed RC, Kroemer G. The central executioner of apoptosis: multiple connections between protease activation and mitochondria in Fas/Apo-1/CD95-and ceramide-induced apoptosis. J. Exp. Med. 1997;186:25. doi: 10.1084/jem.186.1.25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Enari M, Talanian RV, Wong WW, Nagata S. Sequential activation of ICE-like and CPP32-like proteases during Fas-mediated apoptosis. Nature. 1996;380:723. doi: 10.1038/380723a0. [DOI] [PubMed] [Google Scholar]
- 70.Constantini P, Chernyak BV, Petronilli V, Bernardi P. Modulation of the mitochondrial permeability transition pore by pyridine nucleotides and dithiol oxidation at two separate sites. J. Biol. Chem. 1996;271:6746. doi: 10.1074/jbc.271.12.6746. [DOI] [PubMed] [Google Scholar]
- 71.Muzio M, Stockwell BR, Stennicke HR, Salvesen GS, Dixit VM. An induced proximity model for caspase-8 activation. J. Biol. Chem. 1998;273:2926. doi: 10.1074/jbc.273.5.2926. [DOI] [PubMed] [Google Scholar]
- 72.Tewari M, Quan LT, O'Rourke K, Desnoyers S, Zeng Z, Beidler DR, Poirier GG, Salvesen GS, Dixit VM. Yama/CPP32β, a mammalian homolog of CED-3, is a CrmA-inhibitable protease that cleaves the death substrate poly(ADP-ribose) polymerase. Cell. 1995;81:801. doi: 10.1016/0092-8674(95)90541-3. [DOI] [PubMed] [Google Scholar]
- 73.Scaffidi C, Medema JP, Krammer PH, Peter ME. Flice is predominantly expressed as two functionally active isoforms, caspase-8/a and caspase-8/b. J. Biol. Chem. 1997;272:26953. doi: 10.1074/jbc.272.43.26953. [DOI] [PubMed] [Google Scholar]
- 74.Salvesen GS, Dixit VM. Caspases: intracellular signaling by proteolysis. Cell. 1997;91:443. doi: 10.1016/s0092-8674(00)80430-4. [DOI] [PubMed] [Google Scholar]
- 75.Li P, Nijhawan D, Budihardjo I, Srinivasula SM, Ahmad M, Alnemri ES, Wang X. Cytochrome c and dATP-dependent formation of Apaf-1/caspase-9 complex initiates and apoptotic protease cascade. Cell. 1997;91:479. doi: 10.1016/s0092-8674(00)80434-1. [DOI] [PubMed] [Google Scholar]
- 76.Casciola-Rosen LA, Miller DK, Anhalt GJ, Rosen A. Specific cleavage of the 70 kDa protein component of the U1 small nuclear ribonucleo-protein is a characteristic biochemical feature of apoptotic cell death. J. Biol. Chem. 1994;269:30757. [PubMed] [Google Scholar]
- 77.Casciola-Rosen L, Nicholson DW, Chong T, Rowan KR, Thornberry NA, Miller DK, Rosen A. Apopain/CPP32 cleaves proteins that are essential for cellular repair: a fundamental principle of apoptotic cell death. J. Exp. Med. 1996;183:1957. doi: 10.1084/jem.183.5.1957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Enari M, Sakahira H, Yokoyama H, Okawa K, Iwamatsu A, Nagata S. A capase-activated DNase that degrades DNA during apoptosis, and its inhibitor ICAD. Nature. 1998;391:43. doi: 10.1038/34112. [DOI] [PubMed] [Google Scholar]
- 79.Zoratti M, Szabo I. The mitochondrial permeability transition. Biochim. Biophys. Acta. 1995;1241:139. doi: 10.1016/0304-4157(95)00003-a. [DOI] [PubMed] [Google Scholar]
- 80.Salvioli S, Ardizzoni A, Franceschi C, Cossarizza A. JC-1, but not DiOC6 or rhodamine 123, is a reliable fluorescent probe to assess Δψ changes in intact cells: implications for studies on mitochondrial functionality during apoptosis. FEBS Lett. 1997;411:77. doi: 10.1016/s0014-5793(97)00669-8. [DOI] [PubMed] [Google Scholar]
- 81.Adachi S, Gottlieb RA, Babior BM. Lack of release of cytochrome c from mitochondria into cytosol early in the course of Fas-mediated apoptosis of Jurkat cells. J. Biol. Chem. 1998;273:19892. doi: 10.1074/jbc.273.31.19892. [DOI] [PubMed] [Google Scholar]
- 82.Mysler E, Bini P, Drappa J, Ramos P, Friedman SM, Krammer PH, Elkon KB. The apoptosis-1/Fas protein in human systemic lupus erythematosus. J. Clin. Invest. 1994;93:1029. doi: 10.1172/JCI117051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Suthanthiran M, Anderson ME, Sharma VK, Meister A. Glutathione regulates activation-dependent DNA synthesis in highly purified normal human T lymphocytes stimulated via the CD2 and CD3 antigens. Proc. Natl. Acad. Sci. USA. 1990;87:3343. doi: 10.1073/pnas.87.9.3343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Martin DA, Siegel RM, Zheng L, Lenardo MJ. Membrane oligomerization and cleavage activates the caspase-8 (FLICE/MACHα1) death signal. J. Biol. Chem. 1998;273:4345. doi: 10.1074/jbc.273.8.4345. [DOI] [PubMed] [Google Scholar]
- 85.Banki K, Hutter E, Gonchoroff NJ, Perl A. Molecular ordering in HIV-induced apoptosis: oxidative stress, activation of caspases, and cell survival are regulated by transaldolase. J. Biol. Chem. 1998;273:11944. doi: 10.1074/jbc.273.19.11944. [DOI] [PubMed] [Google Scholar]
- 86.Berridge MV, Tan AS, McCoy KD, Kansara M, Rudert F. CD95 (Fas/Apo-1)-induced apoptosis results in loss of glucose transporter function. J. Immunol. 1996;156:4092. [PubMed] [Google Scholar]