

Abstract
Two hallmarks of assembly line polyketide synthases have motivated an interest in these unusual multienzyme systems, their stereospecificity and their capacity for directional biosynthesis. In this review, we summarize the state of knowledge regarding the mechanistic origins of these two remarkable features, using the 6-deoxyerythronolide B synthase as a prototype. Of the 10 stereocenters in 6-deoxyerythronolide B, the stereochemistry of nine carbon atoms is directly set by ketoreductase domains, which catalyze epimerization and/or diastereospecific reduction reactions. The 10th stereocenter is established by the sequential action of three enzymatic domains. Thus, the problem has been reduced to a challenge in mainstream enzymology, where fundamental gaps remain in our understanding of the structural basis for this exquisite stereochemical control by relatively well-defined active sites. In contrast, testable mechanistic hypotheses for the phenomenon of vectorial biosynthesis are only just beginning to emerge. Starting from an elegant theoretical framework for understanding coupled vectorial processes in biology [Jencks, W. P. (1980) Adv. Enzymol. Relat. Areas Mol. Biol. 51, 75–106], we present a simple model that can explain assembly line polyketide biosynthesis as a coupled vectorial process. Our model, which highlights the important role of domain–domain interactions, not only is consistent with recent observations but also is amenable to further experimental verification and refinement. Ultimately, a definitive view of the coordinated motions within and between polyketide synthase modules will require a combination of structural, kinetic, spectroscopic, and computational tools and could be one of the most exciting frontiers in 21st Century enzymology.
More than two decades ago, the discovery that certain polyketide natural products are synthesized by enzymatic assembly lines laid the foundation for a fundamentally new chapter in understanding antibiotic biosynthesis. Since the cloning of the genes encoding the 6-deoxyerythronolide B synthase1,2 [DEBS (Figure 1)], there has been explosive growth in the pace of discovery of assembly line polyketide synthases. According to a recent estimate, the NCBI database now includes sequences of ∼1000 assembly line polyketide synthases, the vast majority of which are “orphans” that synthesize products of as yet unknown structure.3 Whereas most of these synthases are encoded within the genomes of soil bacteria, they can also be found in eukaryotes such as protozoa and nematodes.
Figure 1.

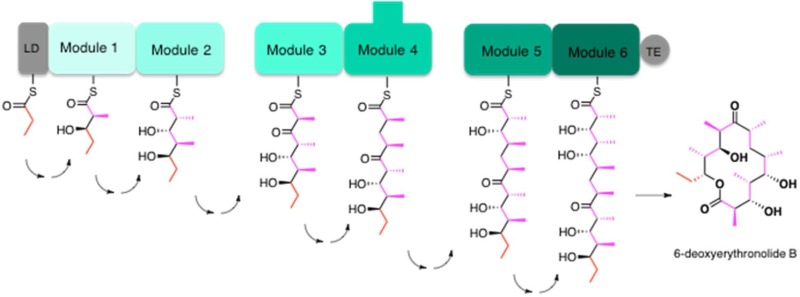
Assembly line organization of the 6-deoxyerythronolide B synthase (DEBS). (A) DEBS is an ∼2 MDa α2β2γ2 protein assembly that harbors six elongation modules (modules 1–6) flanked by a loading didomain (LD) and a thioesterase (TE). It catalyzes the conversion of 1 equiv of propionyl-CoA and 6 equiv of (2S)-methylmalonyl-CoA into 6-deoxyerythronolide B, using 6 equiv of NADPH as a cofactor. Each module harbors the necessary enzymatic activity for one round of chain elongation and associated modifications of the growing polyketide chain. The reaction intermediates shown attached to the ACP domain of each module correspond to the final products of each of the respective modules. (B) Module 3 is a representative catalytic module within the DEBS assembly line. Its active sites are shown, as is the overall transformation catalyzed by this set of active sites. ACP is the acyl carrier protein, AT acyltransferase, KS ketosynthase, and KR0 a ketoreductase homologue that lacks NADPH-dependent reductase activity but retains epimerase activity.
Two fundamental features of assembly line polyketide synthases have motivated an interest in these unusual multienzyme systems for more than 25 years. First, assembly line polyketide synthases are able to program the construction of exceptionally intricate stereochemical patterns on the carbon chain backbones of their products. For example, the macrolide aglycone product of DEBS, 6-deoxyerythronolide B, with 10 stereogenic centers, is generated with striking stereospecificity as only one of 1024 theoretically possible diastereomers. Second, the multimodular architecture of polyketide synthases must allow the precise vectorial channeling of each biosynthetic intermediate with concomitant control of intermediate trafficking and reagent flux. In the vast majority of cases, the product of an assembly line polyketide synthase results from a uniquely defined catalytic cycle in which the active site of each catalytic component operates once and only once on each growing polyketide chain.
Our goal in this review is not simply to review the present-day state of knowledge of the control of reaction stereospecificity and vectorial processing in assembly line synthases. More importantly, we wish to propose testable structural and dynamic hypotheses based on the insights that have already been gained that might lead to general models of polyketide synthase structure and function, with the further possibility of allowing their rational and efficient engineering. We start with a brief overview of DEBS structure and then discuss stereochemical control and vectorial processing separately.
Structure and Catalytic Chemistry of DEBS, the Prototypical Assembly Line Polyketide Synthase
A fundamental understanding of the operation and specificity of any assembly line must be built upon knowledge of its structure. As shown in Figure 1A, DEBS is composed of six multifunctional protein modules, each of which is responsible for a single round of polyketide chain elongation and functional group modification. Each module is in turn composed of specific combinations of catalytic domains that catalyze the individual biochemical steps of chain elongation and processing. The domain organization of module 3 of DEBS is shown in Figure 1B, as is the acyl carrier protein (ACP) domain from the upstream module that supplies module 3 with its substrate and the ketosynthase (KS) domain from the downstream module that receives its product. By now, the atomic structures of one or more prototypical members of every domain family found within DEBS have been determined (Figure 2).4−9 In addition to providing snapshots of the components of the biosynthetic assembly line, these structures also allow deeper analysis of the catalytic chemistry mediated by each domain.
Figure 2.
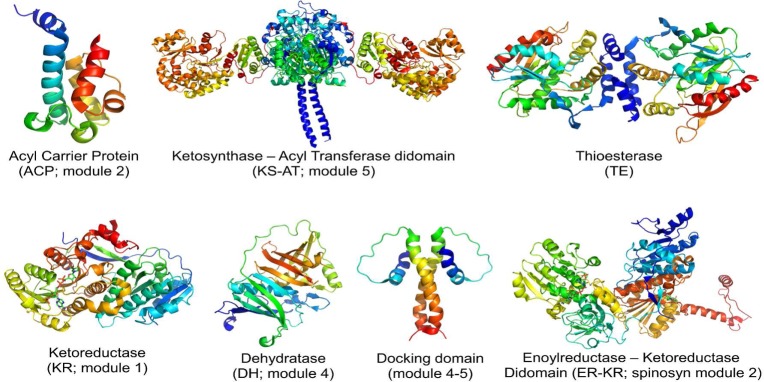
Ribbon diagram representations of atomic structures of prototypical domains and didomains from assembly line polyketide synthases. In figures showing KR and ER domains, the bound NADPH cofactor is also shown. All structures were derived from components of DEBS itself, with the exception of the ER-KR didomain obtained from the spinosyn synthase. For details, see refs (4−9).
While the representative structures of all the component domains have now been established, there are still major gaps in our knowledge of polyketide synthase structural biology. At the most basic level, the three-dimensional relationships among the individual domains and modules of an assembly line polyketide synthase remain largely unknown. The few available X-ray structures of multidomain fragments, exemplified by the ketosynthase–acyltransferase and ketoreductase–enoylreductase fragments shown in Figure 2, highlight the importance of elaborate domain–domain interactions, thus belying an overly simplistic image of these protein assembly lines as merely enzymatic “beads on a string”, in which the individual beads represent autonomously folded and independently functioning domains that dynamically self-assemble to support polyketide chain growth and modification. Although some insight into the possible quaternary architectures of the six elongation modules of DEBS can be derived from the crystal structure of the homologous vertebrate fatty acid synthase,10 such models must be regarded with caution in light of the considerable evolutionary distance between the biochemically distinct polyketide and fatty acid synthase systems. Even once the three-dimensional organization of a single polyketide synthase module is resolved, there is still no experimentally based structural prototype for a bimodular not to mention a multimodular polyketide synthase. Thus, the relative spatial dispositions of adjacent DEBS modules cannot yet be predicted with any confidence. As a first step in addressing these critical gaps in our knowledge, we have analyzed a systematic series of (smaller) structurally characterized and (larger) uncharacterized DEBS fragments using small-angle X-ray scattering (SAXS).11 These inherently low-resolution SAXS data have nonetheless already revealed molecular envelopes of intact modules and bimodules under conditions in which each protein is assuredly active. Other methods such as cryoelectron microscopy may also be applied to this problem, while X-ray crystallography remains the only tool capable of yielding satisfactory atomic-level insights.
Moving beyond the study of static protein structures, however complex, we find a second major structural challenge involves the development of a comprehensive description of the dynamics of an assembly line polyketide synthase as it conducts its unidirectional, multistep catalytic task. Even once there are complete atomic-resolution crystallographic snapshots of the entire DEBS protein, complementary approaches will be essential to visualize and understand the biosynthetically relevant conformational dynamics of this complex biochemical machine. At a minimum, it is already evident that each of the acyl carrier protein (ACP) domains must undergo large translational, rotational, and conformational changes in conjunction with the numerous successive chain elongation, modification, and translocation events (Figure 3). For example, each ACP domain within a homodimeric module of DEBS must interact with one ketosynthase and one acyltransferase domain during the course of polyketide chain elongation; the active sites of these paired catalytic domains are located ∼80 Å from each other on an evidently rigid protein scaffold based on a network of relatively conserved interdomain interactions.5 This same ACP must also then interact with the ketoreductase domain during subsequent α-methyl epimerization and/or β-carbonyl reduction of the tethered polyketide chain elongation substrate and finally be able to interact specifically with the appropriate ketosynthase domain of the immediately downstream module to facilitate high-fidelity intermodular chain translocation. Virtually nothing is known about how these dynamics are mediated by the assembly line polyketide synthase or even by the programmatically much simpler vertebrate fatty acid synthase. It is conceivable that the “DEBS movie” might involve highly coordinated motions within as well as between modules, akin to the movement of robotic arms along multiple way stations of an automobile assembly line! While X-ray crystallography could in principle yield critical snapshots of this versatile assembly line, a definitive view of this movie will undoubtedly require a combination of new kinetic, spectroscopic, and computational tools to understand the workings of this remarkable biosynthetic machinery, representing one of the most exciting frontiers in 21st Century enzymology.
Figure 3.

Individual reactions in the catalytic cycle of DEBS module 3. Each module of DEBS catalyzes a set of reactions that can be categorized as follows: (1) intermodular chain translocation involving transthioesterification from the ACP domain of the upstream module to the KS domain of the target module, (2) transfer of an acyl group from an α-carboxyacyl-CoA extender unit to the ACP domain of the target module, (3) chain elongation involving decarboxylative condensation of the growing polyketide chain onto the extender unit, (4) modification of the newly elongated chain at the α- and β-carbon atoms, and (5) intermodular chain translocation involving transthioesterification from the ACP domain of the target module to the KS domain of the downstream module. In the case of the representative DEBS module 3 shown here, chain modification simply involves epimerization of the α-carbon, a reaction that is catalyzed by KR0. Note that the three proposed states of the ketosynthase are highlighted as KS, KS*, and KS**. For details, see the text.
Stereochemical Programming of the Product Structure
The specific reaction stereochemistry has now been determined for essentially every DEBS catalytic domain. Each module is known to catalyze decarboxylative condensation of (2S)-methylmalonyl-ACP and the incipient polyketide chain with net inversion of configuration at the methyl-bearing carbon to generate the corresponding (2R)-2-methyl-3-ketoacyl-ACP chain elongation intermediate (Figure 3, elongation step).12 This intermediate can then be epimerized and reduced (as occurs in DEBS module 1), reduced without epimerization (as by DEBS modules 2, 5, and 6), or epimerized without reduction (DEBS module 3).
There are 10 stereogenic centers in the final 6-deoxyerythronolide B product. The stereochemistry of nine of these centers is directly set by ketoreductase domains. These domains can catalyze 2-methyl epimerization and/or diastereospecific reduction of their (2R)-2-methyl-3-ketoacyl-ACP substrates.12−14 Notably, reductively inactive ketoreductase domains, such as that found in module 3 of DEBS (Figure 3), harbor an intrinsic epimerase activity, giving rise to the (8S)-methyl-9-keto segment of the eventual polyketide product, 6-deoxyerythronolide B (A. Garg, X. Xie, A. T. Keatinge-Clay, C. Khosla, and D. E. Cane, submitted for publication). For cases in which both reduction and epimerization take place, epimerization occurs immediately after ketosynthase-catalyzed C–C bond formation and precedes β-ketoreduction.12,15 The only stereocenter not set directly by a ketoreductase, corresponding to the (6S)-methyl of 6-deoxyerythronolide B, is established by the sequential action (Figure 4) of three enzymatic domains of DEBS module 4, reduction by ketoreductase, followed by syn dehydration of the resulting β-hydroxy intermediate by a dehydratase,16 and finally reduction of the resulting trans-trisubstituted double bond by an enoylreductase.17 Each enzymatic domain in DEBS exercises complete control of substrate and reaction stereospecificity to ensure the formation of a unique product of each catalytic cycle, before translocation of the transiently formed polyketide intermediate to the next module in the synthase.
Figure 4.
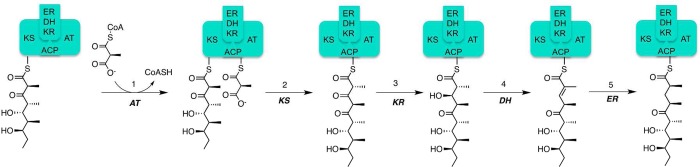
Transformations catalyzed by module 4 of DEBS. Postelongation chain modification reactions include (3) ketoreduction, (4) dehydration, and (5) enoyl reduction.
Although the stereochemistry of the reactions catalyzed by essentially all of the constituent domains of DEBS has now been determined, fundamental gaps in our understanding of the protein structural basis for this exquisite stereochemical control remain. For example, it has been recognized that the observed stereospecificity of ketoreductase domains correlates with the presence or absence of certain conserved sequence motifs.18,19 These structural features are thought to be somehow involved in directing the ACP-tethered substrate into the ketoreductase active site in a single orientation.20 While such models, in principle, can be tested via structure-based protein engineering, the extent to which ketoreductases (or, for that matter, any enzymatic domain of an assembly line polyketide synthase) can be mutagenized without the loss of stereochemical control remains unknown. Further understanding will require structures of ketoreductase domains bound to actual substrates or close analogues. Although the precise origin or sequence of the ACP domain does not seem to significantly influence either the observed ketoreductase-catalyzed reaction rate or stereospecificity, it appears that the polyketide substrate must be covalently tethered to an ACP domain to ensure the intrinsically high level of stereochemical control.12,13,21 Thus, it will be critical to overcome the significant challenges to obtaining crystal structures of noncovalent complexes of ketoreductase domains with ACP-bound substrates to understand the essential protein–protein and protein–substrate interactions that contribute to ketoreductase specificity.
Vectorial Channeling of Biosynthetic Intermediates
What mechanisms does nature use to ensure that an assembly line polyketide synthase invariably catalyzes precisely the same sequence of chemical transformations with control of product structure and stereochemistry? An understanding of the kinetic, thermodynamic, catalytic, and structural principles that govern the operation of these complex biosynthetic assembly lines will have profound implications. On the bioinformatic level, such knowledge would allow decoding of the vast and rapidly growing spectrum of orphan assembly line synthases recorded in the sequence databases, many of which can be presumed to control the biosynthesis of hitherto undiscovered antibiotics in their bacterial or eukaryotic host. Moreover, from the synthetic biology perspective, such conceptual advances might make possible the effective recombination of naturally occurring polyketide synthase modules for the achievement of the longstanding goal of engineering assembly lines that selectively synthesize virtually any imaginable polyketide product.
Polyketide biosynthesis by an enzymatic assembly line is intrinsically a vectorial process, in which each module catalyzes a defined set of reactions before passing its product to a unique downstream module. The demands on this biosynthetic system are highlighted by the fact that each module must conduct a set of biochemical reactions that are superficially similar to those of other modules but in detail completely distinct, in which there is likely a highly coordinated flow of incoming chain elongation substrates and outgoing chain elongation products passing through successive modules. More than 30 years ago, William P. Jencks articulated an elegant theoretical framework for understanding coupled vectorial processes in biology.22 Jencks presented “rules” needed to provide efficient coupling of chemical reactions to work, as in motor protein function or the pumping of ions against a gradient. The same principles formulated by Jencks to differentiate coupled vectorial processes from the more common reversible enzyme-catalyzed metabolic pathways may in fact provide a conceptual thermodynamic and kinetic framework for understanding vectorial channeling of biosynthetic intermediates along assembly line polyketide synthases. Of greatest interest is the argument that enzymes catalyzing coupled vectorial processes must undergo large changes in substrate specificity associated with different conformational states.
As summarized in Figure 3, we have now extended the original analysis of Jencks to propose a simple model that can explain assembly line polyketide biosynthesis as a coupled, three-dimensional vectorial process. Our model, which relates the conformational state of a module to the substrate specificity of its ketosynthase domain, is based on the following rules.
(1) During the catalytic cycle controlled by each polyketide synthase module, the ketosynthase domain can assume three distinct conformational states, depicted as KS, KS*, and KS**. (This model is conceptually analogous to the well-known three-state model for the FoF1 ATP synthase,23,24 although the specific chemistry of each system is completely different.) In the vectorial model for polyketide biosynthesis, each ketosynthase state would exhibit distinct specificity for ACP domains. Thus, KS** has high specificity for the ACP from the same module, whereas KS* prefers the ACP domain of the upstream module. The KS state lacks affinity for either ACP.
(2) KS State. Only when a module is in the KS state can its unoccupied ACP domain undergo acyltransferase-catalyzed loading of its characteristic (methyl)malonyl extender unit. When the module is in the KS state, the ACP-bound polyketide chain is also accessible to the individual ketoreductase, dehydratase, and/or enoylreductase domains as well as to the ketosynthase domain of the downstream module.
(3) KS* State. In the KS* state, both the ketosynthase and the ACP domain of the module must be unoccupied. In this state, the ketosynthase is receptive to intermodular chain translocation from the ACP of the proximal upstream module, while acylation of the intramodular ACP by the extender unit is precluded.
(4) KS** State. In the KS** state, the ketosynthase catalyzes decarboxylative chain elongation. In this state, the growing polyketide chain is covalently bound to the ketosynthase, and the extender unit is tethered to the ACP.
The model described above, which tacitly acknowledges the important role of domain–domain interactions in allowing vectorial polyketide biosynthesis, is consistent with several experimental observations. For example, intermodular docking domains (Figure 1), which buttress ketosynthase-ACP recognition during chain translocation,25,26 can be regarded as recognition motifs whose engagement contributes to the KS* state. Similarly, the ketosynthase has been shown to recognize its upstream ACP partner during chain translocation through interactions (Figure 3, translocation steps) that are distinct from those implicated in intramodular chain elongation with its paired ACP (Figure 3, elongation step),27,28 consistent with the operation of distinct KS* and KS** states, respectively. Although there has been no direct experimental evidence to support the postulated existence of the additional KS* state (which we believe to be the more transient of the two states in which the ketosynthase binds to the ACP), the proposed KS** state is supported by nuclear magnetic resonance detection of specific ketosynthase–ACP interactions that can be observed only when substrates or substrate mimics are covalently tethered to both the ACP and the ketosynthase.29 According to this three-state model, vectorial chain translocation from an ACP to a ketosynthase can occur only if the recipient domain is in the KS* state (Figure 3). Because this KS* state requires that both the ketosynthase and the intramodular paired ACP be unoccupied (rule 3 above), the thermodynamically competitive “back-transfer” reaction of the growing polyketide chain by transthioesterification to the ketosynthase within the same module is precluded. (By contrast, such back-transfer is essential for the operation of a vertebrate fatty acid synthase, in which a single module with the full set of elongation and modification domains must iteratively catalyze repetitive rounds of chain elongation and chain reduction to synthesize the full-length saturated fatty acid product.) It therefore follows that if a mutation were to markedly shift the equilibrium toward the KS* state, it might allow the occupancy of this state even when the polyketide is tethered to the ACP from the same module. Such a mutation would allow back-transfer of the growing chain. Indeed, we recently engineered an ACP domain that was predicted to enhance the affinity of these two domains, based upon our understanding of ketosynthase–ACP recognition principles. This mutation resulted in significant levels of back-transfer in contrast to that of the wild-type system, for which the level of back-transfer was not appreciable.28
Parenthetically, we note that although the model described above is based on conformational changes that occur in the ketosynthase domain, the ACP domain could also undergo additional (perhaps complementary) changes, as suggested for the case of nonribosomal peptides by Marahiel and co-workers.30
There are several experimental approaches that can test and further refine the model for vectorial assembly line polyketide biosynthesis described above. For example, the main difference between an assembly line polyketide synthase and its iterative counterparts should lie in the decarboxylative condensation step. Specifically, we hypothesize that the KS** state harnesses the energy derived from this thermodynamically favorable reaction, coupling it to a conformational exchange that results in the expulsion of the ACP-tethered elongation product from the ketosynthase. By contrast, in an iterative polyketide synthase, the growing polyketide chain is most likely partially retained at all times in the ketosynthase binding pocket. Tight association would favor back-transfer via ACP–ketosynthase thioester exchange before the ACP dissociates from the ketosynthase, thereby initiating another round of chain elongation catalyzed by the same module. Indeed, experiments with the actinorhodin polyketide synthase have established that the growing polyketide chain undergoes back-translocation prior to dissociation of the ACP from the ketosynthase.31 Although it is conceivable that the newly elongated chain must leave the ketosynthase active site along with the ACP prior to reactions involving the ketoreductase, dehydratase, and/or enoylreductase domains, to the best of our knowledge, this point has never been rigorously tested for any “reducing” iterative polyketide synthase or even for a fatty acid synthase under conditions of chain turnover. Experiments involving suitably labeled precursors and ketosynthases could resolve this issue. It must be re-emphasized that an appropriate test of this hypothesis requires robust turnover conditions because, as Jencks pointed out so elegantly, enzyme states that are well populated in a coupled vectorial process may not be evident under conditions that are restricted to only isolated, partial reactions.22
Because DEBS has recently been fully reconstituted under conditions that support robust turnover,32 it should now be possible to test the vectorial model critically using this prototypical assembly line polyketide synthase. For example, the vectorial model requires that, under turnover conditions, no ketosynthase–ACP pair from the same module be simultaneously occupied by two growing polyketide chains. Similarly, the model also predicts that ketosynthase acylation by the incoming polyketide chain will ordinarily precede acylation of the ACP by a cognate methylmalonyl or malonyl extender unit. In principle, further experiments can be designed to verify quantitatively or refute both predictions. For example, a numerical estimate of the fractions of each DEBS module that are occupied by zero, one, or two polyketide chains under steady-state turnover conditions, or the fraction of each DEBS module that has an unoccupied ketosynthase but an extender unit bound to its ACP, should allow a statistically rigorous assessment of this coupled vectorial process. Experiments of this nature have been performed with the first two modules of the yersiniabactin synthetase,33 the first module of DEBS,34 and an iterative polyketide synthase35 and could, in principle, be extended to the entire DEBS assembly line.
Most intriguingly from a biological perspective, the mechanistic model for assembly line polyketide synthase function leads to a provocative prediction about the evolution of these remarkable biosynthetic machines. Comparison of the sequences of numerous assembly line polyketide synthases has already suggested that these multimodular systems arose by multiple gene duplication events.36 Dissimilar systems like DEBS and the rifamycin polyketide synthase would therefore presumably have each evolved through duplications of distinct ancestral modules. If that were indeed the case, then the requirements of the vectorial model suggest that these ancestral modules must already have evolved the salient characteristics summarized in Figure 3. A primordial assembly line polyketide synthase would thus have been fashioned from two or more copies of a homodimeric protein that associated in an ACP → ketosynthase dovetailed manner and to satisfy the set of vectorial rules that we have outlined. If that were the case, then the breathtaking diversity of Nature’s polyketide assembly lines would suggest that such proto-modules may well still exist in modern-day genomes and might themselves be subject to induced duplication, recombination, and mutation to evolve new biosynthetic assembly lines under appropriate selective pressures. We look forward to future reports that might identify, analyze, and engineer such proto-modules.
Assembly Line Biosynthesis of Nonribosomal Peptides and Hybrid Molecules
Many antibiotics, such as the antitumor agent epothilone and the immunosuppressant rapamycin, are of mixed polyketide–nonribosomal peptide origin. The responsible hybrid assembly lines include polyketide synthase modules interspersed with nonribosomal peptide synthetase modules, which can therefore harbor both nonribosomal peptide synthetase → polyketide synthase as well as polyketide synthase → nonribosomal peptide synthetase junctions (Figure 5). These two types of junctions pose different challenges and constraints for chain elongation and translocation and raise analogous questions regarding assembly line mechanisms in nonribosomal peptide synthetases and hybrid systems.
Figure 5.

Key reactions catalyzed by the first three modules of the hybrid assembly line responsible for epothilone biosynthesis. The first three modules of this synthetase comprise a polyketide synthase (PKS) module (light green), followed by a nonribosomal peptide synthetase (NRPS) module (yellow), followed by another PKS module (dark green). The first module harbors a KS0 domain that catalyzes the decarboxylation of malonyl-ACP, yielding acetyl-ACP. (This reaction is not explicitly shown.) The condensation (C) domain of the second module then catalyzes condensation between acetyl-ACP on module 1 and cysteinyl-PCP on module 2; this reaction is accompanied by concomitant translocation of the growing chain from the PKS to the NRPS module. The NRPS module also catalyzes chain modification via cyclization (Cy) and oxidation (Ox), yielding a PCP-bound thiazole moiety. This intermediate then undergoes translocation onto the KS domain of the downstream PKS module in a manner that is entirely analogous to the downstream translocation event shown in Figure 3.
Analogous to polyketide synthases, nonribosomal peptide synthetase modules also have carrier protein domains with phosphopantetheine prosthetic groups and use tethered acyl thioester intermediates. The incoming monomer is attached as an aminoacyl thioester, and the growing chain exists as a peptidyl thioester. The key difference is that the condensing enzyme domains of nonribosomal peptide synthetase modules lack a Cys active site onto which the peptide is transiently translocated. Thus, both the monomeric extender unit and the growing peptide chain are bound to peptidyl carrier (PCP) domains at all times.
At nonribosomal peptide synthetase → polyketide synthase junctions [e.g., between modules 2 and 3 of the epothilone synthetase (Figure 5)], chain elongation is catalyzed by a ketosynthase, which presumably undergoes equivalent conformational transitions to those described in the legend of Figure 3. In contrast, at polyketide synthase → nonribosomal peptide synthetase junctions [e.g., between modules 1 and 2 of the epothilone synthetase (Figure 5)], the attacking nucleophile (which is now the primary amine of an aminoacyl-S-PCP) and its electrophilic partner (i.e., the polyketidyl-S-ACP intermediate) are ligated while being attached to separate modules. The condensation domain must therefore harbor orthogonal registers for these two substrates to catalyze amide bond formation. Indeed, structural analysis has provided clear evidence of such a bisubstrate binding pocket.37 However, it remains unknown whether geometric considerations alone preclude the back-transfer of intermediates from the downstream to the upstream PCP, or if condensation domains also have conformationally driven differential-state recognition of acylated (aminoacyl and peptidyl) and unoccupied PCP domains. The latter case, for which some evidence is already beginning to emerge,38 would require formulation of a new set of rules to relate these conformational states to domain specificity. A particularly relevant recent precedent has been identified in ubiquitin thioester transfer, in which elaborate conformational changes are observed as ubiquitin is transferred from the E1 enzyme to the E2 enzyme.39
Conclusion
Since the discovery of polyketide biosynthetic assembly lines, these modular megasynthases have attracted considerable scientific and engineering interest. In this review, we have summarized the state of knowledge regarding two of their most interesting features, their stereospecificity and their capacity for directional biosynthesis. With the recent identification of individual catalytic domains that control stereochemistry, the former problem has been reduced to a challenge in mainstream enzymology. In contrast, the phenomenon of vectorial biosynthesis is more appropriately considered a systems-level phenomenon for which testable mechanistic hypotheses are only just beginning to emerge. The problem will require substantially deeper insights into the architecture, dynamics, and coupling of polyketide assembly lines. We believe that the formalism developed more than three decades ago by William Jencks offers a particularly powerful conceptual framework for visualizing and analyzing this extraordinarily successful evolutionary strategy for complex molecule biosynthesis.22 In that spirit, we have provided some initial questions and models framed from this perspective and look forward to vigorous discussions in the literature and incisive experiments to further our understanding of the fundamental operational rules for these fascinating enzyme machines.
The authors declare no competing financial interest.
This work was supported by grants from the National Institutes of Health (R01 GM087934 to C.K., R01 GM64798 to D.H., R01 GM022172 to D.E.C., and R01 GM49338 to C.T.W.).
Funding Statement
National Institutes of Health, United States
References
- Cortes J.; Haydock S.; Roberts G. (1990) An unusually large multifunctional polypeptide in the erythromycin-producing polyketide synthase of Saccharopolyspora erythraea. Nature 348, 176–178. [DOI] [PubMed] [Google Scholar]
- Donadio S.; Staver M.; McAlpine J. (1991) Modular organization of genes required for complex polyketide biosynthesis. Science 252, 675–679. [DOI] [PubMed] [Google Scholar]
- O’Brien R. V.; Davis R. W.; Khosla C.; Hillenmeyer M. E. (2014) Computational identification and analysis of orphan assembly-line polyketide synthases. J. Antibiot. 67, 89–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alekseyev V. Y.; Liu C. W.; Cane D. E.; Puglisi J. D.; Khosla C. (2007) Solution structure and proposed domain domain recognition interface of an acyl carrier protein domain from a modular polyketide synthase. Protein Sci. 16, 2093–2107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang Y.; Kim C.-Y.; Mathews I. I.; Cane D. E.; Khosla C. (2006) The 2.7-Angstrom crystal structure of a 194-kDa homodimeric fragment of the 6-deoxyerythronolide B synthase. Proc. Natl. Acad. Sci. U.S.A. 103, 11124–11129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keatinge-Clay A. T.; Stroud R. M. (2006) The structure of a ketoreductase determines the organization of the β-carbon processing enzymes of modular polyketide synthases. Structure 14, 737–748. [DOI] [PubMed] [Google Scholar]
- Keatinge-Clay A. (2008) Crystal structure of the erythromycin polyketide synthase dehydratase. J. Mol. Biol. 384, 941–953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Broadhurst R. W.; Nietlispach D.; Wheatcroft M. P.; Leadlay P. F.; Weissman K. J. (2003) The structure of docking domains in modular polyketide synthases. Chem. Biol. 10, 723–731. [DOI] [PubMed] [Google Scholar]
- Zheng J.; Piasecki S. K.; Keatinge-Clay A. T. (2013) Structural Studies of an A2-Type Modular Polyketide Synthase Ketoreductase Reveal Features Controlling α-Substituent Stereochemistry. ACS Chem. Biol. 8, 1964–1971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maier T.; Leibundgut M.; Ban N. (2008) The crystal structure of a mammalian fatty acid synthase. Science 321, 1315–1322. [DOI] [PubMed] [Google Scholar]
- Edwards A. L.; Matsui T.; Weiss T. M.; Khosla C. (2014) Architectures of Whole-Module and Bimodular Proteins from the 6-Deoxyerythronolide B Synthase. J. Mol. Biol. DOI: 10.1016/j.jmb.2014.03.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valenzano C. R.; Lawson R. J.; Chen A. Y.; Khosla C.; Cane D. E. (2009) The biochemical basis for stereochemical control in polyketide biosynthesis. J. Am. Chem. Soc. 131, 18501–18511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castonguay R.; He W.; Chen A. Y.; Khosla C.; Cane D. E. (2007) Stereospecificity of ketoreductase domains of the 6-deoxyerythronolide B synthase. J. Am. Chem. Soc. 129, 13758–13769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- You Y.-O.; Khosla C.; Cane D. E. (2013) Stereochemistry of reductions catalyzed by methyl-epimerizing ketoreductase domains of polyketide synthases. J. Am. Chem. Soc. 135, 7406–7409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garg A.; Khosla C.; Cane D. E. (2013) Coupled methyl group epimerization and reduction by polyketide synthase ketoreductase domains. Ketoreductase-catalyzed equilibrium isotope exchange. J. Am. Chem. Soc. 135, 16324–16327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valenzano C. R.; You Y.-O.; Garg A.; Keatinge-Clay A.; Khosla C.; Cane D. E. (2010) Stereospecificity of the dehydratase domain of the erythromycin polyketide synthase. J. Am. Chem. Soc. 132, 14697–14699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwan D. H.; Sun Y.; Schulz F.; Hong H.; Popovic B.; Sim-Stark J. C. C.; Haydock S. F.; Leadlay P. F. (2008) Prediction and manipulation of the stereochemistry of enoylreduction in modular polyketide synthases. Chem. Biol. 15, 1231–1240. [DOI] [PubMed] [Google Scholar]
- Reid R.; Piagentini M.; Rodriguez E.; Ashley G.; Viswanathan N.; Carney J.; Santi D. V.; Hutchinson C. R.; McDaniel R. (2003) A model of structure and catalysis for ketoreductase domains in modular polyketide synthases. Biochemistry 42, 72–79. [DOI] [PubMed] [Google Scholar]
- Caffrey P. (2003) Conserved amino acid residues correlating with ketoreductase stereospecificity in modular polyketide synthases. ChemBioChem 4, 654–657. [DOI] [PubMed] [Google Scholar]
- Keatinge-Clay A. (2007) A tylosin ketoreductase reveals how chirality is determined in polyketides. Chem. Biol. 14, 898–908. [DOI] [PubMed] [Google Scholar]
- Chen A. Y.; Cane D. E.; Khosla C. (2007) Structure-based dissociation of a type I polyketide synthase module. Chem. Biol. 14, 784–792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jencks W. P. (1980) The utilization of binding energy in coupled vectorial processes. Adv. Enzymol. Relat. Areas Mol. Biol. 51, 75–106. [DOI] [PubMed] [Google Scholar]
- Boyer P. (1997) The ATP synthase: A splendid molecular machine. Annu. Rev. Biochem. 66, 717–749. [DOI] [PubMed] [Google Scholar]
- Walker J. E. (2013) The ATP synthase: The understood, the uncertain and the unknown. Biochem. Soc. Trans. 41, 1–16. [DOI] [PubMed] [Google Scholar]
- Gokhale R. S.; Tsuji S. Y.; Cane D. E.; Khosla C. (1999) Dissecting and Exploiting Intermodular Communication in Polyketide Synthases. Science 284, 482–485. [DOI] [PubMed] [Google Scholar]
- Tsuji S. Y.; Cane D. E.; Khosla C. (2001) Selective protein-protein interactions direct channeling of intermediates between polyketide synthase modules. Biochemistry 40, 2326–2331. [DOI] [PubMed] [Google Scholar]
- Kapur S.; Chen A.; Cane D.; Khosla C. (2010) Molecular recognition between ketosynthase and acyl carrier protein domains of the 6-deoxyerythronolide B synthase. Proc. Natl. Acad. Sci. U.S.A. 107, 22066–22071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kapur S.; Lowry B.; Yuzawa S.; Kenthirapalan S.; Chen A. Y.; Cane D. E.; Khosla C. (2012) Reprogramming a module of the 6-deoxyerythronolide B synthase for iterative chain elongation. Proc. Natl. Acad. Sci. U.S.A. 109, 4110–4115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charkoudian L. K.; Liu C. W.; Capone S.; Kapur S.; Cane D. E.; Togni A.; Seebach D.; Khosla C. (2011) Probing the interactions of an acyl carrier protein domain from the 6-deoxyerythronolide B synthase. Protein Sci. 20, 1244–1255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strieker M.; Tanović A.; Marahiel M. A. (2010) Nonribosomal peptide synthetases: Structures and dynamics. Curr. Opin. Struct. Biol. 20, 234–240. [DOI] [PubMed] [Google Scholar]
- Dreier J.; Khosla C. (2000) Mechanistic analysis of a type II polyketide synthase. Role of conserved residues in the β-ketoacyl synthase-chain length factor heterodimer. Biochemistry 39, 2088–2095. [DOI] [PubMed] [Google Scholar]
- Lowry B.; Robbins T.; Weng C.-H.; O’Brien R. V.; Cane D. E.; Khosla C. (2013) In vitro reconstitution and analysis of the 6-deoxyerythronolide B synthase. J. Am. Chem. Soc. 135, 16809–16812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McLoughlin S. M.; Kelleher N. L. (2004) Kinetic and regiospecific interrogation of covalent intermediates in the nonribosomal peptide synthesis of yersiniabactin. J. Am. Chem. Soc. 126, 13265–13275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hong H.; Leadlay P. F.; Staunton J. (2009) The changing patterns of covalent active site occupancy during catalysis on a modular polyketide synthase multienzyme revealed by ion-trap mass spectrometry. FEBS J. 276, 7057–7069. [DOI] [PubMed] [Google Scholar]
- Vagstad A. L.; Bumpus S. B.; Belecki K.; Kelleher N. L.; Townsend C. A. (2012) Interrogation of global active site occupancy of a fungal iterative polyketide synthase reveals strategies for maintaining biosynthetic fidelity. J. Am. Chem. Soc. 134, 6865–6877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ridley C. P.; Lee H. Y.; Khosla C. (2008) Evolution of polyketide synthases in bacteria. Proc. Natl. Acad. Sci. U.S.A. 105, 4595–4600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanovic A.; Samel S. A.; Essen L.-O.; Marahiel M. (2008) Crystal structure of the termination module of a nonribosomal peptide synthetase. Science 321, 659–663. [DOI] [PubMed] [Google Scholar]
- Bloudoff K.; Rodionov D.; Schmeing T. M. (2013) Crystal structures of the first condensation domain of CDA synthetase suggest conformational changes during the synthetic cycle of nonribosomal peptide synthetases. J. Mol. Biol. 425, 3137–3150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olsen S. K.; Lima C. D. (2013) Structure of a ubiquitin E1-E2 complex: Insights to E1-E2 thioester transfer. Mol. Cell 49, 884–896. [DOI] [PMC free article] [PubMed] [Google Scholar]