

Abstract
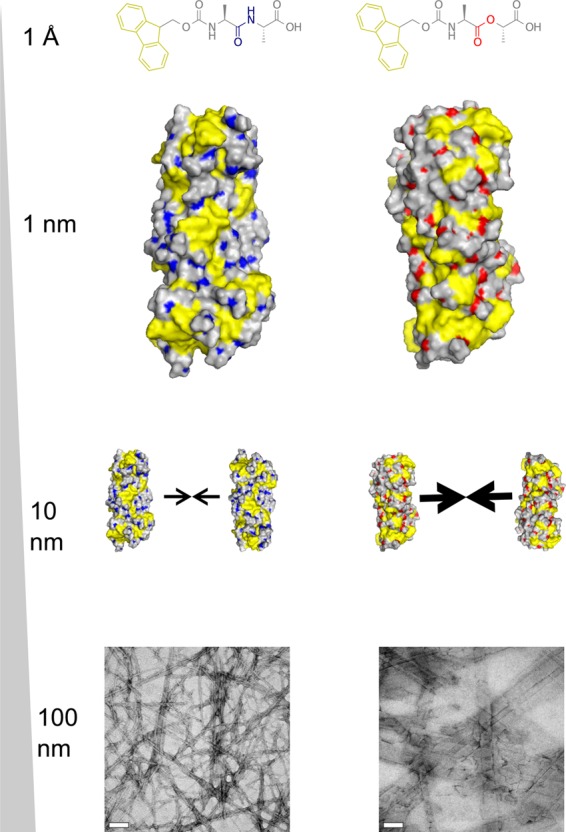
In our work toward developing ester-containing self-assembling peptides as soft biomaterials, we have found that a fluorenylmethoxycarbonyl (Fmoc)-conjugated alanine-lactic acid (Ala-Lac) sequence self-assembles into nanostructures that gel in water. This process occurs despite Fmoc-Ala-Lac’s inability to interact with other Fmoc-Ala-Lac molecules via β-sheet-like amide–amide hydrogen bonding, a condition previously thought to be crucial to the self-assembly of Fmoc-conjugated peptides. Experimental comparisons of Fmoc-Ala-Lac to its self-assembling peptide sequence analogue Fmoc-Ala-Ala using a variety of microscopic, spectroscopic, and bulk characterization techniques demonstrate distinct features of the two systems and show that while angstrom-scale self-assembled structures are similar, their nanometer-scale size and morphological properties diverge and give rise to different bulk mechanical properties. Molecular dynamics simulations were performed to gain more insight into the differences between the two systems. An analysis of the hydrogen-bonding and solvent-surface interface properties of the simulated fibrils revealed that Fmoc-Ala-Lac fibrils are stronger and less hydrophilic than Fmoc-Ala-Ala fibrils. We propose that this difference in fibril amphiphilicity gives rise to differences in the higher-order assembly of fibrils into nanostructures seen in TEM. Importantly, we confirm experimentally that β-sheet-type hydrogen bonding is not crucial to the self-assembly of short, conjugated peptides, and we demonstrate computationally that the amide bond in such systems may act mainly to mediate the solvation of the self-assembled single fibrils and therefore regulate a more extensive higher-order aggregation of fibrils. This work provides a basic understanding for future research in designing highly degradable self-assembling materials with peptide-like bioactivity for biomedical applications.
Introduction
Peptide-based low-molecular-weight gelators (LMWG) hold great promise for use as biomaterials for tissue engineering. These short peptides, typically conjugated to an N-terminal hydrophobic protecting group, can be triggered to self-assemble into gel-forming nanofibrillar structures via a pH change, solvent switch, and/or heating and subsequent cooling.1 Because of this ability to trigger their self-assembly, peptide-conjugate LMWGs may be ideal for placement by injection; many polymer gel systems previously proposed for use as biomaterials require photoinitiators for cross-linking by UV light, which is highly scattered by biological tissues. Peptides also have inherent sequence-specific biological activity, which may be easily leveraged to enhance therapeutic cell signaling or promote cell–biomaterial adhesion. Finally, with recent examples of these peptide-based LMWGs using naturally occurring hydrophobic N-protecting groups (such as DNA bases2) or groups with known biocompatibility (such as ibuprofen3 and naproxen4) these materials are likely to be highly biocompatible and possibly even actively antibacterial and anti-inflammatory.
While peptide-based gels possess many properties favorable for use as biomaterials, they are not readily degradable by nonenzymatic means at physiological pH and temperature. For this reason, our group has been developing synthetic protocols for ester-containing depsipeptides that have side-chain functionality analogous to that of peptides. As esters have a half-life over 1000 times shorter than that of amide bonds,5 depsipeptides are prone to hydrolysis over a time scale relevant to wound healing and tissue remodeling processes. We have developed didepsipeptide units (containing a single N-protected amino acid conjugated to an α-hydroxy acid via an ester linkage) that can be coupled to other amino acids or depsipeptide units using standard solid-phase peptide synthesis chemistries.6
The general structure of a depsipeptide unit is almost identical to that of a fluorenylmethyloxycarbonyl (Fmoc)-protected dipeptide. Several Fmoc-dipeptides have been reported to self-assemble into nanofibrillar structures to form hydrogels, and it has been hypothesized that β-sheet-like hydrogen bonding between amide bonds on adjacent molecules helps to drive self-assembly and stabilize the supramolecular nanostructures.1,7 We recently developed computational simulation techniques to model the self-assembly of Fmoc-Ala-Ala (i.e., Fmoc-AA), a simple Fmoc-dipeptide with uniform side-chain functionality, and we found that amide–amide hydrogen bonding comprises less than 10% of the total number of hydrogen bonds between Fmoc-Ala-Ala molecules in a supramolecular assembly.8 Given these results, we hypothesized that Fmoc-depsipeptides may be able to self-assemble into gel-forming nanostructures under conditions similar to those for an analogous Fmoc-dipeptide.
A depsipeptide analogue of an Fmoc-dipeptide contains an ester bond in place of an amide bond. Because esters lack a hydrogen bond donor, they are unable to interact with adjacent ester bonds via hydrogen bonding. Therefore, a depsipeptide analogue serves as an ideal model for testing whether β-sheet-like amide–amide hydrogen bonding is truly a requirement for the self-assembly and gelation of N-terminal hydrophobic protected peptides. In this article we show that Fmoc-Ala-Lac (i.e., Fmoc-ALac), the depsipeptide analogue of the known LMWG Fmoc-Ala-Ala, indeed undergoes self-assembly and gelation despite its inability to form specific β-sheet-like hydrogen bonds. Morphological characterization of the gels shows striking similarities, whereas spectroscopic and mechanical analyses reveal distinct differences in the two systems that are attributable only to differences in the extent of hydrogen bonding. We also performed computational simulations of the self-assembly process of these molecules to compare the features of Fmoc-Ala-Lac Fmoc-Ala-Ala that are not readily measurable by experimental means, including the number and nature of hydrogen bonds formed, the intrafibril stability, and the fibril–fibril aggregation potential. When combined, the results of the experimental and computational work herein provide critical information needed for the successful design of new degradable, injectable, and biologically active biomaterials based on modified peptides.
Experimental Section
Fmoc-l-Ala-l-Ala-OH
The N-protected dipeptide was purchased from Bachem, Inc. (Torrance, CA) and used without further purification.
Fmoc-l-Ala-l-Lac–OH Synthesis
Fmoc-l-Ala-L-Lac–OH was synthesized using the general strategy for depsipeptide unit synthesis introduced by Nguyen et al.6 Synthesis and purification details as well as identification and purity analysis though NMR and HPLC-MS can be found in the Supporting Information.
Gel Preparation
All gels characterized in this study were prepared using a pH switch method. To a solution of either Fmoc-AA or Fmoc-ALac, 1 equiv of 0.5 M NaOH was added in order to deprotonate and dissolve the molecules. This mixture was vortex mixed and sonicated just until clear and then immediately added to a small volume of 1 equiv of 0.1 M HCl in a separate vial to reduce the gel inhomogeneity. Gelator solutions at high pH were always prepared fresh and gelled immediately to prevent Fmoc removal. Although related work would indicate that ester-containing Fmoc-ALac gels are relatively stable to hydrolysis over the time course of analysis experiments,9 to minimize these effects only freshly prepared gels were analyzed. For 5 mg/mL gels, the final pH of Fmoc-AA gels was between 3.1 and 3.5, and the final pH of Fmoc-ALac gels was between 3.2 and 3.4.
Transmission Electron Microscopy (TEM)
TEM samples were prepared using 5 mg/mL gels. Approximately 10 μL of gel was placed on the shiny side of a non-glow-discharged 300-mesh carbon-coated copper grid (Electron Microscopy Sciences, Hatfield, PA) and allowed to adsorb for 2 min. Excess gel and water were wicked away using filter paper, and the grid was placed shiny side down on top of a 10 μL drop of DI water for 30 s to wash away any salts. After wicking the washing water with filter paper, grids were placed shiny side down on top of a 10 μL drop of 4% uranyl acetate in water for 1 min. Finally, the uranyl acetate solution was wicked away using filter paper, and the grids were allowed to dry completely before imaging. Samples were imaged on the nanometer scale using a FEI Tecnai instrument with an acceleration voltage of 80 kV, and the AMT Advantage HR 1kX1k digital camera associated with the instrument was used to produce digital micrographs.
Wide-Angle X-ray Scattering (WAXS)
All gel samples were prepared at 5 mg/mL as described above, allowed to dry slowly on single-crystal quartz slides over 48 h in a humid environment, and then dried completely in vacuo. Dried gel films were subjected to X-rays operating at a wavelength of 1.54059 Å (Cu Kα1) on a Scintag X1 θ–θ powder diffractometer. Diffraction patterns were analyzed using Jade v9.1.1 (Materials Data, Inc.) software.
Fourier Transform Infrared Absorption (FTIR) Spectroscopy
Gel samples for FTIR were prepared at 5 mg/mL and were prepared by tightly sandwiching gels between two calcium fluoride (CaF2) windows, which were then seated within a flow cell. Solution-phase samples were prepared at 20 mg/mL in ethanol to ensure adequate signal and were loaded into a sample cell composed of two CaF2 windows separated with 50-μm-thick PET spacers. All samples were measured using a transmission configuration. A Bruker Vertex 70 FTIR instrument equipped with a liquid-nitrogen-cooled MCT detector collected and averaged 250 scans (4000 to 400 cm–1) of each sample spectrum at a 2 cm–1 resolution. The ethanol background spectrum was subtracted from all solution-phase sample spectra, and an empty flow cell background spectrum was subtracted from all gel-phase sample data.
Circular Dichroism (CD) Spectroscopy
A Jasco J-815 instrument was used to collect CD spectra. Gels (5 mg/mL) were placed in a cylindrical demountable quartz cuvette with a path length of 0.1 mm for analysis. Background scans consisted of water containing the same concentrations of NaOH and HCl used to induce the gelation of Fmoc-AA and Fmoc-ALac, and all scans were performed at a scan speed of 200 nm/min with a data pitch of 0.5 nm and a response time of 1 s. For each scan, three spectra were taken with background subtraction and then averaged to give the final spectrum. Linear dichroism artifacts were characterized by rotating the cuvette 90, 180, and 270° from the original orientation but were found to be small for both Fmoc-AA and Fmoc-ALac gels.
Rheological Characterization
The storage and loss moduli of Fmoc-AA and Fmoc-ALac gels were determined using an Anton-Paar Physica MCR-101 rheometer with a parallel-plate geometry (top plate diameter of 8 mm). Gels (5 mg/mL) for rheological analysis (n = 4 for each system) were formed within polystyrene cloning cylinders (∼8 mm inner diameter) adhered to a polystyrene Petri dish with silicone grease. Each gel had a volume of 200 μL. For all tests, the operating height of the top plate was 2.5 mm to ensure good contact with the gel’s top surface, which was often concave due to the meniscus formed within the cloning cylinder during gelation. Because the gel diameter matched that of the top plate, gels were not trimmed prior to analysis. The strain amplitude used in the tests was 1%, and the angular frequency was varied from 0.2 to 5 rad/s over eight points. For each gel, the storage and loss moduli were calculated as the average of the eight points.
Molecular Dynamics (MD) Simulations
MD simulations were performed on Fmoc-AA and Fmoc-ALac fibrils. The simulated fibril structures were based on previous results derived for the Fmoc-AA fibril (100 molecules that had already undergone a 160 ns simulation and here proven to be stable).8 MD simulations were performed using GROMACS (version 4.5.4). The atomic force field parameters were transformed from OPLS-AA,10 OPLS-AA/L,11 MM3,12 and our previous work.8 Molecular dynamics simulations were performed on the Texas Advanced Computing Center’s “Lonestar” supercomputer. All simulations were applied periodic boxes with explicit TIP3P water molecules, and all were subjected to steepest-descent energy minimizations with 5000 maximum steps to remove high-energy contacts. In order to keep the system stable at 300 K and 1 bar, 100 ps NVT and 100 ps NPT equilibrations were applied, respectively. The LINCS algorithm was applied to constrain bonds.13 Further details can be found in our previous paper.8 Modeling parameters were derived from previous studies,8,14 and final simulated fibril structures in .pdb format are provided in the Supporting Information.
Melting-Temperature Simulations
For DNA and similar materials stabilized largely by noncovalent intermolecular interactions, the measurement of a melting temperature can be used to quantify the material’s stability and predict sequence-dependent thermodynamic behavior.15−17 In this work, we simulated a similar melting procedure and obtained the melting curves to understand the effect of replacing the amide bond of Fmoc-AA with an ester bond. When the temperature is gradually increased to some point (the so-called melting temperature in this article), the noncovalent interactions between individual molecules within a fibril structure will be disrupted; therefore, the fibril structure breaks apart and the fraction of the molecular surface area exposed to solvent will increase. By measuring the solvent -accessible surface area (SASA) with increasing temperature from 670 to 870 K, we quantitatively estimated the effect of changing the terminal residue (Ala to Lac) on noncovalent intermolecular interactions between Fmoc-dipeptides within a self-assembled fibril structure. During the simulations, for each selected temperature window at least one 50 ns MD simulation was performed and the last 30 ns SASA results were averaged to plot. The standard deviation for each point has been indicated as error bars in Figure 5A.
Figure 5.
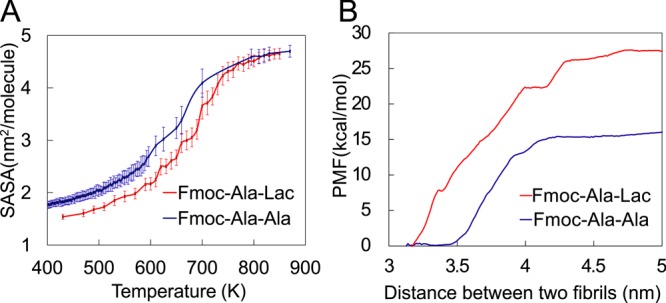
Computational study of the interaction energy between Fmoc-dipeptides and fibrils. Melting curves (A) show that Fmoc-Ala-Lac fibril assemblies are slightly more stable than Fmoc-AA fibrils. (B) The potential of mean force (PMF) calculated at varying fibril–fibril separation distances shows that Fmoc-Ala-Lac fibrils require more force to pull apart, or tend to aggregate more readily than Fmoc-Ala-Ala fibrils.
Potential of Mean Force
In our previous study,8 we observed that the surface of a fibril is composed of both hydrophobic and hydrophilic regions, which may lead to higher-order aggregation behavior.18 Using collective reaction coordinates between two fibril surfaces, we described the assembly procedure via depicting the potential of mean force (PMF) profile.19 With the MD simulated fibril structure (each of which had undergone an MD simulation for at least 100 ns), for each Fmoc-dipeptide system, we investigated the PMF for fibril–fibril assembly. We harmonically restrained two fibrils at increasing center-of mass (COM) distances (so-called sampling windows); therefore, a series of configurations along a single degree of freedom were achieved. With the weighted histogram analysis method (WHAM),20 the interfibril PMF was constructed along the dissociation coordinates.
Results and Discussion
Experimental Analysis
One of the goals of this study was to understand better the role of amide–amide hydrogen bonding in the self-assembly of short, conjugated peptides. To eliminate the ability of Fmoc-AA to hydrogen bond via protein-like amide–amide hydrogen bonding interactions, we synthesized Fmoc-Ala-Lac (Fmoc-ALac), an ester-containing analogue of Fmoc-AA in which the terminal alanine residue is replaced by lactic acid. As lactic acid is the α-hydroxy acid side-chain analogue of alanine, the use of lactic acid in place of alanine results in a change in the molecule at only one site—replacing the −NH group in the backbone with an oxygen atom, as illustrated in Figure 1A. Because the resulting ester group contains no hydrogen atoms, it acts only as a hydrogen bond acceptor rather than a donor and acceptor. Ester groups are therefore unable to form hydrogen bonds with other ester groups on adjacent molecules. This short conjugated depsipeptide thus serves as an ideal system for assessing the importance of hydrogen bonding in the self-assembly of short aromatic N-protected peptides.
Figure 1.
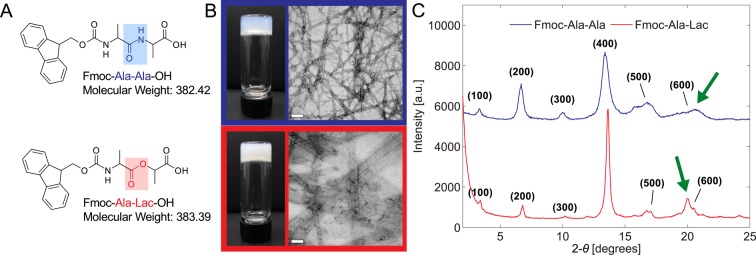
Both Fmoc-Ala-Ala (A, top) and Fmoc-Ala-Lac (A, bottom) self-assemble upon a pH change to form hydrogels (B, left) composed of fibrillar structures seen in TEM micrographs (B, right). Scale bar = 100 nm. X-ray diffraction patterns obtained from dried gel films (C) show similar peak positions and intensities, indicating that characteristic dimensions (such as fibril diameter) are similar between the two systems. The green arrows point out a unique, non-higher-order reflection in each diffraction pattern that may correspond to different Fmoc–Fmoc or strand–strand spacing in each system.
Despite the inability of neighboring ester bonds to interact via hydrogen bonding, Fmoc-ALac (Figure 1B, bottom) self-assembles to form a self-supporting gel (as evidenced by the inverted vial test) under exactly the same conditions as for Fmoc-AA (Figure 1B, top). Furthermore, upon inspection of the gels on the nanoscale by transmission electron micrsocopy (TEM), we find that both Fmoc-AA and Fmoc-ALac gels consist of flat, ribbonlike structures. Fmoc-ALac nanostructures are wider and seemingly more aggregated than Fmoc-AA ribbons. From a purely morphological standpoint on both the macroscale and nanoscale, amide–amide hydrogen bonding (and specifically β-sheet-like hydrogen bonding) appears not to be absolutely crucial to the self-assembly of short peptides into nanofibrillar structures that entrap water to form gels, but the larger structure size and therefore reduced gel clarity in Fmoc-ALac systems are clearly a result of the chemical change.
Wide angle X-ray scattering (WAXS) experiments also reveal important similarities between the two systems. Dried films of Fmoc-AA and Fmoc-ALac were analyzed and found to have strikingly similar diffraction patterns in terms of the reflection d spacing and relative peak intensities (Figure 1C). The majority of the peaks in each pattern are higher-order reflections (presumably attributable to regular fibril stacking during drying) of the leftmost peak, which for Fmoc-AA corresponds to a d spacing of 26.6 Å and for Fmoc-ALac, 26.0 Å. This dimension is thought to be the characteristic unit cell distance, which in this case would be the diameter of the supramolecular fibril structure that, when aggregated, gives rise to the ribbon structures seen in TEM.7,21 The absence of amide–amide hydrogen bonding, whether β-sheet-like or not, therefore seems to have little impact on the characteristic dimensions of the smallest fibrils that form as a result of self-assembly.
Another crucial similarity is apparent. The diffraction patterns for both Fmoc-AA and Fmoc-ALac contain a unique (i.e., not a higher order) reflection corresponding to a d spacing of 4.3–4.4 Å as indicated by the green arrows. Similar dimensions have been previously attributed to the distance between adjacent peptide chains within antiparallel β sheets.7,21 Through a previous computational analysis of Fmoc-AA self-assembly we discovered that this dimension could also correspond to the predominant interaction distance between Fmoc groups toward the center of the fibril.8 The direct experimental evidence presented here supports our computational findings, since Fmoc-ALac molecules’ ester residues preclude them from associating in a conventional β-sheet manner. It is still possible that this dimension corresponds to the spacing between the backbones of adjacent molecules, but our system allows us to rule out this dimension seen in WAXS as specific to and characteristic of β sheets in these supramolecular structures. It should be noted that Adams and colleagues have reported that the drying of gel films may cause supramolecular rearrangements unrepresentative of the gel-phase structure.22 However, for the sake of comparison to previous studies in which dried gel films were subjected to powder X-ray diffraction to measure supramolecular structural dimensions, we opted to use this simple method.
The spectroscopic analysis of solutions and self-assembled gels of Fmoc-AA and Fmoc-ALac reveals distinct differences between the two systems attributable only to their backbone bond chemistry. For reference, the structures of Fmoc-AA and Fmoc-ALac molecules are reproduced in Figure 2A, with bond chemistry and carbonyl bonds highlighted to aid in the interpretation of FTIR spectra in Figure 2B. FTIR spectra of the molecules in solution (20 mg/mL in ethanol, Figure 2B, top) show a vibration present in both systems at 1707– 1709 cm–1 and similar shoulder peaks at ∼1728 cm–1. A spectrum of Fmoc-Ala-OH in ethanol was also included to aid in peak assignment, and in this spectrum we see the same peak at 1706 cm–1 and a shoulder between 1720 and 1733 cm–1. Other readily apparent spectral features are an intense peak in the Fmoc-AA spectrum at 1670 cm–1 and a broad shoulder peak unique to Fmoc-ALac at ∼1750 cm–1.
Figure 2.

Fmoc-Ala-Ala and Fmoc-Ala-Lac are spectrally distinct in both their gelled and nongelled forms. Carbonyl bonds are highlighted in the chemical structures of Fmoc-Ala-Ala (A, top) and Fmoc-Ala-Lac (A, bottom), and the resonance stretching frequencies of these particular bonds are highlighted in the same color in the IR spectra of solutions in ethanol (B, top) and gels in water (B, bottom). The spectral overlap can be correlated with carbonyl features, with the carbamate linker’s carbonyl stretching (purple) and the terminal carboxylic acid carbonyl (gray) common to Fmoc-Ala-Ala, Fmoc-Ala-Lac, and Fmoc-Ala (structure not shown). CD spectra (C) show a marked increase in dichroism in the gelled state (solid lines) relative to that in the solution state (high pH, dashed lines) in both Fmoc-Ala-Ala and Fmoc-Ala-Lac, indicating induced chirality as a result of self-assembly. Large peaks above 280 nm indicate the interaction of aromatic Fmoc groups within a chiral assembly.
Similar feature overlap is also observed in the gel phase (5 mg/mL in D2O, Figure 2B, bottom), where the shared vibration occurs at 1687 cm–1. Fmoc-AA’s unique vibration occurs at 1646 cm–1, and Fmoc-ALac’s, at 1740 cm–1. Though the peaks are likely shifted by solvent interactions, all of these vibrations fall within the carbonyl stretching absorption band (1870–1540 cm–1).23 A reference spectrum of Fmoc-Ala-OH is not included in Figure 2B, as this molecule precipitates and does not gel under the same conditions as for Fmoc-AA and Fmoc-ALac. A non-normalized version of this plot is provided in the Supporting Information (Figure S2).
It is well known that the amide I (C=O) absorption band is centered around 1650 cm–1; therefore, we attribute Fmoc-AA’s unique vibration at 1646 cm–1 in D2O and 1670 cm–1 in ethanol (shaded in light blue in Figure 2A,B) to its amide carbonyl stretch. Furthermore, Fmoc-ALac’s unique absorption at 1740 cm–1 in D2O and shoulder at ∼1750 cm–1 in ethanol (shaded in light red) fall within the range for the carbonyl stretch of saturated aliphatic esters (1750–1735 cm–1);23 therefore, we attribute this absorption to the ester bond between alanine and lactic acid. We attribute the gray-shaded shoulder peak absorptions (shared between Fmoc-AA, Fmoc-ALac, and Fmoc-Ala-OH in the solution phase and Fmoc-AA and Fmoc-ALac in the gel phase) to the carboxylic acid carbonyl stretch, as this peak is misaligned only in the aqueous gel phase, indicating the effect of pH and a potentially different protonation state. The stronger shared absorption (shaded in light purple) therefore must represent the carbamate carbonyl stretching absorption. The shared peak also corresponds to a frequency range consistent with previous demonstrations of the carbamate carbonyl stretching absorption.24−26
Importantly, these data serve as unambiguous evidence to rule out the higher-frequency (1687 cm–1 in D2O) carbonyl absorption as an indicator of the presence of antiparallel β-sheets in supramolecular assemblies of Fmoc-dipeptides. In FTIR spectra of solutions of proteins with high antiparallel β-sheet content, two peaks between 1625–1640 and 1675–1695 cm–1 are typically observed in the amide I range,27 and many papers from another group studying Fmoc-peptide self-assembly have put forth similar-looking spectra in argument for the presence of specific β-sheet-like interactions within self-assembled structures.7,21,28,29 As this peak near 1690 cm–1 is present in β-sheet-incapable Fmoc-ALac systems and overlaps the Fmoc-AA spectrum exactly in both sol and gel phases, one must conclude that this absorption is not due to specific β-sheet-like modulation of the amide I carbonyl stretching frequency and should therefore no longer be assumed to indicate the presence of antiparallel β-sheet structures in Fmoc-dipeptide systems.
Fleming et al. independently discovered that this higher-frequency peak arises from the carbamate carbonyl stretch by showing that the peak disappears from the IR spectrum when Fmoc is replaced with fluorenylmethylcarbonyl (Fmc), which is coupled to the peptide via an amide bond rather than a carbamate group.30 Both of these examples of small structural changes leading to greatly altered IR spectra illustrate that attempts to elucidate supramolecular structural features of short peptide conjugate systems using canonical protein spectroscopic data for comparison may be inappropriate.
CD spectroscopy also reveals important differences between the two systems. As shown in Figure 2C, both Fmoc-AA and Fmoc-ALac systems exhibit marked dichroism in the gelled state (solid line) relative to the solution state (high pH, dotted line), confirming that both molecules self-assemble into nanostructures with supramolecular chirality. The most apparent difference between the two spectra is the feature above 280 nm. Similar fine structure is apparent in both spectra, but Fmoc-AA exhibits far less dichroism than Fmoc-ALac. Transitions in this range are often seen in aromatic amino acids placed in chiral environments,31 and Xu and co-workers previously assigned a peak at 304 nm as the π–π* transition induced by interactions of fluorenyl groups within a supramolecular assembly of both d and l forms of Fmoc-AA.32 We hesitate to make a specific structural conclusions from lower-wavelength spectral features, as we have argued in previous work that such assignments are often inappropriately based on data obtained from protein solutions.8
Viscoelastic analysis by oscillatory rheometry (frequency sweep, Figure 3A) shows a significant difference between the strength of Fmoc-AA and Fmoc-ALac hydrogels directly attributable to differences in the backbone chemistry of the two molecules and the resulting effect on self-assembly. Figure 3B shows the average storage and loss moduli of small Fmoc-AA and Fmoc-ALac hydrogels at a concentration of 5 mg/mL. The storage modulus (G′) of Fmoc-AA is roughly twice that of Fmoc-ALac. Owing to the presence of hydrogen bond-capable amide groups, Fmoc-AA assemblies are potentially held together by many more intermolecular interactions than ester-containing Fmoc-ALac structures. However, it is unclear how this difference translates into differences in bulk mechanical properties. It may be that these self-assembled systems are similar to fibrin gel systems in which gel strength is governed both by fiber size and network branchpoint density, two characteristics that are interconnected.33 Within the context of these findings, it seems that while Fmoc-AA assemblies form thinner fibers/ribbons, these structures are more extensively entangled (as shown in TEM), leading to stronger bulk gels. Under the same conditions, Fmoc-ALac assemblies tend to form larger aggregates that are necessarily less branched and therefore contribute less to the overall gel strength.
Figure 3.

Fmoc-Ala-Ala gels are stiffer than Fmoc-Ala-Lac gels, as evidenced by frequency sweeps (A) in parallel plate rheometry. When averaged, both the storage and loss moduli of Fmoc-Ala-Ala gels are greater than those of Fmoc-Ala-Lac gels but within the same order of magnitude (B).
Several studies on Fmoc-FF systems have found that final gel pH relative to the apparent pKa is important in governing nanoscale feature size and mechanical properties.21,34 Generally speaking, Fmoc-FF forms gels just below its apparent pKa, and gels exhibit thicker fibers and enhanced mechanical strength as the pH drops further below the apparent pKa (up to the point of precipitation). Fmoc-AA has a higher apparent pKa (∼5.1) than does Fmoc-ALac (∼4.3) (titration data in Figure S3), and all else equal, one would expect that at the same final pH (∼3.1–3.5) Fmoc-AA would exhibit larger structure sizes than Fmoc-ALac. As this is opposite to what we have described, it appears that ester replacement has implications for higher-order aggregation beyond pH relative to the apparent pKa. Indeed, Raeburn et al. studied the variability in the reported mechanical properties of Fmoc-dipeptide gels and concluded that the gel formation method and kinetics seem to influence the rheology as much as the final gel pH relative to the apparent pKa of the system.35 We have observed that self-assembly leading to gelation proceeds more slowly for Fmoc-ALac than for Fmoc-AA under the same gelation conditions, as gelation occurs in mere seconds for Fmoc-AA and several minutes for Fmoc-ALac. Therefore, it is possible that kinetics alone may determine the difference in mechanical properties; however, to our knowledge no strong correlation between the self-assembly kinetics and final gel modulus has been established, likely because the gelation kinetics often depend upon the gelation method. It is therefore very difficult to predict and/or rationalize differences in mechanical properties between related gelator systems.
Computational Simulations and Analysis
In this work, we characterized the conformational features of self-assembled Fmoc-ALac and compared the results with our previous observations from molecular dynamics simulations of the Fmoc-AA self-assembly.8 Similar to what we previously observed in Fmoc-AA simulations, Fmoc-ALac molecules form a stable fibril structure, as demonstrated by the plateau of the fibril’s total number of hydrogen bonds (includes H bonds with water) per molecule (Figure 4 A) and solvent-accessible surface area (SASA, Figure 4B) vs time. The number of H bonds (∼3.3/molecule) and SASA (∼1.6 nm2/molecule) of the Fmoc-ALac fibril converge to smaller values than the Fmoc-AA results (∼4 H bonds/molecule and SASA ∼1.7 nm2/molecule); this result is consistent with our hypothesis that substituting lactic acid for alanine to result in an ester linkage would reduce the hydrogen bonding between neighboring molecules within an assembly.
Figure 4.

Computational results characterizing the structural properties of Fmoc-Ala-Ala compared to Fmoc-Ala-Lac. In both systems, the number of hydrogen bonds per molecule (A) and the solvent-accessible surface area (SASA) (B) converge, indicating fibril stability. The non-normalized radial distribution function (RDF) plots of the last 50 ns of each simulation for the distance between the terminal residue’s hydroxyl hydrogen and fibril axis (approximating the radius) (C) and the distance between fluorenyl rings (D) show feature size similarity between Fmoc-Ala-Ala and Fmoc-Ala-Lac systems. For the Fmoc-Ala-Lac fibril assembly, a Ramachandran plot for the alanine (E) during the last 50 ns of simulation shows a large population at (ϕ, ψ) = (−70°, 164°) (indicative of polyproline-II conformation) and a minor population at (ϕ, ψ) = (−70°, −39°). The Ramachandran plot for the terminal Lac residue (F) in Fmoc-Ala-Lac during the last 50 ns of simulation shows a large population at (ϕ, ψ) = (−70°, −39°) (indicative of an α-helix-like conformation) and a minor population at (ϕ, ψ) = (−70°, 148°).
Radial distribution function (RDF) analysis of the fibril structures also indicates similar assembled structures between Fmoc-AA and Fmoc-ALac systems. Figure 4C,D shows the RDF of the peptide terminus–fibril center and the Fmoc–Fmoc centroid distance, respectively. The peak in the former RDF corresponds to the fibril radius. The fibril radius for Fmoc-ALac is virtually indistinguishable from that of Fmoc-AA by RDF analysis, confirming our experimental WAXS results that suggest a very similar fibril size between the two systems. Also, as suggested previously in the discussion of experimental results, the reappeared unique d spacing of 4.3–4.4 Å in WAXS and predominant Fmoc–Fmoc and strand–strand distance (Figure S4) of 4–5 Å from RDF analysis of the computational results for Fmoc-ALac assemblies suggest that this dimension is likely not indicative specifically of β-sheet interactions. Rather, it appears that the strand–strand and Fmoc–Fmoc association distances are quite similar between the two systems and independent of the difference in propensity of hydrogen bonding between adjacent molecules.
Similar to the observations in our previous Fmoc-AA study, the distribution of backbone torsion (ϕ, ψ) angles (Figure 4E,F, respectively) for the alanine residue in Fmoc-ALac suggests that polyproline II-like angles are predominant. Interestingly, the Ramachandran plot for the terminal Lac residues indicates that Lac predominantly adopts torsional angles within the α-helix secondary structure region, which had not been observed in the earlier Fmoc-AA study. Notably, in a previous study of depsipeptide conformational properties in solution, we observed that Lac did show a high population in the α-helix region when adjacent to glycine residues but not next to lysines, suggesting that the Lac residue’s conformation strongly depends on the chemistry of the adjacent residues.14 In the current study, the situation is more complicated; as the peptides aggregate, the environment (water and other peptide molecules) would also have an impact. Also, note that there are some Lac structures near the (−150, 0) region, which is a result of unique intramolecular interactions arising from the ester group.14 The Ramachandran plots (Figure 4E,F) reflect average distributions of individual residues’ torsion angles within the fibrils. Previous studies on free alanine dipeptides36 and depsipeptides14 in solution show similar Ramachandran patterns to those observed in this work, with dominant polyproline-II-like populations that are not present in gas-phase simulations. Thus, we reason that solvation by water is likely the main cause of the observed conformational distribution, with some variation arising from the packing of molecules within the supramolecular assembly.
An ensemble of noncovalent forces including aromatic stacking, hydrogen bonding, and electrostatic and van der Waals interactions drive the self-assembly of low-molecular-weight hydrogelators, including short peptides. These diverse forces stabilize the supramolecular structures resulting from Fmoc-dipeptide self-assembly and likely drive further assembly into higher-order structures. To understand the extent and nature of noncovalent interactions in the Fmoc-AA and Fmoc-ALac systems, we performed melting temperature and potential of mean force (PMF) studies, which are computational tools used to examine the relative stability of self-assembled fibrils upon modification of the peptide backbone chemistry.37,38 To investigate the strength of the intermolecular interaction between Fmoc-dipeptides within fibril aggregation, we computed “melting” curves for both Fmoc-AA and Fmoc-ALac, as shown in Figure 5A. We increased the temperature from 400 to 850 K and performed a 50 ns MD simulation at each selected temperature window for Fmoc-AA and Fmoc-ALac separately. Note that the computational “temperature” has no experimental counterpart, as simulations were performed under high pressures and natural effects such as reaction and decomposition were not accounted for. While not directly experimentally relevant, the use of this technique provides us with a measure of the relative stability between the two systems.
The fibril’s SASA has been calculated as the measurement of melting. In the low-temperature regime (<400 K in Figure 5 A), Fmoc-AA shows a larger SASA than does Fmoc-ALac that persists during heating from 500 to 750 K. At 800 K or above, the assembly is completely “melted” (solvated in water), and thus Fmoc-AA and Fmoc-ALac reached the same SASA. The inflection points around 670–700 K for both Fmoc-AA and Fmoc-ALac suggest a “melting temperature”; Fmoc-ALac’s melting point at roughly 700 K is about 50 K higher than that of Fmoc-AA. This observation suggests that the Fmoc-ALac fibril structure is slightly more stable than that of the Fmoc-AA fibril. Furthermore, the lower SASA values for Fmoc-ALac at most temperatures suggest a more “hydrophobic” character of the fibril, i.e., individual Fmoc-ALac molecules prefer coaggregation over solvation in water to a greater degree than do Fmoc-AA molecules.
We have noted in previous simulations the apparently amphiphilic nature of the surface of stable Fmoc-AA fibrils.8 In this work, we also observed that the simulated Fmoc-ALac fibril’s surface was similarly amphiphilic. Hydrophobicity has been widely discussed as a multifaceted phenomenon observed in self-assembled systems.39 In our system the hydrophobic units exposed on the observed fibril’s amphiphilic surface likely reduce the volume of available configuration space for hydrogen bonding with other fibrils or water but increase the possibility of forming hydrophobic assemblies with other fibrils. On the other hand, the hydrophilic components, mainly the hydroxyl group on terminal alanine or lactic acid, affect the assembly between fibrils by rearranging the adjacent water molecules’ orientations, and such an amphiphilic assembly has already been widely discussed in studies including micelle formation.39,40
In this work, to further determine the potential effects of presenting an amphiphilic surface, we first derived the interfibril association energy from the potential of mean force (PMF) between two fibrils, as shown in Figure 5B. In our simulations, we harmonically restrained two fibrils (each of which had undergone an MD simulation for 100 ns at least to reach a stable structure) at increasing center-of mass (COM) distances (so-called “sampling windows”) using an umbrella biasing potential, with the defined reaction coordinate sampled by multiple windows. The interfibril interaction energies were computed along the dissociation coordinates using WHAM (see Potential of Mean Force). Interestingly, the interaction energy minimum occurred at a COM distance of 3.25–3.5 nm, which is in good agreement with fibril diameters that we determined by WAXS experiments and RDF calculations and which also indicates an energetic preference for fibril–fibril surface contact, i.e., aggregation. As predicted by the PMF calculations, with no covalent bonds breaking or forming, the dissociation energies between two fibrils differ: the Fmoc-AA two-fibril system has a less favorable dissociation energy (or lower barrier to break the association) than the analogous Fmoc-ALac system, indicating that Fmoc-ALac fibrils tend to aggregate more easily. Taken with the melting-temperature calculations, this observation indicates that both the intra- and interfibril interactions in Fmoc-ALac are somewhat stronger than in Fmoc-AA assemblies.
In order to examine the effect of Ala vs Lac within simulated fibril systems more closely, we calculated the residue- and bond-specific solvent accessible surface area as well as the number and specific connectivity of H bonds formed between Fmoc-AA or Fmoc-ALac molecules and water at the solvent interface of the fibril. As suggested in the discussion of the melting temperature and PMF results, Fmoc-ALac fibrils appear to form tighter aggregates than Fmoc-AA fibrils. The SASA and H-bonding values calculated here support that observation and are presented in Tables 1 and 2, respectively.
Table 1. Total and Partially Decomposed SASA of Fmoc-Dipeptidesa.
| Fmoc-AA |
||||||
|---|---|---|---|---|---|---|
| total | Fmoc | Ala-mid-C=O | Ala-term-COOH | Ala-mid-NH | Ala-term-NH | |
| nm2 | 165.3 | 58.5 | 9.2 | 33.1 | 1.4 | 2.5 |
| percentage | 100.0 | 35.4 | 5.5 | 20.0 | 0.9 | 1.5 |
| Fmoc-ALac |
||||||
|---|---|---|---|---|---|---|
| total | Fmoc | Ala-C=O (ester) | Lac-COOH | Ala-NH | Lac-O(ester) | |
| nm2 | 155.7 | 55.9 | 8.1 | 37.5 | 1.9 | 1.1 |
| percentage | 100.0 | 35.9 | 5.2 | 24.1 | 1.2 | 0.7 |
Ala-mid refers to the N-terminal alanine, and Ala-term refers to the C-terminal alanine. Percentages do not add up to 100% because the hydrophobic and hydrophilic groups of interest listed here do not comprise the entire surface area of the fibril.
Table 2. H Bonds between Residues on Fmoc-Dipeptides and Water Moleculesa.
| Fmoc-AA |
||||||
|---|---|---|---|---|---|---|
| total | Fmoc | Ala-mid-C=O | Ala-term-COOH | Ala-mid-NH | Ala-term-NH | |
| number | 408.1 | 100.9 | 70.5 | 168.9 | 20.7 | 47.1 |
| percentage | 100.0 | 24.7 | 17.3 | 41.4 | 5.1 | 11.6 |
| Fmoc-ALac |
||||||
|---|---|---|---|---|---|---|
| total | Fmoc | Ala-C=O (ester) | Lac-COOH | Ala-NH | Lac-O (ester) | |
| number | 331.3 | 93.9 | 44.1 | 165.1 | 26.4 | 1.8 |
| percentage | 100.0 | 28.4 | 13.3 | 49.8 | 8.0 | 0.5 |
Hydrogen bonds were calculated as interactions between water molecules and groups of interest, with an donor–acceptor distance of ≤3.5 Å and an interaction angle of ≤30°.
From these calculations, we observed that the substitution of the amide group with an ester from Fmoc-AA to Fmoc-ALac (1) leads to a decreased total SASA and therefore fewer H bonds with water molecules; (2) reduces the middle alanine carbonyl group’s H-bonding ability with water while only slightly reducing the SASA; (3) slightly enhances the hydrophobic Fmoc groups’ exposure on the surface; and (4) reduces the exposure to water of the between-residue bond, which in the case of Fmoc-AA is an amide bond capable of acting as an H-bond donor. Therefore, the replacement of Ala with Lac leads to a decreased overall fibril hydrophilicity by decreasing the SASA and H bonding with the solvent (water). Remarkably, the average total number of hydrogen bonds between the Fmoc-AA fibril and water is 23% higher than for the Fmoc-ALac fibril. This result suggests that Fmoc-AA structures are more hydrated than Fmoc-ALac fibrils. Combined with the melting and PMF studies, these results corroborate well our TEM images that suggest that under the same gelation conditions Fmoc-ALac gels contain larger structures that result from an increased aggregation of fibrils relative to Fmoc-AA fibrils. As discussed earlier, it is possible that a consequence of this behavior on the macroscale is that Fmoc-ALac forms larger fibrils at the expense of network branchpoint density, resulting in its reduced bulk mechanical strength relative to that of Fmoc-AA, as indicated in Figure 3.
Taken together, these experimental and computational results provide a new perspective of the role of the amide bond in the self-assembly of short peptide-based gelator systems. Whereas previously it was hypothesized that extensive amide–amide hydrogen bonding helped stabilize self-assembled structures, our results suggest that the amide bond plays less of a role in forming and stabilizing the single fibril structure and a greater role in determining the nature of the higher-order aggregation of intermediate fibrils. The idea that fibril amphiphilicity impacts higher-order self-assembly is not a new concept; in 2001, Aggeli et al. suggested that beyond initially assembled β-sheet-like tapes of peptide monomers, such tapes’ amphiphilicity can further drive their formation into higher-order structures, a process the authors referred to as “hierarchical self-assembly”.18 Indeed, many sophisticated functional materials are created through a hierarchical self-assembly process mediated by the balance between the hydrophobicity and hydrophilicity of intermediate structures.41−45 In nature, virus capsids are hierarchically assembled from protomers, pentamers, or hexamers.46 In the study of amyloid fibril formation, with the ability to determine the atomic-resolution structure, researchers proved that amyloids can assemble hierarchically into higher-dimensional structures including protofilaments, filaments, and mature fibrils.44,47,48
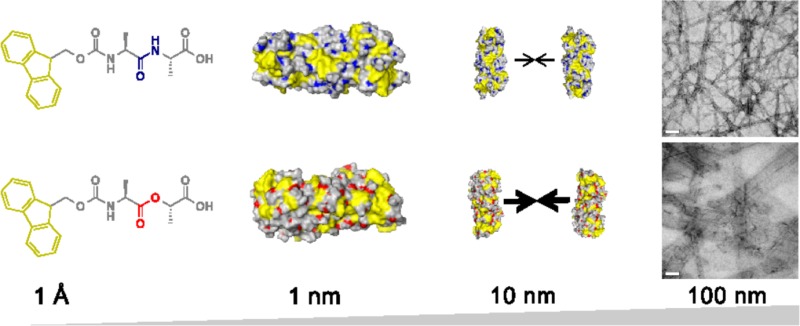
Following the above discussion, we summarize our findings as such in Figure 6: Fmoc-dipeptides have amphiphilic character, which enables them to self-assemble initially into a fibril structure (diameter ∼3 nm). Owing to the amphiphilic nature of the fibril surfaces exposed to water, fibrils further aggregate into higher-order supramolecular aggregates (width ∼20–200 nm), such as fiberlike and ribbonlike structures observed in TEM. The amide-to-ester modification we have made here seems to have little effect in the first stage of the process but greater implications in the second stage of the hierarchical assembly process, leading to observable differences in nanostructure morphology and bulk properties.
Figure 6.
Schematic of the hierarchical assembly of Fmoc-AA and Fmoc-ALac. Both systems self-assemble into similar intermediate single fibrils, but differences in the amphiphilicity of fibrils cause Fmoc-ALac fibrils to associate more strongly and extensively, giving rise to larger nanoscale structures observed in TEM.
Conclusions
We have demonstrated here experimentally what we hypothesized in our previous study: that β-sheet-specific hydrogen bonding between adjacent molecules within a supramolecular structure is likely not as important as hydrophobic or aromatic–aromatic interactions in driving and stabilizing self-assembly of Fmoc-dipeptides. However, while the initial self-assembly may not be completely affected by a chemical change resulting in different hydrogen bonding capabilities (as in the Ala to Lac change), resulting intermediate fibrils may have different surface–solvent interface properties that result in different propensities of fibrils to associate and form higher-order supramolecular aggregates.
Through the combination of experimental and computational molecular dynamics simulations, it is clear that the inclusion of the ester bond (and thereby a lack of extensive amide–amide hydrogen bonding) in this simple Fmoc-dipeptide system still leads to gelation under the same experimental conditions but also has implications for the amphiphilic nature of the self-assembled fibril units, the nature of their higher-order assembly, and ultimately the bulk mechanical properties of the gel. These considerations must be taken into account when designing more structurally complex and potentially bioactive peptide-based gelator systems for use in tissue engineering and regenerative medicine applications.
Acknowledgments
K.M.E. acknowledges the NSF for funding through the Graduate Research Fellowship Program, Dr. S. Swinnea for assistance with WAXS, Dr. D. Romanowicz for assistance with TEM, A. Spangenberg for assistance with NMR, and Dr. L. Webb and members of her research group for training and use of their FTIR instrument. We thank TACC and XSEDE (TG-MCB100057) for high-performance computing resources. X.M. and P.R. acknowledge support by the Welch Foundation (F-1691) and the National Institutes of Health (GM106137). K.M.E. and L.J.S. acknowledge support by the Welch Foundation (F-1695) and the National Institutes of Health (HL102806).
Supporting Information Available
Final simulated fibril structures in .pdb format, NMR and HPLC characterization of Fmoc-ALac, titration curves for Fmoc-AA and Fmoc-ALac, and strand–strand RDF plots for Fmoc-AA and Fmoc-ALac. This material is available free of charge via the Internet at http://pubs.acs.org.
Author Contributions
K.M.E. and X.M. contributed equally to this work. All authors have given approval to the final version of this manuscript.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Adams D. J. Dipeptide and Tripeptide Conjugates as Low-Molecular-Weight Hydrogelators. Macromol. Biosci 2011, 11, 160–173. [DOI] [PubMed] [Google Scholar]
- Li X.; Kuang Y.; Xu B. Molecular Trinity for Soft Nanomaterials: Integrating Nucleobases, Amino Acids, and Glycosides to Construct Multifunctional Hydrogelators. Soft Matter 2012, 8, 2801–2806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhuniya S.; Seo Y. J.; Kim B. H. (S)-(+)-Ibuprofen-Based Hydrogelators: An Approach toward Anti-Inflammatory Drug Delivery. Tetrahedron Lett. 2006, 47, 7153–7156. [Google Scholar]
- Li J.; Kuang Y.; Shi J.; Gao Y.; Zhou J.; Xu B. The Conjugation of Nonsteroidal Anti-Inflammatory Drugs (NSAID) to Small Peptides for Generating Multifunctional Supramolecular Nanofibers/hydrogels. Beilstein J. Org. Chem. 2013, 9, 908–917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mabey W.; Mill T. Critical Review of Hydrolysis of Organic Compounds in Water under Environmental Conditions. J. Phys. Chem. Ref. Data 1978, 7, 383–415. [Google Scholar]
- Nguyen M. M.; Ong N.; Suggs L. A General Solid Phase Method for the Synthesis of Depsipeptides. Org. Biomol. Chem. 2013, 11, 1167. [DOI] [PubMed] [Google Scholar]
- Smith A. M.; Williams R. J.; Tang C.; Coppo P.; Collins R. F.; Turner M. L.; Saiani A.; Ulijn R. V. Fmoc-Diphenylalanine Self Assembles to a Hydrogel via a Novel Architecture Based on Π–π Interlocked β-Sheets. Adv. Mater. 2008, 20, 37–41. [Google Scholar]
- Mu X.; Eckes K. M.; Nguyen M. M.; Suggs L. J.; Ren P. Experimental and Computational Studies Reveal an Alternative Supramolecular Structure for Fmoc-Dipeptide Self-Assembly. Biomacromolecules 2012, 13, 3562–3571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen M. M.; Eckes K. M.; Suggs L. J. Charge and Sequence Effects on the Self-Assembly and Subsequent Hydrogelation of Fmoc-Depsipeptides. Soft Matter 2014, 10, 2693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jorgensen W. L.; Briggs J. M.; Gao J. A Priori Calculations of pKa’s for Organic Compounds in Water. The pKa of Ethane. J. Am. Chem. Soc. 1987, 109, 6857–6858. [Google Scholar]
- Jorgensen W. L.; Schyman P. Treatment of Halogen Bonding in the OPLS-AA Force Field: Application to Potent Anti-HIV Agents. J. Chem. Theory Comput. 2012, 8, 3895–3901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allinger N. L.; Yuh Y. H.; Lii J. H. Molecular Mechanics. The MM3 Force Field for Hydrocarbons. 1. J. Am. Chem. Soc. 1989, 111, 8551–8566. [Google Scholar]
- Hess B.; Bekker H.; Berendsen H. J. C.; Fraaije J. G. E. M. LINCS: A Linear Constraint Solver for Molecular Simulations. J. Comput. Chem. 1997, 18, 1463–1472. [Google Scholar]
- Zhang J.; King M.; Suggs L.; Ren P. Molecular Modeling of Conformational Properties of Oligodepsipeptides. Biomacromolecules 2007, 8, 3015–3024. [DOI] [PubMed] [Google Scholar]
- Blake R. D.; Bizzaro J. W.; Blake J. D.; Day G. R.; Delcourt S. G.; Knowles J.; Marx K. A.; SantaLucia J. Statistical Mechanical Simulation of Polymeric DNA Melting with MELTSIM. Bioinformatics 1999, 15, 370–375. [DOI] [PubMed] [Google Scholar]
- Le Novère N. MELTING, Computing the Melting Temperature of Nucleic Acid Duplex. Bioinformatics 2001, 17, 1226–1227. [DOI] [PubMed] [Google Scholar]
- Khandelwal G.; Bhyravabhotla J. A Phenomenological Model for Predicting Melting Temperatures of DNA Sequences. PLoS One 2010, 5, e12433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aggeli A.; Nyrkova I. A.; Bell M.; Harding R.; Carrick L.; McLeish T. C. B.; Semenov A. N.; Boden N. Hierarchical Self-Assembly of Chiral Rod-like Molecules as a Model for Peptide B-Sheet Tapes, Ribbons, Fibrils, and Fibers. Proc. Natl. Acad. Sci. U.S.A. 2001, 98, 11857–11862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lemkul J. A.; Bevan D. R. Assessing the Stability of Alzheimer’s Amyloid Protofibrils Using Molecular Dynamics. J. Phys. Chem. B 2010, 114, 1652–1660. [DOI] [PubMed] [Google Scholar]
- Kumar S.; Rosenberg J. M.; Bouzida D.; Swendsen R. H.; Kollman P. A. The Weighted Histogram Analysis Method for Free-Energy Calculations on Biomolecules. I. The Method. J. Comput. Chem. 1992, 13, 1011–1021. [Google Scholar]
- Tang C.; Smith A. M.; Collins R. F.; Ulijn R. V.; Saiani A. Fmoc-Diphenylalanine Self-Assembly Mechanism Induces Apparent pKa Shifts. Langmuir 2009, 25, 9447–9453. [DOI] [PubMed] [Google Scholar]
- Houton K. A.; Morris K. L.; Chen L.; Schmidtmann M.; Jones J. T. A.; Serpell L. C.; Lloyd G. O.; Adams D. J. On Crystal versus Fiber Formation in Dipeptide Hydrogelator Systems. Langmuir 2012, 28, 9797–9806. [DOI] [PubMed] [Google Scholar]
- Silverstein R. M.Spectrometric Identification of Organic Compounds, 5th ed.; Wiley: New York, 1991. [Google Scholar]
- Xu L.; Li C.; Ng K. Y. S. In-Situ Monitoring of Urethane Formation by FTIR and Raman Spectroscopy. J. Phys. Chem. A 2000, 104, 3952–3957. [Google Scholar]
- Wu Y.; Sellitti C.; Anderson J. M.; Hiltner A.; Lodoen G. A.; Payet C. R. An FTIR–ATR Investigation of in Vivo Poly (ether Urethane) Degradation. J. Appl. Polym. Sci. 1992, 46, 201–211. [Google Scholar]
- Ni Y.; Becquart F.; Chen J.; Taha M. Polyurea–Urethane Supramolecular Thermo-Reversible Networks. Macromolecules 2013, 46, 1066–1074. [Google Scholar]
- Jackson M.; Mantsch H. H. The Use and Misuse of FTIR Spectroscopy in the Determination of Protein Structure. Crit. Rev. Biochem. Mol. Biol. 1995, 30, 95–120. [DOI] [PubMed] [Google Scholar]
- Zhou M.; Smith A. M.; Das A. K.; Hodson N. W.; Collins R. F.; Ulijn R. V.; Gough J. E. Self-Assembled Peptide-Based Hydrogels as Scaffolds for Anchorage-Dependent Cells. Biomaterials 2009, 30, 2523–2530. [DOI] [PubMed] [Google Scholar]
- Jayawarna V.; Richardson S. M.; Hirst A. R.; Hodson N. W.; Saiani A.; Gough J. E.; Ulijn R. V. Introducing Chemical Functionality in Fmoc-Peptide Gels for Cell Culture. Acta Biomater. 2009, 5, 934–943. [DOI] [PubMed] [Google Scholar]
- Fleming S.; Frederix P. W. J. M.; Ramos Sasselli I.; Hunt N. T.; Ulijn R. V.; Tuttle T. Assessing the Utility of Infrared Spectroscopy as a Structural Diagnostic Tool for B-Sheets in Self-Assembling Aromatic Peptide Amphiphiles. Langmuir 2013, 29, 9510–9515. [DOI] [PubMed] [Google Scholar]
- Nordén B.; Rodger A.; Dafforn T.. Linear Dichroism and Circular Dichroism: A Textbook on Polarized-Light Spectroscopy; Royal Society of Chemistry: Cambridge, U.K., 2010. [Google Scholar]
- Zhang Y.; Gu H.; Yang Z.; Xu B. Supramolecular Hydrogels Respond to Ligand–Receptor Interaction. J. Am. Chem. Soc. 2003, 125, 13680–13681. [DOI] [PubMed] [Google Scholar]
- Ryan E. A.; Mockros L. F.; Weisel J. W.; Lorand L. Structural Origins of Fibrin Clot Rheology. Biophys. J. 1999, 77, 2813–2826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raeburn J.; Pont G.; Chen L.; Cesbron Y.; Lévy R.; Adams D. J. Fmoc-Diphenylalanine Hydrogels: Understanding the Variability in Reported Mechanical Properties. Soft Matter 2012, 8, 1168–1174. [DOI] [PubMed] [Google Scholar]
- Raeburn J.; Cardoso A. Z.; Adams D. J. The Importance of the Self-Assembly Process to Control Mechanical Properties of Low Molecular Weight Hydrogels. Chem. Soc. Rev. 2013, 42, 5143–5156. [DOI] [PubMed] [Google Scholar]
- Parchaňský V.; Kapitán J.; Kaminský J.; Šebestík J.; Bouř P. Ramachandran Plot for Alanine Dipeptide as Determined from Raman Optical Activity. J. Phys. Chem. Lett. 2013, 4, 2763–2768. [DOI] [PubMed] [Google Scholar]
- Yu T.; Lee O.-S.; Schatz G. C. Steered Molecular Dynamics Studies of the Potential of Mean Force for Peptide Amphiphile Self-Assembly into Cylindrical Nanofibers. J. Phys. Chem. A 2013, 117, 7453–7460. [DOI] [PubMed] [Google Scholar]
- Yu T.; Schatz G. C. Free Energy Profile and Mechanism of Self-Assembly of Peptide Amphiphiles Based on a Collective Assembly Coordinate. J. Phys. Chem. B 2013, 117, 9004–9013. [DOI] [PubMed] [Google Scholar]
- Chandler D. Interfaces and the Driving Force of Hydrophobic Assembly. Nature 2005, 437, 640–647. [DOI] [PubMed] [Google Scholar]
- Stillinger F. Structure in Aqueous Solutions of Nonpolar Solutes from the Standpoint of Scaled-Particle Theory. J. Solution Chem. 1973, 2, 141–158. [Google Scholar]
- Whitesides G. M.; Grzybowski B. Self-Assembly at All Scales. Science 2002, 295, 2418–2421. [DOI] [PubMed] [Google Scholar]
- Buehler M. J.; Ackbarow T. Nanomechanical Strength Mechanisms of Hierarchical Biological Materials and Tissues. Comput. Methods Biomech. Biomed. Eng. 2008, 11, 595–607. [DOI] [PubMed] [Google Scholar]
- Buehler M. J.; Yung Y. C. Deformation and Failure of Protein Materials in Physiologically Extreme Conditions and Disease. Nat. Mater. 2009, 8, 175–188. [DOI] [PubMed] [Google Scholar]
- Knowles T. P. J.; Oppenheim T. W.; Buell A. K.; Chirgadze D. Y.; Welland M. E. Nanostructured Films from Hierarchical Self-Assembly of Amyloidogenic Proteins. Nat. Nano 2010, 5, 204–207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ortoleva P.; Singharoy A.; Pankavich S. Hierarchical Multiscale Modeling of Macromolecules and Their Assemblies. Soft Matter 2013, 9, 4319–4335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baschek J.; R Klein H.; Schwarz U. Stochastic Dynamics of Virus Capsid Formation: Direct versus Hierarchical Self-Assembly. BMC Biophys. 2012, 5, 22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fitzpatrick A. W. P.; Debelouchina G. T.; Bayro M. J.; Clare D. K.; Caporini M. A.; Bajaj V. S.; Jaroniec C. P.; Wang L.; Ladizhansky V.; Müller S. A.; et al. Atomic Structure and Hierarchical Assembly of a Cross-B Amyloid Fibril. Proc. Natl. Acad. Sci. U.S.A. 2013, 110, 5468–5473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qin S.-Y.; Pei Y.; Liu X.-J.; Zhuo R.-X.; Zhang X.-Z. Hierarchical Self-Assembly of a [small Beta]-Amyloid Peptide Derivative. J. Mater. Chem. B 2013, 1, 668–675. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.