

Abstract
UVB-induced DNA damage plays a critical role in development of photoimmunosuppression. The purpose of this study was to determine whether repair of UVB-induced DNA damage is regulated by Toll-like receptor-4 (TLR4). When TLR4 gene knockout (TLR4-/-) and TLR4 competent (TLR4+/+) mice were subjected to 90 mJ/cm2 UVB radiation locally, DNA damage in the form of CPD, were repaired more efficiently in the skin and bone marrow dendritic cells (BMDC) of TLR4-/- mice in comparison to TLR4+/+ mice. Expression of DNA repair gene XPA (Xeroderma pigmentosum complementation group A) was significantly lower in skin and BMDC of TLR4+/+ mice than TLR4-/- mice after UVB exposure. When cytokine levels were compared in these strains after UVB exposure, BMDC from UV-irradiated TLR4-/- mice produced significantly more interleukin (IL)-12 and IL-23 cytokines (p<0.05) than BMDC from TLR4+/+ mice. Addition of anti-IL-12 and anti-IL-23 antibodies to BMDC of TLR4-/- mice (before UVB exposure) inhibited repair of CPD, with a concomitant decrease in XPA expression. Addition of TLR4 agonist to TLR4+/+ BMDC cultures decreased XPA expression and inhibited CPD repair. Thus, strategies to inhibit TLR4 may allow for immunopreventive and immunotherapeutic approaches for managing UVB-induced cutaneous DNA damage and skin cancer.
Keywords: Toll like receptor-4, ultraviolet B radiation, DNA damage, nucleotide excision repair
1. Introduction
Ultraviolet (UV) B radiation (290-320 nm) is an important trigger for suppression of immune responses and initiation of non-melanoma skin cancers (Elmets et al., 1983; Kovach et al., 2005; Fisher et al., 1982; Kripke et al. 1974; Toews et al., 1980). The molecular basis for these activities partially resides in the demonstrated ability of such radiation to damage DNA, predominantly through formation of cyclobutane pyrimidine dimers (CPD). When UVB-induced DNA damage occurs in cells, there is a meticulous attempt to repair it through activation of DNA repair enzymes. Incorrect or inefficient repair of these lesions leads to deleterious mutations after DNA replication, and those mutations are major contributors to the initiation of non-melanoma skin cancers (Berneburg et al., 2000). For example, xeroderma pigmentosum (XP), an autosomal recessive disorder of DNA repair, is associated with a greater than 1000-fold increase in susceptibility to UVB-induced skin malignancies. Patients with this disease also have defects in cell mediated immune function (Gaspari et al., 1993). Xeroderma pigmentosum complementation group A (XPA), is the most common type of XP, and a deficiency results in inadequate nucleotide excision repair (NER) (Berneburg et al., 2000). The cytokine IL-12 activates XPA and thus reduces UV-induced DNA damage (Schwarz et al,. 2002). IL-12 also has the capacity to prevent UV-induced immunosuppression (Schwarz et al., 2005). IL-23 reverses the effects of UV radiation on DNA damage as well. IL-23 is capable of reducing CPD both in vitro and in vivo through an effect on NER (Majewski et al., 2009).
Toll-like receptors (TLRs) are a component of innate immunity that influence acquired immune responses. They are abundant within immune system cells and epithelial cells. TLRs, of which 11 have been identified in mammals, are membrane glycoproteins with an extracellular domain with differing numbers of leucine-rich repeats and a cytoplasmic domain similar to the IL-1 receptor. Most TLRs are coupled to a cytoplasmic signaling cascade that includes the adaptor protein MyD88, which ultimately leads to transcription of genes controlling inflammatory responses. TLRs sense pathogen associated molecular patterns (PAMPs), highly conserved sequences in macromolecules expressed primarily by microbial pathogens, and damage-associated molecular patterns (DAMPs), endogenous host-derived danger signals released by cells upon acute damage and/or infection (Takeda et al., 2003). One TLR, TLR4, was first recognized to be activated by lipopolysaccharide from the cell wall of gram negative bacteria. Recent studies have shown that it also recognizes a variety of non-bacterial agonists such as taxol (Byrd-Leifer et al., 2001), fibronectin (Okamura et al., 2001; Termeer et al., 2002), and heat shock protein 60 (Ohashi et al., 2000). In humans, polymorphisms in the TLR4 allele have been associated with both susceptibility and resistance to cancer, depending on the type of tumor examined (Yusuf et al., 2008; Szczepanski et al., 2009; Schroder et al., 2005; Apetoh et al., 2007; Hold et al., 2007).
Previous studies have suggested that TLR4 is required for UV-induced immune suppression (Lewis et al., 2011; Yoshikawa et al., 1990; Kurimoto et al., 1994). Since UVB-induced DNA damage is one of the earliest molecular events in UVB-induced immune suppression and activation of NER is associated with reversal of DNA damage, we evaluated the role of TLR4 in repair of DNA damage.
2. Results
2.1. Enhanced repair of UVB-induced DNA damage in TLR4 deficient mice
In previous studies, we have shown that TLR4-/- mice, when compared with TLR4+/+ mice are resistant to UVB induced immunosuppression (Lewis et al., 2011). Studies were conducted to determine whether there were, after UVB exposure, differences in CPD+ cells as well. The shaved backs of TLR4-/- and TLR4+/+ mice were exposed to UVB (90 mJ/cm2). Mice were sacrificed either immediately (≈30 minutes), 24 hours, or 48 hours later; skin samples were collected and subjected to immunohistochemical detection of CPD+ cells. In skin samples obtained 30 minutes after UV exposure, no differences in CPD staining pattern were observed between the two groups. The number of CPD+ cells decreased progressively in both strains of mice but the decrease was more substantial in the TLR4-/- mice (Fig. 1). The difference in the CPD pattern of the strains was statistically significant (***p<0.001) at 24 and 48 hours. CPD+ cells were not detectable in non-UVB-exposed skin.
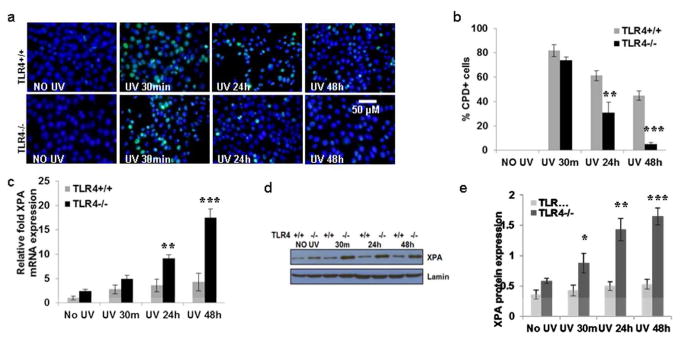
Figure 1. TLR4 deficiency promotes repair of CPD due to increase in XPA expression in mouse skin.

(a) Mice were exposed to acute UVB (90 mJ/cm2) radiation and thereafter sacrificed at 0-48 h. Skin samples were obtained and analyzed for CPD. Frozen sections (5 μm thick) were subjected to immunofluorescent staining to detect CPD+ cells that are green as mentioned in the Methods section. CPD were not detected in non-UV-exposed skin. (b) CPD+ cells were counted as a percentage of all epidermal cells in immunofluorescent stained tissue of TLR4-/- and TLR4+/+ mice. There were significantly fewer CPD at 24h and 48h post UV-exposure in the skin of TLR4-/- mice than in the UV-exposed skin of TLR4+/+ mice. (c) There was an increase in XPA mRNA expression with increasing time post UV exposure, as determined by quantitative real time PCR (qPCR) in the skin of TLR4-/- mice than in the UV-exposed skin of TLR4+/+ mice. (d, e) There was increased XPA protein expression with increase in time post UV exposure as determined by western blot analysis, and this increase was more prominent in the skin of TLR4-/- mice than in the UV-exposed skin of TLR4+/+ mice. Experiments were conducted and repeated separately in 5 animals in each group with identical results. Scale bar = 50 μm. The experiments were repeated twice with similar results. (*p<0.05, **p<0.01, and ***p<0.001).
2.2. TLR4 deficiency enhances UV-induced XPA expression in mouse skin
As TLR4 deficiency reduced UVB-induced DNA damage in skin, the next question was whether repair of DNA damage occurred through stimulation of NER. The XPA protein is involved in recognition of UV damage to DNA, an early event in the DNA repair process (Berneburg et al., 2000). To investigate this question, the shaved backs of TLR4-/- and TLR4+/+ mice were exposed to UVB (90 mJ/cm2). Mice were sacrificed at 30 minutes, 24 hours, and 48h hours after UVB exposure; skin samples were collected, and RNA was isolated and subjected to XPA mRNA expression analysis using real-time PCR. Even without UV exposure, there was somewhat greater XPA mRNA expression in TLR4-/- mice than TLR4+/+ mice (Fig. 1c). Acute exposure to UVB significantly enhanced (*p<0.05-***p<0.001) the disparity in XPA mRNA levels in the skin of TLR4-/- mice compared to TLR4+/+ mice (Fig.1c). The protein level of XPA was also significantly greater (**p<0.01 -***p<0.001) in skin lysates of TLR4-/- mice than in skin lysates of TLR4+/+ mice after UVB-exposure (Fig.1d, e).
2.3 Reduced DNA damage due to increased XPA expression in BMDC of TLR4 deficient mice
DNA damage in antigen presenting cells is a hallmark of UV-induced immune suppression in skin (Vink et al., 1996). We used BMDC as a model to study the effect of UVB on dendritic cells. To determine whether TLR4 deficiency decreased DNA damage in bone marrow-dendritic cells, BMDC were prepared from TLR4-/- and TLR4+/+ mice and DNA damage following exposure to UV-radiation (3 mJ/cm2) was assessed. Immediately after UVB exposure (∼30 minutes), there were equivalent percentages of CPD+ cells in the TLR4+/+ and TLR4-/- panels. However, by 24 hours, there were significantly fewer CPD in BMDC from TLR4-/- mice than TLR4+/+ mice (**p<0.01). CPD were almost completely repaired 48 hours (***p<0.001) after UVB exposure in TLR4-/- mice. In TLR4+/+ mice, a significant number of CPD+ BMDC were still present at 48 hours post UVB exposure (Fig. 2a, b).
Figure 2. TLR4 deficiency enhances repair of CPD in mouse bone marrow-dendritic cells (BMDC) due to increase in XPA expression.
(a) BMDC were prepared as described in the Methods section. BMDC were exposed to acute UVB (3 mJ/cm2) radiation and thereafter harvested at 0-48 h and analyzed for CPD. BMDC were subjected to immunofluorescent staining to detect CPD+ cells that are green as mentioned in the Methods section. CPD were not detected in non-UV-exposed BMDC. (b) CPD+ cells were counted as a percentage of BMDCs from TLR4-/- and TLR4+/+ mice. There were significantly fewer CPD at 24h and 48h post UV-exposure in the BMDC of TLR4-/- mice than in the UV-exposed BMDC of TLR4+/+ mice. (c) There was increased XPA mRNA expression with increase in time post UV exposure as determined by qPCR, and this increase was more prominent in the BMDC of TLR4-/- mice than in the UV-exposed BMDC of TLR4+/+ mice. (d, e) There was increased XPA protein expression with increase in time post UV exposure as determined by western blot analysis, and this increase was more prominent in the BMDC of TLR4-/- mice than in the UV-exposed skin of TLR4+/+ mice. Experiments were conducted and repeated separately in triplicates in each group with identical results. Scale bar = 50 μm. The experiments were repeated twice with similar results. (*p<0.05, **p<0.01 and ***p<0.001)
The expression of XPA mRNA and protein was determined in BMDC after exposure to UVB (Fig. 2c-e). BMDC from TLR4-/- mice had an elevated expression of XPA mRNA even without UVB exposure. In both TLR4-/- and TLR4+/+ mice, UVB exposure increased those levels, but the levels increased much more dramatically in TLR4-/- BMDC than in TLR4+/+ BMDC. The difference was present at 30 minutes, increased at 24 hours (**p<0.01), and increased even further at 48 hours (***p<0.001) (Fig. 2c). Similar results were found with XPA protein expression (Fig. 2d, e).
2.4 TLR4 deficiency augments IL-12 and IL-23 mRNA expression in mouse BMDC
Since both IL-12 and IL-23 have been reported to play an important role in DNA repair through stimulation of NER, BMDC from TLR4-/- and WT mice were exposed to UVB (3 mJ/cm2) and mRNA expression for IL-12p35 and IL-23p19 was determined. BMDC from TLR4-/- mice had higher expression of IL-12p35 and IL-23p19 at 30 minutes (**p<0.01), 24 hours (**p<0.01), and 48 hours (***p<0.001) than TLR4+/+ mice after UVB exposure (Figs. 3a, b). Similar results were obtained for IL-12p35 and IL-23p19 proteins as detected by ELISA (Figs. 3c, d).
Figure 3. TLR4 deficiency augments IL-12 and IL-23 mRNA expression in mouse BMDC.
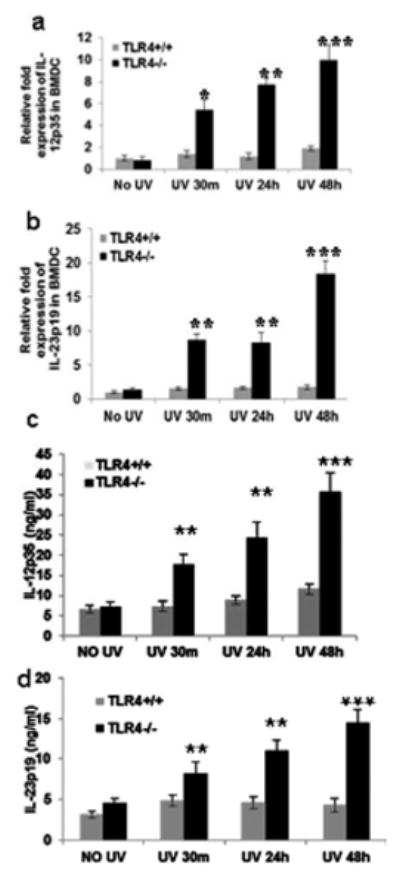
BMDC were prepared as described in the Methods section. BMDC were exposed to acute UVB (3 mJ/cm2) radiation and thereafter harvested at 0-48 h. BMDC were obtained and analyzed for IL-12p35 and IL-23p19 expression as mentioned in the Methods section. There was an increase in (a) IL-12p35 and (b) IL-23p19 mRNA expression with increase in time post UV exposure as determined by quantitative real time PCR (qPCR), and this increase was more prominent in the BMDC of TLR4-/- mice than in the UV-exposed BMDC of TLR4+/+ mice. Similar results were obtained for (c) IL-12p35 and (d) IL-23p19 protein expression by ELISA. Experiments were conducted and repeated separately in triplicates in each group with identical results. Scale bar = 50 μm. The experiments were repeated twice with similar results. (**p<0.01 and ***p<0.001)
Next, BMDC from TLR4-/- mice were pre-treated with anti-IL-12p35 and IL-23p19 antibodies, and then exposed to UVB. There was a modest but significant increase in CPD at 30 minutes (**p<0.01), which was further enhanced at 24 hours (***p<0.001) (Figs. 4a, d) in UV treated TLR4-/- mice after treatment with anti-IL-12p35 and anti-IL-23p19 antibodies. A significant decrease in XPA mRNA expression was also observed at 30 minutes (**p<0.01) and 24 hours (***p<0.001) in UV treated BMDC of TLR4-/- mice after treatment with IL-12p35 and IL-23p19 antibodies (Figs. 4b, e). These results suggest that TLR4 deficiency up-regulates IL-12p35 and IL-23p19 expression which then augments XPA expression in TLR4-/- BMDC.
Figure 4. Neutralization of IL-12 and IL-23 inhibits the repair of BMDC in TLR4 deficient mice.
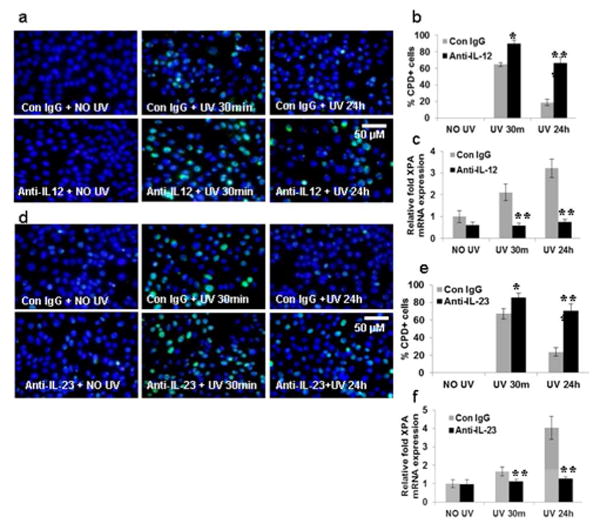
BMDC were prepared as described in the Methods section. BMDC from TLR4 deficient mice were exposed to acute UVB (3 mJ/cm2) radiation and thereafter harvested at 0-24 h. (a) Neutralizing anti-IL-12p35 antibody was added to BMDC cultures before UV exposure. BMDC were exposed to acute UVB (3 mJ/cm2) radiation and thereafter harvested at 0-24 h and analyzed for CPD. BMDC were subjected to immunofluorescent staining to detect CPD+ cells that are green as mentioned in the Methods section. CPD were not detected in non-UV-exposed BMDC. (b) CPD+ cells were counted as a percentage of BMDCs from TLR4-/- mice. There were significantly more CPD at 24h post UV-exposure in the BMDC that were treated with neutralizing anti-IL-12p35 antibody than those treated with control IgG. (c) There was a decrease in XPA mRNA expression after neutralization with anti-IL-12p35 antibody as determined by quantitative real time PCR (qPCR) in the BMDC of TLR4-/- mice. (d) Neutralizing anti-IL-23p19 antibody was added separately to BMDC cultures before UV exposure. BMDC were exposed to acute UVB (3 mJ/cm2) radiation and thereafter harvested at 0-24 h and analyzed for CPD. BMDC were subjected to immunofluorescent staining to detect CPD+ cells that are green as mentioned in the Methods section. CPD were not detected in non-UV-exposed BMDC. (e) CPD+ cells were counted as a percentage of BMDCs from TLR4-/- mice. There were significantly more CPD at 24h post UV-exposure in the BMDC that were treated with neutralizing anti-IL-23p19 antibody than those treated with control IgG. (f) There was a decrease in XPA mRNA expression after neutralization with anti-IL-23p19 antibody as determined by quantitative real time PCR (qPCR) in the BMDC of TLR4-/- mice. Experiments were conducted and repeated separately in triplicates in each group with identical results. Scale bar = 50 μm. The experiments were repeated twice with similar results. (*p<0.05, **p<0.01 and ***p<0.001).
3. Discussion
TLRs are a major component of innate immunity that serve as a first line of defense against a wide range of insults (Takeda et al., 2003). Previous studies have suggested that TLR4 plays an important role in UVB-induced cutaneous immunosuppression (Lewis et al., 2011; Yoshikawa et al. 1990; Kurimoto et al., 1994).
The development of UVB-induced immune suppression is a complex process in which UVB radiation is absorbed by chromophores in the skin, which alter the function of antigen presenting cells. UV-irradiated DCs have a reduced ability to stimulate helper and effector T cells. There is evidence that DNA repair mechanisms are related directly to the function of UVB-irradiated DCs in terms of their stimulation of T cells and the induction of immune reactions (Vink et al., 1997; Vink et al., 1996). Reduction of CPDs via application of DNA repair enzymes prevents UV-induced immunosuppression (Kripke et al., 1992; Stege et al., 2000).
The main objective of our study was to investigate possible mechanisms by which the lack of TLR4 activity promoted resistance to UVB-induced immune suppression, focusing on the cytokines IL-12 and IL-23. IL-12 and IL-23 promote the induction of IFN-γ producing CD8+ TC1 cells and IL-17 CD8+ TC17 cells respectively, both of which are known to serve as effector cells for contact hypersensitivity responses (He et al., 2009; He et al., 2006). When administered to UVB-irradiated mice, IL-12 and IL-23 reversed the immunosuppressive actions of UV radiation, and shift the balance of regulatory and effector T-cells from one in which effector T-cells rather than regulatory T-cells dominate (Majewski et al., 2009; Majewski et al., 2010). Interestingly, these cytokines also stimulate the synthesis of DNA repair enzymes and reduce CPDs via through the NER pathway (Schwarz et al., 2002; Schwarz et al., 2005). Repair of UVB-induced DNA damage is absent in IL-12 deficient mice, indicating a cause and effect relationship. Moreover, the repair effect of IL-12 and IL-23 is absent in mice with a mutated XPA gene which encodes for a protein necessary for NER. This is relevant to UVB-induced immune suppression because DNA damage and the lack of its repair in dendritic cells is essential for the induction of UVB-induced immune suppression. Exogenous administration of IL-12 can break immunotolerance and can reverse the suppressive activity of regulatory T-cells (Majewski et al., 2009) (Gläser et al., 2009). Similarly, IL-23 can reduce UV-induced DNA damage and thereby prevent immunosuppression (Gläser et al., 2009). IL-12 and IL-23 share the unique capacity to restore suppressed immune responses through an effect on regulatory T-cells. IL-12 and IL-23 have been reported to reduce CPDs via induction of NER, as the repair effect was absent in mice with a mutated XPA gene and thus no functional NER.
Our investigation showed that BMDC from TLR4 deficient mice synthesize increased amounts of IL-12 and IL-23 in response to UV radiation, and when IL-12 and IL-23 activity is blocked in mice with normal TLR4 expression, CPDs in BMDC are inefficiently repaired. Since both IL-12 and IL-23 are known stimuli for DNA repair enzyme synthesis, the results suggest that activation of TLR4 signaling by UV radiation initiates a cascade of events which diminishes IL-12 and/or IL-23 synthesis, thereby reducing expression of DNA repair enzymes and ultimately prolonging DNA damage in those cells. The findings thus support the hypothesis that the innate immune system substantially modulates adaptive immune responses to UV radiation.
Several other mediators have been found to contribute to UV-induced immune suppression. These include prostaglandin E2, cis-urocanic acid, platelet activating factor and oxidative stress (Halliday, 2010; Sreevidya, et al, 2010). Several of these act through a common pathway involving increased activation of DNA repair enzymes. It is reasonable to speculate that some of these mediators may have their effects through interactions with TLR4 or its ligands. Previous studies have shown that green tea polyphenols reverse the immunosuppressive effects of UVB radiation (Katiyar et al. 2010; Yusuf et al., 2007; Meeran et al., 2006). As is observed in TLR4-/-mice, administration of green tea polyphenols augments the repair of DNA damage in UVB-irradiated skin, and stimulates IL-12 production (Katiyar, 2011). Because the effects of green tea polyphenols and TLR4 deficiency parallel each other, it is possible that green tea polyphenols provide protection from UV radiation, at least in part, by inhibiting ligands from binding to TLR4, diminishing TLR4 itself, or impairing the signaling pathways that TLR4 activates.
The process by which TLR4 participates in UVB-induced immune suppression most likely involves additional steps that remain to be investigated. For example, several TLR4 ligands are up-regulated by UV radiation, including heat shock proteins (Gaspari et al., 1993), S100 family proteins (Gläser et al., 2009; Grimbaldeston et al., 2003), HMGB1 (Barkauskaite et al., 2007), and hyaluronic acid (Hiramoto et al., 2012). TLR4 itself is also up-regulated in human Langerhans cells post UV exposure (Wang et al., 2011). The ability of these ligands to activate TLR4 could be the proximate target for UVB radiation. In addition, TLR4+/+ mice are known to produce higher levels of IL-10 than TLR4-/- mice upon exposure to UVB (Lewis et al., 2011). Since IL-10 antagonizes IL-12, a reduction in IL-10 levels in TLR4-/- mice may be a possible cause of the increase in IL-12.
Other TLRs have been implicated in UVB-induced immune suppression. Recent studies have shown that TLR3 is also necessary for UVB-induced immune suppression and inflammation (Bernard et al., 2012). UVB-irradiated keratinocytes release damaged self-noncoding RNAs that serve as potent TLR3 agonists. In contrast to the findings with TLR3 and TLR4, other TLRs have been shown to reverse the immunosuppressive effects of UVB radiation. Topical application of the TLR7 agonist imiquimod abolishes UVB-induced suppression of contact hypersensitivity responses and reduces the growth of squamous cell carcinomas in mice through effects on TH1 and TH17 cells (Thatcher et al., 2006; Yokogawa et al., 2012). Imiquimod has also been shown to repair DNA damage in BMDCs (Fishelevich et al., 2011). In addition, an in-silico gene expression study indicated that intraperitoneal injection as well as intranasal inoculation with the TLR9 agonist CpG DNA upregulated DNA repair genes in immune cells (Sommariva et al., 2011). Thus, the relationship between TLRs and UVB-induced immune suppression is complex, and like cytokines, may have immunostimulatory or immunosuppressive effects depending on the TLR examined.
Genetic single-nucleotide polymorphisms (SNPs) may affect the development of human diseases. Two SNPs in the TLR4 gene are largely studied. The +896A/G polymorphism causes an amino acid change of aspartate to glycine at position 299 (Asp299Gly), and the +1196C/T polymorphism causes a switch from threonine to isoleucine at position 399 (Thr399Ile) (Arbour et al., 2000). These two polymorphisms have been linked to different diseases such as gastric cancer, pyelonephritis, and hepatitis C virus-induced hepatocellular carcinoma (de Oliveira et al., 2000; Akil et al., 2012; Agúndez et al., 2012). The frequency of the TLR4 +3725C allele is significantly elevated in breast cancer patients, and is associated with shorter survival time overall (P=0.006), suggesting that TLR4 polymorphisms are associated with increased susceptibility to breast cancer (Yang et al., 2013). The clinical relevance of TLR4 polymorphisms in UV-induced immune suppression could explain genetic susceptibility to dermatologic diseases. This would include non-melanoma skin cancers and melanoma, lupus erythematosus, and polymorphous light eruption, all of which have been reported to have dysregulated immune responses following UV exposure.
In summary, our studies show that innate immunity plays a major role in UVB-induced immune suppression, and implicate the role of TLR4 in DNA damage, as deficiency of TLR4 enhances the repair process by up-regulation of IL-12 and IL-23, which activate the DNA repair gene XPA. These studies provide further evidence of the importance of innate immunity in the development of UVB-induced immune suppression. Thus, targeting TLR4 may become an attractive approach for developing preventive and therapeutic strategies against UVB-induced DNA damage and for prevention of UV-induced dermatological diseases in which the depressed immune system plays a permissive role.
4. Materials and Methods
4.1. Animals and Reagents
TLR4 competent TLR4+/+ female C57BL/6 mice 6-8 weeks of age were purchased from National Cancer Institute (Frederick, MD). TLR4-/- mice on a C57BL/6 background were purchased from Jackson Laboratories (Bar Harbor, ME). All animal procedures were performed according to National Institutes of Health guidelines under protocols approved by the Institutional Animal Care and Use Committee of the University of Alabama at Birmingham.
4.2. UVB Light Source and Irradiation of Mice
The clipper-shaved dorsal skin of mice was exposed to UVB radiation (90 mJ/cm2) from four UVB lamps (Daavlin, UVA/UVB Research Irradiation Unit, Bryan, OH) equipped with an electronic controller to regulate UVB dosage at the fixed distance of 24 cm from the lamps to the dorsal skin surface of mice. Wavelengths <290 nm were filtered out using Kodacel cellulose film (Eastman Kodak Co., Rochester, NY). Most resulting wavelengths were in the UVB (290–320 nm; ∼80%) and UVA (∼20%) ranges, with peak emission at 314 nm as monitored regularly.
4.3. UV-Induced DNA damage
The shaved dorsal skin of TLR4-/- and WT mice was exposed to UVB radiation (90mJ/cm2) to induce DNA damage as described earlier with some modifications (Meeran et al, 2008). Mice were sacrificed at various time points (30 minutes, 24 hours, and 48 hours) after UVB exposure and skin samples were collected for immunostaining, DNA isolation, RNA extraction, and preparation of lysates for western blot. Mice receiving no UVB exposure were used as controls.
4.4. Generation of bone marrow-derived dendritic cells (BMDC)
BMDCs were prepared from TLR4-/- and TLR4++ mice as described with some modifications (Inaba et al, 1992). Briefly, bone marrow cells from femurs and tibias of mice were incubated in RPMI 1640 medium with a cocktail of antibodies against Iak, CD45R/B220, Lyt-2 and GK1.5 (2 μg/106 cells) on ice for 1 hour and washed once with Hanks balanced salt solution [HBSS] after lysis of RBCs. Different cellular populations were removed from the cell suspension by antibody-mediated depletion using sheep anti-rat IgG dynabeads according to the manufacturer's instructions. Cells were washed once with HBSS and cultured in 10% fetal calf serum [FCS] RPMI 1640 media supplemented with recombinant mouse granulocyte macrophage-colony stimulating factor [GM-CSF] (10 ng/ml) and IL-4 (10 ng/ml) in 6-well plates (5×105 cells/well). On day 5, half of the medium was replaced with fresh medium and cells were stimulated on the following day for the experiments. The culture medium was removed and BMDC were exposed to UVB (3mJ/cm2) in PBS. After treatment, the cells were fixed for immunostaining and RNA extraction.
4.5 Immunohistochemical detection of CPD
For immunohistochemical detection of CPD+ cells in skin samples, frozen skin sections (5 μm thick) were thawed and kept in 70 mM NaOH in 70% ethanol for 2 minutes to denature nuclear DNA, followed by neutralization for 1 minute in 100 mM Tris-HCl (pH 7.5) in 70% ethanol. The sections were washed with PBS buffer and incubated with 10% goat serum in PBS to prevent non-specific binding prior to incubation with a monoclonal antibody specific for CPD, or its isotype control (IgG1). Bound anti-CPD antibody was detected by incubation with biotinylated goat-anti-mouse IgG1 followed by Alexa Fluor 488 (Patrick, 1977).
4.6. RNA Extraction and quantitative real-time RT-PCR (qPCR)
Total RNA from skin or BMDC was extracted using Trizol reagent (Invitrogen, Carlsbad, CA) according to the manufacturer's protocol. The concentration of total RNA was determined by measuring absorbance at 260 nm with a BioRad Smart Spec spectrophotometer. Purity of RNA was determined with the ratio of absorbance 260nm/280nm >1.9. cDNA was synthesized from 1 μg RNA using Reverse transcriptase kit (Promega) according to the manufacturer's instructions. Using iQ™ SYBR Green Master Mix (Bio-Rad, Hercules, CA), cDNA was amplified by real-time PCR with a Bio-Rad MyiQ thermocycler and SYBR Green detection system (Bio-Rad, Hercules, CA). The primers used for the XPA, IL-12p35 and IL-23p19 genes have been described earlier (Kang et al., 2010; Zhang et al., 2003). The standard PCR conditions were 95°C for 10 min, then 40 cycles at 95°C for 30 s, 60°C for 30 s, and 72°C for 30 s. The expression levels of genes were normalized to the expression level of GAPDH mRNA in each sample. For mRNA analysis, calculations for determining relative level of gene expression were made using the cycle threshold (Ct) method. The mean Ct values from duplicate measurements were used to calculate the expression of the target gene with normalization to a housekeeping gene used as internal control and using the formulae 2–ΔΔCT.
4.7. Preparation of lysates and western blotting
Either whole cell/tissue lysates or nuclear lysates were prepared from control and UVB-treated skin/BMDC by using the Active Motif (Carlsbad, CA) cytoplasmic and nuclear extraction kit. For western blot analysis, 50-80μg protein was loaded in each well and resolved on 10% SDS-polyacrylamide gel and transferred onto nitrocellulose membranes. Membranes were incubated in blocking buffer for 2 hours, then incubated with the primary antibodies in blocking buffer for 2 hours at room temperature or overnight at 4°C. The membrane was then washed with TBS-T and incubated with secondary antibody conjugated with horseradish peroxidase. Protein bands were visualized using the enhanced chemiluminescence detection system (Amersham Life Science, Inc., Piscataway, NJ). To verify equal protein loading and transfer of proteins from gel to membrane, the blots were stripped and re-probed for β-actin or histone H3. The band density was analyzed using Image J software provided by the National Institutes of Health and the values were normalized to the β-actin band density.
4.8. Statistical Analysis
The differences between experimental groups for immunostaining, qPCR, and western blot were analyzed using the two-way ANOVA test. In all cases, a p<0.05 was considered significant.
Acknowledgments
This work was supported by Department of Defense New Investigator Award W81XWH-10-1-0763, 1R03AR057483 (NIAMS) to NY, and Medical Student grant from American Skin Association to ES.
Footnotes
Conflict of Interest: None of the authors have a potential conflict of interest with this submission.
References
- 1.Agúndez JA, García-Martín E, Devesa MJ, et al. Polymorphism of the TLR4 gene reduces the risk of hepatitis C virus-induced hepatocellular carcinoma. Oncology. 2012;82:35–40. doi: 10.1159/000335606. [DOI] [PubMed] [Google Scholar]
- 2.Akil I, Ozkinay F, Onay H, et al. Assessment of Toll-like receptor-4 gene polymorphism on pyelonephritis and renal scar. Int J Immunogenet. 2012;39:303–7. doi: 10.1111/j.1744-313X.2012.01090.x. [DOI] [PubMed] [Google Scholar]
- 3.Apetoh L, Ghiringhelli F, Tesniere A, et al. Toll-like receptor 4-dependent contribution of the immune system to anticancer chemotherapy and radiotherapy. Nat Med. 2007;13:1050–1059. doi: 10.1038/nm1622. [DOI] [PubMed] [Google Scholar]
- 4.Arbour NC, Lorenz E, Schutte BC, et al. TLR4 mutations are associated with endotoxin hyporesponsiveness in humans. Nat Genet. 2000;25:187–91. doi: 10.1038/76048. [DOI] [PubMed] [Google Scholar]
- 5.Barkauskaite V, Ek M, Popovic K, et al. Translocation of the novel cytokine HMGB1 to the cytoplasm and extracellular space coincides with the peak of clinical activity in experimentally UV-induced lesions of cutaneous lupus erythematosus. Lupus. 2007;16(10):794–802. doi: 10.1177/0961203307081895. [DOI] [PubMed] [Google Scholar]
- 6.Bernard JJ, Cowing-Zitron C, Nakatsuji T, et al. Ultraviolet radiation damages self noncoding RNA and is detected by TLR3. Nat Med. 2012 doi: 10.1038/nm.2861. Epub ahead of print. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Berneburg M, Krutmann J. Photoimmunology, DNA repair and photocarcinogenesis. J Photochem Photobiol B. 2000;54:87–93. doi: 10.1016/s1011-1344(00)00024-5. [DOI] [PubMed] [Google Scholar]
- 8.Byrd-Leifer CA, Block EF, Takeda K, et al. The role of MyD88 and TLR4 in the LPS-mimetic activity of Taxol. Eur J Immunol. 2001;31:2448–2457. doi: 10.1002/1521-4141(200108)31:8<2448::aid-immu2448>3.0.co;2-n. [DOI] [PubMed] [Google Scholar]
- 9.de Oliveira JG, Silva AE. Polymorphisms of the TLR2 and TLR4 genes are associated with risk of gastric cancer in a Brazilian population. World J Gastroenterol. 2000;18:1235–42. doi: 10.3748/wjg.v18.i11.1235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Elmets CA, Bergstresser PR, Tigelaar RE, Wood PJ, Streilein JW. Analysis of mechanism of unresponsiveness produced by haptens painted on skin exposed to low dose ultraviolet radiation. J Exp Med. 1983;158:781–794. doi: 10.1084/jem.158.3.781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Fishelevich R, Zhao Y, Tuchinda, et al. Imiquimod-induced TLR7 signaling enhances repair of DNA damage induced by ultraviolet light in bone marrow-derived cells. J Immunol. 2011;187(4):1664–73. doi: 10.4049/jimmunol.1100755. Epub 2011 Jul 15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Fisher MS, Kripke ML. Suppressor T lymphocytes control the development of primary skin cancers in ultraviolet-irradiated mice. Science. 1982;216:1133–1134. doi: 10.1126/science.6210958. [DOI] [PubMed] [Google Scholar]
- 13.Gaspari AA, Fleisher TA, Kraemer KH. Impaired interferon production and natural killer cell activation in patients with the skin cancer-prone disorder, xeroderma pigmentosum. J Clin Invest. 1993;92(3):1135–42. doi: 10.1172/JCI116682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Thatcher TH, Luzina I, Fishelevich R, et al. Topical imiquimod treatment prevents UV-light induced loss of contact hypersensitivity and immune tolerance. J Invest Dermatol. 2006;126:821–31. doi: 10.1038/sj.jid.5700167. [DOI] [PubMed] [Google Scholar]
- 15.Thatcher TH, Luzina I, Fishelevich R, et al. Topical imiquimod treatment prevents UV-light induced loss of contact hypersensitivity and immune tolerance. J Invest Dermatol. 2006;126:821–31. doi: 10.1038/sj.jid.5700167. [DOI] [PubMed] [Google Scholar]
- 16.Gläser R, Navid F, Schuller W, et al. UV-B radiation induces the expression of antimicrobial peptides in human keratinocytes in vitro and in vivo. J Allergy Clin Immunol. 2009;123(5):1117–23. doi: 10.1016/j.jaci.2009.01.043. [DOI] [PubMed] [Google Scholar]
- 17.Grimbaldeston MA, Geczy CL, Tedla N, et al. 100A8 induction in keratinocytes by ultraviolet A irradiation is dependent on reactive oxygen intermediates. J Invest Dermatol. 2003;121(5):1168–74. doi: 10.1046/j.1523-1747.2003.12561.x. [DOI] [PubMed] [Google Scholar]
- 18.Halliday GM. Common links among the pathways leading to UV-induced immunosuppression. J Invest Dermatol. 2010;130(5):1209–12. doi: 10.1038/jid.2009.374. [DOI] [PubMed] [Google Scholar]
- 19.Hiramoto K, Kobayashi H, Yamate Y, et al. Intercellular pathway through hyaluronic acid in UVB-induced inflammation. Exp Dermatol. 2012;21(12):911–4. doi: 10.1111/exd.12032. [DOI] [PubMed] [Google Scholar]
- 20.He D, Wu L, Kim HK, et al. CD8+ IL-17-producing T cells are important in effector functions for the elicitation of contact hypersensitivity responses. J Immunol. 2006;177:6852–6858. doi: 10.4049/jimmunol.177.10.6852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.He D, Wu L, Kim HK, et al. IL-17 and IFN-gamma mediate the elicitation of contact hypersensitivity responses by different mechanisms and both are required for optimal responses. J Immunol. 2009;183:1463–70. doi: 10.4049/jimmunol.0804108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hold GL, Rabkin CS, Chow WH, et al. A functional polymorphism of toll-like receptor 4 gene increases risk of gastric carcinoma and its precursors. Gastroenterology. 2007;132:905–912. doi: 10.1053/j.gastro.2006.12.026. [DOI] [PubMed] [Google Scholar]
- 23.Inaba K, Inaba M, Romani N, et al. Generation of large numbers of dendritic cells from mouse bone marrow cultures supplemented with granulocyte/macrophage colony-stimulating factor. J Exp Med. 1992;176:1693–1702. doi: 10.1084/jem.176.6.1693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kang TH, Lindsey-Boltz LA, Reardon JT, et al. Circadian control of XPA and excision repair of cisplatin-DNA damage by cryptochrome and HERC2 ubiquitin ligase. Proc Natl Acad Sci U S A. 2010;107(11):4890–5. doi: 10.1073/pnas.0915085107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Katiyar SK, Vaid M, van Steeg H, Meeran SM. Green tea polyphenols prevent UV-induced immunosuppression by rapid repair of DNA damage and enhancement of nucleotide excision repair genes. Cancer Prev Res (Phila) 2010;3:197–89. doi: 10.1158/1940-6207.CAPR-09-0044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Katiyar SK. Green tea prevents non-melanoma skin cancer by enhancing DNA repair. Arch Biochem Biophys. 2011;508(2):152–8. doi: 10.1016/j.abb.2010.11.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kovach BT, Sams HH, Stasko T. Systemic strategies for chemoprevention of skin cancers in transplant recipients. Clin Transplant. 2005;19:726–34. doi: 10.1111/j.1399-0012.2005.00412.x. [DOI] [PubMed] [Google Scholar]
- 28.Kripke ML. Antigenicity of murine skin tumors induced by ultraviolet light. J Natl Cancer Inst. 1974;53:1333–1336. doi: 10.1093/jnci/53.5.1333. [DOI] [PubMed] [Google Scholar]
- 29.Kripke ML, Cox PA, Alas LG, et al. Pyrimidine dimers in DNA initiate systemic immunosuppression in UV-irradiated mice. Proc Natl Acad Sci U S A. 1992;89(16):7516–20. doi: 10.1073/pnas.89.16.7516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kurimoto I, Streilein JW. Characterization of genetic basis of ultraviolet-B light effects on contact hypersensitivity induction. Immunology. 1994;81:352–358. [PMC free article] [PubMed] [Google Scholar]
- 31.Lewis W, Simanyi E, Li H, et al. Regulation of ultraviolet radiation induced cutaneous photoimmunosuppression by toll-like receptor-4. Arch Biochem Biophys. 2011;508(2):171–7. doi: 10.1016/j.abb.2011.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Majewski S, Jantschitsch C, Maeda A, et al. IL-23 Antagonizes UVR-Induced Immunosuppression through Two Mechanisms: Reduction of UVR-Induced DNA Damage and Inhibition of UVR-Induced Regulatory T Cells. J Invest Dermatol. 2009 doi: 10.1038/jid.2009.274. Epub ahead of print. [DOI] [PubMed] [Google Scholar]
- 33.Majewski S, Jantschitsch C, Maeda A, et al. IL-23 Antagonizes UVR-Induced Immunosuppression through Two Mechanisms: Reduction of UVR-Induced DNA Damage and Inhibition of UVR-Induced Regulatory T Cells. J Invest Dermatol. 2010;130(2):554–62. doi: 10.1038/jid.2009.274. [DOI] [PubMed] [Google Scholar]
- 34.Meeran SM, Mantena SK, Katiyar SK. Prevention of ultraviolet radiation induced immunosuppression by (-)-epigallocatechin-3-gallate in mice is mediated through interleukin 12-dependant DNA repair. Clin Cancer Res. 2006;12:2272–80. doi: 10.1158/1078-0432.CCR-05-2672. [DOI] [PubMed] [Google Scholar]
- 35.Meeran SM, Punathil T, Katiyar SK. IL-12 deficiency exacerbates inflammatory responses in UV-irradiated skin and skin tumors. J Invest Dermatol. 2008;128(11):2716–27 d. doi: 10.1038/jid.2008.140. Epub 2008 May 29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Mittal D, Saccheri F, Vénéreau E, et al. TLR4-mediated skin carcinogenesis is dependent on immune and radioresistant cells. EMBO J. 2010;29(13):2242–52. doi: 10.1038/emboj.2010.94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ohashi K, Burkart V, Flohe S, et al. Cutting edge: heat shock protein 60 is a putative endogenous ligand of the Toll-like receptor-4 complex. J Immunol. 2000;164:558–561. doi: 10.4049/jimmunol.164.2.558. [DOI] [PubMed] [Google Scholar]
- 38.Okamura Y, Wataru M, Jerud ES, et al. The extra domain A of fibronectin activates Toll like receptor 4. J Biol Chem. 2001;276:10229–10233. doi: 10.1074/jbc.M100099200. [DOI] [PubMed] [Google Scholar]
- 39.Patrick MH. Studies on thymine-derived UV photoproducts in DNA--I. Formation and biological role of pyrimidine adducts in DNA. Photochem Photobiol. 1977;25(4):357–72. doi: 10.1111/j.1751-1097.1977.tb07355.x. [DOI] [PubMed] [Google Scholar]
- 40.Schroder NW, Schumann RR. Single nucleotide polymorphisms of Toll-like receptors and susceptibility to infectious disease. Lancet Infect Dis. 2005;5:156–164. doi: 10.1016/S1473-3099(05)01308-3. [DOI] [PubMed] [Google Scholar]
- 41.Schwarz A, Ständer S, Berneburg M, et al. Interleukin-12 suppresses ultraviolet radiation-induced apoptosis by inducing DNA repair. Nat Cell Biol. 2002;4:26–31. doi: 10.1038/ncb717. [DOI] [PubMed] [Google Scholar]
- 42.Schwarz A, Maeda A, Kernebeck K, et al. Prevention of UV radiation-induced immunosuppression by IL-12 is dependent on DNA repair. J Exp Med. 2005;201:173–179. doi: 10.1084/jem.20041212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Sommariva M, De Cecco L, De Cesare M, et al. TLR9 agonists oppositely modulate DNA repair genes in tumor versus immune cells and enhance chemotherapy effects. Cancer Res. 2011;71(20):6382–90 d. doi: 10.1158/0008-5472.CAN-11-1285. Epub 2011 Aug 30. [DOI] [PubMed] [Google Scholar]
- 44.Stege H, Roza L, Vink AA, et al. Enzyme plus light therapy to repair DNA damage in ultraviolet-B-irradiated human skin. Proc Natl Acad Sci U S A. 2000;97(4):1790–5. doi: 10.1073/pnas.030528897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Szczepanski MJ, Czystowska M, Szajnik M, et al. Triggering of Toll-like receptor 4 expressed on human head and neck squamous cell carcinoma promotes tumor development and protects the tumor from immune attack. Cancer Res. 2009;69:3105–3113. doi: 10.1158/0008-5472.CAN-08-3838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Takeda K, Kaisho T, Akira S. Toll like receptors. Annu Rev Immunol. 2003;21:335–376. doi: 10.1146/annurev.immunol.21.120601.141126. [DOI] [PubMed] [Google Scholar]
- 47.Termeer C, Benedix F, Sleeman J, et al. Oligosaccharides of hyaluronan activate dendritic cells via toll-like receptor 4. J Exp Med. 2002;195:99–111. doi: 10.1084/jem.20001858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Toews GB, Bergstresser PR, Streilein JW, Sullivan S. Epidermal Langerhans cell density determines whether contact hypersensitivity or unresponsiveness follows skin painting with DNFB. J Immunol. 1980;124:445–453. [PubMed] [Google Scholar]
- 49.Vink AA, Moodycliffe AM, Shreedhar V, et al. The inhibition of antigen-presenting activity of dendritic cells resulting from UV irradiation of murine skin is restored by in vitro photorepair of cyclobutane pyrimidine dimers. Proc Natl Acad Sci USA. 1997;94:5255–5260. doi: 10.1073/pnas.94.10.5255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Vink AA, Strickland FM, Bucana C, et al. Localization of DNA damage and its role in altered antigen-presenting cell function in ultraviolet-irradiated mice. J Exp Med. 1996;183:1491–1500. doi: 10.1084/jem.183.4.1491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Vink AA, Strickland FM, Bucana C, et al. Localization of DNA damage and its role in altered antigen-presenting cell function in ultraviolet-irradiated mice. J Exp Med. 1996;183:1491–1500. doi: 10.1084/jem.183.4.1491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Wang X, Bi Z, Wang Y, et al. Increased MAPK and NF-κB expression of Langerhans cells is dependent on TLR2 and TLR4, and increased IRF-3 expression is partially dependent on TLR4 following UV exposure. Mol Med Rep. 2011;4(3):541–6. doi: 10.3892/mmr.2011.450. [DOI] [PubMed] [Google Scholar]
- 53.Yang CX, Li CY, Feng W. Toll-like receptor-4 genetic variants and prognosis of breast cancer. Tissue Antigens. 2013;81(4):221–6. doi: 10.1111/tan.12096. [DOI] [PubMed] [Google Scholar]
- 54.Yokogawa M, Takaishi M, Nakajima K, et al. Imiquimod attenuates the growth of UVB-induced SCC in mice through Th1/Th17 cells. Mol Carcinog. 2012 doi: 10.1002/mc.21901. Epub ahead of print. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Yoshikawa T, Streilein JW. Genetic basis of the effects of ultraviolet light B on cutaneous immunity. Evidence that polymorphism at the Tnfa and Lps loci governs susceptibility. Immunogenetics. 1990;32:398–405. doi: 10.1007/BF00241633. [DOI] [PubMed] [Google Scholar]
- 56.Yusuf N, Nasti TH, Long JA, et al. Protective Role of Toll-like receptor 4 during the initiation stage of cutaneous chemical carcinogenesis. Cancer Res. 2008;68:615–622. doi: 10.1158/0008-5472.CAN-07-5219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Yusuf N, Irby C, Katiyar SK, Elmets CA. Photoprotective effects of green tea polyphenols. Photodermatol Photoimmunol Photomed. 2007;23:48–56. doi: 10.1111/j.1600-0781.2007.00262.x. [DOI] [PubMed] [Google Scholar]
- 58.Zhang GX, Gran B, Yu S, et al. Induction of experimental autoimmune encephalomyelitis in IL-12 receptor-beta 2-deficient mice: IL-12 responsiveness is not required in the pathogenesis of inflammatory demyelination in the central nervous system. J Immunol. 2003;170(4):2153–60. doi: 10.4049/jimmunol.170.4.2153. [DOI] [PubMed] [Google Scholar]