

Abstract
Background
Uncovering the complex transcriptional regulatory networks (TRNs) that underlie plant and animal development remains a challenge. However, a vast amount of data from public microarray experiments is available, which can be subject to inference algorithms in order to recover reliable TRN architectures.
Results
In this study we present a simple bioinformatics methodology that uses public, carefully curated microarray data and the mutual information algorithm ARACNe in order to obtain a database of transcriptional interactions. We used data from Arabidopsis thaliana root samples to show that the transcriptional regulatory networks derived from this database successfully recover previously identified root transcriptional modules and to propose new transcription factors for the SHORT ROOT/SCARECROW and PLETHORA pathways. We further show that these networks are a powerful tool to integrate and analyze high-throughput expression data, as exemplified by our analysis of a SHORT ROOT induction time-course microarray dataset, and are a reliable source for the prediction of novel root gene functions. In particular, we used our database to predict novel genes involved in root secondary cell-wall synthesis and identified the MADS-box TF XAL1/AGL12 as an unexpected participant in this process.
Conclusions
This study demonstrates that network inference using carefully curated microarray data yields reliable TRN architectures. In contrast to previous efforts to obtain root TRNs, that have focused on particular functional modules or tissues, our root transcriptional interactions provide an overview of the transcriptional pathways present in Arabidopsis thaliana roots and will likely yield a plethora of novel hypotheses to be tested experimentally.
Keywords: Transcriptional regulatory networks, Root, Arabidopsis, Transcription factor, ARACNe
Background
Transcription factors (TFs) play an important role in the regulation of gene expression. The Arabidopsis thaliana (Arabidopsis) TF databases Agris [1,2], RARTF [3,4] or DATF [5,6] contain approximately 1900 entries, that correspond to 6.9% of the 27416 protein coding genes present in the TAIR10 genome release. Forward and reverse genetics, inducible expression systems and, more recently, large scale methods, such as chromatin immunoprecipitation followed by array hybridization or massive parallel sequencing, have provided a vast amount of information regarding target genes and functions of many Arabidopsis TFs. However, obtaining a complete overview of the transcriptional interactions for a given organism or developmental process is still a challenging and expensive task. Brady et al. obtained a stele-enriched root TRN containing protein-DNA and protein-protein interactions identified by Y1H and Y2H assays using stele-enriched TFs, the promoters of these same TFs, and promoters from several miRNA coding genes [7]. However, the low percentage of TF promoters bound by at least one TF and the little overlap in expression enrichment between TFs and their targets suggest that several genes that might be important components of the stele TRN could have been missed in this network.
Gene expression microarrays allow for the rapid quantification of the expression level for a large number of genes in a given biological sample. The most used Arabidopsis gene expression microarray is the Affymetrix ATH1-121501 (ATH1) GeneChip microarray. As of October 2010, there were 686 experiments using the ATH1 chip listed in the EBI ArrayExpress database [8]. All of these experiments provide a quantitative analysis of gene expression in Arabidopsis tissues under a variety of experimental conditions and are therefore a suitable data source for Arabidopsis Transcriptional Regulatory Network (TRN) inference. Although databases such as Genevestigator [9,10], ATTED-II [11,12], or BAR Expression Angler [13,14] have tools for the analysis of Arabidopsis microarray data, they either use a limited set of microarray experiments, the AtGenExpress series [15] (ATTED-II and BAR Expression Angler), or their quality controlled, curated, annotated and normalized data is not publicly available (Genevestigator).
In light of these limitations we decided to create our own curated and annotated Arabidopsis microarray database and use this data to infer TRNs. The 686 microarray experiments indexed in the ArrayExpress database contain over 9000 individual chip hybridizations or CEL files. Preliminary work done in our lab showed that network inference from samples obtained from different tissues, for example whole plant, roots and leaves, yields sub-optimal results and the inferred networks are difficult to interpret in a biological context. We therefore decided to use microarray data obtained from a single organ, namely roots. The Arabidopsis root has several characteristics that make it a suitable organ for our purposes: root anatomy is relatively simple and developmental alterations can be readily observed, there is a vast amount of literature regarding root development and root-expressed TFs and, finally, there is a considerable amount of high quality ATH1 microarray data obtained from root samples. In order to identify the transcriptional interactions occurring in root tissues we used the microarray data as input for the ARACNe algorithm [16].
ARACNe is an information-theoretical method for identifying transcriptional interactions between gene products using microarray expression profile data, which is able to recover non-linear statistical dependencies between variables and has been previously used for TRN reconstruction [17-20]. In this work we show that our database, and the TRNs derived from it, have been able to recover functions and target genes for previously characterized TFs. We further show that the inferred TRNs can accurately predict new TF functions, as exemplified by the predicted role of the MADS-box TF XAL1/AGL12 (AT1G71692) in secondary cell wall formation and its confirmation with loss-of-function mutant root phenotypes for this gene.
Results and discussion
In order to infer the TRNs underlying root development and physiological processes in Arabidopsis, we used two carefully curated datasets obtained from 656 root-specific CEL files from 56 ATH1 microarray experiments (Additional file 1). The first dataset, that we call the TFs-only dataset, is a 656 columns by 2088 rows table that corresponds to our list of 2088 TF probesets. The second dataset, that we call the complete dataset, is a 656 by 22810 table that contains all 22810 probesets present in the ATH1 chip. We used both datasets as input for the ARACNe software [21]. The ARACNe output is a list of interacting probeset pairs ranked through a Mutual Information value and its associated p-value. Details for the theoretical background and practical use of ARACNe can be found in [16] and [21] but, briefly, an interaction between gene A and gene B means that the expression profile of gene A along all 656 experiments explains the expression profile of gene B along those same 656 experiments, and vice versa, as the interactions are not directed. In a biological context, an interaction between gene A and gene B will imply that gene A and gene B participate in the same physiological process and, even further, if gene A is a TF and gene B is a non-TF, the interaction (gene A explains gene B) will suggest that gene A is a transcriptional regulator of gene B.
Network inference was centered on the 2088 TF probesets present in the ATH1 chip and was obtained at three data processing inequality (DPI) values, 0.0, 0.1 and 0.2. DPI is a known information-theoretical property and is explained in the supplementary manual in [21]. Briefly, at DPI 0.0, when a three-node clique (triangle) is present, the interaction with the lowest mutual information will be removed, as this interaction is considered to represent an indirect interaction. At DPI values other than 0.0, three genes loops are allowed and, at DPI 1.0, no interactions are removed. A DPI value of 0.2 (which will preserve triangles if the difference between the mutual information value of its interactions is 20% or less) increases the recovery of true positive interactions while still minimizing the recovery of false positives [16]. After translation of the ARACNe output adjacency files into Cytoscape compatible tables, we obtained the corresponding TFs-only (TFsNet; Additional file 2) and complete (FullNet; Additional file 3) databases. As shown in Table 1, the number of edges increases dramatically from DPI 0.0 to DPI 0.1 to DPI 0.2. For clarity, all graphical representations of the networks in this paper are those obtained at DPI 0.0.
Table 1.
Number of nodes and edges in the TFsNet and FullNet obtained at DPI 0.0, 0.1 and 0.2
|
TFsNet |
|
|
|
DPI |
Nodes |
Edges |
| 0.0 |
2068 |
3574 |
| 0.1 |
2068 |
12049 |
| 0.2 |
2068 |
39113 |
|
FullNet |
|
|
|
DPI |
Nodes |
Edges |
| 0.0 |
22553 |
82059 |
| 0.1 |
22553 |
706043 |
| 0.2 | 22553 | 2570778 |
Nodes and edges present at DPI 0.0 are part of the network obtained at DPI 0.1 and both are part of the network obtained at DPI 0.2.
TFs participating in inferred interactions are expressed in roots
An important question regarding our networks is to determine if the TFs participating in the inferred interactions are actually being expressed in root tissues. The mas5calls function from the affy R package, used to flag microarray expression values as Present, Absent or Marginal, is an unreliable tool to determine if a gene is being expressed or not [22], specially when it involves Arabidopsis TFs [23]. Therefore, in order to determine if the TFs present in our networks are expressed in root tissues, we extracted from both the TFsNet and FullNet obtained at DPI 0.0 all TFs that participate in an interaction and we compared both lists to lists of experimentally determined root-expressed genes (see Methods). Results are presented in Table 2 and Additional file 4. Over 92% of the recovered TFs in the two types of networks have been experimentally determined to be expressed in roots. We are therefore confident that the TFs present in our datasets are indeed root TFs and the interactions that we have recovered represent true in planta transcriptional interactions.
Table 2.
Number of TF from the TFsNet and FullNet whose expression has been experimentally detected in roots
|
TFsNet |
|
|
| |
Number of TFs |
% of total |
|
Detected |
1791 |
92.8 |
|
Not detected |
139 |
7.2 |
|
Total |
1930 |
100.0 |
|
FullNet |
|
|
| |
Number of TFs |
% of total |
|
Detected |
1835 |
92.3 |
|
Not detected |
153 |
7.7 |
| Total | 1988 | 100.0 |
TFs that participate in the same processes are grouped together in the TFsNet
The TFsNet was obtained from a TFs-only dataset that excludes all non-TF genes and constitutes an overview of Arabidopsis roots TFs inferred interactions (Figure 1). TFs participating in the same processes are expected to be grouped together in distinct clusters or modules. Some of these functional modules have been identified and experimentally characterized and serve as probes of the reliability of the inferred networks.
Figure 1.

The TFsNet. (a) Overview of the TFsNet obtained at DPI 0.0. Genes are represented as nodes and inferred interactions as edges. Nodes are colored grey, except genes mentioned in the text that are colored green. Edge width and color intensity is proportional to the Mutual Information (MI) value of the interaction, with higher MI values corresponding to thicker and darker edges. Gene names were omitted for clarity. Zooms on particular TF groups are presented in the subsequent panels. (b-i) Sub-networks of TFs present in the SHR-SCR group (b), the PLT group (c), the vascular development group (d), the AtGRF group (e), the AtHAM group (f), the jasmonate response group (g) the iron-deficiency group (h) and the nitrate-response group (i). Edges are labeled with the p-value of the interaction. Edges from the groups to the rest of the network were omitted for clarity.
Two transcriptional pathways controlling stem-cell niche patterning have been identified [24-28]. The first pathway is composed of the GRAS-family SHORT ROOT (SHR; AT4G37650) and SCARECROW (SCR; AT3G54220) and the C2H2-family, INDETERMINATE DOMAIN (IDD) MAGPIE (MGP; AT1G03840) and JACKDAW (JKD; AT5G03150). As shown in Figure 1b, these four TFs are grouped together with IDD NUTCRACKER (NUC; AT5G44160), the SSXT-domain transcriptional co-activator ANGUSTIFOLIA 3 (AN3; AT5G28640) and the GRAS-family SCARECROW-LIKE 3 (SCL-3; AT1G50420). NUC and SCL-3 are proposed direct transcriptional targets of SHR [29-31]. Note that, as networks obtained at DPI 0.0 cannot contain triangles, the absence of an edge, for example between SHR and NUC, does not imply a lack of interaction between these two genes but merely that both genes have other interactions with better MI scores. Also, interactions between the genes in this module have relatively low MI values, corresponding to p-values of 1e-30 and 1e-40 (relative to the lowest p-value in the dataset, 1e-140). This is probably not surprising since this pathway has a complex mode of molecular interaction [32] that will hinder the ability of the ARACNe algorithm to recover their interaction from microarray data with a higher p-value [21]. Additional IDD genes, AtIDD4 (AT2G02080), AtIDD5 (AT2G02070), AtIDD14 (AT1G68130), AtIDD15 (AT2G01940) and AtIDD16 (AT1G25250), are present in this module. Protein-protein interactions have been reported for SCL-3-NUC, MGP-SCR, MGP-SHR, MGP-JKD, SCR-JKD, and SHR-JKD [33]. On the other hand, the IDD proteins JKD and MGP regulate SHR and SCR expression and movement across root tissues via both transcriptional and protein-protein interactions [27,34]. Finally, movement of the SHR protein is abolished by the substitution of a single threonine residue in its VHIID motif, which is proposed to mediate protein-protein interactions of SHR [35] and its nuclear localization [34]. It is therefore interesting to speculate that AtIDD4, AtIDD5, AtIDD14, AtIDD15 and AtIDD16 could also be involved in root development and patterning via transcriptional regulation of, or protein-protein interactions with SHR and SCR.
The second pathway involves auxin signaling through the activation of as yet unidentified Auxin Response Factors (ARFs) and the PLETHORA (PLT) TFs, of the AP2-EREBP family. The PLT genes, PLT1 (AT3G20840), PLT2 (AT1G51190), PLT3/AIL6 (AT5G10510) and BABY BOOM (BBM; AT5G17430), have overlapping expression profiles and act in a redundant manner [26]. In the TFsNet, the four PLT genes are part of the same group, that also includes the bHLH SPATULA (AT4G36930), ARF5/MONOPTEROS (AT1G19850) and the ERF-family Cytokinin Response Factors CRF2/TMO3 (AT4G23750) and CRF3 (AT5G53290; Figure 1c). Remarkably, the four PLETHORA proteins [26] and ARF5[36] are all expressed in the seedling root stele initials. Root vascular patterning has been shown to be dependent on an auxin-cytokinin cross-talk [37] and the participation in this cross-talk of a few genes, such as SHY2[38], BRX[39] or AHP6[40,41] has been demonstrated. However, a transcriptional network linking the PLETHORA pathway and cytokinin responsive TFs is still missing. The presence of two CRF TFs in this module provides new clues in this direction.
BODENLOS (BDL; AT1G04550), a member of the Aux/IAA family, is a transcriptional inhibitor of ARF5 and its expression is controlled by ARF5 in embryos [42]. Curiously, BDL, as well as two other TARGET OF MONOPTEROS (TMO) genes, ATAIG1/TMO5 (AT3G25710) and TMO6 (AT5G60200), do not group with ARF5 in the TFsNet. Instead, they are part of a group of TFs involved in vascular development that includes genes such as IAA13 (AT2G33310), IAA3/SHY2 (AT1G04240) [39], ATHB-14/PHABULOSA (AT2G34710), ATHB-15/CORONA (AT1G52150) [43], IFL/REVOLUTA (AT5G60690) [44], ATHB-8 (AT4G32880) [45], ATHB9/PHAVOLUTA (AT1G30490) and AtTCP14 (AT3G47620) [46] (Figure 1d). pBDL::GFP expression has been observed in the root stele of 4–5 days-old seedlings (see Figure S6 in [42]), thus pointing to possible novel roles for these auxin-related genes in vascular development.
Other TFs involved in organ development are also grouped together in the TFsNet. For example, the closely related ATHAM1 (AT2G45160), ATHAM2 (AT3G60630) and ATHAM3 (AT4G00150) genes, belonging to the GRAS family, are involved in the maintenance of meristem indeterminacy, and are functionally redundant [47,48]. These three TFs also group in the same module in the TFsNet that we inferred (Figure 1e). Another example concerns the AtGRF genes, of the GRF family, which are expressed in developing tissues, such as shoot tips, flower buds and roots. Single mutants of the AtGRF1 (AT2G22840), AtGRF2 (AT4G37740) or AtGRF3 (AT2G36400) genes have no phenotype and double mutants have minor phenotypes [49], suggesting that these three genes have redundant roles. AtGRF1, AtGRF2 and AtGRF3 group together in the TFsNet put forward here (Figure 1f). Interestingly, our network inference also recovers the interactions AtGRF3-AN3 (p-value 1e-70) and AN3-SCR (p-value 1e-40), suggesting a link between the AtGRF module and the SHR-SCR module during root development.
The TFsNet also recovers transcriptional interactions between genes known to participate in root physiological processes other than development. A first example concerns genes involved in jasmonate response (Figure 1g). This group includes the TIFY domain genes JAZ1 (AT1G19180), JAZ2 (AT1G74950), JAZ5 (AT1G17380), JAZ6 (AT1G72450), JAZ7 (AT2G34600), JAZ8 (AT1G30135), JAZ9 (AT1G70700), JAS1/JAZ10 (AT5G13220), two WRKY genes involved in pathogen response, WRKY18 (AT4G31800) and WRKY40 (AT1G80840) [50], the bHLH-family AIB (AT2G46510) [51] and MYC2 (AT1G32640) [52] and the AP2/ERF RRTF1 (AT4G34410). Interestingly, chromatin immunoprecipitation experiments have shown that WRKY40 binds JAZ8 and RRTF1 regulatory regions [53], while MYC2 was recently shown to be involved in jasmonate-dependent root development inhibition [54].
A second example includes the bHLH TF BHLH038 (AT3G56970), BHLH039 (AT3G56980), BHLH100 (AT2G41240), BHLH101 (AT5G04150), POPEYE (PYE; AT3G47640) and the DNA-binding protein-coding BRUTUS (BTS; AT3G18290), which are involved in iron deficiency stress regulation [55,56]. BHLH039, BHLH101, PYE and BTS are grouped together in the TFsNet (Figure 1h; BHLH038 and BHLH100 are not represented in the ATH1 chip).
A third example involves nitrate response TFs [57]. The earliest TFs to be expressed in response to nitrate stimulus are HRS1 (AT1G13300), LBD37 (AT5G67420), LDB38 (AT3G49940), LBD39 (AT4G37540) and AT3G25790 (cluster 1 in [57]). Four of these five TFs, HRS1, LDB38, LBD39 and AT3G25790 are grouped together in the TFsNet (Figure 1h). Note that the microarray data for Long et al., E-GEOD-21443, and Krouk et al., E-GEOD-20044 in the EBI database, were released a few days after our microarray experiments download and are not part of the data used for our analysis.
Using the FullNet to integrate and analyze high-throughput functional genomics data
The FullNet was obtained from data which included all 22810 probesets present in the ATH1 chip, and was centered on the 2088 TF probesets list (Additional file 3). In this network, TFs will be central nodes, with their interactors, either TFs or non-TFs, as neighboring nodes. Genes participating in the same processes should again be grouped together. For example, the TF groups identified in the TFsNet are still present in the same groups in the FullNet. One must bear in mind that, in this network, non-TF nodes are present. When a non-TF interacts with two TFs, and these interactions have better MI scores than the TF-TF interaction, then the latter interaction will, at DPI 0.0, be considered an indirect interaction, and thus will not appear in the network. However, this does not mean that the TF-TF interaction does not exist, only that it is “masked” by an intermediary non-TF node. When the TF-TF MI value is not the lowest in a triangle it is visible in the DPI 0.0 FullNet. This is the case for the interactions between PLT1, PLT2 and PLT3/AIL6, at p-values of 1e-50, the SCR-SHR interaction at a p-value of 1e-30, the interaction of the early nitrate-responsive TF HRS1 with LBD38, LDB39 and AT3G25790 at p-values of 1e-40 and lower, as well as the interaction of BHLH039 with BHLH101 at a p-value of 1e-60 and with PYE at a p-value of 1e-20. Interaction between AGL71 and AGL72, which was present at a p-value of 1e-20 in the TFsNet, is now recovered with a p-value of 1e-50. These two MADS-box genes have recently been shown to act redundantly in apical and axillary meristems [58].
In the FullNet, interactors of a TF node are potential target genes for that TF. If this is the case, one would expect a significant number of experimentally identified target genes for that TF to be present in the corresponding lists of ARACNe interactors. One example of a TF for which ARACNe-inferred interactions are confirmed experimentally corresponds to VND7/ANAC030 (AT1G71930). VND7 is a NAC-family TF involved in secondary cell wall synthesis and several lists of its putative target genes are available [59-62]. We compared these lists of experimentally identified VND7 target genes with our list of VND7 interactors from the complete dataset at DPI 0.0, 0.1 and 0.2 (Table 3 and Additional file 5). 14 out of 16 genes at DPI 0.0, 24 out of 44 at DPI 0.1 and 24 out of 107 at DPI 0.2 from our VND7 neighbor list are differentially expressed in at least one of the experimental settings. Almost all differentially expressed genes are found at high MI values, corresponding to p-values of 1e-50 and lower. Finally, three of the four differentially expressed TFs identified by Yamaguchi et al. [62], JLO (AT4G00220), MYB46 (AT5G12870) and MYB103 (AT1G63910), are part of the VND7 cluster in the TFsNet, at p-values of 1e-50 and lower. Curiously, a top-ranked VND7 interactor in our dataset, the pinoresinol reductase ATPRR1 (AT1G32100), is not present in any of the experimental VND7 target genes lists. ATPRR1 has, at DPI 0.0, TF interactors with higher MI values than VND7, suggesting that it could instead be regulated by one, or more, of these higher-score TFs. Alternatively, the VND7-ATPRR1 transcriptional interaction could be age-specific and not detectable in any of the above-mentioned experimental settings.
Table 3.
List of ANAC030 / VND7 interactors in the FullNet obtained at DPI 0.0
| Locus | Symbol | Short description | MI | p-value | Target gene |
|---|---|---|---|---|---|
| AT3G62160 |
|
HXXXD-type acyl-transferase family protein |
0.421448 |
1e-70 |
yesa |
| AT5G60720 |
|
Protein of unknown function, DUF547 |
0.376219 |
1e-60 |
yesabcd |
| AT1G32100 |
ATPRR1 |
Pinoresinol reductase 1 |
0.375341 |
1e-60 |
no |
| AT1G01900 |
ATSBT1.1 |
Subtilase family protein |
0.375002 |
1e-60 |
yesbcd |
| AT1G54790 |
|
GDSL-like lipase/acylhydrolase superfamily protein |
0.364787 |
1e-60 |
yesabcd |
| AT2G04850 |
|
Auxin-responsive family protein |
0.363144 |
1e-60 |
yesacd |
| AT3G27200 |
|
Cupredoxin superfamily protein |
0.362569 |
1e-60 |
yesabcd |
| AT2G27740 |
|
Family of unknown function, DUF662 |
0.328449 |
1e-50 |
yesc |
| AT5G01930 |
|
Glycoside hydrolase, family 5 |
0.324593 |
1e-50 |
yesacd |
| AT1G47410 |
|
Unknown protein |
0.293837 |
1e-50 |
yesc |
| AT5G59845 |
|
Gibberellin-regulated family protein |
0.27815 |
1e-50 |
yesac |
| AT5G38610 |
|
Plant invertase/pectin methylesterase inhibitor superfamily protein |
0.27147 |
1e-40 |
yesabcd |
| AT5G26330 |
|
Cupredoxin superfamily protein |
0.252517 |
1e-40 |
yesa |
| AT1G24600 |
|
Unknown protein |
0.243778 |
1e-40 |
yesacd |
| AT4G39320 |
|
Microtubule-associated protein-related |
0.188269 |
1e-30 |
yesc |
| AT2G31085 | CLE6 | CLAVATA3/ESR-RELATED 6 | 0.180814 | 1e-30 | no |
There are also examples of TFs for which there is little overlap between ARACNe-inferred interactors lists and experimental target gene lists. Two examples are the SHR and SCR TFs. SHR and SCR are important genes for root development and several lists of their proposed transcriptional target genes are available [29-31,63]. Sozzani et al. [30] obtained, through microarray data analysis, a comprehensive list of differentially expressed genes during a time-course of SCR or SHR induction, while Cui et al. [31] identified SHR target genes through chromatin inmunoprecipitation (ChIP). A direct comparison of the target gene lists from Sozzani et al., to which we will refer as the Sozzani-SCR and Sozzani-SHR lists, to our ARACNe list of inferred SCR or SHR interactors obtained at DPI 0.0, 0.1 and 0.2, resulted in a low overall overlap: there are 732 ARACNe-SCR and 719 ARACNe-SHR interactors at DPI 0.2, of which 68 (9.2%) and 159 (22%) were found in the corresponding SCR- or SHR-Sozzani lists. In particular, we would expect to find in both the ARACNe and Sozzani lists genes known to participate in the SHR-SCR transcriptional regulation pathway, namely JKD, MGP, NUC and CYCD6;1 (AT4G03270). The first three genes are TFs and they can be found in the same module as SHR and SCR in the TFsNet. CYCD6;1, a non-TF, is present in both the SCR-Sozzani and SHR-Sozzani lists, but is not an ARACNE-inferred interactor of SHR, SCR, JKD, MGP nor NUC. At DPI 0.0 its only interacting TF is AGL92 (AT1G31640), which is not close to the SHR-SCR module in either the TFsNet or FullNet. While disappointing, this result is perhaps not surprising: CYCD6;1 is expressed in very particular wild type root cell types, the cortex/endodermis initial stem cells and lateral root primordium endodermal cells [30,64]. Furthermore, CYCD6;1 participates in a complex regulatory mechanism involving protein-protein interactions, protein phosphorylation and protein degradation [64]. It is likely that these mechanisms are poorly translated into transcript levels of the corresponding genes in whole root samples, which is the input data for ARACNe.
The ability of ARACNe to recover experimentally identified TF target genes will most likely mirror the number and complexity of the regulatory interactions in which that TF participates. VND7 is a TF involved exclusively in secondary cell-wall synthesis (SCWS) [65,66]. As such, we expect VND7 to participate in a very specific transcriptional module, and ARACNe to accurately recover its experimentally identified target genes. On the other hand, SHR and SCR are most likely involved in numerous transcriptional pathways, as mutants for these genes are strongly affected in root development [67,68], and over 200 TFs can be found in the lists of differentially expressed genes for SHR or SCR inductions, which analyzed a specific root cell-type, i.e. ground tissue [30]. Such an important number of differentially expressed TFs (approximately 10% of all Arabidopsis TFs) further suggests that a significant number of these experimentally identified target genes are indirect targets. Additionally, regulation of root development by SCR and SHR involves expression in defined cell types, transport across cell-types, nucleus-cytoplasm translocation, protein-protein interactions and protein phosphorylation [27,30,34,35,64]. In this case, we expect that better results could be obtained by visualizing experimentally identified target genes in the context of the networks where they participate. We therefore decided to retrieve from the FullNet dataset, obtained at DPI 0.0 and with a cutoff p-value of 1e-30, all interactions for which both nodes are present in the list of 2481 differentially expressed genes in the SHR induction kinetic from Sozzani and collaborator’s study [30], to which we added SHR (AT4G37650). The resulting dataset now contains 1668 genes (67% of the original list) and the corresponding network was drawn with Cytoscape [69]. 1647 nodes (66%), including SHR, are grouped together in a single subnetwork (Figures 2a-d). We observe that this subnetwork is clearly divided in two sections, corresponding to genes that, as time progresses in the induction kinetic, switch from an under-expressed to an over-expressed state and vice versa. An analysis of this subnetwork can now help identify relevant nodes, which should play important roles in the SHR transcriptional pathway. For example, three of the main nodes that switch from under- to over-expression are PRMT3 (AT3G12270), KYP (AT5G13960) and HD2A (AT3G44750), which are genes coding for chromatin modification (histone methyl-transferase and histone deacetylase) proteins. An analysis of all genes that switch from under- to over-expression when using David [70,71] and Enrichment Map [72] reveals that this module is enriched, among others, in cell-cycle, microtubule, RNA-processing and putative chromatin modification protein-coding genes (Figure 2e).
Figure 2.

Subnetwork of differentially expressed genes in a SHR induction time-course [30]. Node colors correspond to down-regulated (green) or up-regulated (magenta) genes at 1 hour (a), 3 hours (b), 6 hours (c) and 12 hours (d) after SHR induction. (e) Enrichment Map [72] network of category enrichments calculated with David [70,71] for genes in the subnetwork that switch from a down-regulated to an up-regulated status during the induction kinetic.
ARACNe-inferred networks allow for the prediction of novel genetic interactions for root-expressed TFs: a possible role for SPATULA in the PLETHORA pathway
The TFsNet was obtained from data which included exclusively our list of 2088 TF probesets (see Methods). In this network, TFs that participate in the same biological process should be grouped together. Therefore, we expect higher order mutant plants for genes in a same module to exhibit root phenotypes not observed in single mutant plants. We set to test this hypothesis with genes that are present in the same module, but 1) belong to different TF families, 2) are not immediate neighbors in the TFsNet, and 3) whose mutants have distinct root phenotypes. The genes BABY BOOM (BBM) and SPATULA (SPT) matched these criteria. Both genes are present in the same module (Figure 1c), and mutants of the BBM gene, an AP2-domain TF, have slightly shorter roots [26], while mutants of the SPT gene, a bHLH TF, have slightly longer roots than wild type plants [73]. When grown on vertical plates, the bbm-2/spt-2 double mutant exhibited longer roots than either spt-2 or bbm-2 single mutant seedlings (Figure 3). A previous report showed that PIN4 and DR5::GUS expression is altered in the root meristem of spt-11 mutant seedlings [73]. Taken together, these results point to a possible transcriptional interaction between the PLETHORA pathway and SPATULA in the regulation of auxin transport and/or response in Arabidopsis root meristems.
Figure 3.

Photographs of bbm-2, spt-2 and bbm-2/spt-2 six days-old mutant seedlings grown on vertical plates. Bar represents 0.5 cm.
ARACNe-inferred networks allow for the prediction of novel functions for root-expressed TFs: the case of XAL1/AGL12, a MADS-box TF involved in secondary cell-wall synthesis
Since our ARACNe inferred networks are able to recover known gene associations, we expect them to also be able to predict novel TF functions. As an example of the predictive power of our database, we decided to look for new TFs that could be participating in secondary cell wall synthesis (SCWS). For this aim, our strategy consisted in selecting several genes, both TF and non-TF, known to be involved in SCWS, recover their interactions from the FullNet and draw the resulting network in order to identify new SCWS TFs. Several TFs are known to be involved in SCWS, among which we chose VND6/ANAC101 (AT5G62380), VND7/ANAC030 (AT1G71930) [74], SND2/ANAC073 (AT4G28500) [75], MYB46[76] and IXR11 (AT1G62990) [77]. As SCWS non-TF genes we chose the cellulose synthases CESA4 (AT5G44030), CESA7 (AT5G17420) and CESA8 (AT4G18780) [78], the laccases LAC4 (AT2G38080) and LAC17 (AT5G60020) [79], the cysteine peptidases XCP1 (AT4G35350) and XCP2 (AT1G20850) [80], the chitinase-like ATCTL2 (AT3G16920) [81], the DUF6 domain WAT1 (AT1G75500) [82], TED6 (AT1G43790) [83], the DUF231 domain TBL3 (AT5G01360) [84] and the family 8 glycosyl-transferase GAUT12/IRX8 (AT5G54690) [85]. We then retrieved from the FullNet all interactions involving these genes at DPI 0.0 and a p-value cutoff of 1e-30 and used Cytoscape [69] to visualize the corresponding network (Additional file 6). It immediately appears that these genes are indeed part of a network of SCWS genes that includes our input genes plus several other known, or putative, SCWS genes including MYB83 (AT3G08500) [86], ANAC007/VND4 (AT1G12260) [65] or ATPRR1[87], but also vascular development TFs like ATHB-15[43], ATHB-16 (AT4G40060) [88] and JLO (AT4G00220) a target of VND7 [62].
In a highly connected part of this SCWS network, 22 TFs that were not part of our input gene list are now present (Figure 4). We retrieved from the FullNet all interactions involving these TFs at DPI 0.0, 0.1 and 0.2 and a p-value cutoff of 1e-30. An enrichment analysis, using David, of the lists of interactors for three of the newly identified TFs, XAL1/AGL12 (a MADS-box), BEE2 and AT1G68810 (two bHLH) revealed that they are particularly enriched in SCWS genes (data not shown); the lists of high MI value interactors for each TF are shown in Additional file 7. As these three TFs are present in the highly connected part of the SCWS network, it is not surprising to find that they share several of their interactors. AGL12/XAL1 is a MADS-box transcription factor that is expressed in phloem tissues and is involved in the regulation of both root development and flowering time [89]. BEE2 was first identified as a brassinosteroid-responsive TF [90]. Brassinosteroids promote root growth [91], are essential for the development of the vascular system in Arabidopsis stems [92] and enhance xylem vessel transdifferentiation of Arabidopsis suspension cultures [74]. AT1G68810 is 1) a TF that we found as part of the vascular development cluster in the TFsNet, 2) closely related to ATAIG1/TMO5, which is also part of the TFs-only vascular development cluster and 3) a protein-protein interactor of LONESOME HIGHWAY, a transcriptional activator involved in vascular development [93]. These results predict that XAL1, BEE2 and AT1G68810 are important TFs for SCWS.
Figure 4.

Highly connected part of the SCWS network obtained at DPI 0.0 and a p-value cutoff of 1e-30. Genes are represented as nodes and inferred interactions as edges. TFs not present in the input list are colored yellow. The TFs AT1G68810, BEE2 and AGL12 (XAL1) are further mentioned in the text and colored orange. Edge width is proportional to the Mutual Information (MI) value of the interaction, with higher MI values corresponding to thicker edges.
As MADS-box TFs are not usually associated with SCWS, we decided to look for SCW deposition in xal1-2 loss-of-function mutant roots [89]. Since xal1-2 presents a delay in flowering time, roots from plants of the same chronological age might reveal developmental stage-related SCWS differences rather than a direct SCWS phenotype. Therefore, both Col-0 and xal1-2 roots were collected when the main stem was 29–32 cm in length, which arguably corresponds to plants at the same developmental stage. As predicted by our inferred network, xal1-2 adult roots indeed have altered secondary cell-wall patterns with gaps in the secondary xylem and fiber ring (n = 10/10), a phenotype rarely observed in wild type plants of the same size (n = 1/10; Figure 5). In an intriguing paper, Sibout et al. have shown that xylem expansion in hypocotyls and roots is linked to flowering time [94]. Coincidentally, xal1-2 plants have delayed flowering [89] and altered root SCWS, strongly suggesting that XAL1 could be part of a TRN that connects both processes.
Figure 5.
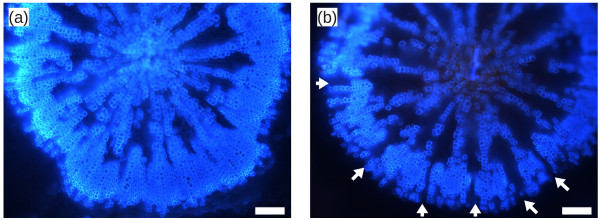
Photographs of wild type (a) and xal1-2 (b) adult root transverse sections observed under UV light. Autofluorescence of lignified tissues is evidenced as a light-blue coloration. White arrows in xal1-2 indicate gaps in the xylem-fiber ring. Bars represent 100 microns.
The confirmation of SCWS alterations in xal1-2 root tissues shows that our bioinformatics methodology to infer TRNs is a successful approach for the accurate prediction of novel functions for root-expressed TFs. This result further strengthens that our networks will likely provide novel hypothesis concerning functional modules involved in root development. As an additional example, the DUF6 protein WAT1 [82] has, at DPI 0.0 and a p-value cutoff of 1e-50, the TF interactors ATHB-15/CNA, AT1G68810, AT4G29100, STH2, ATHB-16 and AT2G28510, all of which are part of the vascular development cluster of the TFsNet (Figure 1d). This suggests, first, that one, or more, of these TFs is the transcriptional regulator of WAT1 in root tissues and, second, that one, or more, of these TFs control vascular development, at least partly, through the direct transcriptional regulation of WAT1. Finally, the DUF6-domain protein-coding genes AT1G43650, AT1G01070, AT3G45870, AT3G18200 and AT4G30420 are interactors of TFs known to be involved in SCWS, suggesting that they might have similar roles to WAT1 in root SCWS.
Conclusions
In this work we show that network inference from multiple compounded, carefully selected and curated microarray datasets allows for the reconstruction of reliable root transcriptional interaction networks. We show that such inferred networks recover both known, functionally characterized TF modules and reliably predict novel components of such modules, as well as novel modules, including unexpected roles for particular TFs. We particularly highlight the discovery of a new module underlying secondary cell wall synthesis that involves the MADS-box TF XAL1/AGL12. Our transcriptional interactions database further provides an overview of the transcriptional pathways present in Arabidopsis roots and will likely yield a plethora of novel hypotheses to be tested experimentally.
Methods
Microarray data
A list of all microarray experiments using the Affymetrix GeneChip ATH1-121501 was downloaded on October 2010 from the EBI ArrayExpress database [8] (Additional file 1). Using the corresponding sample-data relationship files as a guide, all experiments using root tissues were selected and the corresponding CEL files were retrieved. In experiments involving tissue comparisons, for example shoot vs root, particular care was taken to exclude non-root CEL files. Also, in order to obtain a high quality, homogeneous dataset, the arrayQualityMetrics Bioconductor package [95] was ran on each experiment and low quality CEL files were excluded from further analysis, as were CEL files corresponding to samples from ecotypes other than Columbia-0. Finally, in order to avoid possible perturbations of the underlying Gene Regulatory Network [96], all CEL files corresponding to transgenic samples (mutants, overexpressions, promoter constructions) were also excluded. This resulted in 656 CEL files that were normalized using gcRMA [97] under R. The resulting normalized data was used as input for the ARACNe algorithm.
For TFs-only networks, the selected root CEL files were transformed to ASCII format using the celutil utility [98] and the ATH1-121501 array name was replaced with a custom name. A TFs-only CDF file was created using a modified version of the XSpecies [99] use_ME.pl Perl script [100] and the Affymetrix ATH1-121501 probe_tab file was renamed to match the custom CDF name. Both files were packaged for R using the makecdfenv and AnnotationDbi packages, respectively, which allowed us to normalize the modified CEL files with gcRMA [97]. The resulting normalized data was used as input for the ARACNe algorithm.
Transcription factors list
The -e or Data Processing Inequality ARACNe parameter uses a list of TF in order to preserve interactions including one or more TFs [21]. There are three Arabidopsis TF databases, Agris [1,2], RARTF [3,4] and DATF [5,6]. As DATF includes the Aux/IAA family as a TF family, while Agris and DATF do not, and since auxin is a major player in plant development, we decided to create our own TF list by combining all AGI IDs present in the three databases. We further added all AGI IDs from the TAIR10 ATH_GO_GOSLIM file that were annotated with the Gene Ontology entry GO:0003700, “sequence-specific DNA binding transcription factor activity”, as several TFs, like AGL26 or AGL64, were missing from the databases. The final TF list contains 2575 AGI IDs corresponding to 2088 probesets (Additional file 8).
Network inference using ARACNe
Normalized data was used to calculate the config_kernel.txt and config_threshold.txt parameters required by ARACNe using the author provided Matlab scripts. Interactions were inferred for the 2088 TF probesets using the Linux command-line ARACNe 32 bit program at three DPI values, 0.0, 0.1 and 0.2, with the 2088 probeset list as the -l parameter for the complete dataset or without the -l parameter for the TF-only dataset. The command-line execution of the ARACNe program was in the form: aracne –H config_parameters -i normalized_data [-l TF-list] -e 0.0/0.1/0.2 -o output_file.
Adjacency files transformation
The TAIR10 array_elements and aliases tables were combined in order to obtain a single table linking probesets to AGI IDs and, when available, their corresponding symbols. The resulting combined table was used to transform the ARACNe output adjacency files to Cytoscape compatible, tab-delimited tables using a custom Perl script.
List of root-expressed TFs
Root expressed TFs were identified by combining a) the list of TFs detected in 14 days-old seedling roots by real-time RT-PCR [23], b) a list of proteins identified in large scale proteomic screens of root samples, experiments 3332, 15486, 15489, 15517, 15518, 15519, 15525, 15526, 15528 in the EBI PRIDE database [101-103], c) the list of genes annotated as being expressed in roots or whole plant in the TAIR10 Plant Ontology table, i.e. containing a Gene Ontology experimental evidence code, excluding microarray evidence, and d) a list of root expressed genes from RNA-seq experiments, accessions SRR314814 [104] and SRR331219 from the DNA Data Bank of Japan Sequence Read Archive [105]. Primers used by Czechowski et al. [23] were Blasted to the TAIR10 genome to confirm that they were still specific for the intended genes. For RNA-seq data, fastq reads were aligned to the TAIR10 cDNA sequences using bowtie2 (version 2.0.0-beta5; [106]) with the --very-sensitive, -N 1, -k 11 and -S,0,-0.8 parameters in end-to-end mode. Only reads matching a single locus were considered for the identification of expressed genes using a custom Perl script.
bbm-2/spt-2 double mutant
bbm-2 and spt-2 seeds were obtained from the Arabidopsis Biological Resource Center. Adult bbm-2 and spt-2 plants were crossed and homozygous double mutant plants were identified by genotyping of the bbm-2 mutation using the BBM primers 5'-ACTTTAGTGCGGCTAAATCGTAAGC-3′, 5′-CAATAACGAACAAAATGGACCAAAG-3′ and LBb1.3 primer 5′-ATTTTGCCGATTTCGGAAC-3′, and by visual identification of plants exhibiting the spt-2 split carpels phenotype. Seeds for both single mutants and the homozygous double mutant were sown on 0.5X Murashige and Skoog basal medium, 0.5% saccharose, 1% plant agar (PhytoTechnology Laboratories) plates. Plates were placed at 4 °C, and after two days transferred to a growth chamber at 22 °C with a long day light regime (16 hours light, 8 hours dark). Photographs were taken at six days post-germination.
Microscopy
Col-0 and xal1-2[89] plants were grown in soil under standard greenhouse conditions. Plants with a 29–32 cm high main stem were collected, transverse sections of the root below the hypocotyl were hand-cut and autofluorescence of the lignified tissues was immediately observed under UV light with a fluorescence microscope.
Abbreviations
TF: Transcription factor; DPI: Data processing inequality; AGI ID: Arabidopsis Genome Initiative locus identification number.
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
RACM participated in the design of the study, realized all bioinformatics analysis, performed experiments and wrote the manuscript. GC participated in the design of the study and participated in data analysis. KLGA and NMM performed experiments. SdF helped to write the manuscript. ERAB conceived the study, participated in the design of the study, discussed data analyses and results, and helped to write the manuscript. All authors read and approved the final manuscript.
Supplementary Material
Table of ArrayExpress experiments used as microarray data sources for this work.
Cytoscape compatible table for the TFsNet obtained at DPI 0.0.
Cytoscape compatible table for the FullNet obtained at DPI 0.0.
Table of experimental evidence of root expression for all TFs present in the TFsNet and FullNet.
List of ANAC030 / VND7 inferred interactors in the FullNet obtained at DPI 0.0, 0.1 and 0.2.
Figure of the SCWS subnetwork obtained at DPI 0.0 and a p-value cutoff of 1e-30. Genes are represented as nodes and inferred interactions as edges. Nodes corresponding to the input genes mentioned in the text are colored green. Edge width is proportional to the Mutual Information (MI) value of the interaction, with higher MI values corresponding to thicker edges.
Table of XAL1/AGL12, AT1G68810 and BEE2 inferred interactors in the FullNet obtained at DPI 0.0, 0.1 and 0.2. Only interactors with a p-value of 1e-50 or less are shown.
List of Arabidopsis loci considered as TFs in this work.
Contributor Information
Ricardo A Chávez Montes, Email: rchavez@langebio.cinvestav.mx.
Gerardo Coello, Email: gcoello@ifc.unam.mx.
Karla L González-Aguilera, Email: kgonzalez@langebio.cinvestav.mx.
Nayelli Marsch-Martínez, Email: nmarsch@ira.cinvestav.mx.
Stefan de Folter, Email: sdfolter@langebio.cinvestav.mx.
Elena R Alvarez-Buylla, Email: eabuylla@gmail.com.
Acknowledgments
We would like to thank the Arabidopsis Biological Resource Center for seeds. RACM was a recipient of postdoctoral grants from Instituto de Ciencia y Tecnología del Distrito Federal (BI09-570) and Centro de Ciencias de la Complejidad, UNAM. Work in the SdF lab was supported by the Mexican National Council of Science and Technology (CONACyT) grant 177739. Work in ERAB’s lab is supported by grants: CONACyT (81542, 81433, 167705, 152649, 180380,180098), PAPIIT (IN229003-3, IN204011-3, 226510–3, IB201212-2), UC-MEXUS (CN.12-623, CN.12-571), and REDES TEMÁTICAS DE INVESTIGACIÓN CONACyT: Red Complejidad, Ciencia y Sociedad.
References
- Arabidopsis transcription factor database (AtTFDB) http://arabidopsis.med.ohio-state.edu/AtTFDB/
- Yilmaz A, Mejia-Guerra MK, Kurz K, Liang X, Welch L, Grotewold E. AGRIS: the Arabidopsis Gene Regulatory Information Server, an update. Nucleic Acids Res. 2011;39:D1118–D1122. doi: 10.1093/nar/gkq1120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- RIKEN Arabidopsis Transcription Factor database (RARTF) http://rarge.psc.riken.jp/rartf/
- Iida K, Seki M, Sakurai T, Satou M, Akiyama K, Toyoda T, Konagaya A, Shinozaki K. RARTF: database and tools for complete sets of Arabidopsis transcription factors. DNA Res. 2005;12:247–256. doi: 10.1093/dnares/dsi011. [DOI] [PubMed] [Google Scholar]
- The Database of Arabidopsis Transcription Factors (DATF) http://datf.cbi.pku.edu.cn/ [DOI] [PubMed]
- Guo A, He K, Liu D, Bai S, Gu X, Wei L, Luo J. DATF: a database of Arabidopsis transcription factors. Bioinformatics. 2005;21:2568–2569. doi: 10.1093/bioinformatics/bti334. [DOI] [PubMed] [Google Scholar]
- Brady SM, Zhang L, Megraw M, Martinez NJ, Jiang E, Yi CS, Liu W, Zeng A, Taylor-Teeples M, Kim D, Ahnert S, Ohler U, Ware D, Walhout AJM, Benfey PN. A stele-enriched gene regulatory network in the Arabidopsis root. Mol Sys Biol. 2011;7:459. doi: 10.1038/msb.2010.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ArrayExpress. http://www.ebi.ac.uk/arrayexpress/
- Genevestigator. https://www.genevestigator.com/gv/
- Hruz T, Laule O, Szabo G, Wessendorp F, Bleuler S, Oertle L, Widmayer P, Gruissem W, Zimmermann P. Genevestigator v3: a reference expression database for the meta-analysis of transcriptomes. Adv Bioinformatics. 2008;2008:420747. doi: 10.1155/2008/420747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ATTED-II. http://atted.jp.
- Obayashi T, Nishida K, Kasahara K, Kinoshita K. ATTED-II updates: condition-specific gene coexpression to extend coexpression analyses and applications to a broad range of flowering plants. Plant Cell Physiol. 2011;52:213–219. doi: 10.1093/pcp/pcq203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BAR Expression Angler. http://bar.utoronto.ca/ntools/cgi-bin/ntools_expression_angler.cgi.
- Winter D, Vinegar B, Nahal H, Ammar R, Wilson GV, Provart NJ. An “Electronic Fluorescent Pictograph” browser for exploring and analyzing large-scale biological data sets. PLoS One. 2007;2:e718. doi: 10.1371/journal.pone.0000718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- AtGenExpress. http://www.weigelworld.org/resources/microarray/AtGenExpress/
- Margolin AA, Nemenman I, Basso K, Wiggins C, Stolovitzky G, Dalla Favera R, Califano A. ARACNE: an algorithm for the reconstruction of gene regulatory networks in a mammalian cellular context. BMC Bioinformatics. 2006;7(Suppl 1):S7. doi: 10.1186/1471-2105-7-S1-S7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Basso K, Margolin AA, Stolovitzky G, Klein U, Dalla-Favera R, Califano A. Reverse engineering of regulatory networks in human B cells. Nat Genet. 2005;37:382–390. doi: 10.1038/ng1532. [DOI] [PubMed] [Google Scholar]
- Basso K, Saito M, Sumazin P, Margolin AA, Wang K, Lim W-K, Kitagawa Y, Schneider C, Alvarez MJ, Califano A, Dalla-Favera R. Integrated biochemical and computational approach identifies BCL6 direct target genes controlling multiple pathways in normal germinal center B cells. Blood. 2010;115:975–984. doi: 10.1182/blood-2009-06-227017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Agnelli L, Forcato M, Ferrari F, Tuana G, Todoerti K, Walker BA, Morgan GJ, Lombardi L, Bicciato S, Neri A. The reconstruction of transcriptional networks reveals critical genes with implications for clinical outcome of multiple myeloma. Clin Cancer Res. 2011;17:7402–7412. doi: 10.1158/1078-0432.CCR-11-0596. [DOI] [PubMed] [Google Scholar]
- Yu X, Li L, Zola J, Aluru M, Ye H, Foudree A, Guo H, Anderson S, Aluru S, Liu P, Rodermel S, Yin Y. A brassinosteroid transcriptional network revealed by genome-wide identification of BESI target genes in Arabidopsis thaliana. Plant J. 2011;65:634–646. doi: 10.1111/j.1365-313X.2010.04449.x. [DOI] [PubMed] [Google Scholar]
- Margolin AA, Wang K, Lim WK, Kustagi M, Nemenman I, Califano A. Reverse engineering cellular networks. Nat Protoc. 2006;1:662–671. doi: 10.1038/nprot.2006.106. [DOI] [PubMed] [Google Scholar]
- Zilliox MJ, Irizarry RA. A gene expression bar code for microarray data. Nat Methods. 2007;4:911–913. doi: 10.1038/nmeth1102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Czechowski T, Bari RP, Stitt M, Scheible W-R, Udvardi MK. Real-time RT-PCR profiling of over 1400 Arabidopsis transcription factors: unprecedented sensitivity reveals novel root- and shoot-specific genes. Plant J. 2004;38:366–379. doi: 10.1111/j.1365-313X.2004.02051.x. [DOI] [PubMed] [Google Scholar]
- Sabatini S, Heidstra R, Wildwater M, Scheres B. SCARECROW is involved in positioning the stem cell niche in the Arabidopsis root meristem. Gene Dev. 2003;17:354–358. doi: 10.1101/gad.252503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aida M, Beis D, Heidstra R, Willemsen V, Blilou I, Galinha C, Nussaume L, Noh Y-S, Amasino R, Scheres B. The PLETHORA genes mediate patterning of the Arabidopsis root stem cell niche. Cell. 2004;119:109–120. doi: 10.1016/j.cell.2004.09.018. [DOI] [PubMed] [Google Scholar]
- Galinha C, Hofhuis H, Luijten M, Willemsen V, Blilou I, Heidstra R, Scheres B. PLETHORA proteins as dose-dependent master regulators of Arabidopsis root development. Nature. 2007;449:1053–1057. doi: 10.1038/nature06206. [DOI] [PubMed] [Google Scholar]
- Welch D, Hassan H, Blilou I, Immink R, Heidstra R, Scheres B. Arabidopsis JACKDAW and MAGPIE zinc finger proteins delimit asymmetric cell division and stabilize tissue boundaries by restricting SHORT-ROOT action. Gene Dev. 2007;21:2196–2204. doi: 10.1101/gad.440307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Azpeitia E, Benítez M, Vega I, Villarreal C, Alvarez-Buylla ER. Single-cell and coupled GRN models of cell patterning in the Arabidopsis thaliana root stem cell niche. BMC Sys Biol. 2010;4:134. doi: 10.1186/1752-0509-4-134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levesque MP, Vernoux T, Busch W, Cui H, Wang JY, Blilou I, Hassan H, Nakajima K, Matsumoto N, Lohmann JU, Scheres B, Benfey PN. Whole-genome analysis of the SHORT-ROOT developmental pathway in Arabidopsis. PLoS Biol. 2006;4:e143. doi: 10.1371/journal.pbio.0040143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sozzani R, Cui H, Moreno-Risueno MA, Busch W, Van Norman JM, Vernoux T, Brady SM, Dewitte W, Murray JAH, Benfey PN. Spatiotemporal regulation of cell-cycle genes by SHORTROOT links patterning and growth. Nature. 2010;466:128–132. doi: 10.1038/nature09143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cui H, Hao Y, Kovtun M, Stolc V, Deng X-W, Sakakibara H, Kojima M. Genome-wide direct target analysis reveals a role for SHORT-ROOT in root vascular patterning through cytokinin homeostasis. Plant Physiol. 2011;157:1221–1231. doi: 10.1104/pp.111.183178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ogasawara H, Kaimi R, Colasanti J, Kozaki A. Activity of transcription factor JACKDAW is essential for SHR/SCR-dependent activation of SCARECROW and MAGPIE and is modulated by reciprocal interactions with MAGPIE, SCARECROW and SHORT ROOT. Plant Mol Biol. 2011;77:489–499. doi: 10.1007/s11103-011-9826-5. [DOI] [PubMed] [Google Scholar]
- Arabidopsis Interactome Mapping Consortium. Evidence for network evolution in an Arabidopsis interactome map. Science. 2011;333:601–607. doi: 10.1126/science.1203877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gallagher KL, Benfey PN. Both the conserved GRAS domain and nuclear localization are required for SHORT-ROOT movement. Plant J. 2009;57:785–797. doi: 10.1111/j.1365-313X.2008.03735.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gallagher KL, Paquette AJ, Nakajima K, Benfey PN. Mechanisms regulating SHORT-ROOT intercellular movement. Curr Biol. 2004;14:1847–1851. doi: 10.1016/j.cub.2004.09.081. [DOI] [PubMed] [Google Scholar]
- Rademacher EH, Möller B, Lokerse AS, Llavata-Peris CI, van den Berg W, Weijers D. A cellular expression map of the Arabidopsis AUXIN RESPONSE FACTOR gene family. Plant J. 2011;68:597–606. doi: 10.1111/j.1365-313X.2011.04710.x. [DOI] [PubMed] [Google Scholar]
- Bishopp A, Benková E, Helariutta Y. Sending mixed messages: auxin-cytokinin crosstalk in roots. Curr Op Plant Biol. 2011;14:10–16. doi: 10.1016/j.pbi.2010.08.014. [DOI] [PubMed] [Google Scholar]
- Dello Ioio R, Nakamura K, Moubayidin L, Perilli S, Taniguchi M, Morita MT, Aoyama T, Costantino P, Sabatini S. A genetic framework for the control of cell division and differentiation in the root meristem. Science. 2008;322:1380–1384. doi: 10.1126/science.1164147. [DOI] [PubMed] [Google Scholar]
- Scacchi E, Salinas P, Gujas B, Santuari L, Krogan N, Ragni L, Berleth T, Hardtke CS. Spatio-temporal sequence of cross-regulatory events in root meristem growth. PNAS. 2010;107:22734–22739. doi: 10.1073/pnas.1014716108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mähönen AP, Bishopp A, Higuchi M, Nieminen KM, Kinoshita K, Törmäkangas K, Ikeda Y, Oka A, Kakimoto T, Helariutta Y. Cytokinin signaling and its inhibitor AHP6 regulate cell fate during vascular development. Science. 2006;311:94–98. doi: 10.1126/science.1118875. [DOI] [PubMed] [Google Scholar]
- Bishopp A, Help H, El-Showk S, Weijers D, Scheres B, Friml J, Benková E, Mähönen AP, Helariutta Y. A mutually inhibitory interaction between auxin and cytokinin specifies vascular pattern in roots. Curr Biol. 2011;21:917–926. doi: 10.1016/j.cub.2011.04.017. [DOI] [PubMed] [Google Scholar]
- Lau S, De Smet I, Kolb M, Meinhardt H, Jürgens G. Auxin triggers a genetic switch. Nat Cell Biol. 2011;13:611–615. doi: 10.1038/ncb2212. [DOI] [PubMed] [Google Scholar]
- Ochando I, González-Reig S, Ripoll J-J, Vera A, Martínez-Laborda A. Alteration of the shoot radial pattern in Arabidopsis thaliana by a gain-of-function allele of the class III HD-Zip gene INCURVATA4. Int J Dev Biol. 2008;52:953–961. doi: 10.1387/ijdb.072306io. [DOI] [PubMed] [Google Scholar]
- Zhong R, Taylor JJ, Ye ZH. Disruption of interfascicular fiber differentiation in an Arabidopsis mutant. Plant Cell. 1997;9:2159–2170. doi: 10.1105/tpc.9.12.2159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donner TJ, Sherr I, Scarpella E. Regulation of preprocambial cell state acquisition by auxin signaling in Arabidopsis leaves. Development. 2009;136:3235–3246. doi: 10.1242/dev.037028. [DOI] [PubMed] [Google Scholar]
- Tatematsu K, Nakabayashi K, Kamiya Y, Nambara E. Transcription factor AtTCP14 regulates embryonic growth potential during seed germination in Arabidopsis thaliana. Plant J. 2008;53:42–52. doi: 10.1111/j.1365-313X.2007.03308.x. [DOI] [PubMed] [Google Scholar]
- Schulze S, Schäfer BN, Parizotto EA, Voinnet O, Theres K. LOST MERISTEMS genes regulate cell differentiation of central zone descendants in Arabidopsis shoot meristems. Plant J. 2010;64:668–678. doi: 10.1111/j.1365-313X.2010.04359.x. [DOI] [PubMed] [Google Scholar]
- Engstrom EM, Andersen CM, Gumulak-Smith J, Hu J, Orlova E, Sozzani R, Bowman JL. Arabidopsis homologs of the petunia hairy meristem gene are required for maintenance of shoot and root indeterminacy. Plant Physiol. 2011;155:735–750. doi: 10.1104/pp.110.168757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim JH, Choi D, Kende H. The AtGRF family of putative transcription factors is involved in leaf and cotyledon growth in Arabidopsis. Plant J. 2003;36:94–104. doi: 10.1046/j.1365-313X.2003.01862.x. [DOI] [PubMed] [Google Scholar]
- Shen Q-H, Saijo Y, Mauch S, Biskup C, Bieri S, Keller B, Seki H, Ulker B, Somssich IE, Schulze-Lefert P. Nuclear activity of MLA immune receptors links isolate-specific and basal disease-resistance responses. Science. 2007;315:1098–1103. doi: 10.1126/science.1136372. [DOI] [PubMed] [Google Scholar]
- Li H, Sun J, Xu Y, Jiang H, Wu X, Li C. The bHLH-type transcription factor AtAIB positively regulates ABA response in Arabidopsis. Plant Mol Biol. 2007;65:655–665. doi: 10.1007/s11103-007-9230-3. [DOI] [PubMed] [Google Scholar]
- Wang Z, Cao G, Wang X, Miao J, Liu X, Chen Z, Qu L-J, Gu H. Identification and characterization of COI1-dependent transcription factor genes involved in JA-mediated response to wounding in Arabidopsis plants. Plant Cell Rep. 2008;27:125–135. doi: 10.1007/s00299-007-0410-z. [DOI] [PubMed] [Google Scholar]
- Pandey SP, Roccaro M, Schön M, Logemann E, Somssich IE. Transcriptional reprogramming regulated by WRKY18 and WRKY40 facilitates powdery mildew infection of Arabidopsis. Plant J. 2010;64:912–923. doi: 10.1111/j.1365-313X.2010.04387.x. [DOI] [PubMed] [Google Scholar]
- Chen Q, Sun J, Zhai Q, Zhou W, Qi L, Xu L, Wang B, Chen R, Jiang H, Qi J, Li X, Palme K, Li C. The basic helix-loop-helix transcription factor MYC2 directly represses PLETHORA expression during jasmonate-mediated modulation of the root stem cell niche in Arabidopsis. Plant Cell. 2011;23:3335–3352. doi: 10.1105/tpc.111.089870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H-Y, Klatte M, Jakoby M, Bäumlein H, Weisshaar B, Bauer P. Iron deficiency-mediated stress regulation of four subgroup Ib BHLH genes in Arabidopsis thaliana. Planta. 2007;226:897–908. doi: 10.1007/s00425-007-0535-x. [DOI] [PubMed] [Google Scholar]
- Long TA, Tsukagoshi H, Busch W, Lahner B, Salt DE, Benfey PN. The bHLH transcription factor POPEYE regulates response to iron deficiency in Arabidopsis roots. Plant Cell. 2010;22:2219–2236. doi: 10.1105/tpc.110.074096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krouk G, Mirowski P, LeCun Y, Shasha DE, Coruzzi GM. Predictive network modeling of the high-resolution dynamic plant transcriptome in response to nitrate. Genome Biol. 2010;11:R123. doi: 10.1186/gb-2010-11-12-r123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dorca-Fornell C, Gregis V, Grandi V, Coupland G, Colombo L, Kater MM. The Arabidopsis SOC1-like genes AGL42, AGL71 and AGL72 promote flowering in the shoot apical and axillary meristems. Plant J. 2011;67:1006–1017. doi: 10.1111/j.1365-313X.2011.04653.x. [DOI] [PubMed] [Google Scholar]
- Ohashi-Ito K, Oda Y, Fukuda H. Arabidopsis VASCULAR-RELATED NAC-DOMAIN6 directly regulates the genes that govern programmed cell death and secondary wall formation during xylem differentiation. Plant Cell. 2010;22:3461–3473. doi: 10.1105/tpc.110.075036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhong R, Lee C, Ye Z-H. Global analysis of direct targets of secondary wall NAC master switches in Arabidopsis. Mol Plant. 2010;3:1087–1103. doi: 10.1093/mp/ssq062. [DOI] [PubMed] [Google Scholar]
- Yamaguchi M, Ohtani M, Mitsuda N, Kubo M, Ohme-Takagi M, Fukuda H, Demura T. VND-INTERACTING2, a NAC domain transcription factor, negatively regulates xylem vessel formation in Arabidopsis. Plant Cell. 2010;22:1249–1263. doi: 10.1105/tpc.108.064048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamaguchi M, Mitsuda N, Ohtani M, Ohme-Takagi M, Kato K, Demura T. VASCULAR-RELATED NAC-DOMAIN7 directly regulates the expression of a broad range of genes for xylem vessel formation. Plant J. 2011;66:579–590. doi: 10.1111/j.1365-313X.2011.04514.x. [DOI] [PubMed] [Google Scholar]
- Cui H, Levesque MP, Vernoux T, Jung JW, Paquette AJ, Gallagher KL, Wang JY, Blilou I, Scheres B, Benfey PN. An evolutionarily conserved mechanism delimiting SHR movement defines a single layer of endodermis in plants. Science. 2007;316:421–425. doi: 10.1126/science.1139531. [DOI] [PubMed] [Google Scholar]
- Cruz-Ramírez A, Díaz-Triviño S, Blilou I, Grieneisen VA, Sozzani R, Zamioudis C, Miskolczi P, Nieuwland J, Benjamins R, Dhonukshe P, Caballero-Pérez J, Horvath B, Long Y, Mähönen AP, Zhang H, Xu J, Murray JAH, Benfey PN, Bako L, Marée AFM, Scheres B. A bistable circuit involving SCARECROW-RETINOBLASTOMA integrates cues to inform asymmetric stem cell division. Cell. 2012;150:1002–1015. doi: 10.1016/j.cell.2012.07.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kubo M, Udagawa M, Nishikubo N, Horiguchi G, Yamaguchi M, Ito J, Mimura T, Fukuda H, Demura T. Transcription switches for protoxylem and metaxylem vessel formation. Gene Dev. 2005;19:1855–1860. doi: 10.1101/gad.1331305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamaguchi M, Kubo M, Fukuda H, Demura T. Vascular-related NAC-DOMAIN7 is involved in the differentiation of all types of xylem vessels in Arabidopsis roots and shoots. Plant J. 2008;55:652–664. doi: 10.1111/j.1365-313X.2008.03533.x. [DOI] [PubMed] [Google Scholar]
- Benfey PN, Linstead PJ, Roberts K, Schiefelbein JW, Hauser MT, Aeschbacher RA. Root development in Arabidopsis: four mutants with dramatically altered root morphogenesis. Development. 1993;119:57–70. doi: 10.1242/dev.119.Supplement.57. [DOI] [PubMed] [Google Scholar]
- Scheres B, Di Laurenzio L, Willemsen V, Hauser MT, Janmaat K, Weisbeek P, Benfey PN. Mutations affecting the radial organisation of the Arabidopsis root display specific defects throughout the embryonic axis. Development. 1995;121:53–62. [Google Scholar]
- Smoot ME, Ono K, Ruscheinski J, Wang P-L, Ideker T. Cytoscape 2.8: new features for data integration and network visualization. Bioinformatics. 2011;27:431–432. doi: 10.1093/bioinformatics/btq675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang DW, Sherman BT, Lempicki RA. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc. 2009;4:44–57. doi: 10.1038/nprot.2008.211. [DOI] [PubMed] [Google Scholar]
- Huang DW, Sherman BT, Lempicki RA. Bioinformatics enrichment tools: paths toward the comprehensive functional analysis of large gene lists. Nucleic Acids Res. 2009;37:1–13. doi: 10.1093/nar/gkn923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merico D, Isserlin R, Stueker O, Emili A, Bader GD. Enrichment map: a network-based method for gene-set enrichment visualization and interpretation. PLoS One. 2010;5:e13984. doi: 10.1371/journal.pone.0013984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Makkena S, Lamb RS. The bHLH transcription factor SPATULA regulates root growth by controlling the size of the root meristem. BMC Plant Biol. 2013;13:1. doi: 10.1186/1471-2229-13-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamaguchi M, Goué N, Igarashi H, Ohtani M, Nakano Y, Mortimer JC, Nishikubo N, Kubo M, Katayama Y, Kakegawa K, Dupree P, Demura T. VASCULAR-RELATED NAC-DOMAIN6 and VASCULAR-RELATED NAC-DOMAIN7 effectively induce transdifferentiation into xylem vessel elements under control of an induction system. Plant Physiol. 2010;153:906–914. doi: 10.1104/pp.110.154013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhong R, Lee C, Zhou J, McCarthy RL, Ye Z-H. A battery of transcription factors involved in the regulation of secondary cell wall biosynthesis in Arabidopsis. Plant Cell. 2008;20:2763–2782. doi: 10.1105/tpc.108.061325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhong R, Richardson EA, Ye Z-H. The MYB46 transcription factor is a direct target of SND1 and regulates secondary wall biosynthesis in Arabidopsis. Plant Cell. 2007;19:2776–2792. doi: 10.1105/tpc.107.053678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li E, Wang S, Liu Y, Chen J-G, Douglas CJ. OVATE FAMILY PROTEIN4 (OFP4) interaction with KNAT7 regulates secondary cell wall formation in Arabidopsis thaliana. Plant J. 2011;67:328–341. doi: 10.1111/j.1365-313X.2011.04595.x. [DOI] [PubMed] [Google Scholar]
- Atanassov II, Pittman JK, Turner SR. Elucidating the mechanisms of assembly and subunit interaction of the cellulose synthase complex of Arabidopsis secondary cell walls. J Biol Chem. 2009;284:3833–3841. doi: 10.1074/jbc.M807456200. [DOI] [PubMed] [Google Scholar]
- Berthet S, Demont-Caulet N, Pollet B, Bidzinski P, Cézard L, Le Bris P, Borrega N, Hervé J, Blondet E, Balzergue S, Lapierre C, Jouanin L. Disruption of LACCASE4 and 17 results in tissue-specific alterations to lignification of Arabidopsis thaliana stems. Plant Cell. 2011;23:1124–1137. doi: 10.1105/tpc.110.082792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Avci U, Petzold HE, Ismail IO, Beers EP, Haigler CH. Cysteine proteases XCP1 and XCP2 aid micro-autolysis within the intact central vacuole during xylogenesis in Arabidopsis roots. Plant J. 2008;56:303–315. doi: 10.1111/j.1365-313X.2008.03592.x. [DOI] [PubMed] [Google Scholar]
- Hossain MA, Noh H-N, Kim K-I, Koh E-J, Wi S-G, Bae H-J, Lee H, Hong S-W. Mutation of the chitinase-like protein-encoding AtCTL2 gene enhances lignin accumulation in dark-grown Arabidopsis seedlings. J Plant Physiol. 2010;167:650–658. doi: 10.1016/j.jplph.2009.12.001. [DOI] [PubMed] [Google Scholar]
- Ranocha P, Denancé N, Vanholme R, Freydier A, Martinez Y, Hoffmann L, Köhler L, Pouzet C, Renou J-P, Sundberg B, Boerjan W, Goffner D. Walls are thin 1 (WAT1), an Arabidopsis homolog of Medicago truncatula NODULIN21, is a tonoplast-localized protein required for secondary wall formation in fibers. Plant J. 2010;1:469–483. doi: 10.1111/j.1365-313X.2010.04256.x. [DOI] [PubMed] [Google Scholar]
- Endo S, Pesquet E, Yamaguchi M, Tashiro G, Sato M, Toyooka K, Nishikubo N, Udagawa-Motose M, Kubo M, Fukuda H, Demura T. Identifying new components participating in the secondary cell wall formation of vessel elements in Zinnia and Arabidopsis. Plant Cell. 2009;21:1155–1165. doi: 10.1105/tpc.108.059154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bischoff V, Nita S, Neumetzler L, Schindelasch D, Urbain A, Eshed R, Persson S, Delmer D, Scheible W-R. TRICHOME BIREFRINGENCE and its homolog AT5G01360 encode plant-specific DUF231 proteins required for cellulose biosynthesis in Arabidopsis. Plant Physiol. 2010;153:590–602. doi: 10.1104/pp.110.153320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Persson S, Caffall KH, Freshour G, Hilley MT, Bauer S, Poindexter P, Hahn MG, Mohnen D, Somerville C. The Arabidopsis irregular xylem8 mutant is deficient in glucuronoxylan and homogalacturonan, which are essential for secondary cell wall integrity. Plant Cell. 2007;19:237–255. doi: 10.1105/tpc.106.047720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCarthy RL, Zhong R, Ye Z-H. MYB83 is a direct target of SND1 and acts redundantly with MYB46 in the regulation of secondary cell wall biosynthesis in Arabidopsis. Plant Cell Physiol. 2009;50:1950–1964. doi: 10.1093/pcp/pcp139. [DOI] [PubMed] [Google Scholar]
- Nakatsubo T, Mizutani M, Suzuki S, Hattori T, Umezawa T. Characterization of Arabidopsis thaliana pinoresinol reductase, a new type of enzyme involved in lignan biosynthesis. J Biol Chem. 2008;283:15550–15557. doi: 10.1074/jbc.M801131200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishitani C, Demura T, Fukuda H. Primary phloem-specific expression of a Zinnia elegans homeobox gene. Plant Cell Physiol. 2001;42:1210–1218. doi: 10.1093/pcp/pce156. [DOI] [PubMed] [Google Scholar]
- Tapia-López R, García-Ponce B, Dubrovsky JG, Garay-Arroyo A, Pérez-Ruíz RV, Kim S-H, Acevedo F, Pelaz S, Alvarez-Buylla ER. An AGAMOUS-related MADS-box gene, XAL1 (AGL12), regulates root meristem cell proliferation and flowering transition in Arabidopsis. Plant Physiol. 2008;146:1182–1192. doi: 10.1104/pp.107.108647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedrichsen DM, Nemhauser J, Muramitsu T, Maloof JN, Alonso J, Ecker JR, Furuya M, Chory J. Three redundant brassinosteroid early response genes encode putative bHLH transcription factors required for normal growth. Genetics. 2002;162:1445–1456. doi: 10.1093/genetics/162.3.1445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Müssig C, Shin G-H, Altmann T. Brassinosteroids promote root growth in Arabidopsis. Plant Physiol. 2003;133:1261–1271. doi: 10.1104/pp.103.028662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ibañes M, Fàbregas N, Chory J, Caño-Delgado AI. Brassinosteroid signaling and auxin transport are required to establish the periodic pattern of Arabidopsis shoot vascular bundles. PNAS. 2009;106:13630–13635. doi: 10.1073/pnas.0906416106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohashi-Ito K, Bergmann DC. Regulation of the Arabidopsis root vascular initial population by LONESOME HIGHWAY. Development. 2007;134:2959–2968. doi: 10.1242/dev.006296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sibout R, Plantegenet S, Hardtke CS. Flowering as a condition for xylem expansion in Arabidopsis hypocotyl and root. Curr Biol. 2008;18:458–463. doi: 10.1016/j.cub.2008.02.070. [DOI] [PubMed] [Google Scholar]
- Kauffmann A, Gentleman R, Huber W. arrayQualityMetrics–a bioconductor package for quality assessment of microarray data. Bioinformatics. 2009;25:415–416. doi: 10.1093/bioinformatics/btn647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tischler J, Lehner B, Fraser AG. Evolutionary plasticity of genetic interaction networks. Nat Genet. 2008;40:390–391. doi: 10.1038/ng.114. [DOI] [PubMed] [Google Scholar]
- Wu Z, Irizarry RA, Gentleman R, Martinez-Murillo F, Spencer F. A model-based background adjustment for oligonucleotide expression arrays. J Am Stat Assoc. 2004;99:909–917. doi: 10.1198/016214504000000683. [DOI] [Google Scholar]
- Celutil. http://www.bioinformatics.org/celutil/
- XSpecies. http://affymetrix.arabidopsis.info/xspecies/
- Hammond JP, Broadley MR, Craigon DJ, Higgins J, Emmerson ZF, Townsend HJ, White PJ, May ST. Using genomic DNA-based probe-selection to improve the sensitivity of high-density oligonucleotide arrays when applied to heterologous species. Plant Methods. 2005;1:10. doi: 10.1186/1746-4811-1-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- PRoteomics IDEntifications database (PRIDE) http://www.ebi.ac.uk/pride/archive. [DOI] [PubMed]
- Baerenfaller K, Grossmann J, Grobei MA, Hull R, Hirsch-Hoffmann M, Yalovsky S, Zimmermann P, Grossniklaus U, Gruissem W, Baginsky S. Genome-scale proteomics reveals Arabidopsis thaliana gene models and proteome dynamics. Science. 2008;320:938–941. doi: 10.1126/science.1157956. [DOI] [PubMed] [Google Scholar]
- Baerenfaller K, Hirsch-Hoffmann M, Svozil J, Hull R, Russenberger D, Bischof S, Lu Q, Gruissem W, Baginsky S. pep2pro: a new tool for comprehensive proteome data analysis to reveal information about organ-specific proteomes in Arabidopsis thaliana. Integr Biol. 2011;3:225–237. doi: 10.1039/c0ib00078g. [DOI] [PubMed] [Google Scholar]
- Gan X, Stegle O, Behr J, Steffen JG, Drewe P, Hildebrand KL, Lyngsoe R, Schultheiss SJ, Osborne EJ, Sreedharan VT, Kahles A, Bohnert R, Jean G, Derwent P, Kersey P, Belfield EJ, Harberd NP, Kemen E, Toomajian C, Kover PX, Clark RM, Rätsch G, Mott R. Multiple reference genomes and transcriptomes for Arabidopsis thaliana. Nature. 2011;477:419–423. doi: 10.1038/nature10414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DDBJ Sequence Read Archive (DRA) http://trace.ddbj.nig.ac.jp/dra/index_e.shtml.
- Langmead B, Trapnell C, Pop M, Salzberg SL. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 2009;10:R25. doi: 10.1186/gb-2009-10-3-r25. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table of ArrayExpress experiments used as microarray data sources for this work.
Cytoscape compatible table for the TFsNet obtained at DPI 0.0.
Cytoscape compatible table for the FullNet obtained at DPI 0.0.
Table of experimental evidence of root expression for all TFs present in the TFsNet and FullNet.
List of ANAC030 / VND7 inferred interactors in the FullNet obtained at DPI 0.0, 0.1 and 0.2.
Figure of the SCWS subnetwork obtained at DPI 0.0 and a p-value cutoff of 1e-30. Genes are represented as nodes and inferred interactions as edges. Nodes corresponding to the input genes mentioned in the text are colored green. Edge width is proportional to the Mutual Information (MI) value of the interaction, with higher MI values corresponding to thicker edges.
Table of XAL1/AGL12, AT1G68810 and BEE2 inferred interactors in the FullNet obtained at DPI 0.0, 0.1 and 0.2. Only interactors with a p-value of 1e-50 or less are shown.
List of Arabidopsis loci considered as TFs in this work.