

ABSTRACT
Adhesion between cells is established by the formation of specialized intercellular junctional complexes, such as desmosomes. Desmosomes contain isoforms of two members of the cadherin superfamily of cell adhesion proteins, desmocollins (Dsc) and desmogleins (Dsg), but their combinatorial roles in desmosome assembly are not understood. To uncouple desmosome assembly from other cell–cell adhesion complexes, we used micro-patterned substrates of Dsc2aFc and/or Dsg2Fc and collagen IV; we show that Dsc2aFc, but not Dsg2Fc, was necessary and sufficient to recruit desmosome-specific desmoplakin into desmosome puncta and produce strong adhesive binding. Single-molecule force spectroscopy showed that monomeric Dsc2a, but not Dsg2, formed Ca2+-dependent homophilic bonds, and that Dsg2 formed Ca2+-independent heterophilic bonds with Dsc2a. A W2A mutation in Dsc2a inhibited Ca2+-dependent homophilic binding, similar to classical cadherins, and Dsc2aW2A, but not Dsg2W2A, was excluded from desmosomes in MDCK cells. These results indicate that Dsc2a, but not Dsg2, is required for desmosome assembly through homophilic Ca2+- and W2-dependent binding, and that Dsg2 might be involved later in regulating a switch to Ca2+-independent adhesion in mature desmosomes.
KEY WORDS: Desmosome, Adhesion, Desmocollin, Desmoglein
INTRODUCTION
Cell–cell adhesion is essential for the organization and maintenance of complex tissues in multicellular organisms. The cadherin superfamily of cell–cell adhesion proteins is essential for tissue formation throughout the Metazoa (Franke, 2009; Oda and Takeichi, 2011). Disruption of cadherin binding generally results in tissue defects, and is common in metastatic cancers (Chidgey and Dawson, 2007; Garrod, 1995; Runswick et al., 2001; Saito et al., 2012; Simpson et al., 2011). There are four major subfamilies of cadherins: the classical cadherins, desmosomal cadherins, protocadherins and atypical cadherins (Shapiro and Weis, 2009).
Desmosomes comprise two different cadherins, termed desmogleins (Dsg) and desmocollins (Dsc), that form a protein-dense adhesion complex that is required in tissues and organs that resist mechanical stress (Green and Simpson, 2007; Green et al., 1998). For example, the intercalated discs that connect heart muscle cells contain one isoform of each subtype (Dsc2 and Dsg2) (Delva et al., 2009), and mutations in Dsc2 and Dsg2 cause hereditary diseases such as arrhythmogenic right ventricular cardiomyopathy (ARVC) (Bhuiyan et al., 2009; Heuser et al., 2006) and mild striate palmoplantar keratoderma (SPPK) (Simpson et al., 2009). Several of these hereditary mutations have been mapped to the extracellular domain of Dsg2 and Dsc2 (Gaertner et al., 2012; Gehmlich et al., 2011). In the epidermis, different Dsg and Dsc isoforms are expressed in a cell-type-specific manner, and autoimmune diseases such as pemphigus foliaceus and pemphigus vulgaris target extracellular binding of Dsg1 and Dsg3, causing loss of adhesion in the epidermis and mucosal membranes (Amagai and Stanley, 2012; Waschke, 2008). The range of heart and skin defects caused by disruption of desmosomal adhesion suggests that Dsc and Dsg play important roles in cell–cell adhesion and differentiation (Kottke et al., 2006).
Desmosomal cadherins contain evolutionarily conserved domains, with low sequence similarity (∼30%) to classical cadherins, that include five extracellular ‘cadherin’ repeats, a transmembrane domain and a cytoplasmic domain that binds desmosome-specific adaptor proteins (Patel et al., 2003). Desmosomes form punctate structures at cell–cell contacts (Pasdar and Nelson, 1988a; Pasdar and Nelson, 1988b) that comprise an intercellular midline corresponding to sites of trans interactions between opposing cadherins, and a dense plaque structure in the cytoplasm that connects to intermediate filaments (Al-Amoudi et al., 2011; Al-Amoudi et al., 2007; He et al., 2003; North et al., 1999).
Analysis of the structure of classical cadherins bound in trans identified a crucial tryptophan at position 2 (W2) in the first N-terminal extracellular (EC1) repeat that forms a ‘strand swap dimer’ with the opposing cadherin (Boggon et al., 2002). An alanine replacement mutation (W2A), which inhibits strand swap dimerization, revealed a second configuration termed the X-dimer, which involves residues near the EC1–EC2 Ca2+-binding sites, and is thought to be an intermediate that facilitates the formation of the W2 strand-swap dimer (Harrison et al., 2010; Sivasankar et al., 2009). Importantly, homophilic binding of Dsc2 is blocked by mutations in W2 and A80, the latter of which contributes to the hydrophobic pocket into which W2 inserts during strand swapping (Nie et al., 2011), but there is no sequence similarity in desmosomal cadherins to residues required for the X-dimer.
The cytoplasmic domains of Dsc isoforms are derived from two different splicing events (a, b), whereas the cytoplasmic domains of Dsg isoforms are longer and might have additional functions (Brennan and Mahoney, 2009; Brennan et al., 2007; Getsios et al., 2009; Harmon et al., 2013). Biochemical studies indicate that the cytoplasmic domain of both Dsc and Dsg have binding sites for the core desmosomal adaptor protein plakoglobin (Kowalczyk et al., 1996; Troyanovsky et al., 1994b), which binds other desmosome-specific proteins, plakophilin (PKP) and desmoplakin (DP I/II, the two major proteins encoded by DSP), that link the complex to intermediate filaments (cytokeratins) (Bornslaeger et al., 1996; Kowalczyk et al., 1997; Troyanovsky et al., 1994a). There is also evidence that DP I/II can be recruited to Dsc–Dsg complexes in the absence of plakoglobin (Troyanovsky et al., 1994a; Troyanovsky et al., 1994b). The recruitment of these cytoplasmic adaptor proteins and the intermediate filament cytoskeleton is thought to promote Dsc–Dsg clustering and stabilize the structural integrity of the desmosome.
Both Dsg and Dsc are thought to be involved in extracellular trans interactions between adjacent cells (Chitaev and Troyanovsky, 1997; Dusek et al., 2007; Getsios et al., 2004; Marcozzi et al., 1998; Roberts et al., 1998; Runswick et al., 2001). How Dsc and Dsg combine to form adhesive bonds is unclear because different binding properties have been assigned to the most ubiquitously expressed isoforms (Dsg2 and Dsc2) depending on the assay used (Waschke, 2008). Knockdown of Dsc2 (Roberts et al., 1998) or knockout of Dsg2 expression (Eshkind et al., 2002) results in a loss of functional desmosomes. In addition, Dsg2 and Dsc2 can be depleted from junctions by blocking their microtubule (MT)-mediated trafficking, each resulting in weakened adhesive strength (Nekrasova et al., 2011). Cell-free studies showed that the EC1–EC2 domain of Dsc2 can form homophilic bonds and heterophilic bonds with Dsg2, but the EC1–EC2 domain of Dsg2 does not form homophilic bonds (Syed et al., 2002). In contrast, atomic force microscope (AFM) experiments with dimeric Fc fusion proteins have indicated that Dsg2 can also form homophilic bonds (Hartlieb et al., 2013; Schlegel et al., 2010). Cell-based assays in keratinocytes using cross-linking reagents found only homophilic binding for both Dsg2 and Dsc2 (Nie et al., 2011). Thus, the requirement for both Dsg and Dsc in the structural organization and maintenance of the desmosomal complex remains unclear.
To distinguish the roles of Dsc2a and Dsg2, we used three independent assays: a desmosome assembly assay using dual-patterned surfaces containing purified Dsg2, Dsc2a, or a combination of Dsc2a plus Dsg2, and collagen IV; a single-molecule binding assay with wild-type and W2A mutant monomeric Dsg2 and Dsc2a using single-molecule force spectroscopy (SMFS); and a cell-based assay to examine the organization and stability of wild-type and W2A mutant Dsg2 and Dsc2a in desmosomes in situ. Our results identify unique roles of Dsc2a and Dsg2 in desmosome organization.
RESULTS
Different requirements of desmosomal cadherins for the formation of punctate desmosomal structures
To distinguish roles of Dsg2 and Dsc2a in desmosome adhesion and assembly, we adapted a method (Borghi et al., 2010) to analyze cell binding to micro-patterned surfaces of Dsc2a and/or Dsg2, and the extracellular matrix protein collagen IV. This assay uncouples desmosome assembly from other cell adhesion complexes such as E-cadherin. The complete extracellular domain (EC1–5) of Dsg2 and Dsc2a was fused to the constant region of human IgG1 (Fc) (supplementary material Fig. S1A), and Dsg2Fc and Dsc2aFc were expressed in mammalian HEK293T cells to ensure correct post-translational modifications. Fusion proteins were secreted into the medium, purified by affinity chromatography with Protein-A–Sepharose (Drees et al., 2005), and labeled with Cy5 dye to visualize protein patterns on micro-patterned coverslips (supplementary material Fig. S1B).
Patterned 10 µm stripes were contact printed with collagen IV, followed by saturating amounts of Dsg2Fc, Dsc2aFc or Dsg2Fc plus Dsc2aFc (50∶50) so that the collagen IV stripes were juxtaposed to Dsc2a/Dsg2 stripes, with both stripes showing uniform staining of the proteins (supplementary material Fig. S1C). MDCK cells were seeded onto the dual-patterned coverslips and allowed to adhere for at least 2 h before fixation. Total internal reflection fluorescence microscopy (TIRF-M) was used to detect cellular DP I/II on the ventral membrane of MDCK cells attached to stripes containing Dsg2Fc, Dsc2aFc, or Dsg2Fc plus Dsc2aFc (Fig. 1). Given that MDCK cells express both Dsc2a and Dsg2 (Pasdar and Nelson, 1988a; Pasdar and Nelson, 1988b) they could form homophilic and/or heterophilic bonds with Dsc2aFc and Dsg2Fc stripes and recruit other desmosomal complex proteins such as DP I/II, which links the complex to the cytoskeleton.
Fig. 1.

Localization of desmoplakin I/II to surface complexes depends on desmosomal cadherin engagement. MDCK cells were plated at single cell density onto micro-patterned substrates functionalized with collagen IV and either Fc (A), Dsg2Fc (B) or Dsc2aFc (C), or Dsg2Fc plus Dsc2aFc (D). After 4–6 h, cells were fixed and immunostained for the desmosomal adaptor protein desmoplakin (DP I/II). Localization of DP I/II at the cell–substrate interface, and collagen IV stripe location (Cy3), were visualized using TIRF microscopy. Most cells showed this behavior (n>5) and a representative cell was chosen for each condition; merged images show specific recruitment of DP I/II with respect to stripe functionalization and location. Dsc2aFc, Dsg2Fc and Fc control stripes are marked as the non-collagen IV printed surfaces (black stripes). The white line designates the cell outline. Scale bar: 10 µm.
Cellular DP I/II was distributed evenly over alternating stripes of collagen IV and Fc (Fig. 1A). In contrast, cellular DP I/II distribution was restricted over stripes containing Dsg2Fc (Fig. 1B), Dsc2aFc (Fig. 1C) or Dsg2Fc plus Dsc2aFc (Fig. 1D), and generally excluded from areas over the collagen IV stripe. Significantly, the organization of DP I/II was diffuse over the Dsg2Fc stripe (Fig. 1B), but more punctate over the Dsc2aFc stripe (Fig. 1C) and Dsg2Fc plus Dsc2aFc stripe (Fig. 1D). Indeed, the DP I/II puncta over Dsg2Fc plus Dsc2aFc stripes appeared similar to bone fide desmosomes between adherent pairs of MDCK cells (supplementary material Fig. S1D). The punctate localization of DP I/II does not appear to be due to a punctate distribution of Dsc2aFc or Dsg2Fc in the stripes, as both of them were evenly distributed (supplementary material Fig. S1E). These results indicate that Dsc2a is necessary for DP I/II organization into desmosome-like puncta, whereas Dsg2 is not required on at least one of the opposing surfaces.
Desmosomal cadherins have distinct binding properties
The results from MDCK binding to micro-patterned surfaces indicate that formation of punctate desmosome-like cellular structures involved homophilic binding of Dsc2a, but not Dsg2. Heterophilic interactions, if they occurred, were not sufficient to form these puncta. To directly address whether Dsc2a and Dsg2 form homophilic and/or heterophilic interactions, we measured their binding using SMFS with an AFM. We did not use the Fc fusion construct in the SMFS experiments because Fc dimerization positions the cadherins in close proximity in a cis orientation (Zhang et al., 2009); thus, interactions between opposing cadherin pairs cannot be distinguished from single-molecule interactions which might introduce artifacts in data interpretation. Therefore, we designed monomeric Dsc2a and Dsg2 fusion proteins, as described previously for E-cadherin (Rakshit et al., 2012; Sivasankar et al., 2009; Zhang et al., 2009), comprising the extracellular domain of Dsc2a or Dsg2 fused at the C-terminal to a tag comprising an Avi tag, for binding biotin attached to a surface, and a TEV-removable His tag, for purification from HEK293T cell conditioned medium using a Ni-NTA column (pATH; Fig. 2A). The purified proteins had an expected molecular mass of ∼90 kDa, similar to the E-cadherin extracellular domain fused to the same tag (Fig. 2B); a slower migrating protein was detected occasionally and is likely the uncleaved precursor (Fig. 2B, *).
Fig. 2.

Desmosomal cadherins have distinct extracellular trans binding properties. (A) A schematic of the monomeric desmosomal cadherins fused to tags comprising Avadin, Tev and His (pATH). (B). Coomassie-blue stained SDS-PAGE gel of purified Dsg2pATH, Dsc2apATH and E-cadherinpATH (EcadpATH, control), which have similar molecular masses (∼95 kDa); an unprocessed precursor form of the cadherins is present at low levels in the Dsc2apATH sample (*). (C) Schematic of an AFM cantilever tip and coverslip coated with PEG (5% coverage), and functionalized with monomeric Dsg2pATH or Dsc2apATH to measure single-molecule interactions. (D) The AFM cantilever tip was lowered and raised thousands of times to measure hundreds of single-molecule interactions between protein on the AFM tip and protein on the coverslip, and only events showing a single binding event were used for analysis. (E) Single-molecule binding frequency of different combinations of Dsg2pATH and Dsc2apATH was plotted as a percentage of all events and compared to non-specific binding (identically functionalized cantilevers lacking the desmosomal cadherins). Addition of the Ca2+ chelator EGTA was used to test Ca2+-dependence of binding events. (F) The relationship between loading rate (pN/s) and bond rupture force was plotted for both homophilic binding of Dsg2pATH (with or without EGTA) and Dsg2pATH–Dsc2apATH heterophilic binding.
Purified Dsg2pATH and Dsc2apATH monomers were biotinylated and immobilized via polyethylene glycol (PEG) linkers to a glass coverslip and an AFM cantilever tip (Fig. 2C), as described previously for E-cadherin (Rakshit et al., 2012; Sivasankar et al., 2009; Zhang et al., 2009). The AFM tip and coverslip were first brought into contact so that opposing cadherins interacted. The tip was then withdrawn from the surface so that force was applied to the bond. Single-molecule interactions were identified from the freely-jointed chain stretching of the polymer tether that anchored the proteins to the surface (Smith et al., 1992) (Fig. 2D); the contour length of the stretched polymer was used to distinguish specific interactions from non-specific binding (Zhang et al., 2009). Approximately 1000–2000 measurements were carried out at 11 different pulling rates (loading rates), and histograms of the unbinding forces were compiled (supplementary material Fig. S2). Dsg2pATH and Dsc2apATH densities on the AFM tip and coverslip were adjusted to yield binding frequencies in the 5% range (Poisson statistics predicts that more than 95% of the measured events at this unbinding frequency occur due to single-molecule unbinding), and the adjusted densities were kept constant for all experimental conditions.
The force required to rupture the bond between homophilic (Dsg2pATH–Dsg2pATH or Dsc2apATH–Dsc2apATH) or heterophilic (Dsg2pATH–Dsc2apATH) interactions was measured in the presence of Ca2+ or EGTA, to chelate Ca2+ (Fig. 2E). Non-specific binding rates were measured using identically processed AFM cantilevers and coverslips that were not decorated with Dsg2pATH or Dsc2apATH. The binding frequency (percentage) showed that the greatest number of unbinding events involved homophilic interactions between Dsc2apATH (>4%), and that binding was significantly reduced, to <2%, upon addition of EGTA, indicating a Ca2+-dependent binding mechanism. In contrast, the homophilic binding frequency between Dsg2pATH was <1%, which was below that of non-specific binding events (Fig. 2E). We detected heterophilic binding events between Dsc2apATH and Dsg2pATH, and the frequency of those binding events was similar in the presence or absence of Ca2+, indicating a Ca2+-independent binding mechanism (Fig. 2E). Given that Dsg2pATH bound to Dsc2apATH through heterophilic interactions, the lack of homophilic Dsg2pATH binding is not due to inactivity of Dsg2pATH.
Because the unbinding force increases linearly with the natural log of the loading rate (Fig. 2F), we used the Bell–Evans model (Eqn 3) (Bell, 1978; Evans and Ritchie, 1997) to determine intrinsic unbinding rates. Our results showed that the unbinding rates of Ca2+-dependent homophilic binding of Dsc2apATH and Ca2+-independent heterophilic binding between Dsc2apATH and Dsg2pATH monomers were ∼0.23 s−1 and that they were statistically indistinguishable from each other. These results indicate that monomeric Dsc2a, but not Dsg2, forms Ca2+-dependent homophilic interactions, and that Dsc2a forms Ca2+-independent heterophilic interactions with Dsg2.
W2 is required for Dsc2a Ca2+-dependent homophilic binding and recruitment into desmosome puncta
The amino acid sequence of the extracellular domain of classical and desmosomal cadherins contains a tryptophan at position 2 (W2) in the first extracellular domain (EC1). W2 in classical cadherins is crucial for trans binding between opposing extracellular domains (Shapiro et al., 1995) because it forms a strand-swap dimer between opposing cadherins (Boggon et al., 2002). A crystal structure of desmosomal cadherins has not been reported, but it has been suggested that desmosomal cadherins use a similar binding mechanism (Nie et al., 2011). To test for a role of W2, we made alanine substitution mutants in Dsc2a (Dsc2aW2A) and Dsg2 (Dsg2W2A), and monomeric extracellular domain mutants containing the pATH tag (Dsc2aW2ApATH, Dsg2W2ApATH) were used for SMFS (Fig. 3A).
Fig. 3.

A W2A mutation in Dsc2a disrupts single-molecule trans binding. (A) A tryptophan residue at position 2 (W2) in the first extracellular binding repeat (EC1) was mutated to an alanine residue (W2A) in Dsg2W2ApATH and Dsc2aW2ApATH. Coomassie-stained SDS-PAGE shows a major protein band of the expected molecular mass, and low-level contamination by possible dimers (*; ∼200 kDa) and degradation products (**). A lane between the samples has been deleted for simplification purposes (dashed line). (B) SMFS showed low levels of Ca2+-independent homophilic binding of Dsg2W2ApATH and Dsc2aW2ApATH . Ca2+-independent heterophilic binding of Dsg2W2ApATH and Dsc2aW2ApATH was comparable to that in wild type (Fig. 2E).
The W2A mutation resulted in loss of Ca2+-dependent Dsc2apATH homophilic binding measured by SMFS, because the cadherin binding frequency did not change when free Ca2+ was chelated by EGTA (Fig. 3B), unlike the wild-type Dsc2apATH (see Fig. 2E). However, the frequency of Ca2+ independent heterophilic binding between Dsc2aW2ApATH and Dsg2W2ApATH (Fig. 3B) was similar to the wild-type protein binding (Fig. 2E). These results indicate that homophilic Dsc2a–Dsc2a binding is Ca2+ dependent, and requires a strand-swap mechanism involving W2. In contrast, heterophilic binding between Dsc2a and Dsg2 is Ca2+ independent and occurs through a W2-independent mechanism. This is in contrast to E-cadherin, in which mutations of both W2 and K14 residues are required to inhibit binding (Rakshit et al., 2012).
We tested whether the Dsg2W2A and Dsc2aW2A mutants could be incorporated into endogenous desmosome puncta in contacting MDCK cells. C-terminal GFP-tagged mutant protein was transiently expressed in MDCK cells and their distributions (linear versus punctate) were compared to transiently expressed wild-type protein and/or endogenous desmosomal proteins (Fig. 4). Dsg2W2A–GFP was restricted to puncta along cell–cell contacts, similar to wild-type Dsg2–GFP (Fig. 4A). However Dsc2aW2A–GFP had a linear distribution along the plasma membrane at cell-cell contacts (Fig. 4B, C) compared to wild-type Dsc2a–GFP (Fig. 4D,E). Mislocalization of Dsc2aW2A–GFP was not due to overexpression since the transfection efficiency and expression levels were similar to control Dsc2a–GFP levels (Fig. 4F).
Fig. 4.

The W2A mutation disrupts localization of Dsc2a to punctate complexes. (A) MDCK cells were transiently transfected with wild-type GFP-tagged Dsg2 or GFP-tagged Dsg2W2A and fixed with methanol. Wild-type Dsg2–GFP and mutant Dsg2W2A–GFP localized in characteristic desmosome-like punctate spots at cell–cell contacts. Scale bar: 10 µm. (B–E) MDCK cells were transiently transfected with GFP-tagged Dsc2aW2A or wild-type GFP-tagged Dsc2a, with or without RFP-tagged wild-type Dsc2a, fixed and stained for endogenous desmosomal proteins. Top: a wide-field image with the representative contact outlined in white. Middle: zoomed-in images of the representative cell–cell contacts. Bottom: line scans of normalized fluorescence intensity for Dsc2aW2A–GFP, Dsc2a–GFP, Dsc2a–DsRED, plakoglobin (PG) and DP I/II were plotted for the representative cell–cell contact. Intensity was normalized by subtracting the average intensity over the whole contact and dividing by the difference in maximum and minimum values (pixel range). (B,C) Dsc2aW2A–GFP accumulated in a linear, non-punctate distribution along the plasma membrane and did not co-localize with desmosomal proteins (DP I/II, cytokeratin 8/18, and plakoglobin) or co-transfected wild-type Dsc2a_DsRED, all of which had a characteristic desmosome-like punctate distribution (D,E) Wild-type Dsc2a–GFP localized into punctate spots at cell–cell contacts and colocalized with the desmosomal proteins (DP I/II, cytokeratin 8/18 and plakoglobin). Of note, the focal plane was set using the staining of endogenous proteins as the marker, so as not to create any biases between the transfected GFP-tagged proteins. Scale bar: 5 µm. (F) Expression levels of transiently transfected cells (similar efficiencies with >60% transfected) Dsc2aW2A–GFP were ∼1/2 that of endogenous Dsc2a in MDCK cells.
Next we investigated whether the anomalous localization of Dsc2aW2A–GFP along cell–cell contacts affected the recruitment and localization of endogenous components into desmosome puncta. MDCK cells transiently co-expressing Dsc2aW2A–GFP and Dsc2a–DsRED (Fig. 4B,C), or only Dsc2a–GFP (Fig. 4D,E) were fixed and stained for the desmosome proteins DP I/II, the cytokeratin 8 and cytokeratin 18 pair (hereafter cytokeratin 8/18) (Fig. 4B,D) or plakoglobin (Fig. 4C,E). A representative cell–cell contact with an expressing cell is shown for each staining. Line-scans that measured fluorescence intensity for each protein were normalized and used to determine whether endogenous proteins colocalized with the wild-type or W2A mutant Dsc2a, and whether the localization had a linear or punctate distribution (Fig. 4B–E).
In contrast to the linear plasma membrane staining of Dsc2aW2A–GFP, endogenous DP I/II, cytokeratin 8/18, wild-type Dsc2a–DsRED and wild-type Dsc2a–GFP all localized in puncta along the same cell–cell contacts (Fig. 4B,D). Endogenous plakoglobin staining was also punctate, and more similar to wild-type Dsc2a–DsRED and Dsc2a–GFP than mutant Dsc2aW2A–GFP (Fig. 4C,E). These results indicate that both endogenous desmosomal cytoplasmic proteins and cadherins localized normally in puncta at cell–cell contacts in the presence of Dsc2aW2A, suggesting that Dsc2aW2A did not act as a disrupting (dominant-negative) protein and that it was mostly excluded from endogenous desmosome puncta.
Dsc2aW2A mutant is more mobile than wild-type Dsc2a at cell–cell contacts
We speculated that the lack of incorporation into endogenous desmosome puncta would result in a higher mobility of Dsc2aW2A in the membrane than Dsc2a, Dsg2 and Dsg2W2A, all of which were incorporated into desmosomal puncta. Fluorescence recovery after photobleaching (FRAP) was used to measure the mobile fraction and recovery rate of wild-type and mutant proteins based on a single exponential recovery model (Fig. 5). Dsg2W2A had a slightly higher mobile fraction (0.354; P<0.1) and slower recovery rate (t1/2 = 167 s; P<0.05) than wild-type Dsg2 (0.312 and 127 s, respectively) (Fig. 5B; supplementary material Movies 1, 2). Although the recovery rate of Dsc2aW2A was similar to that of Dsg2W2A (174 versus 167 s, respectively; Fig. 5B; supplementary material Movie 3), the Dsc2aW2A mobile fraction was significantly greater (P<0.0001) than that of Dsc2a (0.714 versus 0.351; Fig. 5B; supplementary material Movie 4). Indeed the mobile fraction of Dsc2aW2A was similar to that of Lyn–GFP, a lipid-anchored protein (0.714 and 0.75, respectively; Fig. 5B).
Fig. 5.

Membrane mobility of Dsc2a increases in the W2A mutant. (A) FRAP of GFP-tagged wild-type and W2A mutant Dsc2a and Dsg2 cadherins at cell–cell contacts showed a significant difference in the recovery rate of Dsc2aW2A and wild-type Dsc2 and Dsg2, whereas mutant Dsg2W2A showed only slight differences. Lyn–GFP is used a reference for proteins that are lipid-anchored to the plasma membrane. Plotted as mean±s.d. (B) Mobile factions and recovery rates (t1/2) are compared using a single exponential curve fitted to the data shown in A. Mobile fractions of Dsc2a–GFP and Dsc2aW2A–GFP are significantly different (P<0.0001), whereas Dsg2–GFP and Dsg2W2A–GFP are less significant (P<0.1). The recovery rates of both pairs are significantly different (P<0.05). Significance was calculated using a comparison of fits, extra sum-of-squares F test.
These FRAP data indicate that the recruitment of wild-type Dsc2a and Dsg2 into desmosome puncta results in a reduced mobile fraction that might be due to their incorporation into a dense protein complex with adaptor proteins and intermediate filaments (see Fig. 4). The Dsg2W2A mutant had a reduced mobile fraction indicating that W2 and strand-swap dimers are not required for Dsg2 incorporation into desmosome puncta. In contrast, the Dsc2aW2A mutant was distributed along the plasma membrane and had a mobile fraction similar to a lipid-anchored protein, confirming that W2 and strand-swap dimers are required for Dsc2a incorporation into desmosome puncta and that the W2A mutant was likely either not incorporated or retained in these structures.
Desmosome formation inhibits MDCK cell migration
In addition to testing protein recruitment to different adhesion complexes, dual patterned substrates test the effects of cell–cell adhesion proteins on cell migration on ECM (Borghi et al., 2010). Although E-cadherin had little effect on the rate of cell migration on collagen IV (Borghi et al., 2010), we found that adhesion to desmosomal cadherins had a significant effect. To quantify cell movements, images were recorded for 6–10 h, and changes in nucleus position were tracked to determine the migration velocity (Fig. 6A) and migration direction (Fig. 6B). In addition, the contour of a representative cell was traced over time to examine lamellipodia dynamics (Fig. 6C).
Fig. 6.

Desmosomal cadherin engagement regulates MDCK cell migration on dual-patterned substrates. The movement of single MDCK cells on each dual-patterned substrate condition was tracked by monitoring changes in nuclear position over time. (A) Cell migration rate (v) was normalized to single cell movement on only collagen IV surfaces, and plotted as a tukey box-and-whisker plot on a natural log scale [Ln(v/10 µm/h)]. The box marks the range from the 25th–75th percentile of the data and the band inside the box marks the median. Whiskers show the maximum and minimum values. (B) Directionality of movement was plotted as a tukey box-and -whisker plot on a natural log scale as a function of movement parallel to stripes (x) divided by perpendicular movement (y). *P<0.1; **P<0.01; ****P<0.0001. (C) Membrane activity during migration assays was assessed over time by tracing the cell contour from DIC images and projecting the outlines onto the stripe patterns (C). Statistical analysis used the Mann–Whitney, two-tailed t-test (Dsg2Fc, n = 13; Dsc2aFc, n = 31; Dsg2Fc+Dsc2aFc, n = 27).
Migration velocities of MDCK cells on all surfaces were normalized to 10 µm/h and plotted on a natural log (ln) scale for comparison. The migration rate of cells plated on collagen IV and Dsg2Fc substrate stripes (∼9 µm/h) was similar to that of cells plated on collagen IV and Fc control substrate stripes (Borghi et al., 2010), and was significantly faster than cells on collagen IV and Dsc2aFc substrate stripes (∼5 µm/h; P<0.01). Importantly, the migration rate of cells plated on collagen IV and Dsg2Fc plus Dsc2aFc substrate stripes was in between that of cells on collagen IV and Dsc2aFc stripes or collagen IV and Dsg2Fc stripes (∼7 µm/h; P<0.1).
The directionality of cell movements on the dual-patterned surfaces were plotted as cell velocity in the direction of the stripes divided by the perpendicular velocity (x/y); ln(x/y) is positive if cells preferentially move in the direction of the stripes (x>y). Cell migration on collagen IV and Dsg2Fc stripes showed biased movement in the direction of the stripes (Fig. 6B), as reported previously for cells migrating on collagen IV and E-cadherinFc stripes (Borghi et al., 2010). Directional bias is due to restriction of the migration-based adhesion machinery, such as vinculin–GFP [supplementary material Fig. S3 (see also Borghi et al., 2010)], to the collagen IV stripes. In contrast, cells were almost stationary on collagen IV and Dsc2aFc stripes and the small amount of movement only showed a slightly significant difference from Dsg2Fc (Fig. 6B; P<0.1). The direction of MDCK cell migration on collagen IV and Dsg2Fc plus Dsc2aFc stripes was significantly biased towards the direction of the stripes (P<0.0001; supplementary material Movie 5) compared to Dsc2aFc alone and slightly significant compared to Dsg2Fc alone (P<0.1).
We examined membrane lamellipodia activity by tracing the cell contour over time and superimposing all images onto the dual-patterned substrate (Fig. 6C); quantification of membrane dynamics was not attempted because the cells migrated during the course of experiment. Cells on collagen IV and Dsg2Fc stripes exhibited membrane dynamics over the whole cell contour, but persistent membrane projections occurred predominantly in the direction of cell movement, which was generally along the collagen IV stripes (Fig. 6C; supplementary material Movie 6). Cells on collagen IV and Dsc2aFc stripes exhibited membrane dynamics, but few persistent membrane projections (Fig. 6C; supplementary material Movie 7) consistent with the significant reduction in the overall migration rate (Fig. 6A). Cells on collagen IV and Dsg2Fc plus Dsc2aFc stripes had membrane dynamics similar to cells on Dsg2Fc, and more persistent membrane projections on the collagen IV stripes, which was accompanied by highly processive cell movement (Fig. 6C; supplementary material Movie 5) consistent with a greater directional migration bias than on collagen IV and Dsg2Fc stripes, collagen IV and Dsc2aFc stripes (Fig. 6B; Movies 6, 7) or collagen IV and Fc control substrates (Borghi et al., 2010).
These results indicate that desmosome puncta formed by homophilic Dsc2a binding (see Fig. 1) result in strong cell adhesion to the Dsc2aFc stripe. We suggest that the smaller velocity decrease in the presence of Dsc2aFc plus Dsg2Fc was due to the dilution of Dsc2aFc homophilic interactions by Dsg2Fc, which results in decreased adhesion strength and thereby increased cell migration. Thus, migration direction bias requires both anisotropic adhesion of integrin-based complexes on the collagen IV substrate and reduced adhesion strength from desmosome assembly due to the incorporation of Dsg2.
DISCUSSION
Desmosomes assemble in a Ca2+-dependent manner between opposing cells, and are required to maintain the structural integrity of tissues. Dsg and Dsc are obligate cadherins in desmosomes, and impairment of either Dsc or Dsg extracellular interactions causes heart defects, and epidermal blistering diseases and syndromes (Al-Jassar et al., 2013; Kottke et al., 2006; Thomason et al., 2010). Studies using different in vitro binding and cell-based assays, however, have not identified specific roles for Dsg and Dsc in desmosome assembly and adhesion. Whether there is specificity in the extracellular binding properties of desmosomal cadherins has been difficult to establish owing to the overlapping roles of other cadherins (e.g. E-cadherin) in epithelial cell–cell adhesion, the complexity of proteins in desmosomes and their resistance to dissociation in non-denaturing conditions (Nie et al., 2011), and the lack of high-resolution crystal structures of the two desmosomal cadherin sub-types (Al-Jassar et al., 2013). Here, we used reductionist approaches to distinguish roles of Dsc2a and Dsg2 in desmosome assembly, organization and adhesion in the absence of other cell–cell adhesions. Our results indicate that Dsc2a and Dsg2 have distinct binding properties and functions in desmosome organization and adhesion.
Micro-patterned substrates of purified Dsc2aFc or Dsg2Fc revealed that Dsc2aFc, but not Dsg2Fc, was necessary and sufficient to induce the recruitment of a desmosome-specific cytoplasmic protein (e.g. DP I/II) into punctate cellular structures (Fig. 1). Given that MDCK cells express both Dsc2a and Dsg2 (Pasdar and Nelson, 1989; Pasdar et al., 1991), the exogenous substrate-bound Dsc2a could have initiated the assembly of desmosome puncta by binding cellular Dsc2a, Dsg2 or both on the ventral surface of the cell. Indeed, our results from the SMFS assay indicated that Dsc2a forms homophilic interactions and heterophilic interactions with Dsg2 (Fig. 2). However, SMFS data indicates only heterotypic interactions between Dsg2 and Dsc2a, suggesting that Dsg2Fc surfaces promote heterotypic binding and diffuse recruitment of DP I/II at the membrane. Interestingly, other studies using chemical cross-linkers only found homophilic binding by Dsc2 and Dsg2 (Nie et al., 2011), whereas bulk biochemical assays indicate heterophilic Dsc2a–Dsg2 binding and weak homophilic Dsg2–Dsg2 binding in agreement with our SMFS results (Syed et al., 2002). Differences between other SMFS results (Hartlieb et al., 2013; Schlegel et al., 2010) and our SFMS experiments might be due to our strict definition of a single-molecule interaction or our use of monomeric proteins, which excluded the possibility of induced lateral (cis) clustering of extracellular domains. In addition, our findings of differences in the Ca2+ dependency of homophilic and heterophilic protein binding have revealed differences between single-molecule data and bulk or cellular assays (see below).
Analysis of the crystal structure of classical cadherins revealed that a tryptophan at position 2 (W2) in the N-terminal EC1 domain is crucial for the formation of a strand-swap dimer between opposed extracellular domains (Boggon et al., 2002); an intermediate adhesion structure is also formed by a weak X-dimer structure (Harrison et al., 2010). Although the atomic structures of full-length Dsc2a or Dsg2 have not been solved, both proteins have a tryptophan at position 2 (Nie et al., 2011) but neither have the necessary residues for the formation of an X-dimer. Significantly, the W2A mutation in Dsc2a inhibited Ca2+-dependent homophilic binding (Fig. 3). Furthermore, the Dsc2aW2A mutant appeared to be excluded from endogenous desmosomes in MDCK cells, which we inferred from its linear plasma membrane staining compared to endogenous desmosome puncta in the same cells (Fig. 4), and significantly increased mobile fraction compared to wild-type Dsc2a (Fig. 5). Given that Dsc2aFc alone was necessary and sufficient to induce desmosome-like DP I/II puncta in cells on micro-patterned substrates (Fig. 1), we suggest that Dsc2a homophilic binding through a Ca2+- and W2- (strand-swap dimer) dependent mechanism is required for desmosome assembly. Furthermore, because Dsc2aW2A was mostly excluded from desmosomes and did not affect the punctate localization of desmosome adaptor proteins (Fig. 4), it is likely that the W2 strand-swap mechanism is also necessary for the incorporation of Dsc2a into desmosome puncta, and that binding to cytoplasmic adaptor proteins is not sufficient.
In contrast, the W2A mutation in Dsg2 affected neither Ca2+-independent heterophilic binding to Dsc2a (Fig. 3), colocalization with endogenous desmosome puncta in MDCK cells (Fig. 4) nor its mobile fraction (Fig. 5). These results indicate that Dsg2 incorporation into desmosomes occurs through a Ca2+- and W2- (strand-swap dimer) independent mechanism that relies on heterophilic interactions and/or cytoplasmic interactions with other proteins in the desmosome. Given that Dsg2Fc alone did not induce assembly of desmosomal-like DP I/II puncta in MDCK cells on micro-patterned substrates (Fig. 1), what role could Dsg2-dependent Ca2+-independent heterophilic adhesion play? Differences in Ca2+-dependency of desmosomal adhesion have been identified in tissue- and cell-based studies, and it has been suggested that mature desmosome complexes are in a hyper-adhesive state that is Ca2+ independent (Garrod and Kimura, 2008). Our results raise the possibility that such Ca2+-independent adhesion might be mediated by heterophilic Dsc2a–Dsg2 binding.
To discriminate the effects of desmosome adhesion on cell migration from the contribution of other adhesion mechanisms (e.g. classical cadherins), we examined cell migration on dual-patterned substrates comprising alternating stripes of collagen IV and Dsc2aFc and Dsg2Fc (Figs 1, 2). We can infer the contribution of desmosome assembly on cell adhesion by measuring the rate of cell migration. Significantly, MDCK cell migration was severely inhibited on dual-patterned substrates coated with saturating amounts of Dsc2aFc (Fig. 6A; supplementary material Movie 7); in contrast, Dsg2Fc substrates did not inhibit cell migration compared to controls (Fig. 6A; supplementary material Movie 6). Our previous studies with substrates coated with E-cadherinFc and collagen IV revealed little or no effect of E-cadherin on the migration rate of MDCK cells (Borghi et al., 2010). Therefore, we propose that the formation of desmosome-like structures, unlike E-cadherin adhesion, mechanically resists cell movement by forming strong adhesive bonds to the Dsc2aFc substrate, rather than a downregulation of the Arp2/3 and actomyosin machineries that are involved in membrane dynamics and cell migration (Pollard and Borisy, 2003). Indeed, whereas Dsc2aFc-containing dual substrates inhibited cell migration, we found that they had little effect on overall lamellipodia dynamics (Fig. 6C). Interestingly, addition of Dsg2Fc to the Dsc2aFc substrate partially rescued the reduction in cell migration rate (Figs 6A,C). We suggest that differences in the ratio of Dsc2a and Dsg2 in desmosomes might regulate the strength of cell–cell adhesion and dynamics of the desmosome complex thereby modulating cell migration in epithelial sheets. However, further studies are needed to test this hypothesis in detail.
In summary, our results have uncovered a specific role for extracellular contacts formed by Dsc2a in the structural organization and function of desmosomes. The assembly of desmosome puncta depends on Ca2+- and W2- (strand-swap dimer) dependent homophilic trans-dimerization between Dsc2a proteins on opposing cell surfaces. Dsg2 might be required for the long-term stability of desmosomes, and also perhaps the formation of a Ca2+-independent hyperadhesive state (Garrod and Kimura, 2008). Whereas Dsc2a might have a specific role in adhesion, Dsg2 might have additional roles (Brennan and Mahoney, 2009; Brennan et al., 2007). Thus, differences in the mechanisms of incorporation and function of Dsc and Dsg cadherins might allow a diversification of desmosome functions in cell adhesion, migration and differentiation.
MATERIALS AND METHODS
Protein purification
The cloning design for desmosomal Fc fusion proteins was described previously for E-cadherinFc (Drees et al., 2005). The C-terminal of the Dsg2 or Dsc2a extracellular domains was fused to the constant region of human IgG1 (Fc). Fusion constructs were expressed in HEK293T cells and proteins purified from conditioned medium over Protein A Sepharose (GE Healthcare Life Sciences, Uppsala, Sweden). Proteins were then labeled with Cy5.5 dye (Invitrogen Life Technologies, Carlsbad, CA, USA) for visualization. Monomeric Dsg2 and Dsc2a construct design was also as described previously (Zhang et al., 2009), where the C-terminal of the extracellular domain was fused to the Avi tag, Tev sequence and His tag (pATH). Fusion constructs were expressed in HEK293T cells, and proteins were purified from conditioned medium over Ni-NTA agarose beads, and protein was biotinylated using BirA enzyme for surface functionalization (BirA500 kit, Avidity LLC, Aurora, CO, USA).
Surface immobilization of desmosomal cadherins
Dual-micropatterned surfaces were prepared using a two-step process as reported previously (Borghi et al., 2010). First, the collagen IV was micro-contact printed using standard protocols (Cuvelier et al., 2003; Nishizawa et al., 2002), and then the non-ECM–coated surfaces were functionalized with by fusion with the human IgG1 Dsg2aFc, Dsc2aFc or Dsg2aFc plus Dsc2aFc as described previously (Drees et al., 2005). Polydimethylsiloxane (PDMS) stamps with striped groves were coated with Cy3.5-tagged collagen IV (200 µg/ml) and printed onto silanized coverslips. Stamped coverslips were first incubated with EZ link sulfo-NHS-LC-LC-biotin (50 mM in water; made fresh each time), then neutravidin (5 mg/l in PBS), then biotinylated Protein A (0.3 mg/ml in PBS), each for 1 h. Before incubation with Dsc2aFc or Dsg2aFc protein, coverslips were blocked with a D-biotin solution (15 mM in DMSO) for 30 min. Coverslips were washed with PBS three times between incubations and all incubations and washes were performed at room temperature in the dark. Finally, coverslips were incubated with Cy5.5-tagged Dsg2aFc, Dsc2aFc or Fc protein (control) for 1 h at room temperature. MDCK cells were added to the coverslips at low density.
For single-molecule force spectroscopy (SMFS), AFM cantilevers and glass coverslips were first cleaned with a 25% H2O2∶75% H2SO4 solution and subsequently washed with deionized water, 1 M potassium hydroxide solution, deionized water and acetone. The cleaned AFM cantilevers and coverslips were made amine reactive by functionalizing with 2% v/v solution of 3-aminopropyltriethoxysilane (Sigma-Aldrich, St. Louis, MO, USA) dissolved in acetone. The cantilevers and coverslips were then functionalized with PEG spacers (PEG 5000, Laysan Bio, Arab, AL, USA) containing an amine-reactive N-hydroxysuccinimide ester at one end to bind to the surface. A known fraction of PEG presented biotin at the other end and was kept low in order to measure single-molecule events; for wild-type and mutant monomers, 5% and 7% of the PEG contained biotin, respectively. The biotinylated surfaces were incubated with 0.1 mg/ml BSA for 12 h, to minimize non-specific surface interactions, and then with 0.1 mg/ml streptavidin for 30 min. Biotinylated Dsc2a or Dsg2a monomers were bound to streptavidin on the AFM tip and coverslip surfaces. Following Dsc2a or Dsg2a immobilization, the surfaces were incubated in 10 µM biotin to block free biotin-binding sites on streptavidin.
Single molecule dynamic force spectroscopy experiments
Single molecule dynamic force spectroscopy experiments were performed at different pulling velocities using an Agilent 5500 AFM. Experiments were performed in 10 mM Tris-HCl pH 7.5, 100 mM NaCl, 10 mM KCl in either 2.5 mM CaCl2 or 2 mM EGTA. Single unbinding force curves were selected and fitted with the freely jointed chain (FJC) model using the following equation (Smith et al., 1992):
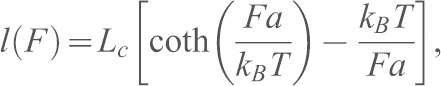 |
(1) |
where 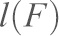 is the end-to-end distance of the PEG tether subjected to a force
is the end-to-end distance of the PEG tether subjected to a force 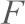 ,
, 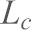 , is the contour length of PEG and a is the Kuhn length. Spring constants of the AFM cantilevers were measured with the thermal fluctuation method (Hutter and Bechhoefer, 1993). In order to measure unbinding forces at different loading rates, the cantilever was mounted on a piezoelectric translator and was moved at different velocities (
, is the contour length of PEG and a is the Kuhn length. Spring constants of the AFM cantilevers were measured with the thermal fluctuation method (Hutter and Bechhoefer, 1993). In order to measure unbinding forces at different loading rates, the cantilever was mounted on a piezoelectric translator and was moved at different velocities (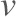 ); the loading rate (
); the loading rate (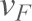 ) was estimated using (Marshall et al., 2006; Ray et al., 2007) the equation:
) was estimated using (Marshall et al., 2006; Ray et al., 2007) the equation:
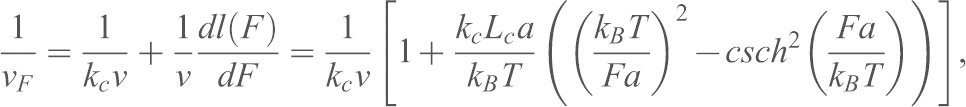 |
(2) |
where 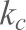 is the spring constant of the cantilever.
is the spring constant of the cantilever.
In order to eliminate hidden multiple unbinding events from visibly indistinguishable single unbinding events, we followed a protocol used for identifying the heterogeneity in chemical bonds or hidden multiple bonds as described previously (Fuhrmann et al., 2012). After eliminating all hidden multiple events, the forces were plotted as a histogram for each retracting velocity with bin width calculated according to a statistical method (Scott, 1979). The histograms were fitted to a Gaussian distribution to obtain the most probable forces (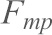 ) for each retracting velocity. Most probable force (
) for each retracting velocity. Most probable force (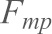 ) vs mean loading rate (
) vs mean loading rate (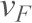 ) data were then fitted (Bell, 1978; Evans and Ritchie, 1997) in order to measure the force-independent off-rate (
) data were then fitted (Bell, 1978; Evans and Ritchie, 1997) in order to measure the force-independent off-rate (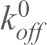 ) and the distance (
) and the distance (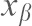 ) from the minimum of the reaction potential to the peak of the potential-barrier using the equation:
) from the minimum of the reaction potential to the peak of the potential-barrier using the equation:
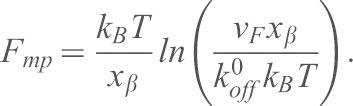 |
(3) |
The statistical significance of parameters obtained from the fits was tested using a Student's t-test.
Cell culture and constructs
Madin–Darby canine kidney (MDCK) type II G cells were grown in DMEM with low glucose and 200 µM G418 for stably expressing tagged proteins of interest (Vin–GFP, E-cadherin–DsRED). Cells stably or transiently expressing fluorescently tagged proteins were monitored on a widefield epifluorescence inverted microscope equipped with TIRF-M and FRAP lasers. Desmosomal cadherin mutants were generated from a Dsg2–GFP construct and a Dsc2a–GFP construct (gift from the laboratory of Sergey M. Troyanovsky, Feinberg School of Medicine, Northwestern University, IL) using site-directed mutagenesis (Aligent Technologies, Stratagene, Santa Clara, CA, USA) with PCR fragments containing the point mutation for monomeric fusion proteins (Dsg2aW2A_pATH, Dsc2aW2A_pATH) and full-length GFP-tagged versions (Dsg2aW2A–GFP and Dsc2aW2A–GFP).
Fluorescence microscopy
Immunofluorescence was performed on MDCK cells fixed with 4% PFA for 20 min at 25°C or 100% methanol for 5 min at −20°C, and extracted with 0.1% Triton X-100 for 5 min before blocking in PBS containing 0.2% BSA, 50 mM NH4Cl2 and 1% normal goat/donkey serum. Cells were incubated with antibodies against the proteins of interest: anti-desmoplakin (DP I/II) (Pasdar and Nelson, 1988a), anti-plakoglobin (clone 15F11 – Thermo Fisher Scientific, Waltham, MA, USA), anti-desmocollin2 (Dsc2a) (ab72792 – Abcam, Cambridge, England), anti-cytokeratin 8/18 [Abcam (Cy90) – ab17151]. Cells were incubated with secondary (Fab′) goat/donkey anti-mouse/rabbit IgG labeled with FITC/Rhodamine Red-X/Cy5 (1∶200; Jackson ImmunoResearch Laboratories, Westgrove, PA, USA) and mounted using Vectashield (Vector Labs, Burlingame, CA, USA) (or PBS for TIRF imaging). Immunofluorescence images were acquired on a Zeiss (Jena, Germany) Axiovert 200 inverted microscope equipped with a Mercury lamp and a 100× objective (Olympus, Tokyo, Japan) and acquired with AxioVision Microscopy Software (Zeiss). Fluorescence live-cell imaging (FRAP) and TIRF images were acquired on a custom-built Zeiss Axiovert 200M inverted widefield epifluorescence microscope [Intelligent Imaging Innovations (3i), Denver, CO, USA] equipped with including a 175 Watt Xenon light source with a dual galvanometric filter changer, a Coolsnap HQ interline CCD camera (Photometrics, Tuscon, AZ, USA), an x,y motorized stage with harmonic drive z-focusing, a quad filter set for DAPI, FITC, Cy3 and Cy5, and a MicroPoint FRAP laser system (Andor Technology, South Widsor, CT, USA), as described previously (Spiliotis et al., 2005; Yamada et al., 2005). Live cells were imaged in DMEM without Phenol Red, supplemented with 25 mM Hepes at 37°C on a 100× 1.4 NA oil objective (FRAP and TIRF) and a 40× air objective for tracking cellular movement; images were acquired using Slidebook (3i) software. The fluorescence signal from cells (GFP) and micro-patterned substrates (Cy3.5 or Cy5.5 dye) was used to assess protein recruitment and cell position at long time scales for measurement of cell migration, whereas DIC images at long time scales were used to monitor cell contour outlines and fluctuations. Image analysis was performed with Image J and statistical analysis was performed using a Mann–Whitney, two-tailed t test. FRAP data was normalized and fitted with a single exponential recovery model as described previously (Borghi et al., 2010). Significant differences between modeled parameters were calculated using a comparison of fits, extra sum-of-squares F test.
Supplementary Material
Acknowledgments
References
- Al-Amoudi A., Díez D. C., Betts M. J., Frangakis A. S. (2007). The molecular architecture of cadherins in native epidermal desmosomes. Nature 450, 832–837 10.1038/nature05994 [DOI] [PubMed] [Google Scholar]
- Al-Amoudi A., Castaño-Diez D., Devos D. P., Russell R. B., Johnson G. T., Frangakis A. S. (2011). The three-dimensional molecular structure of the desmosomal plaque. Proc. Natl. Acad. Sci. USA 108, 6480–6485 10.1073/pnas.1019469108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Al-Jassar C., Bikker H., Overduin M., Chidgey M. (2013). Mechanistic basis of desmosome-targeted diseases. J. Mol. Biol. 425, 4006–4022 10.1016/j.jmb.2013.07.035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amagai M., Stanley J. R. (2012). Desmoglein as a target in skin disease and beyond. J. Invest. Dermatol. 132, 776–784 10.1038/jid.2011.390 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bell G. I. (1978). Models for the specific adhesion of cells to cells. Science 200, 618–627 10.1126/science.347575 [DOI] [PubMed] [Google Scholar]
- Bhuiyan Z. A., Jongbloed J. D. H., van der Smagt J., Lombardi P. M., Wiesfeld A. C. P., Nelen M., Schouten M., Jongbloed R., Cox M. G. P. J., van Wolferen M. et al. (2009). Desmosome mutations in arrhythmogenic right ventricular cardiomyopathy: important insight but only part of the picture. Circ. Cardiovasc. Genet. 2, 415–417 10.1161/CIRCGENETICS.109.909366 [DOI] [PubMed] [Google Scholar]
- Boggon T. J., Murray J., Chappuis-Flament S., Wong E., Gumbiner B. M., Shapiro L. (2002). C-cadherin ectodomain structure and implications for cell adhesion mechanisms. Science 296, 1308–1313 10.1126/science.1071559 [DOI] [PubMed] [Google Scholar]
- Borghi N., Lowndes M., Maruthamuthu V., Gardel M. L., Nelson W. J. (2010). Regulation of cell motile behavior by crosstalk between cadherin- and integrin-mediated adhesions. Proc. Natl. Acad. Sci. USA 107, 13324–13329 10.1073/pnas.1002662107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bornslaeger E. A., Corcoran C. M., Stappenbeck T. S., Green K. J. (1996). Breaking the connection: displacement of the desmosomal plaque protein desmoplakin from cell-cell interfaces disrupts anchorage of intermediate filament bundles and alters intercellular junction assembly. J. Cell Biol. 134, 985–1001 10.1083/jcb.134.4.985 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brennan D., Mahoney M. G. (2009). Increased expression of Dsg2 in malignant skin carcinomas: A tissue-microarray based study. Cell Adhes. Migr. 3, 148–154 10.4161/cam.3.2.7539 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brennan D., Hu Y., Joubeh S., Choi Y. W., Whitaker-Menezes D., O'Brien T., Uitto J., Rodeck U., Mahoney M. G. (2007). Suprabasal Dsg2 expression in transgenic mouse skin confers a hyperproliferative and apoptosis-resistant phenotype to keratinocytes. J. Cell Sci. 120, 758–771 10.1242/jcs.03392 [DOI] [PubMed] [Google Scholar]
- Chidgey M., Dawson C. (2007). Desmosomes: a role in cancer? Br. J. Cancer 96, 1783–1787 10.1038/sj.bjc.6603808 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chitaev N. A., Troyanovsky S. M. (1997). Direct Ca2+-dependent heterophilic interaction between desmosomal cadherins, desmoglein and desmocollin, contributes to cell-cell adhesion. J. Cell Biol. 138, 193–201 10.1083/jcb.138.1.193 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cuvelier D., Rossier O., Bassereau P., Nassoy P. (2003). Micropatterned “adherent/repellent” glass surfaces for studying the spreading kinetics of individual red blood cells onto protein-decorated substrates. Eur. Biophys. J. 32, 342–354 10.1007/s00249--003--0282--2 [DOI] [PubMed] [Google Scholar]
- Delva E., Tucker D. K., Kowalczyk A. P. (2009). The desmosome. Cold Spring Harb. Perspect. Biol. 1, a002543 10.1101/cshperspect.a002543 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drees F., Reilein A., Nelson W. J. (2005). Cell-adhesion assays: fabrication of an E-cadherin substratum and isolation of lateral and Basal membrane patches. Methods Mol. Biol. 294, 303–320 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dusek R. L., Godsel L. M., Green K. J. (2007). Discriminating roles of desmosomal cadherins: beyond desmosomal adhesion. J. Dermatol. Sci. 45, 7–21 10.1016/j.jdermsci.2006.10.006 [DOI] [PubMed] [Google Scholar]
- Eshkind L., Tian Q., Schmidt A., Franke W. W., Windoffer R., Leube R. E. (2002). Loss of desmoglein 2 suggests essential functions for early embryonic development and proliferation of embryonal stem cells. Eur. J. Cell Biol. 81, 592–598 10.1078/0171--9335--00278 [DOI] [PubMed] [Google Scholar]
- Evans E., Ritchie K. (1997). Dynamic strength of molecular adhesion bonds. Biophys. J. 72, 1541–1555 10.1016/S0006--3495(97)78802--7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franke W. W. (2009). Discovering the molecular components of intercellular junction – a historical view. Cold Spring Harb. Perspect. Biol. 1, a003061 10.1101/cshperspect.a003061 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuhrmann A., Getfert S., Fu Q., Reimann P., Lindsay S., Ros R. (2012). Long lifetime of hydrogen-bonded DNA basepairs by force spectroscopy. Biophys. J. 102, 2381–2390 10.1016/j.bpj.2012.04.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaertner A., Klauke B., Stork I., Niehaus K., Niemann G., Gummert J., Milting H. (2012). In vitro functional analyses of arrhythmogenic right ventricular cardiomyopathy-associated desmoglein-2-missense variations. PLoS ONE 7, e47097–e47097 10.1371/journal.pone.0047097 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garrod D. R. (1995). Desmosomes and cancer. Cancer Surv. 24, 97–111 [PubMed] [Google Scholar]
- Garrod D., Kimura T. E. (2008). Hyper-adhesion: a new concept in cell-cell adhesion. Biochem. Soc. Trans. 36, 195–201 10.1042/BST0360195 [DOI] [PubMed] [Google Scholar]
- Gehmlich K., Syrris P., Peskett E., Evans A., Ehler E., Asimaki A., Anastasakis A., Tsatsopoulou A., Vouliotis A-I., Stefanadis C. et al. (2011). Mechanistic insights into arrhythmogenic right ventricular cardiomyopathy caused by desmocollin-2 mutations. Cardiovasc. Res. 90, 77–87 10.1093/cvr/cvq353 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Getsios S., Amargo E. V., Dusek R. L., Ishii K., Sheu L., Godsel L. M., Green K. J. (2004). Coordinated expression of desmoglein 1 and desmocollin 1 regulates intercellular adhesion. Differentiation 72, 419–433 10.1111/j.1432--0436.2004.07208008.x [DOI] [PubMed] [Google Scholar]
- Getsios S., Simpson C. L., Kojima S., Harmon R., Sheu L. J., Dusek R. L., Cornwell M., Green K. J. (2009). Desmoglein 1-dependent suppression of EGFR signaling promotes epidermal differentiation and morphogenesis. J. Cell Biol. 185, 1243–1258 10.1083/jcb.200809044 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Green K. J., Simpson C. L. (2007). Desmosomes: new perspectives on a classic. J. Invest. Dermatol. 127, 2499–2515 10.1038/sj.jid.5701015 [DOI] [PubMed] [Google Scholar]
- Green K. J., Kowalczyk A. P., Bornslaeger E. A., Palka H. L., Norvell S. M. (1998). Desmosomes: integrators of mechanical integrity in tissues. Biol. Bull. 194, 374–376discussion 376-377 10.2307/1543117 [DOI] [PubMed] [Google Scholar]
- Harmon R. M., Simpson C. L., Johnson J. L., Koetsier J. L., Dubash A. D., Najor N. A., Sarig O., Sprecher E., Green K. J. (2013). Desmoglein-1/Erbin interaction suppresses ERK activation to support epidermal differentiation. J. Clin. Invest. 123, 1556–1570 10.1172/JCI65220 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harrison O. J., Bahna F., Katsamba P. S., Jin X., Brasch J., Vendome J., Ahlsen G., Carroll K. J., Price S. R., Honig B. et al. (2010). Two-step adhesive binding by classical cadherins. Nat. Struct. Mol. Biol. 17, 348–357 10.1038/nsmb.1784 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartlieb E., Kempf B., Partilla M., Vigh B., Spindler V., Waschke J. (2013). Desmoglein 2 is less important than desmoglein 3 for keratinocyte cohesion. PLoS ONE 8, e53739 10.1371/journal.pone.0053739 [DOI] [PMC free article] [PubMed] [Google Scholar]
- He W., Cowin P., Stokes D. L. (2003). Untangling desmosomal knots with electron tomography. Science 302, 109–113 10.1126/science.1086957 [DOI] [PubMed] [Google Scholar]
- Heuser A., Plovie E. R., Ellinor P. T., Grossmann K. S., Shin J. T., Wichter T., Basson C. T., Lerman B. B., Sasse-Klaassen S., Thierfelder L. et al. (2006). Mutant desmocollin-2 causes arrhythmogenic right ventricular cardiomyopathy. Am. J. Hum. Genet. 79, 1081–1088 10.1086/509044 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hutter J. L., Bechhoefer J. (1993). Calibration of atomic-force microscope tips. Rev. Sci. Instrum. 64, 1868 10.1063/1.1143970 [DOI] [Google Scholar]
- Kottke M. D., Delva E., Kowalczyk A. P. (2006). The desmosome: cell science lessons from human diseases. J. Cell Sci. 119, 797–806 10.1242/jcs.02888 [DOI] [PubMed] [Google Scholar]
- Kowalczyk A. P., Borgwardt J. E., Green K. J. (1996). Analysis of desmosomal cadherin-adhesive function and stoichiometry of desmosomal cadherin-plakoglobin complexes. J. Invest. Dermatol. 107, 293–300 10.1111/1523--1747.ep12363000 [DOI] [PubMed] [Google Scholar]
- Kowalczyk A. P., Bornslaeger E. A., Borgwardt J. E., Palka H. L., Dhaliwal A. S., Corcoran C. M., Denning M. F., Green K. J. (1997). The amino-terminal domain of desmoplakin binds to plakoglobin and clusters desmosomal cadherin-plakoglobin complexes. J. Cell Biol. 139, 773–784 10.1083/jcb.139.3.773 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marcozzi C., Burdett I. D., Buxton R. S., Magee A. I. (1998). Coexpression of both types of desmosomal cadherin and plakoglobin confers strong intercellular adhesion. J. Cell Sci. 111, 495–509 [DOI] [PubMed] [Google Scholar]
- Marshall B. T., Sarangapani K. K., Wu J., Lawrence M. B., McEver R. P., Zhu C. (2006). Measuring molecular elasticity by atomic force microscope cantilever fluctuations. Biophys. J. 90, 681–692 10.1529/biophysj.105.061010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nekrasova O. E., Amargo E. V., Smith W. O., Chen J., Kreitzer G. E., Green K. J. (2011). Desmosomal cadherins utilize distinct kinesins for assembly into desmosomes. J. Cell Biol. 195, 1185–1203 10.1083/jcb.201106057 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nie Z., Merritt A., Rouhi-Parkouhi M., Tabernero L., Garrod D. (2011). Membrane-impermeable cross-linking provides evidence for homophilic, isoform-specific binding of desmosomal cadherins in epithelial cells. J. Biol. Chem. 286, 2143–2154 10.1074/jbc.M110.192245 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishizawa M., Takoh K., Matsue T. (2002). Micropatterning of HeLa Cells on Glass Substrates and Evaluation of Respiratory Activity Using Microelectrodes. Langmuir 18, 3645–3649 10.1021/la011576k [DOI] [Google Scholar]
- North A. J., Bardsley W. G., Hyam J., Bornslaeger E. A., Cordingley H. C., Trinnaman B., Hatzfeld M., Green K. J., Magee A. I., Garrod D. R. (1999). Molecular map of the desmosomal plaque. J. Cell Sci. 112, 4325–4336 [DOI] [PubMed] [Google Scholar]
- Oda H., Takeichi M. (2011). Evolution: structural and functional diversity of cadherin at the adherens junction. J. Cell Biol. 193, 1137–1146 10.1083/jcb.201008173 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pasdar M., Nelson W. J. (1988a). Kinetics of desmosome assembly in Madin-Darby canine kidney epithelial cells: temporal and spatial regulation of desmoplakin organization and stabilization upon cell-cell contact. I. Biochemical analysis. J. Cell Biol. 106, 677–685 10.1083/jcb.106.3.677 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pasdar M., Nelson W. J. (1988b). Kinetics of desmosome assembly in Madin-Darby canine kidney epithelial cells: temporal and spatial regulation of desmoplakin organization and stabilization upon cell-cell contact. II. Morphological analysis. J. Cell Biol. 106, 687–695 10.1083/jcb.106.3.687 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pasdar M., Nelson W. J. (1989). Regulation of desmosome assembly in epithelial cells: kinetics of synthesis, transport, and stabilization of desmoglein I, a major protein of the membrane core domain. J. Cell Biol. 109, 163–177 10.1083/jcb.109.1.163 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pasdar M., Krzeminski K. A., Nelson W. J. (1991). Regulation of desmosome assembly in MDCK epithelial cells: coordination of membrane core and cytoplasmic plaque domain assembly at the plasma membrane. J. Cell Biol. 113, 645–655 10.1083/jcb.113.3.645 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel S. D., Chen C. P., Bahna F., Honig B., Shapiro L. (2003). Cadherin-mediated cell-cell adhesion: sticking together as a family. Curr. Opin. Struct. Biol. 13, 690–698 10.1016/j.sbi.2003.10.007 [DOI] [PubMed] [Google Scholar]
- Pollard T. D., Borisy G. G. (2003). Cellular motility driven by assembly and disassembly of actin filaments. Cell 112, 453–465 10.1016/S0092--8674(03)00120--X [DOI] [PubMed] [Google Scholar]
- Rakshit S., Zhang Y., Manibog K., Shafraz O., Sivasankar S. (2012). Ideal, catch, and slip bonds in cadherin adhesion. Proc. Natl. Acad. Sci. USA 109, 18815–18820 10.1073/pnas.1208349109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ray C., Brown J. R., Akhremitchev B. B. (2007). Correction of systematic errors in single-molecule force spectroscopy with polymeric tethers by atomic force microscopy. J. Phys. Chem. B 111, 1963–1974 10.1021/jp065530h [DOI] [PubMed] [Google Scholar]
- Roberts G. A., Burdett I. D., Pidsley S. C., King I. A., Magee A. I., Buxton R. S. (1998). Antisense expression of a desmocollin gene in MDCK cells alters desmosome plaque assembly but does not affect desmoglein expression. Eur. J. Cell Biol. 76, 192–203 10.1016/S0171--9335(98)80034--4 [DOI] [PubMed] [Google Scholar]
- Runswick S. K., O'Hare M. J., Jones L., Streuli C. H., Garrod D. R. (2001). Desmosomal adhesion regulates epithelial morphogenesis and cell positioning. Nat. Cell Biol. 3, 823–830 10.1038/ncb0901--823 [DOI] [PubMed] [Google Scholar]
- Saffitz J. E., Jongbloed J. D. H., van der Smagt J., Lombardi P. M., Wiesfeld A. C. P., Nelen M., Schouten M., Jongbloed R., Cox M. G. P. J., van Wolferen M. et al. (2009). Desmosome mutations in arrhythmogenic right ventricular cardiomyopathy: important insight but only part of the picture. Circ. Cardiovasc. Genet. 2, 415–417 10.1161/CIRCGENETICS.109.909366 [DOI] [PubMed] [Google Scholar]
- Saito M., Tucker D. K., Kohlhorst D., Niessen C. M., Kowalczyk A. P. (2012). Classical and desmosomal cadherins at a glance. J. Cell Sci. 125, 2547–2552 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schlegel N., Meir M., Heupel W-M., Holthöfer B., Leube R. E., Waschke J. (2010). Desmoglein 2-mediated adhesion is required for intestinal epithelial barrier integrity. Am. J. Physiol. 298, G774–G783 10.1152/ajpgi.00239.2009 [DOI] [PubMed] [Google Scholar]
- Scott D. W. (1979). On optimal and data-based histograms. Biometrika 66, 605–610 10.1093/biomet/66.3.605 [DOI] [Google Scholar]
- Shapiro L., Weis W. I. (2009). Structure and biochemistry of cadherins and catenins. Cold Spring Harb. Perspect. Biol. 1, a003053–a003053 10.1101/cshperspect.a003053 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shapiro L., Fannon A. M., Kwong P. D., Thompson A., Lehmann M. S., Grübel G., Legrand J. F., Als-Nielsen J., Colman D. R., Hendrickson W. A. (1995). Structural basis of cell-cell adhesion by cadherins. Nature 374, 327–337 10.1038/374327a0 [DOI] [PubMed] [Google Scholar]
- Simpson M. A., Mansour S., Ahnood D., Kalidas K., Patton M. A., McKenna W. J., Behr E. R., Crosby A. H. (2009). Homozygous mutation of desmocollin-2 in arrhythmogenic right ventricular cardiomyopathy with mild palmoplantar keratoderma and woolly hair. Cardiology 113, 28–34 10.1159/000165696 [DOI] [PubMed] [Google Scholar]
- Simpson C. L., Patel D. M., Green K. J. (2011). Deconstructing the skin: cytoarchitectural determinants of epidermal morphogenesis. Nat. Rev. Mol. Cell Biol. 12, 565–580 10.1038/nrm3175 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sivasankar S., Zhang Y., Nelson W. J., Chu S. (2009). Characterizing the initial encounter complex in cadherin adhesion. Structure 17, 1075–1081 10.1016/j.str.2009.06.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith S. B., Finzi L., Bustamante C. (1992). Direct mechanical measurements of the elasticity of single DNA molecules by using magnetic beads. Science 258, 1122–1126 10.1126/science.1439819 [DOI] [PubMed] [Google Scholar]
- Spiliotis E. T., Kinoshita M., Nelson W. J. (2005). A mitotic septin scaffold required for Mammalian chromosome congression and segregation. Science 307, 1781–1785 10.1126/science.1106823 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Syed S-E-H., Trinnaman B., Martin S., Major S., Hutchinson J., Magee A. I. (2002). Molecular interactions between desmosomal cadherins. Biochem. J. 362, 317–327 10.1042/0264--6021:3620317 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomason H. A., Scothern A., McHarg S., Garrod D. R. (2010). Desmosomes: adhesive strength and signalling in health and disease. Biochem. J. 429, 419–433 10.1042/BJ20100567 [DOI] [PubMed] [Google Scholar]
- Troyanovsky S. M., Troyanovsky R. B., Eshkind L. G., Krutovskikh V. A., Leube R. E., Franke W. W. (1994a). Identification of the plakoglobin-binding domain in desmoglein and its role in plaque assembly and intermediate filament anchorage. J. Cell Biol. 127, 151–160 10.1083/jcb.127.1.151 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Troyanovsky S. M., Troyanovsky R. B., Eshkind L. G., Leube R. E., Franke W. W. (1994b). Identification of amino acid sequence motifs in desmocollin, a desmosomal glycoprotein, that are required for plakoglobin binding and plaque formation. Proc. Natl. Acad. Sci. USA 91, 10790–10794 10.1073/pnas.91.23.10790 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waschke J. (2008). The desmosome and pemphigus. Histochem. Cell Biol. 130, 21–54 10.1007/s00418--008--0420--0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamada S., Pokutta S., Drees F., Weis W. I., Nelson W. J. (2005). Deconstructing the cadherin-catenin-actin complex. Cell 123, 889–901 10.1016/j.cell.2005.09.020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y., Sivasankar S., Nelson W. J., Chu S. (2009). Resolving cadherin interactions and binding cooperativity at the single-molecule level. Proc. Natl. Acad. Sci. USA 106, 109–114 10.1073/pnas.0811350106 [DOI] [PMC free article] [PubMed] [Google Scholar]
Footnotes
Competing interests
The authors declare no competing interests.
Author contributions
M.L. designed, performed and interpreted the experiments, assembled the figures, and wrote the manuscript; S.R., O.S. and S.S. performed the SMSF experiments; R.M.H. provided the Dsg-Fc and Dsc2-Fc fusion proteins; N.B. was involved in the initial design of experiments; K.J.G., S.S. and W.J.N. were involved in designing the experiments and writing the manuscript.
Funding
Work in the S.S. laboratory was partially supported by a grant from the American Heart Association (Scientist Development Grant 12SDG9320022). Work in the K.J.G. laboratory was supported by grants from the National Institutes of Health (NIH) [grant numbers R37AR043380, R01 AR41836]. Work in the W.J.N. laboratory was supported by the NIH [grant number GM35527] and Human Frontier Science Program [grant number RGP0040/2012]. M.L. was supported by a Genentech Graduate Fellowship, Cancer Biology Training Grant and Mason Case Graduate Fellowship. R.M.H. was supported by a NIH T32 training grant [grant number GM08061]; and an American Heart Association predoctoral fellowship. Deposited in PMC for release after 12 months.
Supplementary material available online at http://jcs.biologists.org/lookup/suppl/doi:10.1242/jcs.146316/-/DC1
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.