

Background: Allosteric modulators bound to the transmembrane domain (TMD) of the α7 nicotinic acetylcholine receptor (α7nAChR) can potentiate channel function.
Results: ELIC-α7nAChR showed potentiation only when the extracellular-transmembrane domain (ECD-TMD) interface matched that of α7nAChR.
Conclusion: PAM modulation through the TMD requires a more specific ECD-TMD interface than agonist activation.
Significance: The study provides insight into the basis for positive allosteric modulation of α7nAChR.
Keywords: Allosteric Regulation, Ion Channels, Neurotransmitter Receptors, Nicotinic Acetylcholine Receptors, Protein Chimeras, ELIC, α7nAChR, pLGICs, Pentameric Ligand-gated Ion Channels
Abstract
The native α7 nicotinic acetylcholine receptor (α7nAChR) is a homopentameric ligand-gated ion channel mediating fast synaptic transmission and is of pharmaceutical interest for treatment of numerous disorders. The transmembrane domain (TMD) of α7nAChR has been identified as a target for positive allosteric modulators (PAMs), but it is unclear whether modulation occurs through changes entirely within the TMD or changes involving both the TMD and the extracellular domain (ECD)-TMD interface. In this study, we constructed multiple chimeras using the TMD of human α7nAChR and the ECD of a prokaryotic homolog, ELIC, which is not sensitive to these modulators, and for which a high resolution structure has been solved. Functional ELIC-α7nAChR (EA) chimeras were obtained when their ECD-TMD interfaces were modified to resemble either the ELIC interface (EAELIC) or α7nAChR interface (EAα7). Both EAα7 and EAELIC show similar activation response and desensitization characteristics, but only EAα7 retained the unique pharmacology of α7nAChR evoked by PAMs, including potentiation by ivermectin, PNU-120596, and TQS, as well as activation by 4BP-TQS. This study suggests that PAM modulation through the TMD has a more stringent requirement at the ECD-TMD interface than agonist activation.
Introduction
The α7 nicotinic acetylcholine receptor (α7nAChR)2 is a homopentameric acetylcholine-gated cation channel mediating fast synaptic transmission in neuronal cells (1) and calcium signaling in nonneuronal cells (2). The diverse biological functions of α7nAChR have made it a promising therapeutic target for numerous medical conditions, including pain (3, 4), inflammation (5), cardiovascular disease (6), and a variety of psychiatric and neurological disorders (7–9). α7nAChR opens the channel gate in the transmembrane domain (TMD) upon agonist binding to the extracellular domain (ECD), but channel function can also be modulated by ligand binding to allosteric sites distinct from the orthosteric agonist binding-sites in the ECD. Positive allosteric modulators (PAMs), especially those specific for α7nAChR, such as PNU-120596 (10) and TQS (11), are particularly of therapeutic potential (8). Based upon mutational analysis, certain PAMs are thought to act at the ECD or ECD-TMD interface (12, 13), whereas others, such as PNU-120596 and TQS, are thought to act through binding to intrasubunit sites near the middle of the α7nAChR TMD (13–17). The molecular mechanisms of allosteric modulation are not understood.
α7nAChR is a member of the pentameric ligand-gated ion channel (pLGIC) superfamily, including different subtypes of nAChRs, 5-HT3A receptors (5-HT3ARs), glycine receptors (GlyRs), and GABAA receptors (GABAARs). α7nAChR is also homologous to GluCl, and the prokaryotic pLGICs ELIC and GLIC, whose crystal structures have been determined in the absence and presence of ligands (18–28). A wealth of data from decades of biochemical characterization and more recent structural characterization of pLGICs provide a broad understanding of the events relating to agonist binding and channel activation (28–35). Agonist binding to orthosteric sites in the ECD leads to conformational transitions that propagate through the ECD-TMD interface to the TMD and open the channel gate. Upon prolonged exposure to agonist, the channel desensitizes and is no longer sensitive to agonist binding until it returns to the resting state. Among pLGICs, α7nAChR demonstrates particularly rapid desensitization. PAMs such as ivermectin, PNU-120596, and TQS can significantly slow down the desensitization of α7nAChR (10, 11, 36, 37). Several studies have demonstrated involvement of the ECD-TMD interface in desensitization (38–41), but the role of the ECD-TMD interface in allosteric modulation through the TMD is less known.
Chimeras joining the ECD from one pLGIC with the TMD from another provide valuable opportunities to understand how the interplay among different domains/regions underlies pLGIC function. Previous studies of GLIC-α1GlyR (42), α7nAChR-α1GlyR (43), α4β2nAChR-5HT3R (44), and α7nAChR-5HT3R (45) found that functional chimeras could be obtained by splicing the ECD prior to the first transmembrane helix of the TMD, although kinetic parameters could be improved by optimization of the ECD-TMD interface. In contrast, it has proven difficult to express a functional chimera of an α7nAChR TMD (13). A 5HT3R-α7nAChR chimera expressed a small current in only a few oocytes, which was sufficient to show that potentiation by the PAM PNU-120596 could be conferred by the α7nAChR TMD when activated by a suboptimal agonist (13). However, given the high homology between loop 7 and the TM2-3 linker of α7nAChR and 5HT3R (approximately 78% similarity), it is unclear what role the ECD-TMD interface might have played in the observed potentiation.
In this study, we constructed multiple chimeras combining the TMD of human α7nAChR and the ECD of the prokaryotic homolog ELIC (Fig. 1). ELIC is a cation channel activated by a group of primary amines, including cysteamine (46). In contrast to α7nAChR, ELIC is inhibited by the general anesthetic propofol but unaffected by PAMs like ivermectin (21, 47). We show here that ELIC is also unaffected by the α7nAChR-specific PAMs PNU-120596 and TQS. We examined the effect of the ECD-TMD interface on allosteric modulation through the TMD by engineering two classes of ELIC-α7nAChR (EA) chimeras: EAELIC, with an ECD-TMD interface resembling ELIC, and EAα7, with an α7nAChR ECD-TMD interface. Both classes of EA chimeras resulted in functional channels with similar activation and desensitization kinetics. However, only EAα7 retained sensitivity to the PAMs ivermectin, PNU, and TQS as well as the ability to be activated by 4BP-TQS. Thus, allosteric modulation through the TMD requires a specific ECD-TMD interface similar to that of α7nAChR, a more stringent requirement than that needed to activate the channel by agonists.
FIGURE 1.
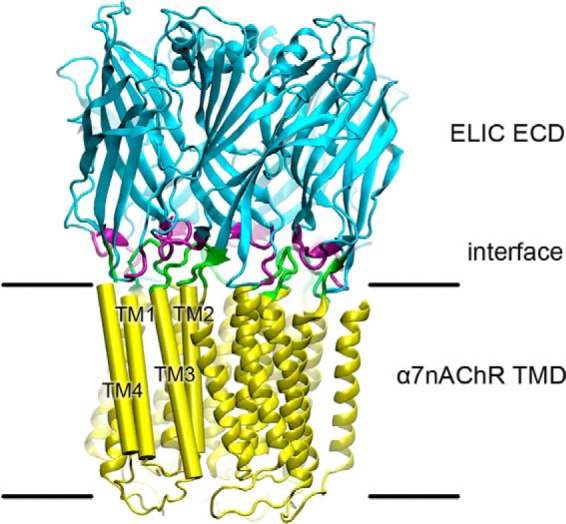
Homology model of ELIC-α7nAChR highlighting the interface between domains. The EA chimera was modeled to the ECD of ELIC (PDB ID code 3RQW) and the TMD of the α7nAChR (PDB ID code 2MAW). Colors depict the ELIC ECD (cyan) and the α7nAChR TMD (yellow). Residues predicted to comprise the ECD-TMD interface are colored purple in the ECD and green in the TMD. For clarity, the TMD helices of one subunit are labeled and depicted as cylinders.
EXPERIMENTAL PROCEDURES
Construction of EA Chimeras
EA chimeras were constructed by fusing the ECD of ELIC ending after pre-TM1 (ELIC-S201) with the beginning of the human α7nAChR TMD (α7-Y210) using overlapping PCR (48). The resulting construct was subcloned into a Xenopus expression vector, pOTV (49) for expression of RNA. Mutations to match the sequence of interface elements between ELIC and α7nAChR were introduced using QuikChange Lightning Site-directed Mutagenesis kits (Agilent). All constructs were confirmed by sequencing in full.
Electrophysiological Recordings in Xenopus Oocytes
Channel function was measured by two-electrode voltage clamp experiments (50) using Xenopus laevis oocytes injected with RNA encoding the indicated constructs as described previously (20). Capped complementary RNA was synthesized with the mMessage mMachine T7 kit (Ambion), purified with the RNeasy kit (Qiagen) and injected (25–50 ng) into X. laevis oocytes (stages 5–6). Oocytes were maintained at 18 °C in modified Barth's solution containing 88 mm NaCl, 1 mm KCl, 2.4 mm NaHCO3, 15 mm HEPES, 0.3 mm Ca(NO3)2, 0.41 mm CaCl2, 0.82 mm MgSO4, 10 μg/ml sodium penicillin, 10 μg/ml streptomycin sulfate, and 100 μg/ml gentamycin sulfate, pH 6.7. After 1–3 days expression, oocytes were clamped to a holding potential of −60 mV with an OC-725C Amplifier (Warner Instruments) in a 20-μl oocyte recording chamber (Automate Scientific). Currents were elicited using cysteamine as an agonist. The recording solutions contained 130 mm NaCl, 0.1 mm CaCl2, 10 mm HEPES, pH 7.0, with the indicated concentrations of cysteamine and other modulators. Data were collected and processed using Clampex 10 software (Molecular Devices). Nonlinear regressions were performed using Prism software (GraphPad).
Modeling of Chimeras
Homology models for EA chimeras were generated using Modeler 9v8 (51, 52) based on the crystal structure of ELIC (Protein Data Bank (PDB) ID code 3RQW) and the NMR structure of the α7nAChR TMD (PDB ID code 2MAW). Five independent models were generated, and the model with the lowest discrete optimized potential energy was selected for presentation.
RESULTS
Engineering Functional EA Chimeras
Multiple EA chimeras were constructed to assess the requirements for efficient coupling at the ECD-TMD interface. The function of each chimera was tested in Xenopus oocytes by two-electrode voltage clamp electrophysiology. The data for agonist response are summarized in Table 1.
TABLE 1.
EA chimeras
The sequences for each ECD-TMD interface element modified in this study and their EC50s for cysteamine are indicated. Residues from α7nAChR are indicated in bold. Residues conserved between the two sequences are italicized. Residues within the TM2-3 linker shown in the structures of ELIC (PDB ID code 3RQW) and α7nAChR (PDB ID code 2MAW) are underlined. A dash indicates no current detected.
| Construct | Loop 2 | Loop 7 | Loop 9 | Pre-TM1 | TM2-3 linker | C terminus | Cysteamine EC50 (mm) |
|---|---|---|---|---|---|---|---|
| ELIC | NTLE | FRL | EEIDEWW | NPS | SNILPRLPYTT | RGITL | 0.4 |
| α7nAChR | DEKN | VRW | IPNGEWD | RTL | AEIMPATSDSVPLIAQY | SAPNFVEAVSKDFA | — |
| EAELIC1 | NTLE | FRL | EEIDEWW | NPS | AEIMPATSDSVPLIAQY | SAPNFVEAVSKDFA | — |
| EAELIC2 | NTLE | FRL | EEIDEWW | NPS | AEIMPATSDSVPLIAQY | RGITL | — |
| EAELIC3 | NTLE | FRL | EEIDEWW | NPS | AEIMPRLPYSVPLIAQY | RGITL | — |
| EAELIC4 | NTLE | FRL | EEIDEWW | NPS | SNILPRLPYTVPLIAQY | RGITL | — |
| EAELIC5 | NTLE | FRL | EEIDEWW | NPS | AEILPRLPYTT----------- | RGITL | 0.4 |
| EAELIC | NTLE | FRL | EEIDEWW | NPS | APRLPYTTDSVPLIAQY | RGITL | 1.0 |
| EAα71 | NTLE | VRW | IPNGEWD | RTL | AEIMPATSDSVPLIAQY | RGITL | 2.2 |
| EA α72 | DEKN | VRW | EEIDEWW | RTL | AEIMPATSDSVPLIAQY | RGITL | 2.6 |
| EA α73 | NTLE | VRW | EEIDEWW | RTL | AEIMPATSDSVPLIAQY | RGITL | — |
| EA α74 | DEKN | FRL | IPNGEWD | RTL | AEIMPATSDSVPLIAQY | RGITL | — |
| EAα7 | DEKN | VRW | IPNGEWD | RTL | AEIMPATSDSVPLIAQY | RGITL | 1.3 |
The original ECD-TMD interfaces for ELIC and α7nAChR are not compatible. A chimera (EAELIC1) simply connecting the ELIC ECD with the α7nAChR TMD at the beginning of TM1 did not show any response to the ELIC agonist cysteamine when the chimera was expressed in Xenopus oocytes (Table 1). This was unexpected. Chimeras between most members of the pLGIC family were found to have at least some degree of function when joined in this manner (13, 42–45), with the exception of the AChBP-5HT3A chimeras (53). To rule out interference by the extended C terminus of α7nAChR, we replaced the last 18 residues of EAELIC1 with the last 4 residues of ELIC. This construct EAELIC2 also failed to exhibit current in response to cysteamine (Table 1).
The sequence, length, and position of the TM2-3 linker vary among the known pLGIC structures. The TM2-3 linker comprises 8 residues beginning at 22′ in ELIC (PDB ID code 3RQU) (20) and 10 residues beginning at 22′ in the NMR structure for the TMD of α7nAChR (PDB ID code 2MAW) (54). The TM2-3 linker in the cryo-EM structure of Torpedo nAChR has only 7 residues in the flexible loop beginning at 29′. If one counts the TM2-3 linker of Torpedo nAChR starting from 22′, it has a total of 14 residues (PDB ID code 2BG9) (55). A recent disulfide trapping experiment with the α subunit of mouse muscle nAChR showed a shift in register between TM2 and TM3, which could not be reconciled with 2BG9 (56), but was consistent with the α7 NMR structure (54).
To improve coupling of the α7nAChR TM2-3 linker to the ELIC ECD without disrupting TMD interactions, we substituted varied lengths of the TM2-3 linker in α7nAChR with their counterparts in ELIC. Substitution of only 4 residues in the middle of the TM2-3 linker (EAELIC3) or more extensive substitution from 19′ to 28′ (EAELIC4) was not functional (Table 1).
A functional EA chimera (EAELIC5) was obtained by replacing all 14 residues from 22′ to 35′ of the TM2-3 region with the 8-residue ELIC TM2-3 linker. A functional EA chimera (EAELIC) was also obtained by substituting the α7nAChR TM2-3 linker from 20′ to 26′ with the ELIC TM2-3 linker from 23′ to 29′. Both EAELIC5 and EAELIC responded efficiently to the ELIC agonist cysteamine, with EC50 values of 0.4 and 1.0 mm, respectively (Table 1 and Fig. 2), comparable with that for ELIC (20, 46). The efficient activation obtained with both EAELIC5 and EAELIC compared with EAELIC3 and EAELIC4 suggests that it is not the length of the TM2-3 linker, but its position at the ECD-TMD interface that is critical for efficient coupling.
FIGURE 2.

Activation of EA chimeras by cysteamine. a and b, representative current traces for EAELIC (a) and EAα7 (b). Bars over the trace indicate the length of application and cysteamine concentrations. Horizontal and vertical scale bars indicate 1 min and 0.1 μA current, respectively. c, cysteamine response curves for the functional EA chimeras. Current is expressed as a fraction of maximal current, n ≥ 5 oocytes. Error bars indicate S.D. Data are fit to Hill equations with the following parameters: ■, EAELIC, EC50 = 1.0 ± 0.02 mm; ●, EAα7, EC50 = 1.3 ± 0.1 mm; ▵, EAELIC5, EC50 = 0.4 ± 0.02 mm; □, EA α71, EC50 = 2.2 ± 0.1 mm; ○, EAα72, EC50 = 2.6 ± 0.1 mm. The corresponding Hill coefficients are 1.7 ± 0.1, 1.3 ± 0.1, 1.5 ± 0.1, 1.9 ± 0.1, and 2.4 ± 0.2, respectively.
To engineer a chimera with a more native α7nAChR ECD-TMD interface, we kept the α7nAChR TM2-3 linker intact but mutated the ELIC ECD to match the sequence of α7nAChR at loops 2, 7, 9, and the pre-TM1 region. This construct, EAα7, is functional with an EC50 of 1.3 mm for cysteamine (Table 1 and Fig. 2). EA chimeras with mismatches at loop 2 (EAα71) or loop 9 (EAα72) still expressed as functional channels, but with a rightward shift in EC50 for cysteamine, suggesting a lower coupling efficiency (Table 1 and Fig. 2c). Mismatches at both loop 2 and loop 9 (EAα73), or at loop 7 alone (EAα74), resulted in chimeras that did not exhibit current in response to cysteamine (Table 1). Taken together, these results suggest that an ensemble of coupling among loop 2, loop 7, loop 9, the pre-TM1 region and the TM2-3 linker is essential for obtaining functional channels. Similar modifications were also needed for the functional AChBP-5HT3 chimera (53).
ELIC (46) and α7nAChR (1) exhibit desensitization with sustained application of agonist, but α7nAChR (0.18 ± 0.05 min, n = 3) desensitizes an order of magnitude faster than ELIC (2.4 ± 0.2 min, n = 3) (Fig. 3). Measuring ensemble currents in Xenopus oocytes, both EAα7 and EAELIC exhibited initial desensitization (1.7 ± 0.2 and 1.6 ± 0.2 min, respectively, n = 3) comparable with ELIC, but then plateaued at 55 ± 11% and 74 ± 16% of the maximal current, respectively (Fig. 3).
FIGURE 3.

EA chimeras desensitize much more slowly than α7nAChR. Representative current traces show a 1.5-min agonist application to Xenopus oocytes injected with the indicated constructs. Acetylcholine at 100 μm was applied to α7nAChR; 30 mm cysteamine was applied to ELIC and EA chimeras. Rate constants for the initial rate of desensitization were calculated (n = 3): α7nAChR = 0.18 ± 0.05; ELIC = 2.4 ± 0.2; EAELIC = 1.6 ± 0.2; and EAα7 = 1.7 ± 0.2 min.
EAα7 Shows Pharmacology Similar to α7nAChR, but EAELIC Does Not
ELIC and α7nAChR have different pharmacological profiles for allosteric modulators. Consistent with previous results (13–16, 57), ivermectin and two α7-specific PAMs, PNU-120596 and TQS, potentiated α7nAChR, but the intravenous general anesthetic propofol had no effects on α7nAChR (Fig. 4). In contrast, ELIC could be inhibited by propofol (21, 47) but was insensitive to ivermectin, PNU-120596, or TQS (Fig. 4). The differences between ELIC and α7 pharmacology present the opportunity to evaluate the role of the TMD and ECD-TMD interface in allosteric modulation. To this end, we compared the functional responses of α7nAChR, ELIC, and EAα7 to the selected allosteric modulators (Fig. 4).
FIGURE 4.

EAα7 resembles α7nAChR pharmacology. Representative traces of ELIC, α7nAChR, and EAα7 show responses to the indicated allosteric modulators. Ivermectin (30 μm), TQS (100 μm), and propofol (100 μm) were applied with agonist cysteamine (ELIC, EAα7) or acetylcholine (α7nAChR) at the EC20 for each construct; PNU-120596 (30 μm) was applied with agonist at the EC70 for each construct. Bars over the traces indicate the length of application. Horizontal and vertical scale bars indicate 1 min and 0.1 μA current, respectively.
EAα7 responded to these modulators similarly to α7nAChR, but distinctly different from ELIC. Resembling α7nAChR, EAα7 was potentiated by ivermectin, PNU-120596, and TQS, and insensitive to propofol (Fig. 4). 4BP-TQS, an allosteric agonist and PAM for α7nAChR (15), could also directly activate EAα7 and potentiate its channel response to agonist (Fig. 5). These results show that EAα7 qualitatively reproduces the pharmacological properties of α7nAChR for these modulators.
FIGURE 5.
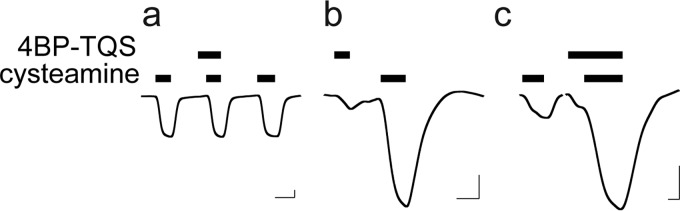
EAα7 is activated and potentiated by 4BP-TQS. Representative traces of ELIC and EAα7 show responses to cysteamine and 4BP-TQS. a, ELIC is insensitive to 4BP-TQS, but EAα7 is activated (b) and potentiated (c) by 4BP-TQS (100 μm). Cysteamine concentrations are at the EC20 for each construct. Horizontal and vertical scale bars indicate 1 min and 0.1 μA current, respectively.
In contrast to EAα7, EAELIC5 and EAELIC were insensitive to ivermectin, TQS, or PNU-120596 (Fig. 6). They were also insensitive to propofol. The lack of sensitivity to propofol for the EA chimeras can be explained by the loss of the binding site within the ELIC TMD. However, EAELIC5, EAELIC, and EAα7 all have the α7nAChR TMD, which contains the binding sites for the tested PAMs. These chimeras differ only in the ECD-TMD interface, suggesting a functional role of the ECD-TMD interface in allosteric potentiation through the TMD.
FIGURE 6.

EAELIC is insensitive to allosteric modulators acting through the TMD. Representative traces of EAELIC (a) and EAELIC5 (b) show responses to the indicated allosteric modulators. Ivermectin (30 μm), TQS (100 μm), and propofol (100 μm) were applied with cysteamine at the EC20; PNU-120596 (30 μm) was applied with cysteamine at the EC70. Bars over the traces indicate length of application. Horizontal and vertical scale bars indicate 1 min and 0.1 μA current, respectively.
DISCUSSION
Unlike other Cys-loop receptor chimeras (42–45), the engineering of functional chimeras between ELIC and α7nAChR requires more extensive optimization of an ensemble of interactions at the ECD-TMD interface. It does not matter how the optimization is achieved: chimeras with either an ELIC interface (EAELIC5, EAELIC) or an α7nAChR interface (EAα7) show similar agonist response and desensitization characteristics. They desensitize much slower than native α7nAChR. PAM modulation through the TMD only occurs with the α7nAChR ECD-TMD interface, suggesting that PAM modulation through the TMD requires a more specific ECD-TMD interface than agonist activation.
One of the primary effects noted for PAMs like PNU-120596 and TQS is a marked decrease in the α7nAChR desensitization rate (10, 11, 36, 37). Our study shows that EAα7 does not reproduce the fast desensitization characteristic of α7nAChR, yet can still be modulated by these PAMs, suggesting that fast desensitization may not be required for potentiation by PNU-120596 or TQS. Because EAα7 and EAELIC show similar desensitization rates, desensitization cannot explain their distinct responses to the tested PAMs.
What may have contributed to the different responses of EAα7 and EAELIC to PAMs? The PAMs tested here bind to the TMD (13–15, 58). The differences between EAα7 and EAELIC/EAELIC5 are localized only to the ECD-TMD interface. One may suspect that lack of modulation in EAELIC5 results from interference with PAMs binding to a structurally distorted TMD due to a shortened TM2-3 linker. However, such a possibility is dismissed by EAELIC, which has a full-length TM2-3 linker that is unlikely to distort the TMD.
Because no high resolution structural data are available, the precise binding sites for ivermectin, PNU-120596, or TQS in α7nAChR are unknown. In the Caenorhabditis elegans GluCl crystal structure, ivermectin contacts both the TM2-3 linker and the residues toward the middle of the TMD (18). Mutational analysis on α7nAChR also suggests that ivermectin interacts with residues near the TM2-3 linker and residues near the middle of the TMD (16, 59). If direct contacts to specific residues in the TM2-3 linker are critical for ivermectin modulation, the ELIC TM2-3 linker may not provide proper contacts in EAELIC and EAELIC5, thereby preventing ivermectin modulation.
In the case of PNU-120596 and TQS, they are much smaller than ivermectin and cannot simultaneously contact residues at both the ECD-TMD interface and near the middle of the TMD. If they directly contact the ECD-TMD interface, the role of the ECD-TMD interface in PAM modulation will be the same as discussed above for ivermectin. However, previous mutation studies suggest an intrasubunit binding site near the middle of the TMD for PNU-120596 and TQS (13–15, 58). In this case, the binding site is remote from the ECD-TMD interface. How can the interface still be important for modulation? It is possible that the PAM binding to the proposed site near the middle of TMD allosterically induces changes to the ECD-TMD interface that are required for potentiation. These changes can be accommodated by the ECD-TMD interface in α7nAChR, but not in ELIC. This hypothesis is supported by a substituted cysteine accessibility study (60), which demonstrates that PNU-120596 binding can independently induce conformational changes to the ECD and ECD-TMD interface. The conformational changes induced by PNU-120596 are similar but not identical to those induced by the agonist acetylcholine (60).
Structural characterization of the precise binding sites for these PAMs is essential for understanding the mechanism of PAM modulation. Future high resolution structural studies of the EA chimeras in the absence and presence of modulators will provide insights into binding of PAMs to the TMD and the role of the ECD-TMD interface in allosteric modulation. Because of the prokaryotic origin of its ECD, the EA chimeras are good candidates for production in quantities suitable for high resolution structural studies (19, 20).
Acknowledgment
We thank Prof. Thomas R. Kleyman's laboratory for providing X. laevis oocytes for electrophysiology experiments.
This work was supported, in whole or in part, by National Institutes of Health Grants R01GM66358 and R01GM56257 (to P. T.) and R37GM049202 (to Y. X.).
- α7nAChR
- α7 nicotinic acetylcholine receptor
- EA
- ELIC-α7nAChR
- ECD
- extracellular domain
- PAM
- positive allosteric modulator
- PDB
- Protein Data Bank
- pLGIC
- pentameric ligand-gated ion channel
- TMD
- transmembrane domain.
REFERENCES
- 1. Couturier S., Bertrand D., Matter J. M., Hernandez M. C., Bertrand S., Millar N., Valera S., Barkas T., Ballivet M. (1990) A neuronal nicotinic acetylcholine receptor subunit (α7) is developmentally regulated and forms a homo-oligomeric channel blocked by α-BTX. Neuron 5, 847–856 [DOI] [PubMed] [Google Scholar]
- 2. Wessler I., Kirkpatrick C. J. (2008) Acetylcholine beyond neurons: the nonneuronal cholinergic system in humans. Br. J. Pharmacol. 154, 1558–1571 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Alsharari S. D., Freitas K., Damaj M. I. (2013) Functional role of α7 nicotinic receptor in chronic neuropathic and inflammatory pain: studies in transgenic mice. Biochem. Pharmacol. 86, 1201–1207 [DOI] [PubMed] [Google Scholar]
- 4. de Jonge W. J., Ulloa L. (2007) The α7 nicotinic acetylcholine receptor as a pharmacological target for inflammation. Br. J. Pharmacol. 151, 915–929 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Wang H., Yu M., Ochani M., Amella C. A., Tanovic M., Susarla S., Li J. H., Wang H., Yang H., Ulloa L., Al-Abed Y., Czura C. J., Tracey K. J. (2003) Nicotinic acetylcholine receptor α7 subunit is an essential regulator of inflammation. Nature 421, 384–388 [DOI] [PubMed] [Google Scholar]
- 6. Egleton R. D., Brown K. C., Dasgupta P. (2008) Nicotinic acetylcholine receptors in cancer: multiple roles in proliferation and inhibition of apoptosis. Trends Pharmacol. Sci. 29, 151–158 [DOI] [PubMed] [Google Scholar]
- 7. Poorthuis R. B., Mansvelder H. D. (2013) Nicotinic acetylcholine receptors controlling attention: behavior, circuits and sensitivity to disruption by nicotine. Biochem. Pharmacol. 86, 1089–1098 [DOI] [PubMed] [Google Scholar]
- 8. Pandya A. A., Yakel J. L. (2013) Effects of neuronal nicotinic acetylcholine receptor allosteric modulators in animal behavior studies. Biochem. Pharmacol. 86, 1054–1062 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Smucny J., Tregellas J. (2013) Nicotinic modulation of intrinsic brain networks in schizophrenia. Biochem. Pharmacol. 86, 1163–1172 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Hurst R. S., Hajós M., Raggenbass M., Wall T. M., Higdon N. R., Lawson J. A., Rutherford-Root K. L., Berkenpas M. B., Hoffmann W. E., Piotrowski D. W., Groppi V. E., Allaman G., Ogier R., Bertrand S., Bertrand D., Arneric S. P. (2005) A novel positive allosteric modulator of the α7 neuronal nicotinic acetylcholine receptor: in vitro and in vivo characterization. J. Neurosci. 25, 4396–4405 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Grønlien J. H., Håkerud M., Ween H., Thorin-Hagene K., Briggs C. A., Gopalakrishnan M., Malysz J. (2007) Distinct profiles of α7 nAChR positive allosteric modulation revealed by structurally diverse chemotypes. Mol. Pharmacol. 72, 715–724 [DOI] [PubMed] [Google Scholar]
- 12. Grønlien J. H., Ween H., Thorin-Hagene K., Cassar S., Li J., Briggs C. A., Gopalakrishnan M., Malysz J. (2010) Importance of M2-M3 loop in governing properties of genistein at the α7 nicotinic acetylcholine receptor inferred from α7/5-HT3A chimera. Eur. J. Pharmacol. 647, 37–47 [DOI] [PubMed] [Google Scholar]
- 13. Bertrand D., Bertrand S., Cassar S., Gubbins E., Li J., Gopalakrishnan M. (2008) Positive allosteric modulation of the α7 nicotinic acetylcholine receptor: ligand interactions with distinct binding sites and evidence for a prominent role of the M2-M3 segment. Mol. Pharmacol. 74, 1407–1416 [DOI] [PubMed] [Google Scholar]
- 14. Young G. T., Zwart R., Walker A. S., Sher E., Millar N. S. (2008) Potentiation of α7 nicotinic acetylcholine receptors via an allosteric transmembrane site. Proc. Natl. Acad. Sci. U.S.A. 105, 14686–14691 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Gill J. K., Savolainen M., Young G. T., Zwart R., Sher E., Millar N. S. (2011) Agonist activation of α7 nicotinic acetylcholine receptors via an allosteric transmembrane site. Proc. Natl. Acad. Sci. U.S.A. 108, 5867–5872 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Collins T., Millar N. S. (2010) Nicotinic acetylcholine receptor transmembrane mutations convert ivermectin from a positive to a negative allosteric modulator. Mol. Pharmacol. 78, 198–204 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Collins T., Young G. T., Millar N. S. (2011) Competitive binding at a nicotinic receptor transmembrane site of two α7-selective positive allosteric modulators with differing effects on agonist-evoked desensitization. Neuropharmacology 61, 1306–1313 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Hibbs R. E., Gouaux E. (2011) Principles of activation and permeation in an anion-selective Cys-loop receptor. Nature 474, 54–60 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Hilf R. J., Dutzler R. (2008) X-ray structure of a prokaryotic pentameric ligand-gated ion channel. Nature 452, 375–379 [DOI] [PubMed] [Google Scholar]
- 20. Pan J., Chen Q., Willenbring D., Yoshida K., Tillman T., Kashlan O. B., Cohen A., Kong X. P., Xu Y., Tang P. (2012) Structure of the pentameric ligand-gated ion channel ELIC cocrystallized with its competitive antagonist acetylcholine. Nat. Commun. 3, 714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Spurny R., Ramerstorfer J., Price K., Brams M., Ernst M., Nury H., Verheij M., Legrand P., Bertrand D., Bertrand S., Dougherty D. A., de Esch I. J., Corringer P. J., Sieghart W., Lummis S. C., Ulens C. (2012) Pentameric ligand-gated ion channel ELIC is activated by GABA and modulated by benzodiazepines. Proc. Natl. Acad. Sci. U.S.A. 109, E3028–E3034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Bocquet N., Nury H., Baaden M., Le Poupon C., Changeux J. P., Delarue M., Corringer P. J. (2009) X-ray structure of a pentameric ligand-gated ion channel in an apparently open conformation. Nature 457, 111–114 [DOI] [PubMed] [Google Scholar]
- 23. Hilf R. J., Dutzler R. (2009) Structure of a potentially open state of a proton-activated pentameric ligand-gated ion channel. Nature 457, 115–118 [DOI] [PubMed] [Google Scholar]
- 24. Nury H., Van Renterghem C., Weng Y., Tran A., Baaden M., Dufresne V., Changeux J. P., Sonner J. M., Delarue M., Corringer P. J. (2011) X-ray structures of general anaesthetics bound to a pentameric ligand-gated ion channel. Nature 469, 428–431 [DOI] [PubMed] [Google Scholar]
- 25. Spurny R., Billen B., Howard R. J., Brams M., Debaveye S., Price K. L., Weston D. A., Strelkov S. V., Tytgat J., Bertrand S., Bertrand D., Lummis S. C., Ulens C. (2013) Multisite binding of a general anesthetic to the prokaryotic pentameric Erwinia chrysanthemi ligand-gated ion channel (ELIC). J. Biol. Chem. 288, 8355–8364 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Gonzalez-Gutierrez G., Lukk T., Agarwal V., Papke D., Nair S. K., Grosman C. (2012) Mutations that stabilize the open state of the Erwinia chrysanthemi ligand-gated ion channel fail to change the conformation of the pore domain in crystals. Proc. Natl. Acad. Sci. U.S.A. 109, 6331–6336 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Gonzalez-Gutierrez G., Cuello L. G., Nair S. K., Grosman C. (2013) Gating of the proton-gated ion channel from Gloeobacter violaceus at pH 4 as revealed by x-ray crystallography. Proc. Natl. Acad. Sci. U.S.A. 110, 18716–18721 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Sauguet L., Shahsavar A., Poitevin F., Huon C., Menny A., Nemecz À., Haouz A., Changeux J. P., Corringer P. J., Delarue M. (2014) Crystal structures of a pentameric ligand-gated ion channel provide a mechanism for activation. Proc. Natl. Acad. Sci. U.S.A. 111, 966–971 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Dacosta C. J., Baenziger J. E. (2013) Gating of pentameric ligand-gated ion channels: structural insights and ambiguities. Structure 21, 1271–1283 [DOI] [PubMed] [Google Scholar]
- 30. Bouzat C. (2012) New insights into the structural bases of activation of Cys-loop receptors. J. Physiol. 106, 23–33 [DOI] [PubMed] [Google Scholar]
- 31. Corringer P. J., Poitevin F., Prevost M. S., Sauguet L., Delarue M., Changeux J. P. (2012) Structure and pharmacology of pentameric receptor channels: from bacteria to brain. Structure 20, 941–956 [DOI] [PubMed] [Google Scholar]
- 32. Smart T. G., Paoletti P. (2012) Synaptic neurotransmitter-gated receptors. Cold Spring Harb. Perspect. Biol. 4, a009662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Auerbach A. (2010) The gating isomerization of neuromuscular acetylcholine receptors. J. Physiol. 588, 573–586 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Sine S. M., Engel A. G. (2006) Recent advances in Cys-loop receptor structure and function. Nature 440, 448–455 [DOI] [PubMed] [Google Scholar]
- 35. Chang Y., Huang Y., Whiteaker P. (2010) Mechanism of allosteric modulation of the Cys-loop receptors. Pharmaceuticals 3, 2592–2609 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Krause R. M., Buisson B., Bertrand S., Corringer P. J., Galzi J. L., Changeux J. P., Bertrand D. (1998) Ivermectin: a positive allosteric effector of the α7 neuronal nicotinic acetylcholine receptor. Mol. Pharmacol. 53, 283–294 [DOI] [PubMed] [Google Scholar]
- 37. Bertrand D., Gopalakrishnan M. (2007) Allosteric modulation of nicotinic acetylcholine receptors. Biochem. Pharmacol. 74, 1155–1163 [DOI] [PubMed] [Google Scholar]
- 38. Zhang J., Xue F., Whiteaker P., Li C., Wu W., Shen B., Huang Y., Lukas R. J., Chang Y. (2011) Desensitization of α7 nicotinic receptor is governed by coupling strength relative to gate tightness. J. Biol. Chem. 286, 25331–25340 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Bouzat C., Bartos M., Corradi J., Sine S. M. (2008) The interface between extracellular and transmembrane domains of homomeric Cys-loop receptors governs open-channel lifetime and rate of desensitization. J. Neurosci. 28, 7808–7819 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Wang Q., Lynch J. W. (2011) Activation and desensitization induce distinct conformational changes at the extracellular-transmembrane domain interface of the glycine receptor. J. Biol. Chem. 286, 38814–38824 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Keramidas A., Lynch J. W. (2013) An outline of desensitization in pentameric ligand-gated ion channel receptors. Cell. Mol. Life Sci. 70, 1241–1253 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Duret G., Van Renterghem C., Weng Y., Prevost M., Moraga-Cid G., Huon C., Sonner J. M., Corringer P. J. (2011) Functional prokaryotic-eukaryotic chimera from the pentameric ligand-gated ion channel family. Proc. Natl. Acad. Sci. U.S.A. 108, 12143–12148 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Grutter T., de Carvalho L. P., Dufresne V., Taly A., Edelstein S. J., Changeux J. P. (2005) Molecular tuning of fast gating in pentameric ligand-gated ion channels. Proc. Natl. Acad. Sci. U.S.A. 102, 18207–18212 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Cooper S. T., Harkness P. C., Baker E. R., Millar N. S. (1999) Up-regulation of cell-surface α4β2 neuronal nicotinic receptors by lower temperature and expression of chimeric subunits. J. Biol. Chem. 274, 27145–27152 [DOI] [PubMed] [Google Scholar]
- 45. Eiselé J. L., Bertrand S., Galzi J. L., Devillers-Thiéry A., Changeux J. P., Bertrand D. (1993) Chimaeric nicotinic-serotonergic receptor combines distinct ligand binding and channel specificities. Nature 366, 479–483 [DOI] [PubMed] [Google Scholar]
- 46. Zimmermann I., Dutzler R. (2011) Ligand activation of the prokaryotic pentameric ligand-gated ion channel ELIC. PLoS Biol. 9, e1001101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Thompson A. J., Alqazzaz M., Ulens C., Lummis S. C. (2012) The pharmacological profile of ELIC, a prokaryotic GABA-gated receptor. Neuropharmacology 63, 761–767 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Higuchi R., Krummel B., Saiki R. K. (1988) A general method of in vitro preparation and specific mutagenesis of DNA fragments: study of protein and DNA interactions. Nucleic Acids Res. 16, 7351–7367 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Arriza J. L., Fairman W. A., Wadiche J. I., Murdoch G. H., Kavanaugh M. P., Amara S. G. (1994) Functional comparisons of three glutamate transporter subtypes cloned from human motor cortex. J. Neurosci. 14, 5559–5569 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Dascal N. (2001) in Current Protocols in Neuroscience (Crawley J. N., McKay R. A., Rogawski M. A., eds) Chapter 6, Unit 6.12, John Wiley and Sons, Hoboken, NJ [Google Scholar]
- 51. Eswar N., Marti-Renom M. A., Webb B., Madhusudhan M. S., Eramian D., Shen M., Pieper U., Sali A. (2006) Current Protocols in Bioinformatics, Supplement 15, 5.6.1–5.6.30, John Wiley and Sons, Hoboken, NJ: [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Fiser A., Do R. K., Sali A. (2000) Modeling of loops in protein structures. Protein Sci. 9, 1753–1773 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Bouzat C., Gumilar F., Spitzmaul G., Wang H. L., Rayes D., Hansen S. B., Taylor P., Sine S. M. (2004) Coupling of agonist binding to channel gating in an ACh-binding protein linked to an ion channel. Nature 430, 896–900 [DOI] [PubMed] [Google Scholar]
- 54. Bondarenko V., Mowrey D. D., Tillman T. S., Seyoum E., Xu Y., Tang P. (2014) NMR structures of the human α7 nAChR transmembrane domain and associated anesthetic binding sites. Biochim. Biophys. Acta 1838, 1389–1395 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Unwin N. (2005) Refined structure of the nicotinic acetylcholine receptor at 4 Å resolution. J. Mol. Biol. 346, 967–989 [DOI] [PubMed] [Google Scholar]
- 56. Mnatsakanyan N., Jansen M. (2013) Experimental determination of the vertical alignment between the second and third transmembrane segments of muscle nicotinic acetylcholine receptors. J. Neurochem. 125, 843–854 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Flood P., Ramirez-Latorre J., Role L. (1997) α4β2 neuronal nicotinic acetylcholine receptors in the central nervous system are inhibited by isoflurane and propofol, but α7-type nicotinic acetylcholine receptors are unaffected. Anesthesiology 86, 859–865 [DOI] [PubMed] [Google Scholar]
- 58. daCosta C. J., Free C. R., Corradi J., Bouzat C., Sine S. M. (2011) Single-channel and structural foundations of neuronal α7 acetylcholine receptor potentiation. J. Neurosci. 31, 13870–13879 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Sattelle D. B., Buckingham S. D., Akamatsu M., Matsuda K., Pienaar I., Jones A. K., Sattelle B. M., Almond A., Blundell C. D. (2009) Comparative pharmacology and computational modelling yield insights into allosteric modulation of human α7 nicotinic acetylcholine receptors. Biochem. Pharmacol. 78, 836–843 [DOI] [PubMed] [Google Scholar]
- 60. Barron S. C., McLaughlin J. T., See J. A., Richards V. L., Rosenberg R. L. (2009) An allosteric modulator of α7 nicotinic receptors, N-(5-chloro-2,4-dimethoxyphenyl)-N′-(5-methyl-3-isoxazolyl)-urea (PNU-120596), causes conformational changes in the extracellular ligand binding domain similar to those caused by acetylcholine. Mol. Pharmacol. 76, 253–263 [DOI] [PMC free article] [PubMed] [Google Scholar]