

Background: Base excision repair is an important pathway for cytosine demethylation at the CpG dinucleotide for epigenetic control.
Results: A deaminated 5-methylcytosine (thymine) and an adjacent oxidized guanine (7,8-dihydro-8-oxoguanine) retard base excision repair of each lesion.
Conclusion: Altered in-tandem CpG dinucleotide is a poor substrate for base excision repair.
Significance: Oxidized guanine in a CpG dinucleotide interferes with active DNA demethylation.
Keywords: Base Excision Repair, DNA Damage, DNA Methylation, DNA Polymerase, Oxidative Stress, 8-Oxoguanine, DNA Demethylation, DNA Glycosylase
Abstract
Cytosine methylation and demethylation in tracks of CpG dinucleotides is an epigenetic mechanism for control of gene expression. The initial step in the demethylation process can be deamination of 5-methylcytosine producing the TpG alteration and T:G mispair, and this step is followed by thymine DNA glycosylase (TDG) initiated base excision repair (BER). A further consideration is that guanine in the CpG dinucleotide may become oxidized to 7,8-dihydro-8-oxoguanine (8-oxoG), and this could affect the demethylation process involving TDG-initiated BER. However, little is known about the enzymology of BER of altered in-tandem CpG dinucleotides; e.g. Tp8-oxoG. Here, we investigated interactions between this altered dinucleotide and purified BER enzymes, the DNA glycosylases TDG and 8-oxoG DNA glycosylase 1 (OGG1), apurinic/apyrimidinic (AP) endonuclease 1, DNA polymerase β, and DNA ligases. The overall TDG-initiated BER of the Tp8-oxoG dinucleotide is significantly reduced. Specifically, TDG and DNA ligase activities are reduced by a 3′-flanking 8-oxoG. In contrast, the OGG1-initiated BER pathway is blocked due to the 5′-flanking T:G mispair; this reduces OGG1, AP endonuclease 1, and DNA polymerase β activities. Furthermore, in TDG-initiated BER, TDG remains bound to its product AP site blocking OGG1 access to the adjacent 8-oxoG. These results reveal BER enzyme specificities enabling suppression of OGG1-initiated BER and coordination of TDG-initiated BER at this tandem alteration in the CpG dinucleotide.
Introduction
There is strong interest in understanding the special features of DNA base lesions and base excision repair (BER)3 in the context of the CpG dinucleotide in genomic DNA. The CpG dinucleotide is subjected to a range of modifications, including methylation of cytosine and oxidation of guanine. For example, methylation of cytosine to 5-methylcytosine (5-mC) at CpG dinucleotides is common in higher eukaryotes and is used as an epigenetic feature in regulation of gene expression (1–3). In mammalian cells, 3–6% of cytosines are methylated, and 70–80% of CpG dinucleotides in tracks within promoters undergo methylation (4–6). DNA methylation affects gene transcription by directly interfering with the binding of transcription factors at promoter regulatory sequences or indirectly mediating histone modification via recruitment of methyl-CpG binding proteins (7, 8). Maintenance of proper methylation patterns is essential for embryonic development, and aberrant DNA methylation has been associated with disease progression.
It is known that 5-mC is spontaneously deaminated to form the T:G mispair in DNA (9). This mispair can induce the C to T transition mutation that results in mutational hotspots in human genes, such as the tumor suppressor p53 (10–13). In addition, 5-mC at CpG dinucleotides is deaminated by cellular deaminases as part of the enzymatic demethylation process. Thus, the repair of deaminated 5-mC in the context of the CpG dinucleotide is important not only for preventing mutagenic events but also for maintaining the proper methylation status of the genome.
Thymine DNA glycosylase (TDG) is a mismatch-specific DNA glycosylase that excises thymine from the T:G mismatch creating an abasic site in DNA (14). The enzyme has been implicated in the restoration of G:C base pair from 5-mC:G utilizing the BER pathway, and this is considered a component of the “active DNA demethylation” system for 5-mCpG dinucleotides. This DNA demethylation system is initiated by deamination of 5-mC to T by activation-induced deaminases (AID) and the apolipoprotein B mRNA-editing catalytic polypeptides (APOBECs) proteins (15). It is also known that DNA methyltransferases DNMT3a/b can deaminate 5-mC (16). TDG interacts with AID and DNMT3a/b (17–19), suggesting a two-step mechanism for active DNA demethylation: deamination of 5-mC followed by restoration of normal cytosine by TDG-initiated BER. Other possible mechanisms for TDG-mediated demethylation also have been suggested, involving hydroxylation followed by deamination or oxidation of 5-mC (i.e. 5-hydroxymethyluracil or 5-formylcytosine and 5-carboxylcytosine) (20). Consistent with the importance of these mechanisms, a deficiency of TDG results in an embryonic lethal phenotype and reveals altered genomic methylation patterns or accumulation of the modified cytosine at promoter regions (17, 21, 22). TDG also has been found to protect the genome from mutagenic consequences of spontaneously deaminated 5-mC; thus, loss of TDG in mismatch-repair deficient tumor cells leads to an increase of C to T mutations at CpG dinucleotides (23).
Whereas the cytosine in CpG dinucleotides is a target of 5-methylation, the neighboring guanine is susceptible to oxidation to 7,8-dihydro-8-oxoguanine (8-oxoG). Due to the altered hydrogen-bonding pattern of 8-oxoG, 8-oxoG can base pair with adenine, and if 8-oxoG remains in the genome during replication, DNA polymerases frequently misincorporate dAMP opposite 8-oxoG, resulting in the G to T transversion. In mammalian cells, 8-oxoG in the genome is processed by 8-oxoguanine DNA glycosylase (OGG1)-initiated BER (24). OGG1 is a lesion-specific DNA glycosylase that recognizes 8-oxoG paired with cytosine in double-stranded DNA and excises the damaged base (25, 26). OGG1 contributes to suppression of G to T transversions in human cells (27, 28), and a deficiency of OGG1 in mouse models exhibits an elevated level of 8-oxoG in genomic DNA and a predisposition to tumorigenesis (29, 30), illustrating the importance of OGG1 in maintaining the genetic integrity in vivo.
The 8-oxoG lesion is thought to contribute not only to mutagenesis but also to the epigenetic status of cells. For example, oxidation of guanine at a methylated or unmethylated CpG dinucleotide inhibits the binding of methyl-CpG binding protein and transcription factors AP-1 and Sp1, respectively (31–33), indicating that 8-oxoG could modulate gene transcription. Likewise, 8-oxoG formed at CpG dinucleotides inhibits the methylation of the adjacent cytosine (34–36), and this could result in an altered methylation pattern. However, the influence of 8-oxoG on the demethylation process at CpG dinucleotide remains to be determined.
The activity of TDG for removing thymine depends on the base pair located 3′ to the substrate T:G mispair (37). This sequence context-dependent affect on TDG activity has been assessed both structurally and kinetically and found to be distinct with several different substrates, including 3,N4-ethenocytosine, uracil, 5-chlorouracil, 5-fluorouracil, and 5-bromouracil (38, 39). Interestingly, the 5-mCpG dinucleotide can be altered in-tandem under oxidative stress conditions (40). Therefore, it is conceivable that 8-oxoG base damage in the CpG dinucleotide context could influence the activity of TDG. Here, probing the CpG dinucleotide context, we assessed the effect of the 3′-flanking 8-oxoG lesion on the activity of TDG and subsequent BER steps using model DNA substrates containing the T:G mispair. We find that a 3′-flanking 8-oxoG decreases TDG excision of the mispaired thymine.
We recently reported that 8-oxoG removal by OGG1 is decreased in the presence of the 5′-adjacent T:G mispair (41). Thus, both TDG and OGG1 are sensitive to these CpG dinucleotide tandem alterations. Because these glycosylases function in BER along with apurinic/apyrimidinic endonuclease 1 (APE1) and other downstream enzymes, we further characterized the glycosylase effects on associated BER activities. Surprisingly, the T:G mispair strongly retards APE1-mediated incision of the adjacent AP site, thereby diminishing overall OGG1-initiated BER. When TDG first processes the T:G mispair, TDG remains bound to its product AP site and prevents subsequent 8-oxoG excision by OGG1. Furthermore, for TDG-initiated BER, the presence of the adjacent 8-oxoG reduces the completion of BER. These results are discussed and indicate key DNA sequence context features of BER at the tandem alteration of the methylated CpG dinucleotide.
EXPERIMENTAL PROCEDURES
Materials
Human OGG1 was overexpressed and purified as described previously (42). Human APE1, DNA polymerase β (pol β), DNA ligase I (Lig I), and DNA ligase III-β (Lig III-β) were purified as described previously (43–45). T4 DNA ligase (T4 Lig) was purchased from New England Biolabs (Beverly, MA). 3′-[α-32P]Deoxyadenosine (Cordycepin) (5000 Ci/mmol) and [α-32P]dCTP (6000 Ci/mmol) were from PerkinElmer Life Sciences.
Oligonucleotides
5′- or 3′-6-Carboxyfluorescein (6-FAM)-labeled oligonucleotides with or without 8-oxoG, and corresponding templates were synthesized and PAGE-purified by Eurofins MWG Operon (Huntsville, AL). The substrates were constructed by annealing 6-FAM-labeled oligonucleotides to their template strand at a molar ratio 1:1.2. The sequences of the oligonucleotides are shown in Tables 1 and 3.
TABLE 1.
Single turnover rate constant for DNA glycosylase base excision
| Name | Substrate DNA sequencea |
kobs |
|
|---|---|---|---|
| TDG | OGG1 | ||
| min−1 | |||
| C/G-oxG/C | FAM-5′-CATGGGCGGCATGAACCXGAGGCCCATCCTCACC-3′ | NSb | 36 ± 3.5 |
| 3′-GTACCCGCCGTACTTGGCCTCCGGGTAGGAGTGG-5′ | |||
| T/G-G/C | FAM-5′-CATGGGCGGCATGAACTGGAGGCCCATCCTCACC-3′ | 2.6 ± 0.29 | NS |
| 3′-GTACCCGCCGTACTTGGCCTCCGGGTAGGAGTGG-5′ | |||
| T/G-oxG/C | FAM-5′-CATGGGCGGCATGAACTXGAGGCCCATCCTCACC-3′ | 0.58 ± 0.12 | 2.0 ± 0.025 |
| 3′-GTACCCGCCGTACTTGGCCTCCGGGTAGGAGTGG-5′ | |||
| T/G-oxG/A | FAM-5′-CATGGGCGGCATGAACTXGAGGCCCATCCTCACC-3′ | 0.028 ± 0.0016 | NS |
| 3′-GTACCCGCCGTACTTGGACTCCGGGTAGGAGTGG-5′ | |||
| T/G-T/A | FAM-5′-CATGGGCGGCATGAACTTGAGGCCCATCCTCACC-3′ | 0.023 ± 0.00096 | NS |
| 3′-GTACCCGCCGTACTTGGACTCCGGGTAGGAGTGG-5′ | |||
| T/G-THF/C | FAM-5′-CATGGGCGGCATGAACTFGAGGCCCATCCTCACC-3′ | <0.002c | NS |
| 3′-GTACCCGCCGTACTTGGCCTCCGGGTAGGAGTGG-5′ | |||
| THF/G-oxG/C | FAM-5′-CATGGGCGGCATGAACFXGAGGCCCATCCTCACC-3′ | NS | 7.8 ± 0.23 |
| 3′-GTACCCGCCGTACTTGGCCTCCGGGTAGGAGTGG-5′ | |||
a X + 8-oxoG. Underlined are mispaired base-pair. F + tetrahydrofuran. FAM indicates the presence of fluorescence tag.
b Not a substrate.
c Rate is too slow to determine. The values represent means and S.D. of at least two independent experiments.
TABLE 3.
Observed steady-state rate for APE1 incision
| Name | DNA Sequencea | kss |
|---|---|---|
| s−1 | ||
| C/G-THF/C | FAM-5′-CATGGGCGGCATGAACCFGAGGCCCATCCTCACC-3′ | 0.49 ± 0.077 |
| 3′-GTACCCGCCGTACTTGGCCTCCGGGTAGGAGTGG-5′ | ||
| T/G-THF/C | FAM-5′-CATGGGCGGCATGAACTFGAGGCCCATCCTCACC-3′ | 0.17 ± 0.068 |
| 3′-GTACCCGCCGTACTTGGCCTCCGGGTAGGAGTGG-5′ | ||
| THF/G-G/C | FAM-5′-CATGGGCGGCATGAACFGGAGGCCCATCCTCACC-3′ | 0.48 ± 0.13 |
| 3′-GTACCCGCCGTACTTGGCCTCCGGGTAGGAGTGG-5′ | ||
| THF/G-oxG/C | FAM-5′-CATGGGCGGCATGAACFXGAGGCCCATCCTCACC-3′ | 0.89 ± 0.38 |
| 3′-GTACCCGCCGTACTTGGCCTCCGGGTAGGAGTGG-5′ |
a F + tetrahydrofuran; underlined bases are a mispair (G:T); X + 8-oxoG. FAM indicates the 5′-fluorescence tag. The values represent the means and S.D. of at least two independent experiments.
Expression and Purification of Human Thymine DNA Glycosylase
The cDNA encoding human TDG was amplified from pCMV6-XL5-TDG (Origene Technologies) with primers TDG-BamHI-F, 5′-GATCGGATCCATGGAAGCGGAGAACGCGGGC-3′, and TDG-EcoRI-R, 5′-GATCGAATTCTCAAGCATGGCTTTCTTCTTCCTG-3′. The amplified DNA was digested with BamHI and EcoRI restriction enzymes, and the digested fragment was gel-purified and ligated with BamHI-EcoRI digested pGEX-6p-1. The resulting overexpression plasmid pGEX-6p-TDG was introduced into BL21(DE3) strain by transformation. A freshly transformed colony was inoculated with LB medium and agitated overnight at 30 °C. The overnight culture was diluted in 2 liters of fresh medium (1 liter of medium in a 2.8 liters of Fernbach flask) and was further agitated at 30 °C until the culture reached an absorbance of 0.5–0.7 at 595 nm. TDG expression was induced by the addition of 2 mm isopropyl-1-thio-β-d-galactopyranoside and shaking for 15 h at 18 °C. The harvested cells were resuspended in Buffer A (50 mm Tris-HCl, pH 7.5, 500 mm NaCl, 1 mm EDTA, 1 mm dithiothreitol (DTT), 0.1 μm 4-(2-aminoethyl)benzenesulfonyl fluoride hydrochloride, 1 mm benzamidine, 1 μg/ml pepstatin A, and 1 μg/ml leupeptin) and lysed by sonication. Cell lysates were subjected to centrifugation at 40,000 rpm for 60 min at 4 °C to separate soluble proteins from cell debris. Soluble proteins were incubated with glutathione-Sepharose 4B (GE Healthcare) for 4 h at 4 °C. Unbound proteins were flushed from the beads with a wash buffer (50 mm Tris-HCl, pH 7.5, 1 m NaCl, 1 mm EDTA, 10 mm β-mercaptoethanol, 0.01% Nonidet P-40, 0.1 μm 4-(2-aminoethyl) benzenesulfonyl fluoride hydrochloride, 1 mm benzamidine, 1 μg/ml pepstatin A, and 1 μg/ml leupeptin), and GST fusion TDG was eluted with 50 mm glutathione. The eluted fraction was incubated with PreScission Protease (GE Healthcare) at 4 °C overnight to cleave the GST tag from the fusion protein. The proteins were then loaded onto Mono Q HR 10/10 column (GE Healthcare). After washing the column with low salt buffer (20 mm Hepes, pH 7.5, 100 mm NaCl, 10 mm β-mercaptoethanol, 10% glycerol), TDG was eluted with a linear gradient of NaCl (100–1000 mm). The purity of the preparation was assessed by SDS-PAGE and judged to be >95% pure.
Determination of the Glycosylase Activity
Active concentration of TDG or OGG1 was determined using the T:G mismatch-containing duplex DNA T/G-G/C or 8-oxoG:C base pair-containing duplex DNA C/G-oxG/C (Table 1), respectively, as described previously (46). The TDG and OGG1 preparations were determined to be 34 and 38% active, respectively. The glycosylase activity was determined under single-turnover conditions where the substrate DNA is saturated with enzyme. A KinTek model RQF-3 rapid quench-flow apparatus (KinTek Corp., Austin, TX) was employed when reactions were too rapid to measure manually. All concentrations refer to the final concentration after mixing. The 5′-6-FAM-labeled DNA substrates (50 nm) were incubated with 250 nm active TDG or OGG1 at 37 °C in reaction buffer (50 mm Hepes, pH 7.5, 100 mm KCl, 0.5 mm EDTA, and 0.1% BSA). At timed intervals, 10-μl aliquots were withdrawn and quenched with 100 mm NaOH (5 min at 90 °C) to inactivate the enzyme and to cleave the resulting AP sites. An equal volume of DNA gel loading buffer (10 m urea, 100 mm EDTA, 0.02% bromphenol blue, and 0.02% xylene cyanol) was added. After incubation at 95 °C for 2 min, the reaction products were separated by electrophoresis in a 15% denaturing polyacrylamide gel. A Typhoon PhosphorImager was used for gel scanning and imaging, and the data were analyzed with ImageQuant software. The first-order rate constants (kobs) were determined by fitting the data with a single exponential equation (Equation 1); A0 represents the amplitude of the exponential phase.
Determination of the Rate of DNA Glycosylase Product Release
Substrate DNA (200 nm) was equilibrated at 37 °C in 50 mm Hepes, pH 7.5, 20 or 100 mm KCl, 0.5 mm EDTA, and 0.1% BSA. When necessary, the reaction mixture contained 5 mm MgCl2. Reactions were started by the addition of 40 nm TDG or 10 nm OGG1 with or without 200 nm (for TDG) or 100 nm (for OGG1) APE1. Aliquots were withdrawn at time intervals and quenched with 100 mm NaOH (5 min at 90 °C). The reaction products were separated on 15% denaturing polyacrylamide gel and analyzed as described below. Time courses for TDG were determined by fitting the data with a rising exponential and linear terms (Equation 2), providing the first-order rate constant for the first turnover (kobs), the amplitude of the burst of product formation (A0, y intercept), and the rate (i.e. slope) of the linear steady-state phase (vss).
From this analysis, the steady-state rate (kss) was calculated as (kss = vss/A0). For OGG1, kss was determined by fitting the data to a linear equation (Equation 3).
TDG Inhibition of OGG1
Substrate DNA was 3′-labeled with terminal deoxynucleotidyltransferase and [α-32P]cordycepin according to the manufacturer's instructions. The 3′-labeled substrate DNA (50 nm) was preincubated with or without 250 nm TDG at 37 °C for 10 min in 50 mm Hepes, pH 7.5, 100 mm KCl, 0.5 mm EDTA, and 0.1% BSA. After preincubation, 0–250 nm OGG1 was added to the reaction mixture and incubated at 37 °C for 5 min. The reaction was terminated by the addition of 100 mm NaOH and heating as described above.
Determination of the APE1 Steady-state Rate
A rapid quench-flow system was employed to measure the steady-state rate of APE1. The 5′-6-FAM-labeled DNA substrate (200 nm) was incubated with 30 nm APE1 at 37 °C in reaction buffer (50 mm Hepes, pH 7.5, 100 mm KCl, 0.5 mm EDTA, and 0.1% BSA). At timed intervals, reactions were terminated by mixing the reaction mixture with 100 mm NaOH. An equal volume of DNA gel loading buffer (10 m urea, 100 mm EDTA, 0.02% bromphenol blue, and 0.02% xylene cyanol) was added. After incubation at 95 °C for 2 min, the reaction products were separated by 15% denaturing polyacrylamide gel and analyzed as described above. For substrates C/G-THF/C, THF/G-G/C, and THF/G-oxoG/C (Table 3), data were fit to a linear equation (Equation 3) to determine kss. For the substrate T/G-THF/C, data were fit to a linear equation with a zero intercept, providing the value for vss. Subsequently, kss was calculated by dividing vss by the y intercept that was determined from the control substrate C/G-THF/C.
In Vitro TDG Initiated BER Assays
The BER assays were performed in a 20-μl reaction mixture as previously described (47, 48). The reaction mixture contained 50 mm Hepes, pH 7.5, 100 mm KCl, 5 mm MgCl2, 0.5 mm EDTA, 0.1% BSA, 2 mm DTT, 4 mm ATP, 2.3 μm [α-32P]dCTP, and 34-mer DNA substrate. The reactions were started by the addition of 10 nm TDG, 20 nm APE1, and 20 nm pol β with or without 100 nm Lig I and incubated at 37 °C. Aliquots (5 μl) were withdrawn at timed intervals and mixed with an equal volume of gel loading buffer. After heating at 95 °C for 2 min, reaction products were separated by electrophoresis in a 15% denaturing polyacrylamide gel and analyzed as described above.
In Vitro DNA Ligase Assays
To examine the activities of DNA ligases, a 34-mer nicked DNA substrate was prepared by annealing a 17-mer oligo A (5′-CATGGGCGGCATGAACC-3′), 17-mer oligo B (5′-XGAGGCCCATCCTCACC-3′, where X = G or 8-oxoG), and 34-mer complementary DNA (5′-GGTGAGGATGGGCCTCCGGTTCATGCCGCCCATG-3′). The 5′ and 3′ termini of oligo B were phosphorylated and conjugated with 6-FAM, respectively. The nicked DNA substrate (250 nm) was incubated with 200 nm Lig I, 200 nm Lig III-β, or 1 unit of T4 Lig in reaction buffer (50 mm Hepes, pH 7.5, 100 mm KCl, 5 mm MgCl2, 0.5 mm EDTA, 0.1% BSA, 2 mm DTT, and 4 mm ATP) at 37 °C. Aliquots (5 μl) were withdrawn at timed intervals and mixed with an equal volume of gel loading buffer. After heating at 95 °C for 2 min, reaction products were separated by electrophoresis in a 15% denaturing polyacrylamide gel and analyzed as described above.
Preparation of 5′-Deoxyribose Phosphate Containing Oligonucleotide
A uracil-containing double-stranded DNA substrate with an 8-oxoG nucleotide was pretreated with uracil DNA glycosylase to generate an AP site. The DNA substrate was prepared by annealing 17-mer (5′-CATGGGCGGCATGAACC-3′) and 18-mer oligonucleotides (5′-UXGAGGCCCATCCTCACC-3′, where X = 8-oxoG) with 34-mer complementary DNA (5′-GGTGAGGATGGGCCTCCGGTTCATGCCGCCCATG-3′). The 5′ and 3′ termini of the 18-mer oligonucleotides were phosphorylated and conjugated with 6-FAM, respectively. The reaction mixture (50 μl) contained 50 mm Hepes, pH 7.4, 20 mm KCl, 0.5 mm EDTA, 2 mm DTT, 20 nm uracil DNA glycosylase, 200 nm DNA, and incubated 37 °C for 30 min. Due to the unstable nature of uracil DNA glycosylase-treated DNA, the DNA substrates were treated just before deoxyribose phosphate (dRP) lyase assay described below.
Pol β dRP Lyase Activity
The dRP lyase assay was performed manually under single-turnover conditions using uracil DNA glycosylase-pretreated DNA substrate that has 8-oxoG immediately downstream (i.e. 3′) of the dRP moiety. The dRP lyase activity was determined as described previously (49). The reaction mixture (10 μl) contained 50 mm Hepes, pH 7.5, 20 mm KCl, 0.5 mm EDTA, 2 mm DTT, and 140 nm DNA substrate. The reaction was initiated by the addition of 500 nm pol β. The reaction mixture was incubated at 37 °C. The dRP lyase reaction was terminated at timed intervals by the addition of freshly prepared 1 m NaBH4 to a final concentration of 100 mm. Because the dRP lyase activity proceeds by a β-elimination reaction (50), the covalently cross-linked complexes between the dRP-containing BER intermediate and pol β is produced by NaBH4 reduction (51). The reaction products were then further incubated on ice for 30 min and separated in a 15% polyacrylamide gel, and data were analyzed as described above.
RESULTS
The aim of this study was to systematically examine BER in the tandem alteration of a methylated CpG dinucleotide to yield Tp8-oxoG dinucleotide. Initially, we measured the activities of purified TDG and OGG1 on variations of the altered CpG dinucleotide. With each DNA glycosylase, rate constants were measured under both single-turnover and steady-state conditions, reflecting the chemistry step of the glycosylase and the DNA product release step, respectively. Finally, a reconstituted BER system with purified enzymes was evaluated to identify obstacles to the repair of tandem lesions.
TDG Activity Is Sensitive to a 3′-Neighboring 8-OxoG and AP Site
First, to examine the effect of a 3′-neighboring 8-oxoG on TDG activity, we used single-turnover conditions to measure the intrinsic rate constant for base excision; experiments were conducted using an excess of enzyme over DNA substrate. With a T:G mispair adjacent to the normal G:C base pair (Table 1), the excision rate (kobs) was 2.6 min−1; however, when T:G was adjacent to the 8-oxoG:C, excision was ∼5-fold lower (kobs = 0.58 min−1, Fig. 1A and Table 1). Excision of mismatched T from the T:G mispair adjacent to the 8-oxoG:A base pair was much lower (100-fold; kobs = 0.028 min−1), and this rate was similar to that determined for T removal from the T:G mispair adjacent to a T:A base pair (Fig. 1B). We next examined the influence of an adjacent AP site, arising for example from OGG1 excision of 8-oxoG adjacent to the T:G mispair. A substrate containing the AP site analog tetrahydrofuran (THF) opposite C (T/G-THF/C) was used. Interestingly, TDG excision on this substrate was too low to measure (Table 1). The results indicate that TDG is sensitive to the 3′-neighboring base. These results suggest that if 8-oxoG is removed before TDG activity on altered in-tandem substrate, BER of thymine in the T:G mispair 5′ to an AP site will be blocked.
FIGURE 1.

Single turnover determination of thymine excision by TDG. A, single-turnover time courses for the removal of T from the T:G mispair where the 3′-base pair is G:C (○) or 8-oxoG:C (□). B, single-turnover time courses for the removal of T from the T:G mispair where the 3′-base pair is T:A (○) or 8-oxoG:A (□). TDG (250 nm) was incubated with 50 nm DNA substrate (T/G-G/C, T/G-oxG/C, T/G-oxG/A, or T/G-T/A) at 37 °C. Products were isolated, quantified, and analyzed as described under “Experimental Procedures.” The data were fit to an exponential time course (N = G, solid lines; N = 8-oxoG, dashed lines). The results from several independent determinations are tabulated in Table 1.
TDG Activity under Steady-state Conditions
Next, we examined the effect of a 3′-neighboring 8-oxoG on TDG activity. In this case, steady-state conditions were employed to ascertain whether the binding affinity of TDG to an AP site might be affected. With the T:G mispair adjacent to the normal G:C base pair (Fig. 2), the steady-state rate (kss) was 0.0043 min−1, a rate similar to when T:G was adjacent to the 8-oxoG:C pair (kss = 0.0075 min−1). Thus, the steady-state rate was much slower than the pre-steady-state rate, as reflected by the pronounced burst in product formation during the reaction time course (Fig. 2). The slower steady-state rate is consistent with a relatively slow DNA product release step for TDG on both of these substrates (52, 53).
FIGURE 2.

Multiple turnover determination of thymine excision by TDG. Steady-state time courses for the removal of T from the T:G mispair where the 3′-base pair is G:C or 8-oxoG:C. TDG (40 nm) was incubated with 200 nm DNA substrate (T/G-G/C or T/G-oxG/C) in the presence of 100 mm KCl and 5 mm MgCl2 at 37 °C. Products were isolated, quantified, and analyzed as described under “Experimental Procedures.” The time courses were fit to an equation with a rising exponential and linear terms (N = G, solid line; N = 8-oxoG, dashed line). The observed rate constant for the burst was similar to that determined by single-turnover analyses (Table 1). The means and S.D. of the steady-state rates from at least two independent experiments were 0.0043 ± 0.0015 and 0.0075 ± 0.0010 s−1 for the G and 8-oxoG substrates, respectively.
TDG Inhibition of OGG1 through Product AP Site Binding
As suggested from the kinetic experiments described above, TDG remains bound to its product AP site even when the adjacent guanine is replaced with 8-oxoG. This suggests that TDG bound to its AP site product could influence OGG1 excision of a neighboring 8-oxoG. To assess the influence of TDG on OGG1 activity, we preincubated DNA substrates with and without TDG and then added OGG1 to the reaction mixtures. Reaction products were examined by gel electrophoresis (Fig. 3). When a control substrate with the normal C:G base pair adjacent to the 8-oxoG:C base pair was preincubated with TDG, OGG1 activity was not altered by the presence of TDG (Fig. 3A). In contrast with the Tp8-oxoG substrate, the amount of OGG1 product was strongly suppressed in the presence of TDG (Fig. 3B). To confirm that the block in OGG1 activity was due to TDG binding to its AP site product, a DNA substrate containing THF 5′ to 8-oxoG was used. As shown in Fig. 3C, TDG again inhibited excision of 8-oxoG with this substrate, indicating that OGG1 was blocked by TDG bound to the synthetic AP site-containing DNA.
FIGURE 3.

Inhibition of OGG1 activity by TDG binding to an adjacent AP site. The DNA substrate (50 nm) was preincubated without or with 250 nm TDG at 37 °C for 10 min. Then OGG1 (0–250 nm, as indicated) was added to the reaction mixture, and the mixture was further incubated for 5 min. The DNA substrates used are illustrated in the figure. A, 5′-C:G (i.e. C/G-8-oxoG/C). B, 5′-T:G (i.e. T/G-8-oxoG/C). C, 5′-THF:G (i.e. THF/G-8-oxoG/C). TDG was added to reaction mixtures as indicated (+). Products were isolated and quantified as described under “Experimental Procedures”.
Taken together, these results suggest that the glycosylase activities of OGG1 and TDG can regulate one another at AP site formation stage of BER. In one case this is because TDG does not use the OGG1 product as a substrate; TDG activity on the substrate T/G-THF/C was negligible (Table 1). In the other case, it is because OGG1 is not able to gain access to the TDG product (Fig. 3, B and C). For these AP site products to be processed through BER, they must be passed to APE1 for incision during the next step of BER. The implication of the reciprocal regulation is that only one of the lesions in the altered in-tandem substrate will be processed through the BER pathway once initiated.
Effect of APE1 and a 3′-8-OxoG on the Steady-state Rate of TDG
As shown above, TDG binds to its AP site product and exhibits a slow steady-state rate, reflecting tight product binding. Yet the addition of APE1 to the reaction mixture strongly increases the steady-state rate of TDG (52, 53). The results are consistent with the idea that APE1 is able to gain access to the AP site and incise it, thereby leading to a faster TDG catalytic cycling. We examined the influence of an adjacent 8-oxoG on the APE1-based stimulation of the steady-state rate of TDG. In the absence of APE1, the steady-state rates of TDG on the substrates with the adjacent G:C base pair and 8-oxoG:C base were 0.0043 and 0.0075 min−1, respectively (Figs. 2 and 4). In the presence of APE1, the steady-state rates were increased by 37- and 5.7-fold with the adjacent G:C and 8-oxoG:C base-pairs (0.16 and 0.043 min−1), respectively. These results indicate that the neighboring 8-oxoG:C base pair reduced the stimulatory effect of APE1 on TDG cycling. In addition, the results in Fig. 4 show that APE1 does not alter the pre-steady-state rate of TDG excision of thymine.
FIGURE 4.

Effect of a 3′-neighboring 8-oxoG:C base pair on APE1-mediated stimulation of the steady-state rate of TDG. Steady-state time courses for the removal of T from the T:G mispair, where the identity of the 3′-neighboring base pair is G:C (A) or 8-oxoG:C (B). TDG (40 nm) was incubated with 200 nm substrate DNA in the presence of 100 mm KCl and 5 mm MgCl2 without (○) or with 200 nm APE1 (□) at 37 °C. Products were isolated and quantified as described under “Experimental Procedures.” The time courses were fit to an equation with a rising exponential and linear terms (−APE1, solid line; +APE1, dashed line). The means and S.D. of the steady-state rates from at least two independent experiments were 0.16 ± 0.034 and 0.043 ± 0.013 s−1 for the G and 8-oxoG substrates, respectively.
OGG1 Activity and the 5′-Neighboring T:G Mispair or AP Site
Kinetic studies with OGG1 had indicated that the single-turnover rate constant is faster than that for TDG against the respective mono-lesion CpG dinucleotides (41). In the present study we evaluated OGG1 activity under similar conditions to those used for evaluation of TDG. Against the altered in-tandem CpG dinucleotide, Tp8-oxoG, the single-turnover rate constants were 0.58 and 2.0 min−1 for TDG and OGG1, respectively (Table 1). OGG1 was sensitive to the presence of the 5′-neighboring T:G mispair, as the rate was ∼20-fold lower compared with that with the C:G base pair (Table 1). The 5′-neighboring AP site reduced the OGG1 activity by ∼5-fold.
Effect of APE1 and 5′-T:G Mispair on the Steady-state Rate of OGG1
As shown above and previously reported (41), 8-oxoG excision by OGG1 is decreased by a 5′-neighboring T:G mispair. Here, we assessed the influence of APE1 and the 5′-T:G mispair on the steady-state rate of OGG1 (Fig. 5 and Table 2). The rates with three 8-oxoG:C-containing substrates were compared (5′-C:G, 5′-5-mC:G, and 5′-T:G). By adding APE1 and 5 mm MgCl2 to the reaction mixture, the OGG1 activity for the 5′-C:G substrate was increased 6-fold, suggesting that the OGG1 product binding was weaker in the presence of APE1. In contrast to a recent report (54), an adjacent 5-mC:G base pair did not affect the APE1-mediated stimulation of OGG1 (Fig. 5B). The contrast with the previous study might be due to the differing reaction conditions or DNA sequence contexts. Importantly, for the OGG1 substrate with a 5′-T:G, APE1 failed to stimulate OGG1 activity (Fig. 5C). This result suggested that APE1 was not able to displace OGG1 tightly bound to the AP site or, alternatively, was not able to incise the AP site with the 5′-adjacent T:G mispair.
FIGURE 5.

Effect of a 5′-neighboring base pair on APE1-mediated stimulation of OGG1. Steady-state time courses for the removal of 8-oxoG where the 5′-base pair was C:G (A), 5-mC:G (B), or T:G (C). OGG1 (10 nm) was incubated with 200 nm DNA substrate in the presence of 20 mm KCl (○) or 20 mm KCl/5 mm MgCl2/100 nm APE1 (□) at 37 °C. Products were isolated and quantified as described in “Experimental Procedures.” Time courses were fit to a linear equation, and the steady-state kinetic parameters from several determinations are tabulated in Table 2.
TABLE 2.
Steady-state rate constants for 8-oxoG excision by OGG1
| 5′-Base pair |
kssa |
-Fold increase | |
|---|---|---|---|
| −APE1b | +APE1c | ||
| min−1 | |||
| C:G | 0.46 ± 0.046 | 2.6 ± 0.10 | 5.7 |
| mC:G | 0.59 ± 0.044 | 2.2 ± 0.066 | 3.7 |
| T:G | 0.37 ± 0.037 | 0.44 ± 0.14 | 1.2 |
a Steady-state rate determined from the linear steady-state phase.
b Rate constants measured in the absence of APE1 and MgCl2.
c Rate constants measured in the presence of 100 nm APE1 and 5 mm MgCl2. The values represent the means and S.D. of at least two independent experiments.
APE1 Activity on Alternate Glycosylase Repair Intermediates in Tandem Lesions
A partnership between APE1 and repair DNA glycosylases seems critical for efficient BER. We decided to directly characterize the activity of APE1 on substrates representing the products of TDG and OGG1 activity on the altered in-tandem substrate. The AP site analog THF was used in each case, and the influence of the neighboring 5′-T:G or 3′-8-oxoG:C (T/G-THF/C or THF/G-oxG/C, respectively) was assessed by comparing the activities with those of the control substrates (C/G-THF/C or THF/G-G/C). We analyzed both the pre-steady-state and steady-state phases of the reaction time courses. With the C/G-THF/C substrate (Fig. 6 and Table 3), a rapid product accumulation phase occurred before the first time point (100 ms), and this was followed by a slower steady-state phase (kss = 0.49 s−1). The rapid phase was too fast to measure (≫700 s−1) (55). In contrast, a rapid phase of product formation was not observed with T/G-THF/C substrate, suggesting that the rapid chemistry step observed with the normal substrate was significantly reduced and became rate-determining. From the slope of the linear fit, the rate of AP site cleavage for the T/G-THF/C substrate was 0.13 s−1, which is ≫5400-fold lower than that for a THF substrate without the mismatch (56). This strongly reduced APE1 activity with the T/G-THF/C substrate is consistent with the lack of APE1 stimulation of OGG1 activity on the corresponding substrate, as suggested above (Fig. 5B).
FIGURE 6.

Effect of a neighboring lesion on the strand incision activity of APE1. Steady-state time courses for the strand incision activity of APE1 on THF-containing DNA are shown. A, product formation was followed to examine the effect of a 5′ C:G base pair (○) or T:G mispair (□). B, product formation was followed to examine the effect of 3′ G:C base pair (○) or 8-oxoG:C base pair (□). APE1 (30 nm) was incubated with 200 nm DNA substrate (C/G-THF/C, T/G-THF/C, THF/G-G/C, or THF/G-oxG/C) at 37 °C. Products were isolated, quantified, and analyzed as described under “Experimental Procedures.” The steady-state kinetic parameters from several independent determinations are tabulated in Table 3.
If TDG initiates repair of the tandem lesion, then the AP site will be adjacent (3′) to 8-oxoG. For the substrates THF/G-G/C and THF/G-8-oxG/C, APE1 rapidly incises the bound THF residue before the first time point (100 ms) followed by the slower steady-state phase as it cycles. The values for kss with the THF/G-G/C and THF/G-8oxG/C substrates were 0.48 and 0.89 s−1, respectively (Table 2). These results indicate that the influence of the 3′-neighboring 8-oxoG:C base pair on APE1 activity was minimal.
A 3′-8-oxoG:C Base Pair Reduces TDG-initiated BER in Vitro
To examine the influence of a 3′-neighboring 8-oxoG:C base pair on TDG-initiated BER, we reconstituted an in vitro BER system with purified enzymes. The substrates included the T:G mispair adjacent to G:C or 8-oxoG:C base pair. These substrates were incubated with TDG, APE1, and pol β with or without DNA ligase I. In the absence of ligase, the unligated gap-filled BER intermediate increased in a time-dependent manner with both G:C and 8-oxoG:C containing substrates (Fig. 7, lanes 1–3 and 7–9). The amount of BER intermediates was comparable between the G:C- and 8-oxoG:C-containing substrates. The addition of DNA ligase resulted in ligated BER product in the reaction with the neighboring G:C base pair, as expected (Fig. 7, lanes 4–6); ∼90% of the un-ligated BER intermediates were converted to the ligated products at each reaction time. Yet ligated product formation in the reaction mixtures with the neighboring 8-oxoG:C base pair was less than that for the G:C base pair (Fig. 7, lanes 7–9); 17, 29, and 36% of the un-ligated BER intermediates were converted to the ligated products at 5, 10, and 15 min, respectively. The reduction in ligated product was not due to a deficiency in TDG, APE1, or pol β based on the amount of gap-filled intermediate formed (Fig. 7, lanes 7–9) and an assessment of the pol β dRP lyase activity (see below). Therefore, we assessed the influence of the 8-oxoG:C base pair on the activities of both mammalian ligases believed to participate in BER, Lig I, and Lig III-β. T4 Lig was included as a reference enzyme (Fig. 8). The activities of the enzymes were compared with those for the normal G:C base pair at the 5′-margin of the nick. The presence of 8-oxoG suppressed ligation and promoted accumulation of the adenylated abortive intermediate with all three enzymes. Thus, DNA ligase deficiency appeared to be responsible for the reduced BER activity observed in Fig. 7 with the 8-oxoG-containing substrate. Pol β can remove the 5′-dRP intermediate from the 8-oxoG at the 5′ terminus (Fig. 9). Hence, rates of the 5′-dRP removal from either the 8-oxoG or G were too fast too measure under single-turnover conditions, suggesting that the inefficient DNA ligation during BER was not due to failure of the pol β dRP lyase activity. We also examined BER with both G:C and 8-oxoG:C substrates where the cytosine in the strand opposite guanine was methylated; methylation of cytosine in the opposite strand did not alter BER efficiency (data not shown).
FIGURE 7.

Effect of the 3′-8-oxoG:C base pair on TDG-initiated BER activity. A schematic representation of the substrate and the reaction scheme are shown in the top panel. A DNA substrate (250 nm) with G:C (T/G-G/C) or 8-oxoG:C (T/G-oxG/C) positioned 3′ to T:G was incubated with 10 nm TDG, 20 nm APE1, and 20 nm pol β with or without 100 nm Lig I at 37 °C for 5, 10, or 15 min, as indicated. Reaction products representing incorporation of labeled dCMP were isolated as described under “Experimental Procedures.”
FIGURE 8.

Effect of 8-oxoG at the 5′-margin of a nick on DNA ligation. The 3′-6-FAM labeled nicked DNA substrate containing an unmodified G or 8-oxoG at the 5′-end of a nick was prepared as described under “Experimental Procedures.” DNA substrate (250 nm) were incubated with 200 nm Lig I, 200 nm Lig III, or 1 unit of T4 Lig at 37 °C. The unligated 17-mer, 5′-adenylated 17-mer, and ligated 34-mer DNA were labeled as 17, 17-AMP, and 34 on the right side of the image of the gel, respectively. Reaction products were isolated as described under “Experimental Procedures.”
FIGURE 9.
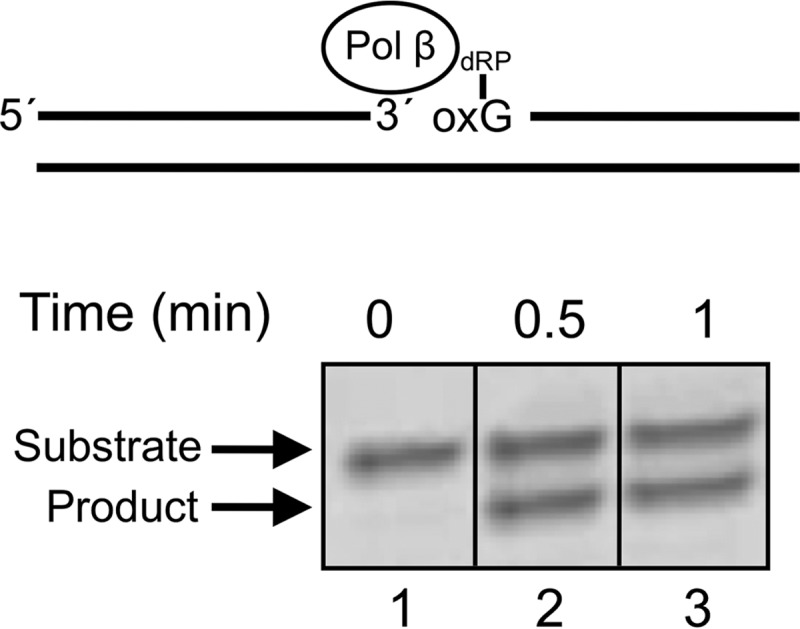
Removal of a 5′-dRP moiety attached to 8-oxoG by pol β. Removal of the 5′-dRP group from the 8-oxoG at the 5′ terminus of a nicked BER substrate. The DNA substrate is illustrated at the top. Pol β (500 nm) was incubated with 140 nm DNA substrate at 37 °C. The product was isolated and quantified as described under “Experimental Procedures.”
DISCUSSION
In higher eukaryotes methylation of cytosine is crucial for regulating gene expression during normal development and processes such as genome imprinting, X chromosome inactivation, tumorigenesis, and aging. The TDG-mediated base excision repair of the modified 5-mC (i.e. T, 5-hydroxymethyluracil, 5-formylcytosine, or 5-carboxylcytosine) at CpG dinucleotides is important for active DNA demethylation during cell development (17, 21, 57). Critically, G to T transversions occur under oxidative stress conditions in the sequence context of methylated CpG dinucleotides (40), implying formation of unrepaired 8-oxoG at these sites. A recent study also discussed the possibility of reduced repair efficiency of 8-oxoG at the methylated CpG dinucleotide (54). It is also noted that reactive oxygen species-induced G to T transversions have been found in the CpG sequence contexts in carcinoma cells (58). In the current study we assessed the influence of 8-oxoG and the tandem C to T changes in the CpG dinucleotide sequence context on the BER activities of TDG, OGG1 and downstream BER factors. Importantly, the two glycosylases strongly influence repair efficiency. There is a strong effect on BER efficiency that is dependent on the identity of the glycosylase that initiates repair of the modified in-tandem substrate; the altered in-tandem CpG dinucleotide will not be processed simultaneously. Instead, complete BER occurs at one lesion before processing of the other lesion.
In the case of TDG initiation of BER, our results revealed that the 3′-flanking 8-oxoG:C base pair decreases the rate of thymine removal from the T:G mispair (Fig. 1A and Table 1). This could be due to a clash between the C8-oxygen of 8-oxoG and the phosphate backbone for the 8-oxoG in an anti-conformation (59). Based on structures of the TDG-DNA complex with an AP site or 5-carboxylcytosine (60, 61), the side chain of Arg-275 inserts into the DNA minor groove to replace the flipped-out modified nucleotide. This arginine interacts with the phosphate backbone immediately 3′ to the substrate nucleotide. These interactions have been suggested to contribute to nucleotide flipping by increasing DNA binding affinity. Thus, in the presence of the 3′ 8-oxoG:C base pair, the 3′-flanking phosphate of the substrate T nucleotide might be altered due to a potential clash with the C8-oxygen of 8-oxoG. As shown in Fig. 1B and Table 1, the substitution of the normal 3′-flanking G:C base pair with 8-oxoG:A resulted in a large decrease (∼100-fold) in the activity of TDG. This result may also be explained by consideration of the crystallographic structure. To ensure the CpG sequence specificity for recognition of the target thymine nucleotide, TDG makes a contact with the N7 atom of 3′-flanking guanine in the anti-conformation (62). In addition, TDG makes interactions via Ala-277 with the phosphodiester backbone and via Glu-278 with the N2 of the 3′-flanking guanine base. Therefore, 8-oxoG in the syn-conformation paired with adenine would be expected to disrupt these interactions. Similarly, TDG interactions with substrates containing alternate 3′-neighboring bases such as adenine would be disrupted, and this is consistent with our results.
Earlier studies had revealed that TDG excises its target base but then fails to dissociate from the product AP site (52, 63). This pattern of tight product DNA binding is also seen with several other base excision repair enzymes but is especially strong in the case of TDG compared with the other DNA glycosylases (64–67). The slow product release could be important in protecting the cell from harmful BER intermediates with strand breaks. Our results suggest that the rate of product release by TDG is extremely slow: kss for T/G-G/C or T/G-8-oxoG/C was 0.0043 and 0.0075 min−1, respectively. In the presence of APE1, on the other hand, time courses of TDG product formation for both T/G-G/C and T/G-8-oxoG/C were increased by 37- and 5.7-fold, respectively (Fig. 4). Nevertheless, once the T is excised by TDG, the adjacent 8-oxoG will not be repaired until this neighboring lesion is restored to a C:G base pair.
If the deaminated 5-mC:G base pair, i.e. T:G mispair, is not repaired, it is interesting to consider how this will influence BER of the adjacent 8-oxoG. We had previously shown that for OGG1-initiated BER, overall BER activity was strongly decreased by the 5′-neighboring T:G mismatch (41). This diminished BER activity could be explained by the reduced rate of 8-oxoG removal by OGG1 and the inefficient extension of a mismatched terminus by pol β (41, 68). In addition to those effects, we now show that APE1 does not stimulate kss of OGG1 on the T/G-8-oxoG/C substrate (Fig. 5). The 5′-T:G mispair greatly reduces APE1-mediated AP site cleavage; the rate is >3 orders of magnitude lower than the incision rate for normal duplex DNA. This appears to be consistent with a previous report that APE1 failed to stimulate the product release of OGG1 for the hairpin substrate containing A:A mismatches (69), and it is known that activity of APE1 is decreased 10-fold by a 5′-neighboring C:C mismatch (70). More recently, it has been reported that the 5′-A:A mispair decreases the rate of AP site cleavage by APE1 ∼10-fold (56).
Reconstituted TDG-initiated BER using TDG, APE1, pol β, and Lig I was decreased by a 3′-neighboring 8-oxoG:C base pair. Consistent with this result, we found that the strand joining activities of Lig I, Lig III-β, and T4 Lig were decreased by the neighboring 8-oxoG (Fig. 8). It is known that the activity of T4 Lig is affected by DNA lesions at the terminus of the nick (71). Furthermore, 8-oxoG at the 5′ terminus promoted accumulation of the abortive adenylated intermediate with all three DNA ligases (Fig. 8). It also has been reported that 8-oxoG at the 3′ terminus in a nick inhibits ligation and results in an abortive ligation intermediate (i.e. 5′-adenylation) (72). In the cell, the adenylated intermediates can be removed by aprataxin (73–75). Therefore, aprataxin would be expected to improve ligation of 5′-8-oxoG terminus by providing additional opportunities to complete successful ligation. Alternatively, long patch BER involving strand displacement synthesis by pol β and excision of the displaced strand by flap endonuclease 1 could also remove an adenylated 5′-8-oxoG to produce a clean nick (76).
When the guanine opposite thymine in the T:G mispair is replaced with 8-oxoG, the rate of T removal by TDG was only moderately decreased (by 2-fold, data not shown). When TDG excises T in the case of the T:8-oxoG mismatch, the following BER steps might be similar to those of a misincorporated adenine opposite 8-oxoG where BER is initiated by adenine DNA glycosylase MutY. APE1 can cleave the AP site product in this case and produce a one-nucleotide gap opposite 8-oxoG, and then pol β will insert dCMP or dAMP opposite 8-oxoG (77). The strand-joining activities of DNA ligases I and III, however, are strongly reduced by the 3′-cytosine terminus that is opposite 8-oxoG (77). Therefore, the oxidation of the guanine opposite a modified 5-mC is potentially detrimental for TDG-mediated BER to restore the C:G base pair.
In conclusion, the presence of the 8-oxoG lesion in a CpG dinucleotide can reduce the active DNA demethylation process by reducing TDG-initiated BER. The results described here are summarized in the scheme shown Fig. 10. First, TDG excises its target base thymine at a reduced rate from the oxidized Tp8-oxoG dinucleotide, but this inhibits the 8-oxoG excision by OGG1. APE1 releases TDG from its product hastening BER. However, the DNA ligase step also is reduced by the presence of the 8-oxoG lesion, as illustrated in Fig. 10. If OGG1 binds and excises 8-oxoG before TDG binds to the altered in-tandem substrate, the T:G mispair will suppress the activity of APE1 and pol β (68). In addition, TDG will not excise its target base because of the absence of an adjacent base pair. In that situation, other repair factors could intervene; e.g. mismatch repair might function to restore the CpG dinucleotide (78). Until now, several possible mechanisms have been suggested as the first step of the active DNA demethylation process: deamination of 5-mC to T by AID/APOBECs; hydroxylation of 5-mC to 5-hydroxymethylcytosine by Ten-eleven translocation (Tet) protein followed by deamination of 5-hydroxymethylcytosine to 5-hydroxymethyluracil, and Tet-mediated oxidation of 5-mC to form 5-formylcytosine and 5-carboxylcytosine. Because all of these altered bases are substrates for TDG (79, 80), we suggest that 8-oxoG in the CpG dinucleotide may reduce TDG-initiated BER for these substrates as well, thereby influencing the active DNA demethylation process. At present, it remains to be seen how the enzymatic activities of AID/APOBECs and/or Tet to modify 5-mC are affected by structural abnormalities in the CpG dinucleotide. Therefore, further studies will be required for a fuller understanding of the cellular impact of the oxidative DNA damage and CpG methylation status.
FIGURE 10.

Scheme for BER of tandem modifications in a CpG dinucleotide. The dotted lines represent a reduced rate of activity of the respective BER enzyme during repair of tandem lesions. Arrows with a cross indicate a strongly inhibited reaction. As illustrated on the left, once OGG1 excises an 8-oxoG adjacent to the T:G mispair, APE1 fails to stimulate OGG1 activity. The subsequent AP site cleavage by APE1 is also retarded by the neighboring T:G mispair. TDG fails to process the T:G mispair due to loss of the adjacent base pair. As illustrated on the right, once TDG processes the T:G mispair adjacent to the 8-oxoG:C base pair, TDG remains bound to the product AP site, inhibiting the excision of the neighboring 8-oxoG by OGG1. TDG can be displaced by APE1 at the AP site adjacent to 8-oxoG hastening repair of the T:G mispair. After APE1 incision of the AP site, pol β-dependent dRP lyase and gap-filling DNA synthesis occurs rapidly; however, the strand joining activity by DNA ligase is reduced by the resulting 5′-8-oxoG in the nick. This is associated with accumulation of an abortive adenylated ligation intermediate that must be processed by aprataxin or long-patch BER.
Acknowledgments
We thank Rajendra Prasad for helpful discussions, Julie K. Horton for critical reading of the manuscript, Bret D. Freudenthal for assistance with the purification of human TDG, and David D. Shock for assistance with kinetic experiments.
This work was supported, in whole or in part, by National Institutes of Health Grants Z01-ES050158 and Z01-ES050159 (NIEHS).
- BER
- base excision repair
- TDG
- thymine DNA glycosylase
- 8-oxoG
- 7,8-dihydro-8-oxoguanine
- OGG1
- 8-oxoguanine DNA glycosylase
- AP
- apurinic-apyrimidinic
- AID
- activation-induced deaminase
- 5-mC
- 5-methylcytosine
- APE1
- apurinic/apyrimidinic endonuclease 1
- pol β
- DNA polymerase β
- Lig I
- DNA ligase I
- Lig III-β
- DNA ligase III-β
- T4 Lig
- T4 DNA ligase
- 6-FAM
- 6-carboxyfluorescein
- APOBEC
- apolipoprotein B mRNA-editing catalytic polypeptide
- dRP
- deoxyribose phosphate.
REFERENCES
- 1. Bird A. (1992) The essentials of DNA methylation. Cell 70, 5–8 [DOI] [PubMed] [Google Scholar]
- 2. Kass S. U., Landsberger N., Wolffe A. P. (1997) DNA methylation directs a time dependent repression of transcription initiation. Curr. Biol. 7, 157–165 [DOI] [PubMed] [Google Scholar]
- 3. Siegfried Z., Cedar H. (1997) DNA methylation: a molecular lock. Curr. Biol. 7, R305–R307 [DOI] [PubMed] [Google Scholar]
- 4. Vanyushin B. F., Tkacheva S. G., Belozersky A. N. (1970) Rare bases in animal DNA. Nature 225, 948–949 [DOI] [PubMed] [Google Scholar]
- 5. Antequera F., Bird A. (1993) Number of CpG islands and genes in human and mouse. Proc. Natl. Acad. Sci. U.S.A. 90, 11995–11999 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Bird A. P. (1995) Gene number, noise reduction, and biological complexity. Trends Genet. 11, 94–100 [DOI] [PubMed] [Google Scholar]
- 7. Clark S. J., Harrison J., Molloy P. L. (1997) Sp1 binding is inhibited by(m) Cp(m) CpG methylation. Gene 195, 67–71 [DOI] [PubMed] [Google Scholar]
- 8. Hendrich B., Bird A. (1998) Identification and characterization of a family of mammalian methyl-CpG binding proteins. Mol. Cell. Biol. 18, 6538–6547 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Shen J. C., Rideout W. M., 3rd, Jones P. A. (1994) The rate of hydrolytic deamination of 5-methylcytosine in double-stranded DNA. Nucleic Acids Res. 22, 972–976 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Cooper D. N., Youssoufian H. (1988) The CpG dinucleotide and human genetic disease. Hum. Genet. 78, 151–155 [DOI] [PubMed] [Google Scholar]
- 11. Gonzalgo M. L., Jones P. A. (1997) Mutagenic and epigenetic effects of DNA methylation. Mutat. Res. 386, 107–118 [DOI] [PubMed] [Google Scholar]
- 12. Pfeifer G. P., Tang M., Denissenko M. F. (2000) Mutation hotspots and DNA methylation. Curr. Top. Microbiol. Immunol. 249, 1–19 [DOI] [PubMed] [Google Scholar]
- 13. Jones P. A., Baylin S. B. (2002) The fundamental role of epigenetic events in cancer. Nat. Rev. Genet. 3, 415–428 [DOI] [PubMed] [Google Scholar]
- 14. Wiebauer K., Jiricny J. (1990) Mismatch-specific thymine DNA glycosylase and DNA polymerase β mediate the correction of G.T mispairs in nuclear extracts from human cells. Proc. Natl. Acad. Sci. U.S.A. 87, 5842–5845 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Morgan H. D., Dean W., Coker H. A., Reik W., Petersen-Mahrt S. K. (2004) Activation-induced cytidine deaminase deaminates 5-methylcytosine in DNA and is expressed in pluripotent tissues: implications for epigenetic reprogramming. J. Biol. Chem. 279, 52353–52360 [DOI] [PubMed] [Google Scholar]
- 16. Métivier R., Gallais R., Tiffoche C., Le Péron C., Jurkowska R. Z., Carmouche R. P., Ibberson D., Barath P., Demay F., Reid G., Benes V., Jeltsch A., Gannon F., Salbert G. (2008) Cyclical DNA methylation of a transcriptionally active promoter. Nature 452, 45–50 [DOI] [PubMed] [Google Scholar]
- 17. Cortellino S., Xu J., Sannai M., Moore R., Caretti E., Cigliano A., Le Coz M., Devarajan K., Wessels A., Soprano D., Abramowitz L. K., Bartolomei M. S., Rambow F., Bassi M. R., Bruno T., Fanciulli M., Renner C., Klein-Szanto A. J., Matsumoto Y., Kobi D., Davidson I., Alberti C., Larue L., Bellacosa A. (2011) Thymine DNA glycosylase is essential for active DNA demethylation by linked deamination-base excision repair. Cell 146, 67–79 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Li Y. Q., Zhou P. Z., Zheng X. D., Walsh C. P., Xu G. L. (2007) Association of Dnmt3a and thymine DNA glycosylase links DNA methylation with base-excision repair. Nucleic Acids Res. 35, 390–400 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Boland M. J., Christman J. K. (2008) Characterization of Dnmt3b:thymine-DNA glycosylase interaction and stimulation of thymine glycosylase-mediated repair by DNA methyltransferase(s) and RNA. J. Mol. Biol. 379, 492–504 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Tahiliani M., Koh K. P., Shen Y., Pastor W. A., Bandukwala H., Brudno Y., Agarwal S., Iyer L. M., Liu D. R., Aravind L., Rao A. (2009) Conversion of 5-methylcytosine to 5-hydroxymethylcytosine in mammalian DNA by MLL partner TET1. Science 324, 930–935 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Cortázar D., Kunz C., Selfridge J., Lettieri T., Saito Y., MacDougall E., Wirz A., Schuermann D., Jacobs A. L., Siegrist F., Steinacher R., Jiricny J., Bird A., Schär P. (2011) Embryonic lethal phenotype reveals a function of TDG in maintaining epigenetic stability. Nature 470, 419–423 [DOI] [PubMed] [Google Scholar]
- 22. Song C. X., Szulwach K. E., Dai Q., Fu Y., Mao S. Q., Lin L., Street C., Li Y., Poidevin M., Wu H., Gao J., Liu P., Li L., Xu G. L., Jin P., He C. (2013) Genome-wide profiling of 5-formylcytosine reveals its roles in epigenetic priming. Cell 153, 678–691 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Vasovcak P., Krepelova A., Menigatti M., Puchmajerova A., Skapa P., Augustinakova A., Amann G., Wernstedt A., Jiricny J., Marra G., Wimmer K. (2012) Unique mutational profile associated with a loss of TDG expression in the rectal cancer of a patient with a constitutional PMS2 deficiency. DNA Repair 11, 616–623 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Nilsen H., Krokan H. E. (2001) Base excision repair in a network of defence and tolerance. Carcinogenesis 22, 987–998 [DOI] [PubMed] [Google Scholar]
- 25. Rosenquist T. A., Zharkov D. O., Grollman A. P. (1997) Cloning and characterization of a mammalian 8-oxoguanine DNA glycosylase. Proc. Natl. Acad. Sci. U.S.A. 94, 7429–7434 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Zharkov D. O., Rosenquist T. A., Gerchman S. E., Grollman A. P. (2000) Substrate specificity and reaction mechanism of murine 8-oxoguanine-DNA glycosylase. J. Biol. Chem. 275, 28607–28617 [DOI] [PubMed] [Google Scholar]
- 27. Yamane A., Shinmura K., Sunaga N., Saitoh T., Yamaguchi S., Shinmura Y., Yoshimura K., Murakami H., Nojima Y., Kohno T., Yokota J. (2003) Suppressive activities of OGG1 and MYH proteins against G:C to T:A mutations caused by 8-hydroxyguanine but not by benzo[a]pyrene diol epoxide in human cells in vivo. Carcinogenesis 24, 1031–1037 [DOI] [PubMed] [Google Scholar]
- 28. Sunaga N., Kohno T., Shinmura K., Saitoh T., Matsuda T., Saito R., Yokota J. (2001) OGG1 protein suppresses G:C→T:A mutation in a shuttle vector containing 8-hydroxyguanine in human cells. Carcinogenesis 22, 1355–1362 [DOI] [PubMed] [Google Scholar]
- 29. Minowa O., Arai T., Hirano M., Monden Y., Nakai S., Fukuda M., Itoh M., Takano H., Hippou Y., Aburatani H., Masumura K., Nohmi T., Nishimura S., Noda T. (2000) Mmh/Ogg1 gene inactivation results in accumulation of 8-hydroxyguanine in mice. Proc. Natl. Acad. Sci. U.S.A. 97, 4156–4161 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Sakumi K., Tominaga Y., Furuichi M., Xu P., Tsuzuki T., Sekiguchi M., Nakabeppu Y. (2003) Ogg1 knockout-associated lung tumorigenesis and its suppression by Mth1 gene disruption. Cancer Res. 63, 902–905 [PubMed] [Google Scholar]
- 31. Ghosh R., Mitchell D. L. (1999) Effect of oxidative DNA damage in promoter elements on transcription factor binding. Nucleic Acids Res. 27, 3213–3218 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Zawia N. H., Lahiri D. K., Cardozo-Pelaez F. (2009) Epigenetics, oxidative stress, and Alzheimer disease. Free Radic. Biol. Med. 46, 1241–1249 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Valinluck V., Tsai H. H., Rogstad D. K., Burdzy A., Bird A., Sowers L. C. (2004) Oxidative damage to methyl-CpG sequences inhibits the binding of the methyl-CpG binding domain (MBD) of methyl-CpG binding protein 2 (MeCP2). Nucleic Acids Res. 32, 4100–4108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Weitzman S. A., Turk P. W., Milkowski D. H., Kozlowski K. (1994) Free radical adducts induce alterations in DNA cytosine methylation. Proc. Natl. Acad. Sci. U.S.A. 91, 1261–1264 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Turk P. W., Laayoun A., Smith S. S., Weitzman S. A. (1995) DNA adduct 8-hydroxyl-2′-deoxyguanosine (8-hydroxyguanine) affects function of human DNA methyltransferase. Carcinogenesis 16, 1253–1255 [DOI] [PubMed] [Google Scholar]
- 36. Maltseva D. V., Baykov A. A., Jeltsch A., Gromova E. S. (2009) Impact of 7,8-dihydro-8-oxoguanine on methylation of the CpG site by Dnmt3a. Biochemistry 48, 1361–1368 [DOI] [PubMed] [Google Scholar]
- 37. Waters T. R., Swann P. F. (1998) Kinetics of the action of thymine DNA glycosylase. J. Biol. Chem. 273, 20007–20014 [DOI] [PubMed] [Google Scholar]
- 38. Abu M., Waters T. R. (2003) The main role of human thymine-DNA glycosylase is removal of thymine produced by deamination of 5-methylcytosine and not removal of ethenocytosine. J. Biol. Chem. 278, 8739–8744 [DOI] [PubMed] [Google Scholar]
- 39. Morgan M. T., Bennett M. T., Drohat A. C. (2007) Excision of 5-halogenated uracils by human thymine DNA glycosylase. Robust activity for DNA contexts other than CpG. J. Biol. Chem. 282, 27578–27586 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Lee D. H., O'Connor T. R., Pfeifer G. P. (2002) Oxidative DNA damage induced by copper and hydrogen peroxide promotes CG→TT tandem mutations at methylated CpG dinucleotides in nucleotide excision repair-deficient cells. Nucleic Acids Res. 30, 3566–3573 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Sassa A., Beard W. A., Prasad R., Wilson S. H. (2012) DNA sequence context effects on the glycosylase activity of human 8-oxoguanine DNA glycosylase. J. Biol. Chem. 287, 36702–36710 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Kovtun I. V., Liu Y., Bjoras M., Klungland A., Wilson S. H., McMurray C. T. (2007) OGG1 initiates age-dependent CAG trinucleotide expansion in somatic cells. Nature 447, 447–452 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Beard W. A., Wilson S. H. (1995) Purification and domain-mapping of mammalian DNA polymerase β. Methods Enzymol. 262, 98–107 [DOI] [PubMed] [Google Scholar]
- 44. Chen X., Pascal J., Vijayakumar S., Wilson G. M., Ellenberger T., Tomkinson A. E. (2006) Human DNA ligases I, III, and IV-purification and new specific assays for these enzymes. Methods Enzymol. 409, 39–52 [DOI] [PubMed] [Google Scholar]
- 45. Strauss P. R., Beard W. A., Patterson T. A., Wilson S. H. (1997) Substrate binding by human apurinic/apyrimidinic endonuclease indicates a Briggs-Haldane mechanism. J. Biol. Chem. 272, 1302–1307 [DOI] [PubMed] [Google Scholar]
- 46. Sassa A., Beard W. A., Shock D. D., Wilson S. H. (2013) Steady-state, pre-steady-state, and single-turnover kinetic measurement for DNA glycosylase activity. J. Vis. Exp. 78, e50695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Singhal R. K., Prasad R., Wilson S. H. (1995) DNA polymerase β conducts the gap-filling step in uracil-initiated base excision repair in a bovine testis nuclear extract. J. Biol. Chem. 270, 949–957 [DOI] [PubMed] [Google Scholar]
- 48. Prasad R., Singhal R. K., Srivastava D. K., Molina J. T., Tomkinson A. E., Wilson S. H. (1996) Specific interaction of DNA polymerase β and DNA ligase I in a multiprotein base excision repair complex from bovine testis. J. Biol. Chem. 271, 16000–16007 [DOI] [PubMed] [Google Scholar]
- 49. Prasad R., Beard W. A., Strauss P. R., Wilson S. H. (1998) Human DNA polymerase β deoxyribose phosphate lyase: Substrate specificity and catalytic mechanism. J. Biol. Chem. 273, 15263–15270 [DOI] [PubMed] [Google Scholar]
- 50. Matsumoto Y., Kim K. (1995) Excision of deoxyribose phosphate residues by DNA polymerase β during DNA repair. Science 269, 699–702 [DOI] [PubMed] [Google Scholar]
- 51. Deterding L. J., Prasad R., Mullen G. P., Wilson S. H., Tomer K. B. (2000) Mapping of the 5′-2-deoxyribose-5-phosphate lyase active site in DNA polymerase β by mass spectrometry. J. Biol. Chem. 275, 10463–10471 [DOI] [PubMed] [Google Scholar]
- 52. Waters T. R., Gallinari P., Jiricny J., Swann P. F. (1999) Human thymine DNA glycosylase binds to apurinic sites in DNA but is displaced by human apurinic endonuclease 1. J. Biol. Chem. 274, 67–74 [DOI] [PubMed] [Google Scholar]
- 53. Fitzgerald M. E., Drohat A. C. (2008) Coordinating the initial steps of base excision repair. Apurinic/apyrimidinic endonuclease 1 actively stimulates thymine DNA glycosylase by disrupting the product complex. J. Biol. Chem. 283, 32680–32690 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Kasymov R. D., Grin I. R., Endutkin A. V., Smirnov S. L., Ishchenko A. A., Saparbaev M. K., Zharkov D. O. (2013) Excision of 8-oxoguanine from methylated CpG dinucleotides by human 8-oxoguanine DNA glycosylase. FEBS Lett. 587, 3129–3134 [DOI] [PubMed] [Google Scholar]
- 55. Maher R. L., Bloom L. B. (2007) Pre-steady-state kinetic characterization of the AP endonuclease activity of human AP endonuclease 1. J. Biol. Chem. 282, 30577–30585 [DOI] [PubMed] [Google Scholar]
- 56. Schermerhorn K. M., Delaney S. (2013) Transient-state kinetics of apurinic/apyrimidinic (AP) endonuclease 1 acting on an authentic AP site and commonly used substrate analogs: the effect of diverse metal ions and base mismatches. Biochemistry 52, 7669–7677 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Kohli R. M., Zhang Y. (2013) TET enzymes, TDG and the dynamics of DNA demethylation. Nature 502, 472–479 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Agar N. S., Halliday G. M., Barnetson R. S., Ananthaswamy H. N., Wheeler M., Jones A. M. (2004) The basal layer in human squamous tumors harbors more UVA than UVB fingerprint mutations: a role for UVA in human skin carcinogenesis. Proc. Natl. Acad. Sci. U.S.A. 101, 4954–4959 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Uesugi S., Ikehara M. (1977) Carbon-13 magnetic resonance spectra of 8-substituted purine nucleosides. Characteristic shifts for the syn conformation. J. Am. Chem. Soc. 99, 3250–3253 [DOI] [PubMed] [Google Scholar]
- 60. Maiti A., Morgan M. T., Pozharski E., Drohat A. C. (2008) Crystal structure of human thymine DNA glycosylase bound to DNA elucidates sequence-specific mismatch recognition. Proc. Natl. Acad. Sci. U.S.A. 105, 8890–8895 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Zhang L., Lu X., Lu J., Liang H., Dai Q., Xu G. L., Luo C., Jiang H., He C. (2012) Thymine DNA glycosylase specifically recognizes 5-carboxylcytosine-modified DNA. Nat. Chem. Biol. 8, 328–330 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Schärer O. D., Kawate T., Gallinari P., Jiricny J., Verdine G. L. (1997) Investigation of the mechanisms of DNA binding of the human G/T glycosylase using designed inhibitors. Proc. Natl. Acad. Sci. U.S.A. 94, 4878–4883 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Hardeland U., Bentele M., Jiricny J., Schär P. (2000) Separating substrate recognition from base hydrolysis in human thymine DNA glycosylase by mutational analysis. J. Biol. Chem. 275, 33449–33456 [DOI] [PubMed] [Google Scholar]
- 64. Hill J. W., Hazra T. K., Izumi T., Mitra S. (2001) Stimulation of human 8-oxoguanine-DNA glycosylase by AP-endonuclease: potential coordination of the initial steps in base excision repair. Nucleic Acids Res. 29, 430–438 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Porello S. L., Leyes A. E., David S. S. (1998) Single-turnover and pre-steady-state kinetics of the reaction of the adenine glycosylase MutY with mismatch-containing DNA substrates. Biochemistry 37, 14756–14764 [DOI] [PubMed] [Google Scholar]
- 66. Liu X., Roy R. (2002) Truncation of amino-terminal tail stimulates activity of human endonuclease III (hNTH1). J. Mol. Biol. 321, 265–276 [DOI] [PubMed] [Google Scholar]
- 67. O'Neill R. J., Vorob'eva O. V., Shahbakhti H., Zmuda E., Bhagwat A. S., Baldwin G. S. (2003) Mismatch uracil glycosylase from Escherichia coli: a general mismatch or a specific DNA glycosylase? J. Biol. Chem. 278, 20526–20532 [DOI] [PubMed] [Google Scholar]
- 68. Beard W. A., Shock D. D., Wilson S. H. (2004) Influence of DNA structure on DNA polymerase β active site function: extension of mutagenic DNA intermediates. J. Biol. Chem. 279, 31921–31929 [DOI] [PubMed] [Google Scholar]
- 69. Jarem D. A., Wilson N. R., Schermerhorn K. M., Delaney S. (2011) Incidence and persistence of 8-oxo-7,8-dihydroguanine within a hairpin intermediate exacerbates a toxic oxidation cycle associated with trinucleotide repeat expansion. DNA Repair 10, 887–896 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Wilson D. M., 3rd, Takeshita M., Grollman A. P., Demple B. (1995) Incision activity of human apurinic endonuclease (Ape) at abasic site analogs in DNA. J. Biol. Chem. 270, 16002–16007 [DOI] [PubMed] [Google Scholar]
- 71. Zhao X., Muller J. G., Halasyam M., David S. S., Burrows C. J. (2007) In vitro ligation of oligodeoxynucleotides containing C8-oxidized purine lesions using bacteriophage T4 DNA ligase. Biochemistry 46, 3734–3744 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Harris J. L., Jakob B., Taucher-Scholz G., Dianov G. L., Becherel O. J., Lavin M. F. (2009) Aprataxin, poly-ADP ribose polymerase 1 (PARP-1) and apurinic endonuclease 1 (APE1) function together to protect the genome against oxidative damage. Hum. Mol. Genet. 18, 4102–4117 [DOI] [PubMed] [Google Scholar]
- 73. Lavin M. F., Gueven N., Grattan-Smith P. (2008) Defective responses to DNA single- and double-strand breaks in spinocerebellar ataxia. DNA Repair 7, 1061–1076 [DOI] [PubMed] [Google Scholar]
- 74. Rass U., Ahel I., West S. C. (2007) Actions of aprataxin in multiple DNA repair pathways. J. Biol. Chem. 282, 9469–9474 [DOI] [PubMed] [Google Scholar]
- 75. Rass U., Ahel I., West S. C. (2007) Defective DNA repair and neurodegenerative disease. Cell 130, 991–1004 [DOI] [PubMed] [Google Scholar]
- 76. Kim K., Biade S., Matsumoto Y. (1998) Involvement of flap endonuclease 1 in base excision DNA repair. J. Biol. Chem. 273, 8842–8848 [DOI] [PubMed] [Google Scholar]
- 77. Hashimoto K., Tominaga Y., Nakabeppu Y., Moriya M. (2004) Futile short-patch DNA base excision repair of adenine:8-oxoguanine mispair. Nucleic Acids Res. 32, 5928–5934 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Brown T. C., Jiricny J. (1987) A specific mismatch repair event protects mammalian cells from loss of 5-methylcytosine. Cell 50, 945–950 [DOI] [PubMed] [Google Scholar]
- 79. Bennett M. T., Rodgers M. T., Hebert A. S., Ruslander L. E., Eisele L., Drohat A. C. (2006) Specificity of human thymine DNA glycosylase depends on N-glycosidic bond stability. J. Am. Chem. Soc. 128, 12510–12519 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Maiti A., Drohat A. C. (2011) Thymine DNA glycosylase can rapidly excise 5-formylcytosine and 5-carboxylcytosine: potential implications for active demethylation of CpG sites. J. Biol. Chem. 286, 35334–35338 [DOI] [PMC free article] [PubMed] [Google Scholar]