

Abstract
Prostate cancer (PCa) is the second most common malignancy among men in the world. Castration-resistant prostate cancer (CRPC) is the lethal form of the disease, which develops upon resistance to first line androgen deprivation therapy (ADT). Emerging evidence demonstrates a key role for the PI3K-AKT-mTOR signaling axis in the development and maintenance of CRPC. This pathway, which is deregulated in the majority of advanced PCas, serves as a critical nexus for the integration of growth signals with downstream cellular processes such as protein synthesis, proliferation, survival, metabolism and differentiation, thus providing mechanisms for cancer cells to overcome the stress associated with androgen deprivation. Furthermore, preclinical studies have elucidated a direct connection between the PI3K-AKT-mTOR and androgen receptor (AR) signaling axes, revealing a dynamic interplay between these pathways during the development of ADT resistance. Thus, there is a clear rationale for the continued clinical development of a number of novel inhibitors of the PI3K pathway, which offer the potential of blocking CRPC growth and survival. In this review, we will explore the relevance of the PI3K-AKT-mTOR pathway in PCa progression and castration resistance in order to inform the clinical development of specific pathway inhibitors in advanced PCa. In addition, we will highlight current deficiencies in our clinical knowledge, most notably the need for biomarkers that can accurately predict for response to PI3K pathway inhibitors.
Keywords: androgen receptor, CRPC, kinase inhibitors, mTOR, prostate cancer, PI3K, resistance
INTRODUCTION
Prostate cancer (PCa) is the second most common malignancy in men worldwide, accounting for 903 500 newly diagnosed cancer cases and 258 400 cancer deaths in 2008.1 The incidence of disease has increased in recent years in part due to widespread screening for prostate-specific antigen (PSA), which allows early detection of tumors that might otherwise remain undetected.1 In the United States, 90% of PCa patients present with localized disease at diagnosis, which is treated with curative intent and has an excellent prognosis.2 However, by 5 years, nearly 30% of definitively treated patients exhibit a rise in PSA levels and evidence of recurrent disease.3,4 Most PCas require androgens for growth and are exquisitely sensitive to androgen deprivation therapy (ADT).5 However, this response is temporary and the majority of patients inevitably develop resistance to androgen deprivation, leading to castration-resistant prostate cancer (CRPC). CRPC is characterized by persistent tumor growth despite castrate levels of serum testosterone, which leads to significant patient mortality.6 At a cellular level, the development of CRPC represents a major compensatory response to androgen deprivation-induced stress, allowing cancer cells to survive and subsequently thrive in a low testosterone environment. A thorough understanding of the molecular mechanisms that drive this process is critical towards targeting CRPC initiation and progression to impact patient survival.
An important mechanism that promotes castration resistance is persistent androgen receptor (AR) signaling.7,8,9,10 A number of contributing mechanisms involving genetic alterations to the AR locus have been identified, including mutations in the ligand-binding domain,11,12 amplification of the AR gene,13 and expression of AR splice variants,14 all of which may promote AR signaling in the setting of low serum testosterone. Another key mechanism is the intracellular upregulation of genes that convert adrenal androgens to highly potent dihydrotestosterone, thus providing alternative ligand sources for hormone-deprived tumors.15 Recently, a gain-of-function mutation in a rate-limiting enzyme responsible for dihydrotestosterone synthesis was reported, demonstrating for the first time a mechanism by which the steroid synthesis enzymatic process itself could be altered at the genomic level to drive the development of castration resistance.16 Together, these findings have led to a series of inhibitors targeting the AR or adrenal androgen synthesis, which have resulted in some survival benefit in patients with CRPC.17,18,19,20 However, advanced PCa remains uniformly fatal, highlighting the dire need for additional therapeutics that move the field past the AR signaling axis to stem the development and progression of CRPC.
There is a growing appreciation that compensation through signal transduction pathways represents another important mechanism to drive CRPC development.21 The phosphoinositide 3-kinase (PI3K)-AKT-mammalian target of rapamycin or mechanistic target of rapamycin (mTOR) signaling pathway is clearly emerging as a very important node that directs ADT resistance and stimulates tumor growth in the setting of castrate levels of testosterone. In fact, this pathway is altered at the genomic and transcriptional level in nearly all advanced PCas.22 The importance of this pathway in PCa progression is founded on its ability to integrate many intra- and extracellular growth signals with critical cellular processes.23,24,25 Thus, cancer cells utilize this pathway to adapt to the cellular stress brought about by ADT. Moreover, recent studies have demonstrated a direct link between PI3K-AKT-mTOR and AR signaling, revealing a dynamic interplay between these pathways during the development of androgen insensitivity.26,27 Most excitingly, a variety of drugs that specifically inhibit the PI3K-AKT-mTOR signaling pathway are currently in clinical development. In this review, we will explore the importance of the PI3K-AKT-mTOR pathway in castration resistance in order to inform the clinical development and use of specific pathway inhibitors in advanced PCa.
PI3K-AKT-mTOR SIGNALING AND FUNCTION
The PI3K-AKT-mTOR signaling pathway is an ancient signal transduction pathway, conserved from worms to humans, that has evolved into an essential regulator of catabolic and anabolic processes in a cell. It provides a critical nexus that connects nutrient and growth factor sensing with a variety of vital cellular processes, including protein synthesis, proliferation, survival, metabolism and differentiation.23,24,25 This diverse range of functions is achieved by signaling through a number of effectors that modulate the phosphorylation, transcription and translation of downstream targets necessary for these processes. Importantly, the PI3K pathway is significantly deregulated in PCa.22 However, to better appreciate its relevance in PCa, it is important to understand the pathway's function and role in normal cellular physiology. Here we will highlight a few of the key PI3K signaling nodes implicated in PCa pathogenesis and some of the downstream cellular processes they regulate (Figure 1a).
Figure 1.
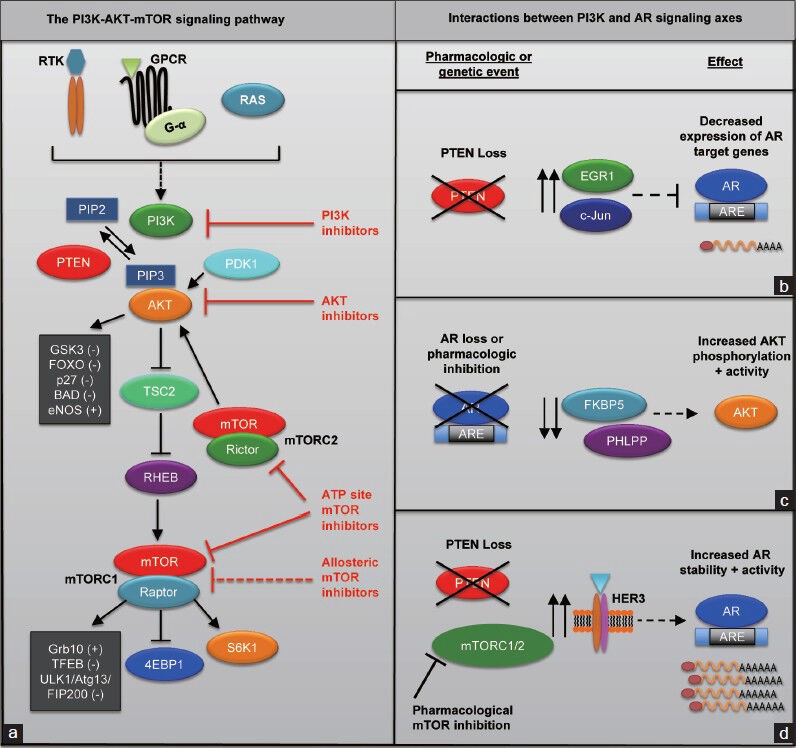
The PI3K-AKT-mTOR signaling pathway and therapeutic opportunities. (a) A simplified schematic of PI3K-AKT-mTOR signaling and therapeutic targets. (b-d) The molecular interplay between the PI3K and AR signaling axes. ARE: androgen response element; mTOR: mammalian target of rapamycin; PI3K: phosphoinositide 3-kinase; AR: androgen receptor; RTK: receptor tyrosine kinase; GPCR: G-protein coupled receptor.
The PI3K family of lipid kinases forms an important interface between upstream growth signals and the downstream signal transduction machinery. PI3Ks are grouped into three classes (I–III) according to their substrate preferences and sequence homology. Their primary function is to phosphorylate the 3’-hydroxyl group of phosphatidylinositol and phosphoinositides. Most relevant to cancer is the class IA PI3K, which is comprised of two functional subunits that form a heterodimer: a catalytic subunit (p110α, p110β or p110δ) and a regulatory subunit (p85α, p55α, p50α, p85β or p85γ). A variety of signals stimulate PI3K activity primarily through receptor tyrosine kinases (RTKs),28,29 but also through G-protein-coupled receptors30 and oncogenes such as Ras that directly bind p110.31,32,33 Upon stimulation, the catalytic subunit of PI3K phosphorylates phosphatidylinositol-4,5-bisphosphate (PI-4,5-P2) to phosphatidylinositol-3,4,5-triphosphate (PI-3,4,5-P3), which acts as a secondary messenger to recruit a variety of pleckstrin homology domain-containing proteins to the cell membrane.34 This process is reversed by the tumor suppressor PTEN (phosphatase and tensin homolog) and INPP4B (inositol polyphosphate 4-phosphatase type II), which collectively dephosphorylate PI-3,4,5-P3 to PI-3-P.35,36,37 One of the targets recruited by PI3K activity is the AKT family of serine/threonine protein kinases, which have been shown to drive PCa formation in vivo.38 Membrane recruitment of AKT and subsequent phosphorylation leads to its activation.39,40 Activated AKT phosphorylates a number of important effectors, including tuberous sclerosis complex 2 (TSC2), glycogen synthase kinase 3 (GSK3), forkhead box O (FOXO) transcription factors, p27, BAD and eNOS, which regulate a variety of processes that coordinate cell growth, survival, proliferation, metabolism and angiogenesis.23 The requirement for each of these downstream nodes of AKT signaling in PCa initiation and progression is an interesting but outstanding question in the field.
One major downstream effector of AKT signaling necessary for prostate tumorigenesis (see below) is the serine/threonine protein kinase mTOR that forms the catalytic subunit of two distinct complexes: mTORC1 and mTORC2. mTORC1 consists of mTOR, Raptor, PRAS40, mLST8, DEPTOR and tti1/tel225 and assembles following AKT phosphorylation of TSC2, which allows for the accumulation of the GTP bound form of Rheb, an mTORC1 activator.23 In addition, AKT phosphorylates and inhibits the repressor of the mTORC1 complex PRAS40, which is also a component of mTORC1.41,42,43,44 mTORC1 substrates include the regulators of protein synthesis S6K1 and 4EBP1,45,46 the autophagy inducing complex ULK1/Atg13/FIP200,47,48,49 the lysosome biogenesis regulator TFEB50 and the negative regulator of RTK signaling Grb10.51,52
The mTORC2 complex is composed of Rictor, mSin1, mLST8, DEPTOR, PROTOR1/2 and tti1/tel2.25 mTORC2 activity appears to be regulated by shared and distinct mechanisms compared to mTORC1. For instance, while TSC1/2 can regulate both mTORC1 and mTORC2 function,53,54 S6K1 has been shown to direct mTORC2 activity.55,56 Importantly, mTORC2 substrates are unique from mTORC1 substrates and include: AKT, SGK1 and PKCα.25 As such, the unique composition of each mTOR complex as well as the distinct downstream substrates position the PI3K-AKT-mTOR signaling pathway to direct a complex network of vital cellular processes.
It is intriguing to speculate why the PI3K-AKT-mTOR signaling pathway is so frequently deregulated in human PCa. Given the significant stresses that a prostate epithelial cell endures during the process of transformation, tumor growth, invasion and hormone deprivation, one possibility is that cancer cells require hyperactivation of the pathway and its downstream networks to overcome the significant cellular stresses that burden a cell during cancer progression. Therefore, an important question is which normal cellular processes controlled by PI3K-AKT-mTOR signaling can be usurped to drive cancer pathogenesis? Here we will briefly highlight some of these cellular processes. PI3K, for example, is a major regulator of metabolism through its role as a critical downstream effector of the insulin receptor. It has been shown in knockout and transgenic mouse models that class IA PI3K is necessary for effective insulin signaling and glucose uptake.24 Loss of the PTEN tumor suppressor in embryonic stem cells increases cell proliferation through an accelerated G1/S transition, which is associated with a decrease in the levels of the cell cycle inhibitor p27.57 AKT has been shown to play a critical role in cell survival. In particular, it phosphorylates critical proapoptotic targets such as BAD leading to binding by 14-3-3 proteins, which triggers release of BAD from its target proteins, such as Bcl-2. This has been shown to promote survival in neurons and other cell types.58,59,60 mTOR coordinates the maturation of multiple hematopoietic lineages, demonstrating a critical role in cellular differentiation.61 The downstream targets of mTORC1, 4EBP1 and S6K1 are major regulators of mRNA translation and have been shown to control cell size and proliferation through regulation of the translation of select mRNAs.62 The cross-section of pathway components described above and the cellular processes they impact illustrate the vital role of PI3K-AKT-mTOR signaling in cellular homeostasis and set a possible mechanistic precedence for why it is frequently deregulated in PCa.
THE PI3K-AKT-mTOR PATHWAY IN PCa PATHOGENESIS
Given the critical role of the PI3K-AKT-mTOR pathway in normal cell physiology, it is not surprising that the pathway is deregulated in a vast array of cancers. In fact, genetic alterations have been identified in nearly every member of the pathway.63 In PCa, the PI3K-AKT-mTOR signaling pathway is deregulated in 42% of localized disease and 100% of advanced-stage disease,22 which implies that alterations in this pathway may be a pre-requisite for the development of CRPC. The functional importance of the mutations, gene amplifications and changes in mRNA expression of PI3K signaling pathway components are highlighted by their significant correlation with PCa patient outcomes. For example, reduced expression of PTEN, a negative regulator of the pathway, is associated with high Gleason score,64 biochemical recurrence after prostatectomy,65,66,67 and shorter time to metastasis.68 Furthermore, high phospho-4EBP1 and eI4E levels are associated with increased patient mortality from PCa, indicating that even the most downstream effectors of the pathway are predictive of disease progression.69
The role of PI3K-AKT-mTOR pathway deregulation towards PCa development has been clearly demonstrated in knockout and transgenic mouse models. In particular, overexpression of the oncogene AKT or biallelic loss of the tumor suppressor PTEN in prostate epithelial cells leads to hyperactivation of the pathway and is sufficient for the development of PCa in vivo.38,70,71 Conditional knockout of mTOR in a mouse model of PCa caused by deletion of PTEN inhibits prostate tumorigenesis, demonstrating the requirement for an intact signaling axis to drive cellular transformation in prostate epithelial cells.72 Interestingly, others have demonstrated that concurrent loss of PTEN and RICTOR, a defining component of the mTORC2 complex, reduces the incidence of PCa formation in mice.73 Thus, PI3K-AKT-mTOR hyperactivation is sufficient to induce PCa formation, and both mTORC1 and mTORC2 are necessary to facilitate this process in vivo.
While these genetic studies demonstrate that PI3K-AKT-mTOR hyperactivity is sufficient to initiate PCa formation, they do not prove that aberrant PI3K pathway signaling is required for PCa progression. To address this issue, inhibitors of the PI3K-AKT-mTOR pathway have been utilized in preclinical models of PCa after the development of tumors. For example, combined PI3K and mTOR inhibition with the dual kinase inhibitor BEZ235 reduced tumor volumes in a mouse model of PCa mediated by PTEN loss, demonstrating a continued requirement for pathway hyperactivity to maintain established tumors.27 Moreover, others have demonstrated that allosteric inhibitors of mTOR (rapalogues) such as rapamycin and everolimus also exhibit antitumor efficacy in established murine PCas.72,74,75 As such, these studies prompted significant efforts to determine the clinical role of allosteric inhibitors of mTOR in advanced human PCa. Despite initial optimism about their potential efficacy, rapalogues have performed poorly in phase I-II clinical trials in PCa patients, raising questions about the utility of targeting the mTOR kinase in this disease.76,77,78
This seeming paradox between the murine preclinical studies and the human clinical studies raises the important question of whether the mTOR pathway is a suboptimal therapeutic target in human PCa or if it has been poorly targeted with allosteric mTOR inhibitors. Given the genetic and pharmacologic studies reported to date, as well as the significant positive correlation between mTOR hyperactivation and poor patient outcomes, the former seems unlikely. Instead, the unique characteristics of first generation mTOR inhibitors (rapalogues) may explain why PCa clinical trials with these agents have demonstrated limited clinical efficacy. Unlike many kinase inhibitors, rapalogues do not directly bind to and inhibit the catalytic core of the mTOR kinase. Instead, rapamycin and associated analogs are partial inhibitors of mTOR, which bind to FKBP12 to allosterically inhibit mTORC1 function.79,80 As a result, these agents only inhibit the phosphorylation of a specific subset of mTORC1 substrates, providing a mechanistic rationale for their poor clinical performance in cancer.81,82
The importance of complete inhibition of mTOR kinase activity towards PCa progression has recently been highlighted by the advent of second generation mTOR inhibitors, which directly target the ATP binding site, such as MLN0128 (previously known as INK128), Torin1/2, CC-223, OSI-027, AZD8055, AZD2014 and Palomid 529.83 These agents potently inhibit all mTOR kinase activity and appear to exhibit significantly more antitumor efficacy over allosteric inhibitors of mTOR in preclinical trials. For example, MLN0128 outperformed everolimus by decreasing tumor burden in a murine model of PCa. Interestingly, in this study, MLN0128 and not everolimus induced apoptosis, suggesting that rapamycin-resistant mTOR substrates may be necessary for the survival of PCa cells in vivo.84 Given the ability of ATP site inhibitors of mTOR to target both mTORC1 and mTORC2 activity, it has been assumed that mTORC2 inhibition is required for the improved therapeutic response. Surprisingly, mechanistic studies have revealed that the therapeutic efficacy of ATP site inhibitors of mTOR is mediated through the inhibition of mTORC1 rapamycin-resistant substrates such as 4EBP1 and less so through its effects on mTORC2.85,86,87 As such, these pharmacogenetic studies and others have greatly improved our understanding of the role of PI3K-AKT-mTOR hyperactivation towards PCa maintenance and progression. In addition, they provide a mechanistic foundation for continued efforts to drug this signaling node in human PCa.
THE DYNAMIC INTERPLAY BETWEEN PI3K-AKT-mTOR AND AR SIGNALING IN RESISTANCE TO ANDROGEN DEPRIVATION THERAPY
It has been shown that PI3K-AKT-mTOR pathway deregulation resulting from PTEN loss is associated with androgen insensitivity and the development of CRPC.70,88 However, the mechanism by which this occurs has been elusive until recently. A series of studies in murine models of PCa have shed light on the in vivo mechanisms by which these two signaling axes regulate each other to promote CRPC. Strikingly, these studies have demonstrated a fundamental relationship between the PI3K and AR signaling axes in the development of CRPC. In particular, it has been shown that loss of PTEN in prostate epithelial cells leads to a decrease in transcription of AR target genes26,27 through de-repression of negative regulators of AR activity, EGR1 and c-Jun (Figure 1b).26 These findings demonstrate that deregulation of the PI3K-AKT-mTOR signaling pathways rewires the AR signaling requirements and thereby decreases the intrinsic need for androgens to fuel PCa growth, which can contribute to castration resistance.88
In addition, genetic and pharmacologic studies have demonstrated that inhibition of either the PI3K-AKT-mTOR or AR signaling axes can drive reciprocal activation of the other pathway. Genetic loss of AR or treatment with the AR inhibitor enzalutamide in a mouse model of PCa driven by PTEN loss enhances AKT signaling through downregulation of FKBP5, leading to a reduction in levels of PHLPP, a negative regulator of AKT signaling. (Figure 1c).26,27 Thus, a decrease in AR levels or activity reciprocally enhances AKT signaling through downregulation of PHLPP. On the other hand, mTOR inhibition in the background of PTEN loss leads to an increase in AR levels through upregulation of HER3, which increases AR stability (Figure 1d).27 These finding demonstrate multiple avenues by which the PI3K-AKT-mTOR signaling pathway interacts with the androgen signaling axis in response to pathway inhibition. The clinical application of these findings is that the PI3K-AKT-mTOR and AR signaling pathways can compensate for each other in the setting of therapeutic inhibition of either pathway alone in PCa. Thus, the PI3K-AKT-mTOR signaling pathway drives a resistance mechanism to ADT that is remarkably wired into its association with the AR signaling axis. This concept is supported by the findings that androgen inhibition actually accelerates progression to invasive PCa in PTEN-deficient mice.89 As such, these studies support combinatorial inhibition of AR and PI3K-AKT-mTOR signaling as a therapeutic modality to prevent castration resistance. To this end, it has been shown in murine models that by targeting both signaling axes, progression to castration resistance is significantly delayed.90
TARGETING THE PI3K-AKT-mTOR SIGNALING AXIS
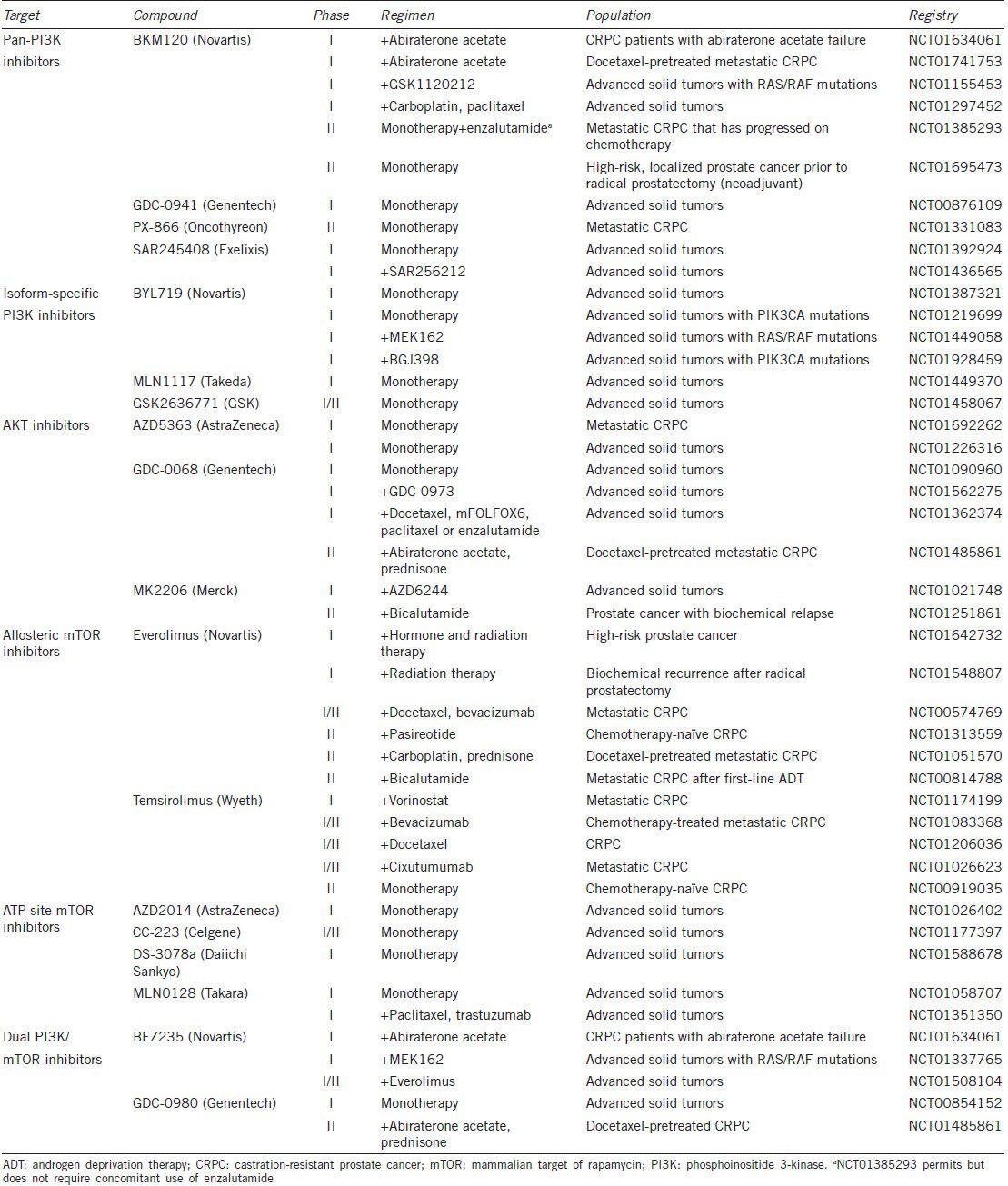
A number of pharmacologic agents have shown efficacy in CRPC over the past decade, including the immunotherapy sipuleucel-T,91 the taxane cabazitaxel,92 the targeted radiotherapy radium-223 dichloride,93 the adrenal androgen synthesis inhibitor abiraterone acetate17,19 and the second generation AR inhibitor enzalutamide.94 Despite these advances, long-term survival rates remain low for patients with CRPC, highlighting the need to consider alternative approaches. As described above, the PI3K-AKT-mTOR pathway plays an important role in the development of ADT resistance. Multiple clinical trials are underway to determine the efficacy of specific pathway inhibitors alone and in combination with inhibitors of androgen signaling. It remains to be seen whether monotherapy and combination therapy will be effective against metastatic CRPC. Here we will discuss the therapeutic profiles and potential use of a number of PI3K-AKT-mTOR pathway inhibitors currently being tested in the clinic in advanced solid cancers as well as PCa (Table 1).
Table 1.
Active trials with PI3K pathway inhibitors in prostate cancer and solid tumor malignancies
PI3K inhibitors
There are two types of PI3K inhibitors: pan-PI3K and isoform-specific inhibitors. Pan-PI3K inhibitors target the catalytic subunits of all isoforms of class IA PI3K as well as class IB p110γ. In preclinical studies, pan-PI3K inhibitor BKM120 exhibited cytostatic effects against a variety of cell lines and blocked tumor growth in xenograft mice harboring androgen-insensitive PC3 PCa cells.95 In a first-in-human, phase I trial of BKM120, the maximum tolerated dose was defined as 100 mg per day, with dose-limiting side effects including mood alteration, rash, hyperglycemia, diarrhea, nausea and fatigue.96 Out of 31 evaluable patients, one patient with triple-negative breast cancer had a confirmed partial response, and seven patients, including one with PCa, remained on treatment for ≥ 8 months.96 A phase I trial of another pan-PI3K inhibitor, PX-866, demonstrated signs of possible activity against advanced solid tumors in combination with docetaxel, including two out of 32 evaluable patients with a confirmed partial response and 22 with stable disease.97 Two other pan-PI3K inhibitors, GDC-0941 and SAR245408 have also been found to be well-tolerated and exhibit potential antitumor activity.98,99 Further studies are necessary to determine whether these agents are efficacious in patients with advanced CRPC. One limitation of pan-PI3K inhibitors is the possibility of a compensatory increase in AR signaling after stringent PI3K-mTOR inhibition, as has been observed in preclinical models.27 Therefore, it will be necessary to assess the efficacy of such drugs both in monotherapy as well as in combination therapy with second-line agents that target the AR signaling axis, such as abiraterone acetate and enzalutamide. Accordingly, BKM120 in combination with abiraterone acetate is currently in phase I clinical trials for men with metastatic CPRC (NCT01634061 and NCT01741753) and a phase II clinical trial that permits concomitant treatment with enzalutamide (NCT01385293).
In contrast to pan-PI3K inhibitors, isoform-specific PI3K inhibitors only target one p110 isoform. This greater specificity offers the possibility of reducing side effects, which would allow for a higher tolerated dose. Despite a narrower spectrum of drug activity, isoform-specific PI3K inhibitors are still promising given that 16% of metastatic PCas exhibit alterations to PIK3CA, the gene that encodes for p110α, at the gene and transcript levels.22 In preclinical studies, p110α isoform-specific PI3K inhibitors BYL719100 and MLN1117101 demonstrated antiproliferative and antitumor activity in cell line and xenograft models harboring PIK3CA mutations. In a phase I clinical trial in patients with advanced solid tumors containing PIK3CA mutations (none of which were PCa patients), BYL719 was well-tolerated up to 400 mg per day, with side effects including hyperglycemia, nausea, diarrhea, decreased appetite, vomiting and fatigue.102 Of the 39 patients on trial, seven exhibited partial responses and 17 remained on study for > 17 weeks.102 Despite these results, the efficacy of p110α inhibitors in PCa remains in question, due to data demonstrating that genetic ablation of p110β but not p110α inhibits tumorigenesis in a mouse model of PCa driven by PTEN loss.103 As such, p110β and not p110α may be the more relevant PI3K target in human PCa. Inhibitors of p110β, such as GSK2636771, are in clinical development, and a phase I/IIa, first-in-human study is currently underway in patients with PTEN-deficient advanced solid tumors (NCT01458067). The relevance of isoform-specific PI3K inhibitors will depend on whether individual isoforms of the p110 subunit serve as drivers of advanced PCa maintenance and whether unique isoforms can compensate for each other.
AKT inhibitors
As a critical signaling junction downstream of PI3K, AKT provides another clear target for blocking PI3K signaling. In this regard, a number of allosteric and ATP site inhibitors have been developed. Despite preclinical studies demonstrating reduced proliferation and increased apoptosis following treatment with the allosteric AKT inhibitor perifosine in PCa cell lines, the drug showed no benefit in clinical trials with CRPC patients.104,105 Though not yet tested clinically, there is a growing body of work demonstrating that ATP site inhibitors of AKT may have more potent antitumor effects. The active site inhibitor AZD5363 was found to inhibit proliferation and induce apoptosis in PCa cell lines and a LNCaP xenograft model.90 Furthermore, co-treatment with the AR inhibitor bicalutamide significantly delayed CRPC tumor progression in castrated mice with LNCaP xenografts, demonstrating possible efficacy of ATP site AKT inhibitors in combination therapy.90 One potential caveat of utilizing AKT inhibitors in CRPC is the finding that both allosteric and ATP site inhibitors of AKT can relieve feedback inhibition and activate multiple RTKs, attenuating their antitumor activity.106 It remains to be seen what effects this will have on the clinical efficacy of this drug class. Currently, two AKT inhibitors, MK2206 and GDC-0068, are in early phase clinical trials in combination with either bicalutamide or abiraterone acetate, respectively (NCT01251861 and NCT01485861).
Allosteric and ATP site mTOR inhibitors
As discussed above, allosteric mTOR inhibitors have demonstrated limited clinical efficacy in advanced PCa clinical trials.76,77,78 This lack of efficacy has been attributed to their inability to target rapamycin-resistant substrates such as 4EBP1. In addition, rapalogues also relieve S6K-mediated feedback inhibition of IRS-1 leading to activation of AKT, which is hypothesized to represent another mechanism that can dampen their therapeutic efficacy.107 A newer class of mTOR inhibitors now in clinical development, ATP site inhibitors, completely block mTORC1 and mTORC2 activity, preventing feedback induction of AKT and the phosphorylation of rapamycin-resistant substrates. In cell lines and a mouse model of PCa, the ATP site inhibitor MLN0128 and not the rapalogue everolimus resulted in a profound reduction in tumor size and invasive potential, demonstrating the potential therapeutic benefit of this class of drugs.84 The potency of MLN0128 was mediated in part by its ability to target the 4EBP1/eIF4E axis and modulate the translation of specific mRNAs involved in critical cellular processes important for tumor initiation and progression, including cell proliferation, metabolism and invasion.84 MLN0128, along with other ATP site inhibitors AZD2014, AZD8055, CC-223, DS-3078a and OSI-027, are currently in early-stage clinical trials in advanced solid tumors (NCT01026402, NCT00731263, NCT01177397, NCT01588678, NCT01058707, NCT01351350 and NCT00698243).
Dual PI3K/mTORC1/2 inhibitors
Dual PI3K/mTORC1/2 inhibitors target the ATP site of all p110 isoforms of PI3K as well as both mTOR complexes, thereby more completely inhibiting the PI3K-AKT-mTOR signaling axis. In preclinical studies, dual kinase inhibitors BEZ235 and GDC-0980 blocked proliferation in a variety of cancer cell lines, with the most notable inhibition in breast, prostate and lung cancer cells.108,109 In cell lines with hyperactivated PI3K signaling caused by PIK3CA mutation or PTEN loss, GDC-0980 also led to profound apoptosis.109 Accordingly, GDC-0980 significantly decreased tumor burden in PTEN null PC3 xenografts,109 and BEZ235 reduced tumor volume in a PTEN loss-driven murine model of PCa.27 In the same model, BEZ235 induced an even more striking effect in tumors when used in combination with the AR antagonist enzalutamide.27 These findings demonstrate potential synergy through co-targeting the AR, PI3K, and mTOR signaling pathways in PCa. In phase I clinical trials of patients with advanced solid tumors, both GDC-0980 and BEZ235 have been well-tolerated with side effects including nausea, diarrhea, vomiting, hyperglycemia and fatigue.110,111 Of the 51 evaluable patients on trial with BEZ235, two demonstrated partial responses and 14 had stable disease for ≥ 4 months.110 Currently, BEZ235 and GDC-0980 are both in phase I/II clinical trials for patients with metastatic CRPC, both as single agents as well as in combination with abiraterone acetate (NCT01634061 and NCT01485861).
THERAPEUTIC IMPLICATIONS AND FUTURE CONSIDERATIONS
The PI3K-AKT-mTOR signaling pathway is seated at a critical interface where intra- and extracellular signals directly impact vital cellular processes, which can be hijacked in the development of castration resistance. Despite initial challenges with targeting this signaling node in advanced PCa, the current movement to test a new arsenal of highly specific pathway inhibitors is warranted based on our understanding of PCa pathogenesis. However, there are important considerations to take into account if the PI3K-AKT-mTOR pathway is to be properly exploited in the treatment of men with PCa.
Perhaps the most significant impediment to accurately targeting the PI3K-AKT-mTOR signaling pathway is the paucity of companion biomarkers that can identify patients who will respond to these types of therapies. For years, genetic mutations, gene expression signatures and phosphorylation of pathway constituents have been studied in this context, but have been met with limited success. For example, phosphorylation of ribosomal protein S6 has been frequently utilized as a read out of mTOR activity as a correlative measure of pathway inhibition in many rapalogue-based clinical trials. However, it was shown in PCa patients that despite achieving significant inhibition of ribosomal protein S6 phosphorylation, there was no association with any effect on tumor proliferation and apoptosis.77 This example highlights the need for new biomarkers. One consideration is that the field needs to move beyond DNA, RNA and phosphorylation-based markers. This is particularly relevant to the PI3K-AKT-mTOR signaling pathway because of its central role in regulating protein synthesis, the end product of gene expression. There are emerging technologies such as ribosome profiling that can now be employed to determine at a genome-wide level changes in mRNA translation.84,112 Ribosome profiling provides codon-based resolution of mRNA translation, which represents a significant advancement over first generation technologies for assessing global changes in protein synthesis such as microarray-based polysome profiling. This new technology has already been used to identify a functionally important translationally regulated gene signature downstream of mTOR that promotes PCa invasion and metastasis.84 Importantly, this signature would not have been identified through traditional DNA and RNA-based whole-genome sequencing platforms. Thus, the protein levels of functionally important translationally regulated genes may represent a yet untapped repository of companion biomarkers for PI3K-AKT-mTOR inhibitors which remain to be tested clinically.
In addition to the need for biomarkers, another issue is to identify the optimal clinical setting to apply PI3K pathway inhibitors in PCa. Currently, most clinical trials with these agents are targeted for patients who have already developed castration resistance (Table 1). However, the preclinical evidence suggests that the PI3K-AKT-mTOR signaling pathway may be required for the development of CRPC26 and that co-targeting the AR and the PI3K pathway may delay the development of ADT resistance.90 Thus, if the toxicity profiles are tolerable, it is worthwhile considering studies in metastatic hormone-sensitive PCa patients to determine if these agents can delay or even prevent CRPC development.
Another important consideration in targeting the PI3K-AKT-mTOR signaling pathway is the issue of resistance mechanisms, which may compensate for the inhibitory effects of these agents. For instance, it has been shown that ATP site inhibition of mTOR relieves feedback inhibition of upstream receptor tyrosine kinases leading to subsequent PI3K activity and partial AKT reactivation.113 Moreover, others have shown that the cellular context of a cancer cell can represent a resistance mechanism to PI3K pathway inhibition. In particular, cancer cells that are attached to extracellular matrix as opposed to those that are not may be specifically protected from the deleterious effects of PI3K-AKT-mTOR pathway inhibition through compensatory signaling mechanisms associated with attachment to the extracellular matrix.114 However, the clinical relevance of these feedback mechanisms in PCa patients remains to be determined, and an effort should be made to incorporate correlative studies into current clinical trials to address these concerns.
Lastly, in the era of highly potent AR and adrenal androgen synthesis inhibitors, there is evidence that selective pressures placed on PCa cells by these agents are leading to a fundamental change in the phenotype of PCa in some patients. In particular, we are witnessing the emergence of treatment-related neuroendocrine PCa (t-NEPC) in patients treated with highly active AR-based therapeutics.115 The mechanisms that govern t-NEPC development remain to be determined; however, it is currently hypothesized that t-NEPCs are prostate adenocarcinomas that have differentiated to exhibit neuroendocrine features.116 Unlike adenocarcinoma, t-NEPC is typically AR-negative and highly refractory to extreme androgen deprivation. Platinum and taxane based agents remain the primary therapeutics against this form of PCa, which is uniformly fatal. Given the role of PI3K-AKT-mTOR signaling in cellular differentiation, it is interesting to speculate about the impact that targeting the PI3K signaling pathway will have on the development of this emerging PCa phenotype.
The PI3K signaling pathway plays an important role in PCa progression and the development of castration resistance. The clinical studies described here will be important in ultimately determining the efficacy of targeting aberrant PI3K-AKT-mTOR signaling in PCa progression. As outlined above, significant challenges remain; however, beyond these challenges is the future possibility of clinically oriented scientific breakthroughs that could impact prostate cancer patient survival.
COMPETING INTERESTS
Authors declare no competing interests.
ACKNOWLEDGEMENTS
We thank Dr. Davide Ruggero, Dr. Lawrence Fong, Dr. Rahul Aggarwal, Dr. John T Cunningham, and Morgan Truitt for input and/or critical reading of this review. We also thank Dr. Jennifer M Shen for editing the manuscript. We apologize to the many scientists whose work we were unable to cite. A.C.H. is supported by a Burroughs Wellcome Fund Career Award for Medical Scientists and by NIH 1K08CA175154-01.
REFERENCES
- 1.Jemal A, Bray F, Center MM, Ferlay J, Ward E, et al. Global cancer statistics. CA Cancer J Clin. 2011;61:69–90. doi: 10.3322/caac.20107. [DOI] [PubMed] [Google Scholar]
- 2.Siegel R, DeSantis C, Virgo K, Stein K, Mariotto A, et al. Cancer treatment and survivorship statistics, 2012. CA Cancer J Clin. 2012;62:220–41. doi: 10.3322/caac.21149. [DOI] [PubMed] [Google Scholar]
- 3.Antonarakis ES, Feng Z, Trock BJ, Humphreys EB, Carducci MA, et al. The natural history of metastatic progression in men with prostate-specific antigen recurrence after radical prostatectomy: long-term follow-up. BJU Int. 2012;109:32–9. doi: 10.1111/j.1464-410X.2011.10422.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Caire AA, Sun L, Ode O, Stackhouse DA, Maloney K, et al. Delayed prostate-specific antigen recurrence after radical prostatectomy: how to identify and what are their clinical outcomes? Urology. 2009;74:643–7. doi: 10.1016/j.urology.2009.02.049. [DOI] [PubMed] [Google Scholar]
- 5.Eisenberger MA, Blumenstein BA, Crawfore ED, Miller G, MacLeod DG, et al. Bilateral orchiectomy with or without flutamide for metastatic prostate cancer. N Engl J Med. 1998;339:1036–42. doi: 10.1056/NEJM199810083391504. [DOI] [PubMed] [Google Scholar]
- 6.Tannock IF, de Wit R, Berry WR, Horti J, Pluzanska A, et al. Docetaxel plus Prednisone or Mitoxantrone plus Prednisone for advanced prostate cancer. N Engl J Med. 2004;351:1502–12. doi: 10.1056/NEJMoa040720. [DOI] [PubMed] [Google Scholar]
- 7.Chen CD, Welsbie DS, Tran C, Baek SH, Chen R, et al. Molecular determinants of resistance to antiandrogen therapy. Nat Med. 2004;10:33–9. doi: 10.1038/nm972. [DOI] [PubMed] [Google Scholar]
- 8.Wang Q, Li W, Zhang Y, Yuan X, Xu K, et al. Androgen receptor regulates a distinct transcription program in androgen-independent prostate cancer. Cell. 2009;138:245–56. doi: 10.1016/j.cell.2009.04.056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Yang X, Guo Z, Sun F, Li W, Alfano A, et al. Novel membrane-associated androgen receptor splice variant potentiates proliferative and survival responses in prostate cancer cells. J Biol Chem. 2011;286:36152–60. doi: 10.1074/jbc.M111.265124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lunardi A, Ala U, Epping MT, Salmena L, Clohessy JG, et al. A co-clinical approach identifies mechanisms and potential therapies for androgen deprivation resistance in prostate cancer. Nat Genet. 2013;45:747–55. doi: 10.1038/ng.2650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Matias PM, Carrondo MA, Coelho R, Thomaz M, Zhao XY, et al. Structural basis for the glucocorticoid response in a mutant human androgen receptor (ARccr) derived from an androgen-independent prostate cancer. J Med Chem. 2002;45:1439–46. doi: 10.1021/jm011072j. [DOI] [PubMed] [Google Scholar]
- 12.Veldscholte J, Ris-Stalpers C, Kuiper GG, Jenster G, Berrevoets C, et al. A mutation in the ligand binding domain of the androgen receptor of human LNCaP cells affects steroid binding characteristics and response to anti-androgens. Biochem Biophys Res. 1990;173:534–40. doi: 10.1016/s0006-291x(05)80067-1. [DOI] [PubMed] [Google Scholar]
- 13.Edwards J, Krishna NS, Grigor KM, Bartlett JM. Androgen receptor gene amplification and protein expression in hormone refractory prostate cancer. Br J Cancer. 2003;89:552–6. doi: 10.1038/sj.bjc.6601127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zhang X, Morrissey C, Sun S, Ketchandji M, Nelson PS, et al. Androgen receptor variants occur frequently in castration resistant prostate cancer metastases. PLoS One. 2011;6:e27970. doi: 10.1371/journal.pone.0027970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Stanbrough M, Bubley GJ, Ross K, Golub TR, Rubin MA, et al. Increased expression of genes converting adrenal androgens to testosterone in androgen-independent prostate cancer. Cancer Res. 2006;66:2815–25. doi: 10.1158/0008-5472.CAN-05-4000. [DOI] [PubMed] [Google Scholar]
- 16.Chang KH, Li R, Kuri B, Lotan Y, Roehrborn CG, et al. A gain-of-function mutation in DHT synthesis in castration-resistant prostate cancer. Cell. 2013;154:1074–84. doi: 10.1016/j.cell.2013.07.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ryan CJ, Smith MR, de Bono JS, Molina A, Logothetis CJ, et al. Abiraterone in metastatic prostate cancer without previous chemotherapy. N Engl J Med. 2013;368:138–48. doi: 10.1056/NEJMoa1209096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Tran C, Ouk S, Clegg NJ, Chen Y, Watson PA, et al. Development of a second-generation antiandrogen for treatment of advanced prostate cancer. Science. 2009;324:787–90. doi: 10.1126/science.1168175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.de Bono JS, Logothetis CJ, Molina A, Fizazi K, North S, et al. Abiraterone and increased survival in metastatic prostate cancer. N Engl J Med. 2011;364:1995–2005. doi: 10.1056/NEJMoa1014618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Rathkopf DE, Morris MJ, Fox JJ, Danila DC, Slovin SF, et al. Phase I study of ARN-509, a novel antiandrogen, in the treatment of castration-resistant prostate cancer. J Clin Oncol. 2013;31:3525–30. doi: 10.1200/JCO.2013.50.1684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bitting RL, Armstrong AJ. Targeting the PI3K/Akt/mTOR pathway in castration-resistant prostate cancer. Endocr Relat Cancer. 2013;20:R83–99. doi: 10.1530/ERC-12-0394. [DOI] [PubMed] [Google Scholar]
- 22.Taylor BS, Schultz N, Hieronymus H, Gopalan A, Xiao Y, et al. Integrative genomic profiling of human prostate cancer. Cancer Cell. 2010;18:11–22. doi: 10.1016/j.ccr.2010.05.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Manning BD, Cantley LC. AKT/PKB signaling: navigating downstream. Cell. 2007;129:1261–74. doi: 10.1016/j.cell.2007.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Engelman JA, Luo J, Cantley LC. The evolution of phosphatidylinositol 3-kinases as regulators of growth and metabolism. Nat Rev Genet. 2006;7:606–19. doi: 10.1038/nrg1879. [DOI] [PubMed] [Google Scholar]
- 25.Laplante M, Sabatini DM. mTOR signaling in growth control and disease. Cell. 2012;149:274–93. doi: 10.1016/j.cell.2012.03.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Mulholland DJ, Tran LM, Li Y, Cai H, Morim A, et al. Cell autonomous role of PTEN in regulating castration-resistant prostate cancer growth. Cancer Cell. 2011;19:792–804. doi: 10.1016/j.ccr.2011.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Carver BS, Chapinski C, Wongvipat J, Hieronymus H, Chen Y, et al. Reciprocal feedback regulation of PI3K and androgen receptor signaling in PTEN-deficient prostate cancer. Cancer Cell. 2011;19:575–86. doi: 10.1016/j.ccr.2011.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Skolnik EY, Margois B, Mohammadi M, Lowenstein E, Fischer R, et al. Cloning of PI3 kinase-associated p85 utilizing a novel method for expression/cloning of target proteins for receptor tyrosine kinases. Cell. 1991;65:83–90. doi: 10.1016/0092-8674(91)90410-z. [DOI] [PubMed] [Google Scholar]
- 29.Carpenter CL, Auger KR, Chanudhuri M, Yoakim M, Schaffhausen B, et al. Phosphoinositide 3-Kinase is activated by phosphopeptides that bind to the SH2 domains of the 85-kDa subunit. J Biol Chem. 1993;268:9478–83. [PubMed] [Google Scholar]
- 30.Stephens L, Smrcka A, Cooke FT, Jackson TR, Sternweis PC, et al. A novel phosphoinositide 3 kinase activity in myeloid-derived cells is activated by G protein beta-gamma subunits. Cell. 1994;77:83–93. doi: 10.1016/0092-8674(94)90237-2. [DOI] [PubMed] [Google Scholar]
- 31.Chan TO, Rodeck U, Chan AM, Kimmelman AC, Rittenhouse SE, et al. Small GTPases and tyrosine kinases coregulate a molecular switch in the phosphoinositide 3-kinase regulatory subunit. Cancer Cell. 2002;1:181–91. doi: 10.1016/s1535-6108(02)00033-8. [DOI] [PubMed] [Google Scholar]
- 32.Rodriguez-Viciana P, Warne PH, Dhand R, Vanhaesebroeck B, Gout I, et al. Phosphatidylinositol-3-OH kinase as a direct target of Ras. Nature. 1994;370:527–32. doi: 10.1038/370527a0. [DOI] [PubMed] [Google Scholar]
- 33.Rodriguez-Viciana P, Warne PH, Vanhaesebroeck B, Waterfield MD, Downward J. Activation of phosphoinositide 3-kinase by interaction with Ras and by point mutation. EMBO J. 1996;15:2442–51. [PMC free article] [PubMed] [Google Scholar]
- 34.Lemmon MA. Pleckstrin homology (PH) domains and phosphoinositides. Biochem Soc Symp. 2007;74:81–93. doi: 10.1042/BSS0740081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Gewinner C, Wang ZC, Richardson A, Teruya-Feldstein J, Etemadmoghadam D, et al. Evidence that inositol polyphosphate 4-phosphatase type II is a tumor suppressor that inhibits PI3K signaling. Cancer Cell. 2009;16:115–25. doi: 10.1016/j.ccr.2009.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Maehama T, Dixon JE. The tumor suppressor, PTEN/MMAC1, dephosphorylates the lipid second messenger, phosphatidylinositol 3,4,5-trisphosphate. J Biol Chem. 1998;273:13375–8. doi: 10.1074/jbc.273.22.13375. [DOI] [PubMed] [Google Scholar]
- 37.Li J, Yen C, Liaw D, Podsypanina K, Bose S, et al. PTEN, a putative protein tyrosine phosphatase gene mutated in human brain, breast, and prostate cancer. Science. 1997;275:1943–7. doi: 10.1126/science.275.5308.1943. [DOI] [PubMed] [Google Scholar]
- 38.Majumder PK, Yeh JJ, George DJ, Febbo PG, Kum J, et al. Prostate intraepithelial neoplasia induced by prostate restricted Akt activation: The MPAKT model. Proc Natl Acad Sci U S A. 2003;100:7841–6. doi: 10.1073/pnas.1232229100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sarbassov DD, Guertin DA, Ali SM, Sabatini DM. Phosphorylation and regulation of Akt/PKB by the rictor-mTOR complex. Science. 2005;307:1098–101. doi: 10.1126/science.1106148. [DOI] [PubMed] [Google Scholar]
- 40.Alessi DR, James SR, Downes CP, Holmes AB, Gaffney PR, et al. Characterization of a 3-phosphoinositide-dependent protein kinase which phosphorylates and activates protein kinase B alpha. Curr Biol. 1997;7:261–9. doi: 10.1016/s0960-9822(06)00122-9. [DOI] [PubMed] [Google Scholar]
- 41.Kovacina KS, Park GY, Bae SS, Guzzetta AW, Schaefer E, et al. Identification of a proline-rich Akt substrate as a 14-3-3 binding partner. J Biol Chem. 2003;278:10189–94. doi: 10.1074/jbc.M210837200. [DOI] [PubMed] [Google Scholar]
- 42.Sancak Y, Thoreen CC, Peterson TR, Lindquist RA, Kang SA, et al. PRAS40 is an insulin-regulated inhibitor of the mTORC1 protein kinase. Mol Cell. 2007;25:903–15. doi: 10.1016/j.molcel.2007.03.003. [DOI] [PubMed] [Google Scholar]
- 43.Vander Haar E, Lee SI, Bandhakavi S, Griffin TJ, Kim DH. Insulin signalling to mTOR mediated by the Akt/PKB substrate PRAS40. Nat Cell Biol. 2007;9:316–23. doi: 10.1038/ncb1547. [DOI] [PubMed] [Google Scholar]
- 44.Oshiro N, Takahashi R, Yoshino K, Tanimura K, Nakashima A, et al. The proline-rich Akt substrate of 40 kDa (PRAS40) is a physiological substrate of mammalian target of rapamycin comple×1. J Biol Chem. 2007;282:20329–39. doi: 10.1074/jbc.M702636200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Gingras AC, Kennedy SG, O’Leary MA, Sonenberg N, Hay N. 4E-BP1, a repressor of mRNA translation, is phosphorylated and inactivated by the Akt (PKB) signaling pathway. Genes Dev. 1998;12:502–13. doi: 10.1101/gad.12.4.502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Brown EJ, Beal PA, Keith CT, Chen J, Shin TB, et al. Control of p70 S6 kinase by kinase activity of FRAP in vivo. Nature. 1995;377:441–6. doi: 10.1038/377441a0. [DOI] [PubMed] [Google Scholar]
- 47.Jung CH, Jun CB, Ro SH, Kim YM, Otto NM, et al. ULK-Atg13-FIP200 complexes mediate mTOR signaling to the autophagy machinery. Mol Biol Cell. 2009;20:1992–2003. doi: 10.1091/mbc.E08-12-1249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Hosokawa N, Hara T, Kaizuka T, Kishi C, Takamura A, et al. Nutrient-dependent mTORC1 association with the ULK1-Atg13-FIP200 complex required for autophagy. Mol Biol Cell. 2009;20:1981–91. doi: 10.1091/mbc.E08-12-1248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ganley IG, Lam du H, Wang J, Ding X, Chen S, et al. ULK1-ATG13-FIP200 complex mediates mTOR signaling and is essential for autophagy. J Biol Chem. 2009;284:12297–305. doi: 10.1074/jbc.M900573200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Settembre C, Zoncu R, Medina DL, Vetrini F, Erdin S, et al. A lysosome-to-nucleus signalling mechanism senses and regulates the lysosome via mTOR and TFEB. EMBO J. 2012;31:1095–108. doi: 10.1038/emboj.2012.32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Hsu PP, Kang SA, Rameseder J, Zhang Y, Ottina KA, et al. The mTOR-regulated phosphoproteome reveals a mechanism of mTORC1-mediated inhibition of growth factor signaling. Science. 2011;332:1317–22. doi: 10.1126/science.1199498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Yu Y, Yoon SO, Poulogiannis G, Yang Q, Ma XM, et al. Phosphoproteomic analysis identifies Grb10 as an mTORC1 substrate that negatively regulates insulin signaling. Science. 2011;332:1322–6. doi: 10.1126/science.1199484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Huang J, Dibble CC, Matsuzaki M, Manning BD. The TSC1-TSC2 complex is required for proper activation of mTOR comple×2. Mol Cell Biol. 2008;28:4104–5. doi: 10.1128/MCB.00289-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Huang J, Wu S, Wu CL, Manning BD. Signaling events downstream of mammalian target of rapamycin comple×2 are attenuated in cells and tumors deficient for the tuberous sclerosis complex tumor suppressors. Cancer Res. 2009;69:6107–14. doi: 10.1158/0008-5472.CAN-09-0975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Dibble CC, Asara JM, Manning BD. Characterization of Rictor phosphorylation sites reveals direct regulation of mTOR comple×2 by S6K1. Mol Cell Biol. 2009;29:5657–70. doi: 10.1128/MCB.00735-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Julien LA, Carriere A, Moreau J, Roux PP. mTORC1-activated S6K1 phosphorylates Rictor on threonine 1135 and regulates mTORC2 signaling. Mol Cell Biol. 2010;30:908–21. doi: 10.1128/MCB.00601-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Sun H, Lesche R, Li D, Liliental J, Zhang H, et al. PTEN modulates cell cycle progression and cell survival by regulating phosphatidylinositol 3,4,5,-triphosphate and Akt/protein kinase B signaling pathway. Proc Natl Acad Sci U S A. 1999;96:6199–204. doi: 10.1073/pnas.96.11.6199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Datta SR, Dudek H, Tao X, Masters S, Fu H, et al. Akt phosphorylation of BAD couples survival signals to the cell-intrinsic death machinery. Cell. 1997;91:231–41. doi: 10.1016/s0092-8674(00)80405-5. [DOI] [PubMed] [Google Scholar]
- 59.Humbert S, Bryson EA, Cordelieres FP, Connors NC, Datta SR, et al. The IGF-1/Akt pathway is neuroprotective in Huntington's Disease and involves Huntingtin phosphorylation by Akt. Developmental Cell. 2002;2:831–7. doi: 10.1016/s1534-5807(02)00188-0. [DOI] [PubMed] [Google Scholar]
- 60.Datta SR, Katsov A, Hu L, Petros A, Fesik SW, et al. 14-3-3 proteins and survival kinases cooperate to inactivate BAD by BH3 domain phosphorylation. Mol Cell. 2000;6:41–51. [PubMed] [Google Scholar]
- 61.Kalaitzidis D, Sykes SM, Wang Z, Punt N, Tang Y, et al. mTOR comple×1 plays critical roles in hematopoiesis and Pten-loss-evoked leukemogenesis. Cell Stem Cell. 2012;11:429–39. doi: 10.1016/j.stem.2012.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Dowling RJ, Topisirovic I, Alain T, Bidinosti M, Fonseca BD, et al. mTORC1-mediated cell proliferation, but not cell growth, controlled by the 4E-BPs. Science. 2010;328:1172–6. doi: 10.1126/science.1187532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Yuan TL, Cantley LC. PI3K pathway alterations in cancer: variations on a theme. Oncogene. 2008;27:5497–510. doi: 10.1038/onc.2008.245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.McMenamin ME, Soung P, Perera S, Kaplan I, Loda M, et al. Loss of PTEN expression in paraffin-embedded primary prostate cancer correlates with high gleason score and advanced stage. Cancer Res. 1999;59:4291–6. [PubMed] [Google Scholar]
- 65.Ayala G, Thompson T, Yang G, Frolov A, Li R, et al. High levels of phosphorylated form of Akt-1 in prostate cancer and non-neoplastic prostate tissues are strong predictors of biochemical recurrence. Clin Cancer Res. 2004;10:6572–8. doi: 10.1158/1078-0432.CCR-04-0477. [DOI] [PubMed] [Google Scholar]
- 66.Kreisber JI, Malik SN, Prihoda TJ, Bedolla RG, Troyer DA, et al. Phosphorylation of Akt (Ser473) is an excellent predictor of poor clinical outcome in prostate cancer. Cancer Res. 2004;64:5232–6. doi: 10.1158/0008-5472.CAN-04-0272. [DOI] [PubMed] [Google Scholar]
- 67.Yoshimoto M, Cunha IW, Coudry RA, Fonseca FP, Torres CH, et al. FISH analysis of 107 prostate cancers shows that PTEN genomic deletion is associated with poor clinical outcome. Br J Cancer. 2007;97:678–85. doi: 10.1038/sj.bjc.6603924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Lotan TL, Gurel B, Sutcliffe S, Esopi D, Liu W, et al. PTEN protein loss by immunostaining: Analytic validation and prognostic indicator for a high risk surgical cohort of prostate cancer patients. Clin Cancer Res. 2011;17:6563–73. doi: 10.1158/1078-0432.CCR-11-1244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Graff JR, Konicek BW, Lynch RL, Dumstorf CA, Dowless MS, et al. eIF4E activation is commonly elevated in advanced human prostate cancers and significantly related to reduced patient survival. Cancer Res. 2009;69:3866–73. doi: 10.1158/0008-5472.CAN-08-3472. [DOI] [PubMed] [Google Scholar]
- 70.Wang S, Gao J, Lei Q, Rozengurt N, Pritchard C, et al. Prostate-specific deletion of the murine Pten tumor suppressor gene leads to metastatic prostate cancer. Cancer Cell. 2003;4:209–21. doi: 10.1016/s1535-6108(03)00215-0. [DOI] [PubMed] [Google Scholar]
- 71.Trotman LC, Niki M, Dotan ZA, Koutcher JA, Di Cristofano A, et al. Pten dose dictates cancer progression in the prostate. PLoS Biol. 2003;1:E59. doi: 10.1371/journal.pbio.0000059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Nardella C, Carracedo A, Alimonti A, Hobbs RM, Clohessy JG, et al. Differential requirement of mTOR in postmitotic tissues and tumorigenesis. Sci Signal. 2009;2:ra2. doi: 10.1126/scisignal.2000189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Guertin DA, Stevens DM, Saitoh M, Kinkel S, Crosby K, et al. mTOR comple×2 is required for the development of prostate cancer induced by Pten loss in mice. Cancer Cell. 2009;15:148–59. doi: 10.1016/j.ccr.2008.12.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Kinkade CW, Castillo-Martin M, Puzio-Kuter A, Yan J, Foster TH, et al. Targeting AKT/mTOR and ERK MAPK signaling inhibits hormone-refractory prostate cancer in a preclinical mouse model. J Clin Invest. 2008;118:3051–64. doi: 10.1172/JCI34764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Majumder PK, Febbo PG, Bikoff R, Berger R, Xue Q, et al. mTOR inhibition reverses Akt-dependent prostate intraepithelial neoplasia through regulation of apoptotic and HIF-1-dependent pathways. Nat Med. 2004;10:594–601. doi: 10.1038/nm1052. [DOI] [PubMed] [Google Scholar]
- 76.Nakabayashi M, Werner L, Courtney KD, Buckle G, Oh WK, et al. Phase II trial of RAD001 and bicalutamide for castration-resistant prostate cancer. BJU Int. 2012;110:1729–35. doi: 10.1111/j.1464-410X.2012.11456.x. [DOI] [PubMed] [Google Scholar]
- 77.Armstrong AJ, Netto GJ, Rudek MA, Halabi S, Wood DP, et al. A pharmacodynamic study of rapamycin in men with intermediate- to high-risk localized prostate cancer. Clin Cancer Res. 2010;16:3057–66. doi: 10.1158/1078-0432.CCR-10-0124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Templeton AJ, Dutoit V, Cathomas R, Rothermundt C, Bartschi D, et al. Phase 2 trial of single-agent everolimus in chemotherapy-naive patients with castration-resistant prostate cancer (SAKK 08/08) Eur Urol. 2013;64:150–8. doi: 10.1016/j.eururo.2013.03.040. [DOI] [PubMed] [Google Scholar]
- 79.Yip CK, Mrata K, Walz T, Sabatini DM, Kang SA. Structure of the human mTOR complex I and its implications for rapamycin inhibition. Mol Cell. 2010;38:768–74. doi: 10.1016/j.molcel.2010.05.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Yang H, Rudge DG, Koos JD, Vaidialingam B, Yang HJ, et al. mTOR kinase structure, mechanism and regulation. Nature. 2013;497:217–23. doi: 10.1038/nature12122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Choo AY, Yoon SO, Kim SG, Roux PP, Blenis J. Rapamycin differentially inhibits S6Ks and 4E-BP1 to mediate cell-type-specific repression of mRNA translation. Proc Natl Acad Sci U S A. 2008;105:17414–9. doi: 10.1073/pnas.0809136105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Kang SA, Pacold ME, Cervantes CL, Lim D, Lou HJ, et al. mTORC1 phosphorylation sites encode their sensitivity to starvation and rapamycin. Science. 2013;341:1236566. doi: 10.1126/science.1236566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Benjamin D, Colombi M, Moroni C, Hall MN. Rapamycin passes the torch: A new generation of mTOR inhibitors. Nat Rev Drug Discov. 2011;10:868–80. doi: 10.1038/nrd3531. [DOI] [PubMed] [Google Scholar]
- 84.Hsieh AC, Liu Y, Edlind MP, Ingolia NT, Janes MR, et al. The translational landscape of mTOR signalling steers cancer initiation and metastasis. Nature. 2012;485:55–61. doi: 10.1038/nature10912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Feldman ME, Apsel B, Uotila A, Loewith R, Knight ZA, et al. Active-site inhibitors of mTOR target rapamycin-resistant outputs of mTORC1 and mTORC2. PLoS Biol. 2009;7:e38. doi: 10.1371/journal.pbio.1000038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Thoreen CC, Kang SA, Chang JW, Liu Q, Zhang J, et al. An ATP-competitive mammalian target of rapamycin inhibitor reveals rapamycin-resistant functions of mTORC1. J Biol Chem. 2009;284:8023–32. doi: 10.1074/jbc.M900301200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Hsieh AC, Costa M, Zollo O, Davis C, Feldman ME, et al. Genetic dissection of the oncogenic mTOR pathway reveals druggable addiction to translational control via 4EBP-eIF4E. Cancer Cell. 2010;17:249–61. doi: 10.1016/j.ccr.2010.01.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Jiao J, Wang S, Qiao R, Vivanco I, Watson PA, et al. Murine cell lines derived from Pten null prostate cancer show the critical role of PTEN in hormone refractory prostate cancer development. Cancer Res. 2007;67:6083–91. doi: 10.1158/0008-5472.CAN-06-4202. [DOI] [PubMed] [Google Scholar]
- 89.Jia S, Gao X, Lee SH, Maira SM, Wu X, et al. Opposing effects of androgen deprivation and targeted therapy on prostate cancer prevention. Cancer Discov. 2013;3:44–51. doi: 10.1158/2159-8290.CD-12-0262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Thomas C, Lamoureux F, Crafter C, Davies BR, Beraldi E, et al. Synergistic targeting of PI3K/AKT-pathway and androgen-receptor axis significantly delays castration-resistant prostate cancer progression in vivo. Mol Cancer Ther. 2013;12:2342–55. doi: 10.1158/1535-7163.MCT-13-0032. [DOI] [PubMed] [Google Scholar]
- 91.Small EJ, Schellhammer PF, Higano CS, Redfern CH, Nemunaitis JJ, et al. Placebo-controlled phase III trial of immunologic therapy with sipuleucel-T (APC8015) in patients with metastatic, asymptomatic hormone refractory prostate cancer. J Clin Oncol. 2006;24:3089–94. doi: 10.1200/JCO.2005.04.5252. [DOI] [PubMed] [Google Scholar]
- 92.de Bono JS, Oudard S, Ozguroglu M, Hansen S, Machiels JP, et al. Prednisone plus cabazitaxel or mitoxantrone for metastatic castraction-resistant prostate cancer progressing after docetaxel treatment: a randomised open-label trial. Lancet. 2010;376:1147–54. doi: 10.1016/S0140-6736(10)61389-X. [DOI] [PubMed] [Google Scholar]
- 93.Parker C, Nilsson S, Heinrich D, Helle SI, O’Sullivan JM, et al. Alpha emitter radium-223 and survival in metastatic prostate cancer. N Engl J Med. 2013;369:213–23. doi: 10.1056/NEJMoa1213755. [DOI] [PubMed] [Google Scholar]
- 94.Scher HI, Fizazi K, Saad F, Taplin ME, Sternberg CN, et al. Increased survival with enzalutamide in prostate cancer after chemotherapy. N Engl J Med. 2012;367:1187–97. doi: 10.1056/NEJMoa1207506. [DOI] [PubMed] [Google Scholar]
- 95.Maira SM, Pecchi S, Huang A, Burger M, Knapp M, et al. Identification and characterization of NVP-BKM120, an orally available pan-class I PI3-kinase inhibitor. Mol Cancer Ther. 2012;11:317–28. doi: 10.1158/1535-7163.MCT-11-0474. [DOI] [PubMed] [Google Scholar]
- 96.Bendell JC, Rodon J, Burris HA, de Jonge M, Verweij J, et al. Phase I, dose-escalation study of BKM120, an oral pan-Class I PI3K inhibitor, in patients with advanced solid tumors. J Clin Oncol. 2012;30:282–90. doi: 10.1200/JCO.2011.36.1360. [DOI] [PubMed] [Google Scholar]
- 97.Jimeno A, Senzer NN, Rudin CM, Ma WW, Halmos B, et al. PX-866 and docetaxel in patients with advanced solid tumors. J Clin Oncol. 2012;30 abstract 3024. [Google Scholar]
- 98.Edelman G, Bedell C, Shapiro G, Pandya SS, Kwak EL, et al. A phase I dose-escalation study of XL147 (SAR245408), a PI3K inhibitor administered orally to patients (pts) with advanced malignancies. J Clin Oncol. 2010;28 abstract 3004. [Google Scholar]
- 99.Von Hoff DD, LoRusso P, Demetri GD, Weiss GJ, Shapiro G, et al. A phase I dose-escalation study to evaluate GDC-0941, a pan-PI3K inhibitor, administered QD or BID in patients with advanced or metastatic solid tumors. J Clin Oncol. 2011;29 abstract 3052. [Google Scholar]
- 100.Fritsch CM, Schnell C, Chatenay-Rivauday C, Guthy DA, De Pover A, et al. NVP-BYL719, a novel PI3Kalpha selective inhibitor with all the characteristics required for clinical development as an anti-cancer agent. Cancer Res. 2012;72 abstract 3748. [Google Scholar]
- 101.Jessen K, Kessler L, Kucharski J, Xin G, Staunton J, et al. A potent and selective PI3K inhibitor, INK1117, targets human cancers harboring oncogenic PIK3CA mutations. Mol Cancer Ther. 2011;10 abstract A171. [Google Scholar]
- 102.Gonzalez-Angulo AM, Juric D, Argiles G, Schellens JH, Burris HA, et al. Safety, pharmacokinetics, and preliminary activity of the a-specific PI3K inhibitor BYL719: results from the first-in-human study. J Clin Oncol. 2013;31 abstract 2531. [Google Scholar]
- 103.Jia S, Liu Z, Zhang S, Liu P, Zhang L, et al. Essential roles of PI(3)K-p110beta in cell growth, metabolism and tumorigenesis. Nature. 2008;454:776–9. doi: 10.1038/nature07091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Posadas EM, Gulley J, Arlen PM, Trout A, Parnes HL, et al. A phase II study of perifosine in androgen independent prostate cancer. Cancer Biol Ther. 2005;4:1133–7. doi: 10.4161/cbt.4.10.2064. [DOI] [PubMed] [Google Scholar]
- 105.Chee KG, Longmate J, Quinn DI, Chatta G, Pinski J, et al. The AKT inhibitor perifosine in biochemically recurrent prostate cancer: a phase II California/Pittsburgh cancer consortium trial. Clin Genitourin Cancer. 2007;5:433–7. doi: 10.3816/CGC.2007.n.031. [DOI] [PubMed] [Google Scholar]
- 106.Chandarlapaty S, Sawai A, Scaltriti M, Rodrik-Outmezguine V, Grbovic-Huezo O, et al. AKT inhibition relieves feedback suppression of receptor tyrosine kinase expression and activity. Cancer Cell. 2011;19:58–71. doi: 10.1016/j.ccr.2010.10.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.O’Reilly KE, Rojo F, She QB, Solit D, Mills GB, et al. mTOR inhibition induces upstream receptor tyrosine kinase signaling and activates Akt. Cancer Res. 2006;66:1500–8. doi: 10.1158/0008-5472.CAN-05-2925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Maira SM, Stauffer F, Brueggen J, Furet P, Schnell C, et al. Identification and characterization of NVP-BEZ235, a new orally available dual phosphatidylinositol 3-kinase/mammalian target of rapamycin inhibitor with potent in vivo antitumor activity. Mol Cancer Ther. 2008;7:1851–63. doi: 10.1158/1535-7163.MCT-08-0017. [DOI] [PubMed] [Google Scholar]
- 109.Wallin JJ, Edgar KA, Guan J, Berry M, Prior WW, et al. GDC-0980 is a novel class I PI3K/mTOR kinase inhibitor with robust activity in cancer models driven by the PI3K pathway. Mol Cancer Ther. 2011;10:2426–36. doi: 10.1158/1535-7163.MCT-11-0446. [DOI] [PubMed] [Google Scholar]
- 110.Burris H, Rodon J, Sharma S, Herbst RS, Tabernero J, et al. First-in-man phase I study of the oral dual PI3K and mTORC1/2 inhibitor BEZ235 in patients with advanced solid tumors. J Clin Oncol. 2010;28 abstract 3005. [Google Scholar]
- 111.Wagner AJ, Bendell JC, Dolly S, Morgan JA, Ware JA, et al. A first-in-human phase I study to evaluate GDC-0980, an oral PI3K/mTOR inhibitor, admistered QD in patients with advanced solid tumors. J Clin Oncol. 2011;29 abstract 3020. [Google Scholar]
- 112.Ingolia NT, Ghaemmaghami S, Newman JR, SWeissman JS. Genome-wide analysis in vivo of translation with nucleotide resolution using ribosome profiling. Science. 2009;324:218–23. doi: 10.1126/science.1168978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Rodrik-Outmezguine VS, Chandarlapaty S, Pagano NC, Poulikakos PI, Scaltriti M, et al. mTOR kinase inhibition causes feedback-dependent biphasic regulation of AKT signaling. Cancer Discov. 2011;1:248–59. doi: 10.1158/2159-8290.CD-11-0085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Muranen T, Selfors LM, Worster DT, Iwanicki MP, Song L, et al. Inhibition of PI3K/mTOR leads to adaptive resistance in matrix-attached cancer cells. Cancer Cell. 2012;21:227–39. doi: 10.1016/j.ccr.2011.12.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Beltran H, Tagawa ST, Park K, MacDonald T, Milowsky MI, et al. Challenges in recognizing treatment-related neuroendocrine prostate cancer. J Clin Oncol. 2012;30:e386–9. doi: 10.1200/JCO.2011.41.5166. [DOI] [PubMed] [Google Scholar]
- 116.Beltran H, Rickman DS, Park K, Chae SS, Sboner A, et al. Molecular characterization of neuroendocrine prostate cancer and identification of new drug targets. Cancer Discovery. 2011;1:487–95. doi: 10.1158/2159-8290.CD-11-0130. [DOI] [PMC free article] [PubMed] [Google Scholar]