

Abstract
BACKGROUND
Mutations in the PITX2 homeobox gene are known to contribute to Axenfeld-Rieger syndrome (ARS), an autosomal-dominant developmental disorder. Although most mutations are in the homeodomain and result in a loss of function, there is a growing subset in the C-terminal domain that has not yet been characterized. These mutations are of particular interest because the C-terminus has both inhibitory and stimulatory activities.
METHODS
In this study we used a combination of in vitro DNA binding and transfection reporter assays to ask the fundamental question of whether C-terminal mutations result in gain or loss of function at a cellular level.
RESULTS
We report a new frameshift mutation in the PITX2 allele that predicts a truncated protein lacking most of the C-terminal domain (D122FS). This newly reported mutant and another ARS C-terminal mutant (W133Stop) both have greater binding than wildtype to the bicoid element. Of interest, the mutants yielded ~5-fold greater activation of the prolactin promoter in CHO cells, even though the truncated proteins were expressed at lower levels than the wildtype protein. The truncated proteins also had greater than wildtype activity in 2 other cell lines, including the LS8 oral epithelial line that expresses the endogenous Pitx2 gene.
CONCLUSIONS
These results indicate that the PITX2 C-terminal domain has inhibitory activity and support the possibility that ARS might also be caused by gain-of-function mutations.
Keywords: Axenfeld-Rieger Syndrome, PITX2, homeodomain, transcription factor
INTRODUCTION
Axenfeld-Rieger syndrome (ARS) is an autosomal dominant disorder characterized by ocular anterior chamber, dental, and umbilical abnormalities (Semina et al., 1996; Amendt et al., 2000; Lines et al., 2002). Other occasional features associated with this syndrome include abnormal cardiac, limb, and pituitary development. Mutations in the transcription factors PITX2 and FOXC1 have been identified in patients who have ARS (Semina et al., 1996; Amendt et al., 2000; Lines et al., 2002). PITX2 is a bicoid type homeodomain protein. Three isoforms of PITX2 have been identified that differ only in their N-terminus and have been reported to have different or similar activities in various developmental and cellular systems (Tremblay et al., 2000; Schweickert et al., 2000; Essner et al. 2000; Smidt et al. 2000; Yu et al., 2001; Cox et al., 2002). In this study we focused on the PITX2a isoform.
To date, all PITX2 mutations delete the entire gene or occur in the homeodomain or C-terminal region, which we define as encompassing the entire coding region 3′ of the homeodomain (Amendt et al., 2000; Wang et al., 2002; Lines et al., 2004). Most of the PITX2 homeodomain mutations result in complete loss of function, although some mutants retain residual function (Kozlowski and Walter, 2000; Espinoza et al., 2002), and at least 1 mutant has dominant negative properties (Saadi et al., 2001). Of interest, a valine→leucine (V45L) homeodomain mutation yielded decreased DNA binding, yet increased transactivation in a cell-based assay (Priston et al., 2001). This observation first raised the possibility that increased PITX2 activity might underlie some cases of ARS. Within the C-terminal region, there are reports of 5 nonsense mutations and 4 missense mutations (Semina et al., 1996; Perveen et al., 2000; Philips, 2002; Brooks et al., 2004; Lines et al., 2004; Espinoza et al., 2005). However, there is only 1 reported characterization of a C-terminal mutant protein, which was a frameshift mutation in the distal C-terminus that removes 34 amino acids and yielded decreased activity (Espinoza et al., 2005).
The C-terminal region of PITX2 has multiple activities. Truncation of the C-terminal 39 amino acids resulted in increased DNA binding, yet reduced transactivation of target promoters in COS-7 and HeLa cells (Amendt et al., 1999). The C-terminal region is required for interaction with the Pit-1 transcription factor (Amendt et al., 1999), and PITX2 can synergistically activate the prolactin promoter with Pit-1 (Amendt et al., 1998). However, a physical interaction is not necessary for synergism (Saadi et al., 2001). PITX2 can form dimers (Amendt et al., 1999; Cox et al., 2002; Saadi et al., 2003), and the C-terminal region along with the homeodomain was required for dimerization in a yeast 2-hybrid assay (Saadi et al., 2003). The role of the C-terminal region in dimerization is illustrated by the enhancement of dimerization in the presence of Pit-1 or a peptide containing the C-terminal 39 residues (Amendt et al., 1999). Dimerization appears to involve both the C-terminal region and the homeodomain, as exemplified by the dominant negative PITX2 K88E protein, which cannot bind DNA, but does dimerize more strongly (Saadi et al., 2003). This dual role of DNA binding and protein interactions by the homeodomain has been documented for other proteins (Yuan et al., 1996; Burz and Hanes, 2001). To account for these activities, we have proposed that the PITX2 C-terminal tail loops back and inhibits DNA binding and that the inhibition can be relieved by protein-protein interactions (Amendt et al., 1999). In addition, the recent finding that the PITX2 C-terminus is phosphorylated raises another potential level of regulation of C-terminal activities (Espinoza et al., 2005). Thus, given the multifunctional nature of the C-terminal tail, we were intrigued as to whether C-terminal ARS mutations would be gain- or loss-of-function mutants.
In this study we characterized the DNA binding, transactivation, and dimerization abilities of two PITX2 ARS C-terminal truncation mutants. W133Stop eliminates the C-terminal 139 amino acids and D122FS introduces a shift in the reading frame that eliminates the C-terminal 150 amino acids. Unexpectedly, the mutants had greater than wildtype activity on the prolactin promoter. These results suggest that the C-terminal PITX2 mutants may have a gain of function on certain promoters.
MATERIALS AND METHODS
Cell culture and reporter gene assays
CHO (Chinese Hamster Ovary) cells were cultured in α-MEM, 10% fetal bovine serum, 300 µg/ml glutamine, 100 U/ml penicillin, and 100 µg/ml streptomycin. The mouse oral epithelial cell line LS8 was kindly provided by Dr. Malcolm Snead (University of Southern California, Los Angeles, CA). COS7 and LS8 cells were grown in DMEM, 10% fetal bovine serum, 100 units/ml penicillin, and 100 µg/ml streptomycin.
Approximately 1 × 105 CHO, COS7 or LS8 cells were cultured in 12-well plates and transfected with a total of 2 µg of DNA including 0.5 µg of prolactin-luciferase, and 0.5 µg of each expression vector (as indicated) using 4 µl of Lipofectamine 2000 (Invitrogen, Carlsbad, CA) for CHO and LS8 cells or 3 µl of Fugene 6 (Roche, Indianapolis, IN) for COS7 cells. The wildtype PITX2a, W133Stop and D122FS (including the encoding of 31 novel amino acids) cDNAs were cloned into pcDNA3.1 MycHisC expression vector (Invitrogen), which contains the cytomegalovirus (CMV) and T7 promoters and an in-frame C-terminal c-Myc epitope. The prolactin-luciferase reporter contains 2500 bp of the rat prolactin promoter (Maurer and Notides, 1987). CMV5 DNA (empty expression vector) was used to bring the total amount of DNA to 2 µg. An SV40 β-galactosidase plasmid (0.1 µg of pSV-βgal) (BD-Clontech, Palo Alto, CA) was used as a control. The DNA-transfection reagent mixture was added to cells growing in antibiotic-free media in the presence of serum and incubated for 16–20 h (CHO) or 32–48 h (LS8, COS7). Following incubation, the cells were scraped, lysed and assayed using luciferase assay reagents (Promega, Madison, WI) and β-galactosidase assay reagents (Tropix Inc., Bedford, MA). Luciferase activities were corrected for transfection efficiency. Statistical significance was calculated using a Student t test for means of 2 samples.
Electrophoretic mobility shift assay (EMSA)
The probe was generated by annealing complementary oligonucleotides containing the sequence gatccGCACGGCCCATCTAATCCCGTGggatc and end-filled using Klenow polymerase with 32P-dATP as described (Tverberg and Russo, 1993). Binding assays were carried out by incubating the probe (1.0 pmol) in a 20-µl reaction containing binding buffer (20 mM HEPES, pH 7.5, 5% glycerol, 50 mM NaCl, 1 mM dithiothreitol), 0.1 µg of poly (dI-dC), and 2–10 µl of TNT products or 0.5–2.0 µg of cell lysate (3–12 µl), or 100–300 ng of glutathione–S transferase (GST)–fusion proteins on ice for 15 min. In vitro synthesized proteins were generated by transcription and translation (TNT) reactions using 0.5–1 µg DNA, according to the manufacturer (Promega). Mammalian cell lysates were prepared as described above. GST-fusion proteins were prepared, and the GST moiety was removed by protease cleavage as previously described (Saadi et al., 2003). The samples were electrophoresed for 2.5 h at 280 V on 8% polyacrylamide gels as described (Amendt et al., 1998). Autoradiographs of EMSAs were scanned and band intensities were calculated with NIH Image software. The amount bound was normalized to the intensity of an aliquot of the input protein. Protein was measured by Bradford assays (BioRad, Hercules, CA) with a bovine serum albumin standard.
GST-pulldown and Western blots
For preparation of mammalian cell lysates, transfections were done using Lipofectamine 2000 (10 µl) in 60-mm dishes with 5 × 106 cells and 5 µg of Myc-PITX2a, Myc-W133Stop, or Myc-D122FS expression plasmids. Cells were incubated for 48 h after transfection, washed with PBS, and lysed with 1× lysis buffer (1% Triton X-100, 150 mM NaCl, 10 mM Tris pH 7.4, 1 mM EDTA, 0.2 mM PMSF, 0.1% NP-40, and 2% protease inhibitor cocktail (Sigma, St. Louis, MO) for 30 min at 4°C. Following lysis, cells were scraped, centrifuged, and the supernatant collected. Protein was measured by Bradford assays (BioRad) with a bovine serum albumin standard.
GST-alone or GST-PITX2a immobilized on glutathione sepharose beads (Amersham Pharmacia Biotech, Piscataway, NJ) were incubated with equal amounts of total lysate from CHO cells transfected with Myc-tagged PITX2a, W133Stop, or D122FS plasmid in a 100-µl volume in the presence of PBS and 300 ng/µl ethidium bromide for 15 min. Following incubation, the beads were centrifuged and washed 3 times with buffer containing 50 mM Tris (pH 7.0), and 140 mM NaCl. The beads were resuspended in 1 SDS sample buffer, boiled for 5 min, and loaded on a 12.5% SDS-polyacrylamide gel for electrophoresis. Proteins were transferred to polyvinylidine difluoride filters (Millipore, Bedford, MA). Western blotting was carried out using a c-Myc antibody (9E10, Santa Cruz Labs, Santa Cruz, CA) and ECL reagents (Amersham Pharmacia Biotech, Piscataway, NJ). Autoradiographs were scanned and band intensities were calculated with NIH Image software. The signal from the bound protein was normalized to the intensity of the input protein from the same blot.
Results
Identification of the D122FS mutation
We have identified a new PITX2 mutation in a classic ARS patient (family 707) (Fig. 1A). The proband at age 5 presented the characteristic ARS features, including absent incisors, peg-shaped teeth, maxillary hypoplasia, an elongated umbilicus with umbilical hernia, asymmetric pupils and megalocornea, and hearing loss. The father, who carries the same mutation, has glaucoma, maxillary hypoplasia, and partial absence of permanent teeth. Sequence analysis of the proband identified a deletion of a C at position 366 that shifts the reading frame (Fig. 1B). This results in a change in codon 122, which encodes an aspartate in wildtype. The numbering is relative to PITX2a (Semina et al., 1996). The mutant cDNA encodes a truncated protein of 153 amino acids that contains 121 amino acids of PITX2a (Fig. 1C). This frameshift mutant will be referred to as D122FS.
Figure 1. Identification of the D122FS mutation in an ARS patient.
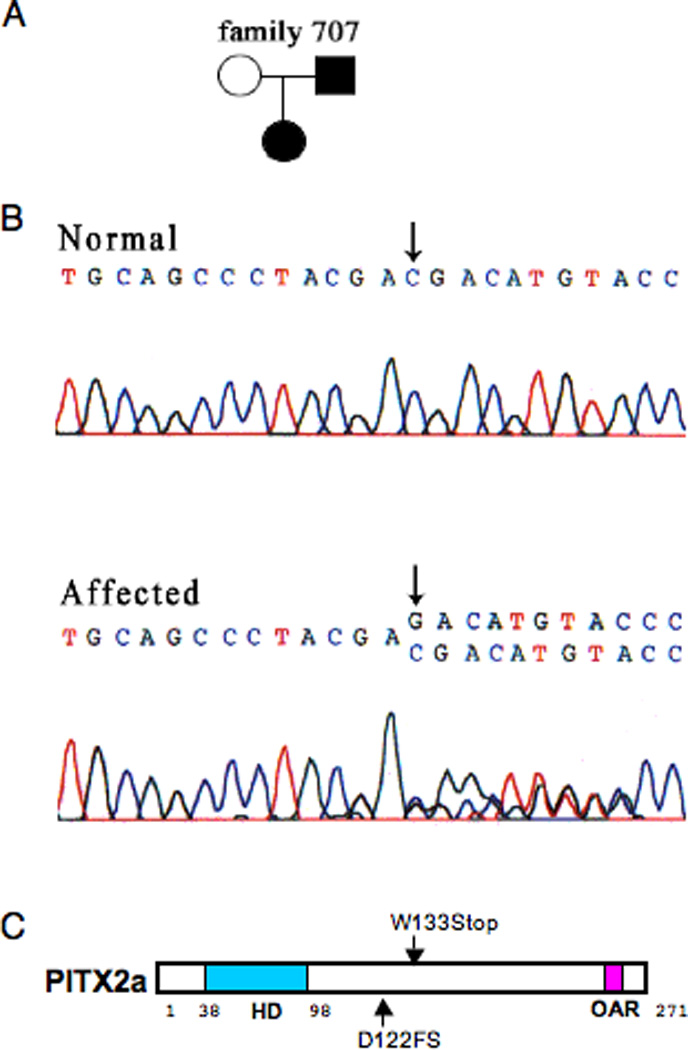
A. Pedigree of the affected proband showing the D122FS mutation. B. Sequence of the PITX2 fragment from genomic DNA of unaffected and affected members of the family. Deletion of a single base (C) results in a codon frameshift that predicts a truncated protein. C. Schematic representation of the human PITX2a gene showing the relative positions of the homeodomain (HD), the 2 C-terminal mutations, and the 14 amino acid Otp and aristaless homologous region (OAR) (Cox et al., 2002).
The properties of a family with the PITX2 W133Stop mutation (family 704) have previously been reported (Semina et al., 1996). The affected individuals had considerable umbilical involvement with omphalocele in 1 individual. The codon at position 133 (tryptophan in wildtype) contains a mutation that generates a stop codon. The PITX2 W133Stop cDNA encodes a truncated protein of 132 amino acids (Fig. 1C).
W133Stop and D122FS mutants bind DNA more strongly than wildtype PITX2
We first characterized the DNA-binding properties of W133Stop and D122FS by EMSA. PITX2 proteins were prepared by 3 different means. CHO cells were transfected with expression vectors encoding Myc-tagged wildtype or truncated PITX2a proteins. We used lysates of the transfected cells and a probe containing the bicoid consensus binding site (TAATCC) that is known to bind PITX2a (Amendt et al., 1998). Both the W133Stop and D122FS proteins had comparable levels of increased DNA binding compared to wildtype (Fig. 2A). Densitometric analysis revealed a 3.8- to 6.7-fold increase in DNA binding of the W133Stop and D1222FS proteins compared to wildtype PITX2a when normalized to the wildtype and truncated protein levels. The relative amounts of wildtype and truncated PITX2a proteins in the CHO lysates were measured by use of Western blots (Fig. 2B). A similar increase in binding was observed with W133Stop and D122FS proteins that had been synthesized in vitro using reticulocyte lysates (Fig. 2C). Densitometric analysis revealed a 2.3- to 5.6-fold increase in binding of the W133Stop and D1222FS proteins compared to wildtype PITX2a when normalized to the synthesized protein levels. There were equivalent levels of the Myc-tagged wildtype and truncated PITX2a proteins in the in vitro preparations (Fig. 2D). To further substantiate the increased binding of the W133Stop truncated protein relative to wildtype, an EMSA was performed with multiple concentrations of proteins prepared by a third technique. The wildtype and W133Stop proteins were synthesized as GST-fusion proteins in bacteria, and the GST moiety was removed by protease cleavage. W133Stop binding was at least 3-fold greater at all concentrations tested (Fig. 2E).
Figure 2. W133Stop and D122FS mutant proteins show increased binding to the bicoid DNA element.
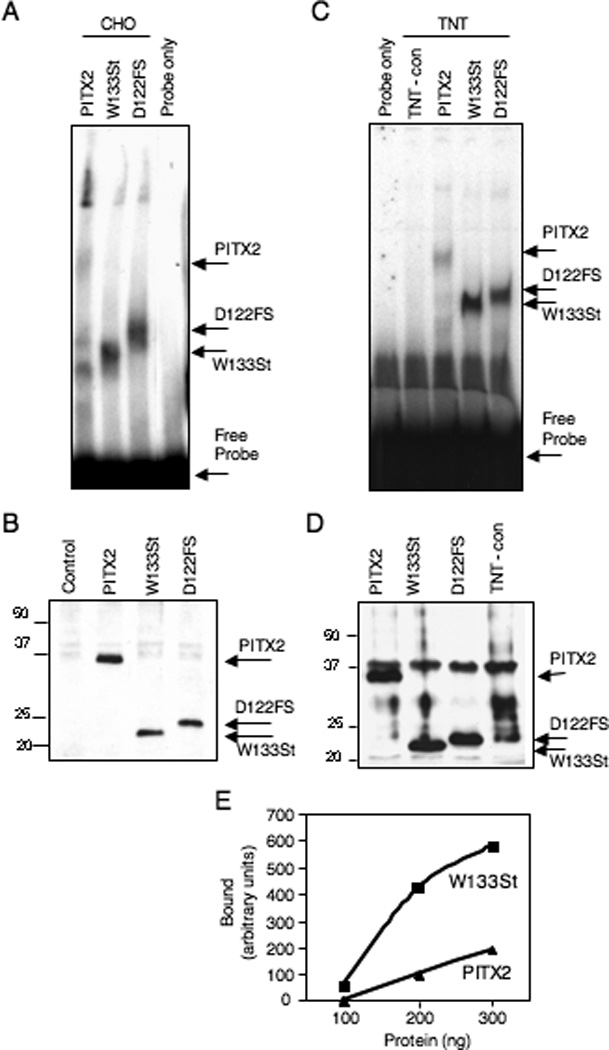
A. CHO cells transfected with wildtype PITX2, W133Stop, or D122FS expression vectors (as indicated) were lysed using a modified RIPA buffer. Approximately equal amounts of the resulting cell lysate were incubated with a probe containing the bicoid consensus sequence (TAATCC) in an EMSA. The bicoid probe only is shown as a control. The wildtype PITX2a, W133Stop, and D122FS complexes and free probe are indicated. The identities of the extra bands are not known. The image is representative of 4 independent experiments with lysates showing 3.8-, 4.3-, 4.4-, and 6.7-fold greater binding of W133Stop relative to wildtype and 2 independent experiments showing 4.3- and 4.4-fold greater binding of D122FS. B. Expression levels of the Myc-tagged PITX2a, W133Stop and D122FS proteins were determined for the CHO lysates in panel A with an anti-Myc Western blot. C. EMSA of 5 µl of wildtype PITX2a, W133Stop, and D122FS in vitro transcription and translation (TNT) products and an unprogrammed TNT-negative control incubated with the bicoid probe. The wildtype, W133Stop, and D122FS complexes are indicated. The image is representative of 3 independent experiments with TNT products showing 2.6-, 2.8- and 5.6-fold greater binding of W133Stop relative to wild type and a single experiment showing 2.3-fold greater binding for D122FS. D. An anti-Myc Western blot of the TNT products used in panel C. The wildtype PITX2a, W133Stop, D122FS proteins and prestained size standards are indicated. The identity of the band just above PITX2 is not known. E. EMSA measurement of bound wildtype and W133Stop PITX2a proteins. Increasing amounts (100, 200, 300 ng) of protease-cleaved GST-fusion proteins were incubated with a probe containing the bicoid consensus sequence. The band intensities were calculated from densitometric scans and the relative amount of bound protein is shown.
Dimerization of W133Stop and D122FS mutants with wildtype PITX2a
As a further characterization of the C-terminal mutant PITX2 proteins, we tested their ability to dimerize with wildtype PITX2a. We have reported the ability of PITX2a to form dimers independently of DNA binding (Saadi et al., 2003). To assess dimerization ability, we performed GST-pulldown assays using total cell lysates from CHO cells transfected with wildtype or truncated PITX2a mutants (Fig. 3). The truncated mutants could indeed bind better than wildtype PITX2a to the immobilized GST-PITX2a protein. Based on densitometric scans of the input and bound protein signals from 2 different experiments, we found that 6–10% of wildtype and 20–38% of W133Stop and 19–33% of D122FS PITX2a proteins were bound. As a control, there was no detectable binding to agarose beads containing only GST.
Figure 3. Increased binding of W133Stop and D122FS with wildtype PITX2a.
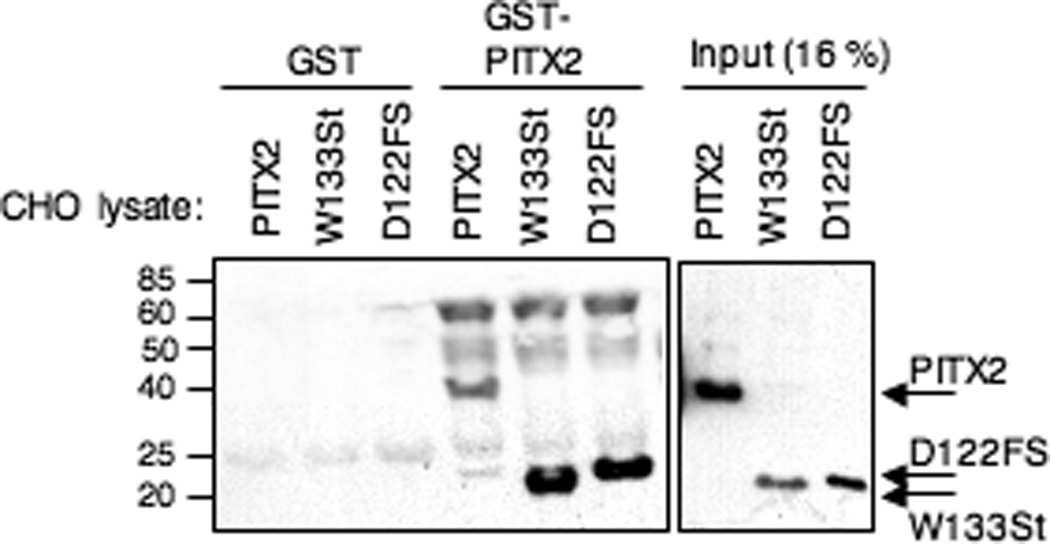
Approximately 90 µg of total lysate from CHO cells transfected with Myc-tagged PITX2a, W133Stop, or D122FS was incubated with glutathione sepharose beads containing GST alone or GST-PITX2a. Bound protein was detected by a Western blot using anti-Myc antibody; 15 µg of total lysate (16% of the input) is shown for comparison. The wildtype PITX2a, W133Stop, and D122FS proteins and prestained size standards are indicated.
Increased transactivation of the prolactin promoter in CHO, LS8 and COS7 cells by the W133Stop and D122FS mutants
We were interested to see if the increased DNA binding of the C-terminal mutants would result in a concomitant increase in transactivation. As an assay promoter, we used the prolactin promoter, which is likely to be a physiological target of PITX2 (Gage et al., 1999). Transfection of wildtype PITX2a activated the prolactin promoter ~20-fold in CHO cells (Fig. 4A). In contrast, when the W133Stop or D122FS mutants were expressed in CHO cells, there was a striking ~100-fold transactivation of the prolactin promoter (Fig. 4A).
Figure 4. Increased transactivation by the D122FS and W133Stop mutant proteins in mammalian cells.
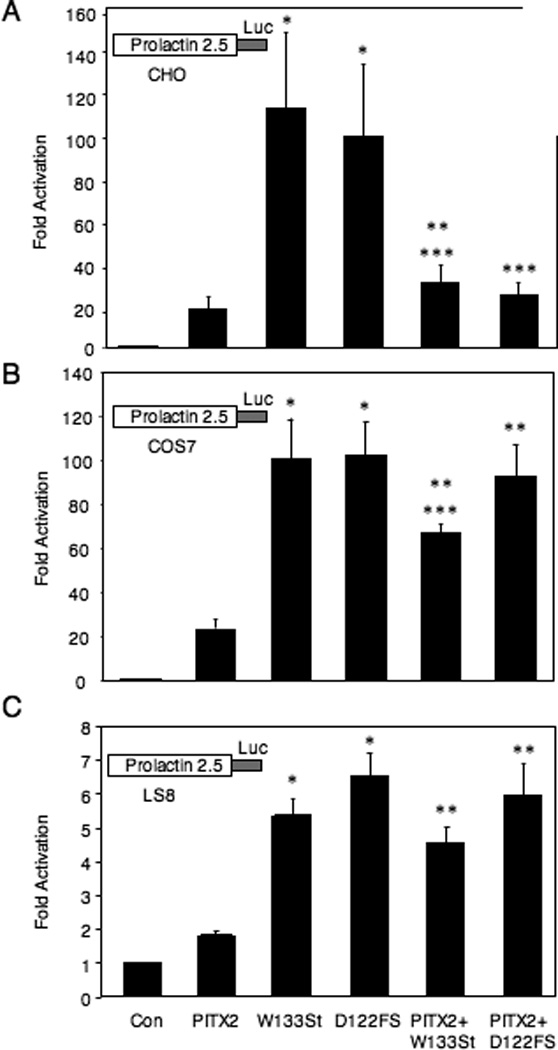
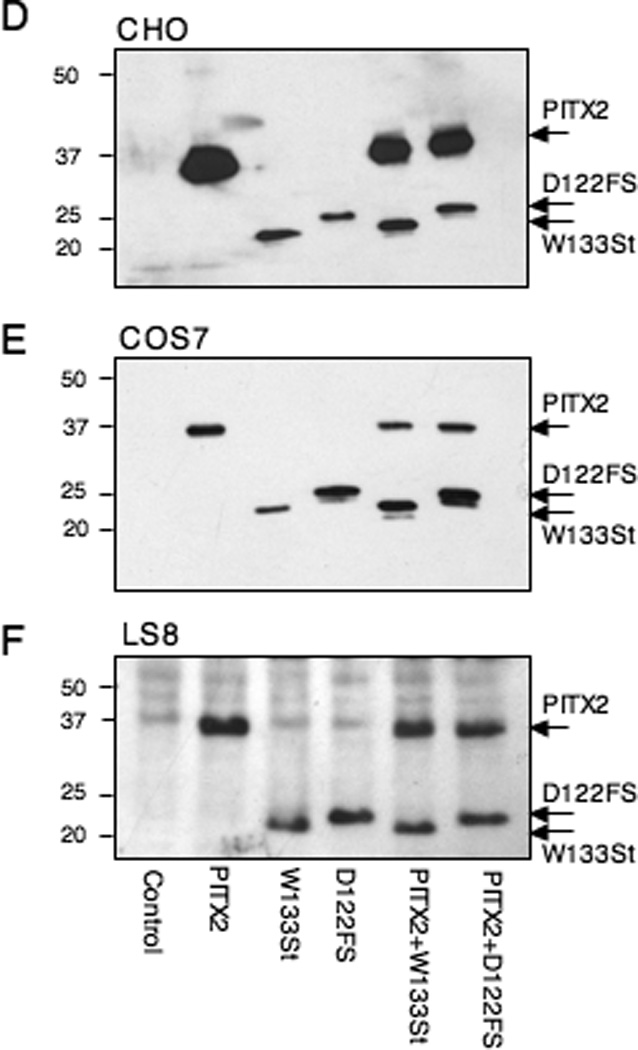
A–C. CHO, COS7 and LS8 cells were transfected with a prolactin-luciferase reporter alone (Con), or with PITX2a, W133Stop, D122FS, or combinations, as indicated. The luciferase activity is shown as the mean fold-activation compared to reporter alone +/− SEM from at least 4 independent experiments. *= P < .05 PITX2 vs. W133Stop or D122FS, **= P < .05 PITX2 vs. PITX2+W133Stop or PITX2+D122FS, ***= P < .05 W133Stop vs. PITX2+W133Stop or D122FS vs. PITX2+D122FS. D–F. Western blots were performed to show the relative expression level of the Myc-tagged wildtype and truncated proteins in the CHO, COS7, and LS8 cells as indicated. The wildtype PITX2a, W133Stop, and D122FS proteins and prestained size standards are indicated.
We then asked if the increased activity seen with the C-terminal mutants was an intrinsic property of the truncated protein or was it dependent on the cell type. We examined 2 additional cell lines, COS7 and LS8, in parallel with the CHO cells. Like the CHO cells, COS7 cells are heterologous, while LS8 cells express the endogenous Pitx2 gene (Green et al., 2001). Similar to what was seen in CHO cells (Fig. 4A), we found that both PITX2 mutants showed increased activation compared to wildtype PITX2 in both COS7 and LS8 cells (Fig. 4B, C).
The increased activity of the truncated proteins in CHO cells is even more noteworthy in light of the reproducibly greater expression of wildtype over mutant proteins in CHO cells (Figs. 2B and 4D). There were generally similar levels of wildtype and truncated proteins in the COS7 and LS8 cells (Fig. 4E, F). The expression vector used for the wildtype and truncated proteins encoded a C-terminal Myc tag. To address the possibility that the Myc tag might confer stability to the truncated proteins, we replaced the C-terminal tag with an N-terminal FLAG tag. The relative levels of FLAG-tagged wildtype and truncated proteins were the same as seen with the C-terminal Myc tag in CHO and COS7 cells (data not shown).
Because ARS is an autosomal dominant syndrome, we wanted to mimic the genotype by cotransfection of equal amounts of wildtype and mutant protein expression vectors. Unexpectedly, in CHO cells, cotransfection of wildtype with the W133Stop or D122FS mutant vectors reduced activity to similar or slightly greater (<2-fold) levels than wildtype (Fig. 4A). We have previously shown that transfection with twice as much wildtype PITX2a expression vector DNA yielded essentially the same level of activity, indicating that PITX2a is not limiting under these conditions in CHO or LS8 cells (Saadi et al., 2001; Saadi. 2003). Furthermore, since the truncated mutants bind DNA more effectively than wildtype, this indicates that the suppression of activity by the wildtype protein is not due to competition for DNA-binding sites. In contrast to CHO cells, cotransfection of wildtype and mutant proteins in both COS7 and LS8 cells yielded activity that was more like that observed with the mutants alone (Fig 4B, C). This suggests some cell specificity when the genotype of ARS patients is mimicked. As a control, there was relatively equivalent expression of the transfected proteins in COS7 and LS8 cells (Fig. 4 E, F).
DISCUSSION
ARS is an autosomal dominant disorder that is caused by mutations in the FOXC1 and PITX2 genes (Semina et al., 1996; Amendt et al., 2000; Lines et al., 2002). In the case of PITX2, the disorder is considered to be due to haploinsufficiency (Schinzel et al., 1997; Flomen et al., 1998). Indeed, until now, among the PITX2 mutations that have been characterized at a cellular level, there has been only 1 other report of a gain-of-function mutation (Priston et al., 2001). The V45L mutation in the homeodomain generated a protein with decreased DNA binding, but it unexpectedly increased transactivation activity in HeLa and COS7 cells (Priston et al., 2001). The 13 known C-terminal domain mutations, including the D122FS mutant described in this report, comprise a growing subset of the 34 reported ARS mutations (Semina et al., 1996; Perveen et al., 2000; Priston et al., 2001; Saadi et al., 2001; Wang et al., 2002; Philips, 2002; Borges et al., 2002; Brooks et al., 2004; Lines et al., 2004; Espinoza et al., 2005). Only 1 of these mutants, which lacks the C-terminal 34 amino acids, has been characterized at a cellular level (Espinoza et al., 2005). In this report, we have characterized the molecular properties of 2 mutants that lack most of the C-terminal domain.
In contrast to all other PITX2 mutations, the C-terminal truncations result in increased DNA binding. Both the W133Stop and D122FS truncation mutants bound DNA more strongly than wildtype PITX2a prepared from cell lysates or in vitro. Both W133Stop and D122FS could also bind wildtype PITX2a in the GST-pulldown assay to a greater extent than wildtype homodimers. We had previously reported that the homeodomain alone can form a dimer on DNA (Amendt et al., 1999). In contrast, the C-terminal region and homeodomain are both required for dimerization in a yeast 2-hybrid assay (Saadi et al., 2003). The reason for this difference is not known, but it may be due to higher levels of PITX2a protein in the EMSA and GST-pulldown assays relative to the yeast assay. The observation that both DNA binding and dimerization is increased in the absence of the C-terminal region is consistent with our model that predicts an inhibitory cis interaction between the C-terminus and homeodomain (Amendt et al., 1999).
From our previous studies with synthetic C-terminal mutations, we expected that the increased DNA binding would not result in a concomitant increase in transactivation ability (Amendt et al., 1999). However, the surprising result was that there was increased transactivation by the W133Stop and D122FS truncated proteins. In CHO cells there was about a 5-fold greater transactivation of the prolactin promoter than seen with the wildtype PITX2 protein. In the COS7 and LS8 cells, there was also greater activity with the mutants. The increased transactivation was observed even though the mutant proteins were expressed at lower levels than wildtype in CHO cells and at comparable levels in COS7 and LS8 cells. Of note, both C-terminal truncations described in this work are still more active than wildtype despite the elimination of 5 phosphorylation sites that have been attributed to increased PITX2 transactivational activity (Espinoza et al., 2005). One possible explanation is that phosphorylation of the C-terminal sites results in a conformational change that unmasks the homeodomain, analogous to what is thought to happen when Pit-1 binds to the C-terminal portion of PITX2 (Amendt et al., 1999).
To mimic the autosomal dominant state of the ARS patient, we coexpressed the wildtype PITX2a allele with the W133Stop and D122FS truncation mutants. In CHO cells this meant that the large gain of function relative to wildtype was reduced to only a minimal change upon cotransfection with wildtype PITX2a. This effect appears to be cell specific because in the COS7 and LS8 cells, coexpression of wildtype and either truncation mutant showed comparable activity to that seen with just the truncated mutants. The mechanism behind this apparently cell-specific dampening of activity is not known, but it could at least partly involve differences in wildtype-mutant dimer protein-protein interactions with cellular cofactors and/or the phosphorylation state of wildtype PITX2 in the different cell types.
An extrapolation from the cell-based assays in this report is that C-terminal PITX2 mutants might have a gain-of-function phenotype in ARS patients. This challenges the view that ARS is caused only by haploinsufficiency but is in accord with the gain-of-function mutant reported by Priston et al. (Priston et al., 2001). One caveat of this interpretation is that although the truncated proteins are expressed in cell lines, the mutants may not expressed in the patients because of nonsense-mediated RNA degradation or protein degradation. Nonetheless, there is precedence of dosage-dependent birth defects. In particular, it is intriguing that duplication of the FOXC1 chromosomal region has been linked to ocular developmental defects (Lehmann et al., 2000; Nishimura et al., 2001) and that ARS can also be caused by mutations in the FOXC1 gene (Mirzayans et al., 2000). Importantly, Holmberg et al. (2004) have recently shown that overexpression of PITX2 in the developing mouse eye can also cause phenotypes consistent with ARS, which provides further evidence for gain of function potentially underlying ARS. These results underscore the complexity and potential ramifications of mutations in the PITX2 C-terminal tail in ARS patients.
ACKNOWLEDGMENTS
We thank Dr. K. Keppler for identifying family 707 with the D122FS mutation, Dr. B. Amendt for discussions, N. Rorick for assistance, and Dr. M. Snead for the LS8 cell line
This work was supported by National Institutes of Health grants DE13076 and DE016511 to AFR and EY12384 to JCM.
REFERENCES
- Amendt BA, Sutherland LB, Semina EV, Russo AF. The molecular basis of Rieger syndrome. Analysis of Pitx2 homeodomain protein activities. J Biol Chem. 1998;273:20066–20072. doi: 10.1074/jbc.273.32.20066. [DOI] [PubMed] [Google Scholar]
- Amendt BA, Sutherland LB, Russo AF. Multifunctional role of the Pitx2 homeodomain protein C-terminal tail. Mol Cell Biol. 1999;19:7001–7010. doi: 10.1128/mcb.19.10.7001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amendt BA, Semina EV, Alward WL. Rieger syndrome: a clinical, molecular, and biochemical analysis. Cell Mol Life Sci. 2000;57:1652–1666. doi: 10.1007/PL00000647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borges AS, Susanna R, Jr, Carani JC, Betinjane AJ, Alward WL, Stone EM, Sheffield VC, Nishimura DY. Genetic analysis of PITX2 and FOXC1 in Rieger Syndrome patients from Brazil. J Glaucoma. 2002;11:51–56. doi: 10.1097/00061198-200202000-00010. [DOI] [PubMed] [Google Scholar]
- Brooks BP, Moroi SE, Downs CA, Wiltse S, Othman MI, Semina EV, Richards JE. A novel mutation in the PITX2 gene in a family with Axenfeld-Rieger syndrome. Ophthalmic Genet. 2004;25:57–62. doi: 10.1076/opge.25.1.57.29002. [DOI] [PubMed] [Google Scholar]
- Burz DS, Hanes SD. Isolation of mutations that disrupt cooperative DNA binding by the Drosophila bicoid protein. J Mol Biol. 2001;305:219–230. doi: 10.1006/jmbi.2000.4287. [DOI] [PubMed] [Google Scholar]
- Cox CJ, Espinoza HM, McWilliams B, Chappell K, Morton L, Hjalt TA, Semina EV, Amendt BA. Differential regulation of gene expression by PITX2 isoforms. J Biol Chem. 2002;12:25001–25010. doi: 10.1074/jbc.M201737200. [DOI] [PubMed] [Google Scholar]
- Espinoza HM, Cox CJ, Semina EV, Amendt BA. A molecular basis for differential developmental anomalies in Axenfeld-Rieger syndrome. Hum Mol Genet. 2002;11:743–753. doi: 10.1093/hmg/11.7.743. [DOI] [PubMed] [Google Scholar]
- Espinoza HM, Ganga M, Vadlamudi U, Martin DM, Brooks BP, Semina EV, Murray JC, Amendt BA. Protein kinase C phosphorylation modulates N- and C-terminal regulatory activities of the PITX2 homeodomain protein. Biochemistry. 2005;44:3942–3954. doi: 10.1021/bi048362x. [DOI] [PubMed] [Google Scholar]
- Essner JJ, Branford WW, Zhang J, Yost HJ. Mesendoderm and left-right brain, heart and gut development are differentially regulated by pitx2 isoforms. Development. 2000;127:1081–1093. doi: 10.1242/dev.127.5.1081. [DOI] [PubMed] [Google Scholar]
- Flomen RH, Vatcheva R, Gorman PA, Baptista PR, Groet J, Barisic I, Ligutic I, Nizetic D. Construction and analysis of a sequence-ready map in 4q25: Rieger syndrome can be caused by haploinsufficiency of RIEG, but also by chromosome breaks approximately 90 kb upstream of this gene. Genomics. 1998;47:409–413. doi: 10.1006/geno.1997.5127. [DOI] [PubMed] [Google Scholar]
- Gage PJ, Suh H, Camper SA. Dosage requirement of Pitx2 for development of multiple organs. Development. 1999;126:4643–4651. doi: 10.1242/dev.126.20.4643. [DOI] [PubMed] [Google Scholar]
- Green PD, Hjalt TA, Kirk DE, Sutherland LB, Thomas BL, Sharpe PT, Snead ML, Murray JC, Russo AF, Amendt BA. Antagonistic regulation of Dlx2 expression by PITX2 and Msx2: implications for tooth development. Gene Expr. 2001;9:265–281. doi: 10.3727/000000001783992515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holmberg J, Liu CY, Hjalt TA. PITX2 gain-of-function in Rieger syndrome eye model. Am J Pathol. 2004;165:1633–1641. doi: 10.1016/S0002-9440(10)63420-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kozlowski K, Walter MA. Variation in residual PITX2 activity underlies the phenotypic spectrum of anterior segment developmental disorders. Hum Mol Genet. 2000;9:2131–2139. doi: 10.1093/hmg/9.14.2131. [DOI] [PubMed] [Google Scholar]
- Lehmann OJ, Ebenezer ND, Jordan T, Fox M, Ocaka L, Payne A, Leroy BP, Clark BJ, Hitchings RA, Povey S, Khaw PT, Bhattacharya SS. Chromosomal duplication involving the forkhead transcription factor gene FOXC1 causes iris hypoplasia and glaucoma. Am J Hum Genet. 2000;67:1129–1135. doi: 10.1016/s0002-9297(07)62943-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lines MA, Kozlowski K, Walter MA. Molecular genetics of Axenfeld-Rieger malformations. Hum Mol Genet. 2002;11:1177–1184. doi: 10.1093/hmg/11.10.1177. [DOI] [PubMed] [Google Scholar]
- Lines MA, Kozlowski K, Kulak SC, Allingham RR, Heon E, Ritch R, Levin AV, Shields MB, Damji KF, Newlin A, Walter MA. Characterization and prevalence of PITX2 microdeletions and mutations in Axenfeld-Rieger malformations. Invest Ophthalmol Vis Sci. 2004;45:828–833. doi: 10.1167/iovs.03-0309. [DOI] [PubMed] [Google Scholar]
- Maurer RA, Notides AC. Identification of an estrogen-responsive element from the 5'-flanking region of the rat prolactin gene. Mol Cell Biol. 1987;7:4247–4254. doi: 10.1128/mcb.7.12.4247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mirzayans F, Gould DB, Heon E, Billingsley GD, Cheung JC, Mears AJ, Walter MA. Axenfeld-Rieger syndrome resulting from mutation of the FKHL7 gene on chromosome 6p25. Eur J Hum Genet. 2000;8:71–74. doi: 10.1038/sj.ejhg.5200354. [DOI] [PubMed] [Google Scholar]
- Nishimura DY, Searby CC, Alward WL, Walton D, Craig JE, Mackey DA, Kawase K, Kanis AB, Patil SR, Stone EM, Sheffield VC. A spectrum of FOXC1 mutations suggests gene dosage as a mechanism for developmental defects of the anterior chamber of the eye. Am J Hum Genet. 2001;68:364–372. doi: 10.1086/318183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perveen R, Lloyd IC, Clayton-Smith J, Churchill A, van Heyningen V, Hanson I, Taylor D, McKeown C, Super M, Kerr B, Winter R, Black GC. Phenotypic variability and asymmetry of Rieger syndrome associated with PITX2 mutations. Invest Opthal Mol Vis Sci. 2000;41:2456–2460. [PubMed] [Google Scholar]
- Phillips JC. Four novel mutations in the PITX2 gene in patients with Axenfeld-Rieger syndrome. Ophthalmic Res. 2002;34:324–326. doi: 10.1159/000065602. [DOI] [PubMed] [Google Scholar]
- Priston M, Kozlowski K, Gill D, Letwin K, Buys Y, Levin AV, Walter MA, Heon E. Functional analyses of two newly identified PITX2 mutants reveal a novel molecular mechanism for Axenfeld-Rieger syndrome. Hum Mol Genet. 2001;10:1631–1638. doi: 10.1093/hmg/10.16.1631. [DOI] [PubMed] [Google Scholar]
- Saadi I, Semina EV, Amendt BA, Harris DJ, Murphy KP, Murray JC, Russo AF. Identification of a dominant negative homeodomain mutation in Rieger syndrome. J Biol Chem. 2001;276:23034–23041. doi: 10.1074/jbc.M008592200. [DOI] [PubMed] [Google Scholar]
- Saadi I, Kuburas A, Engle JJ, Russo AF. Dominant negative dimerization of a mutant homeodomain protein in Axenfeld-Rieger syndrome. Mol Cell Biol. 2003;23:1968–1982. doi: 10.1128/MCB.23.6.1968-1982.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schinzel A, Brecevic L, Dutly F, Baumer A, Binkert F, Largo RH. Multiple congenital anomalies including the Rieger eye malformation in a boy with interstitial deletion of (4) (q25-->q27) secondary to a balanced insertion in his normal father: evidence for haplotype insufficiency causing the Rieger malformation. J Med Genet. 1997;34:1012–1014. doi: 10.1136/jmg.34.12.1012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schweickert A, Campione M, Steinbeisser H, Blum M. Pitx2 isoforms: involvement of Pitx2c but not Pitx2a or Pitx2b in vertebrate left-right asymmetry. Mech Dev. 2000;90:41–51. doi: 10.1016/s0925-4773(99)00227-0. [DOI] [PubMed] [Google Scholar]
- Semina EV, Reiter R, Leysens NJ, Alward WL, Small KW, Datson NA, Siegel-Bartelt J, Bierke-Nelson D, Bitoun P, Zabel BU, Carey JC, Murray JC. Cloning and characterization of a novel bicoid-related homeobox transcription factor gene, RIEG, involved in Rieger syndrome. Nat Genet. 1996;14:392–399. doi: 10.1038/ng1296-392. [DOI] [PubMed] [Google Scholar]
- Smidt MP, Cox JJ, van Schaick HS, Coolen M, Schepers J, van der Kleij AM, Burbach JP. Analysis of three Ptx2 splice variants on transcriptional activity and differential expression pattern in the brain. J Neurochem. 2000;75:1818–1825. doi: 10.1046/j.1471-4159.2000.0751818.x. [DOI] [PubMed] [Google Scholar]
- Tremblay JJ, Goodyear CG, Drouin J. Transcriptional properties of Ptx1 and Ptx2 isoforms. Neuroendocrinology. 2000;71:277–286. doi: 10.1159/000054547. [DOI] [PubMed] [Google Scholar]
- Tverberg LA, Russo AF. Regulation of the calcitonin/calcitonin gene-related peptide gene by cell-specific synergy between helix-loop-helix and octamer-binding transcription factors. J Biol Chem. 1993;268:15965–15973. [PubMed] [Google Scholar]
- Wang Y, Zhao H, Zhang X, Feng H. Novel identification of a four-base-pair deletion mutation in PITX2 in a Rieger syndrome family. J Dent Res. 2002;82:1008–1012. doi: 10.1177/154405910308201214. [DOI] [PubMed] [Google Scholar]
- Yu X, St. Amand TR, Wang S, Li G, Zhang Y, Hu YP, Nguyen L, Qiu MS, Chen YP. Differential expression and functional analysis of Pitx2 isoforms in regulation of heart looping in the chick. Development. 2001;128:1005–1013. doi: 10.1242/dev.128.6.1005. [DOI] [PubMed] [Google Scholar]
- Yuan D, Ma X, Ma J. Sequences outside the homeodomain of bicoid are required for protein-protein interaction. J Biol Chem. 1996;271:21660–21665. [PubMed] [Google Scholar]