

Abstract
Malaria remains a major health concern for a large percentage of the world’s population. While great strides have been made in reducing mortality due to malaria, new strategies and therapies are still needed. Therapies that are capable of blocking the transmission of Plasmodium parasites are particularly attractive, but only primaquine accomplishes this, and toxicity issues hamper its widespread use. In this study, we describe a series of pyrazolopyrimidine- and imidazopyrazine-based compounds that are potent inhibitors of PfCDPK4, which is a calcium-activated Plasmodium protein kinase that is essential for exflagellation of male gametocytes. Thus, PfCDPK4 is essential for the sexual development of Plasmodium parasites and their ability to infect mosquitos. We demonstrate that two structural features in the ATP-binding site of PfCDPK4 can be exploited in order to obtain potent and selective inhibitors of this enzyme. Furthermore, we demonstrate that pyrazolopyrimidine-based inhibitors that are potent inhibitors of the in vitro activity of PfCDPK4 are also able to block P. falciparum exflagellation with no observable toxicity to human cells. This medicinal chemistry effort serves as a valuable starting point in the development of safe, transmission-blocking agents for the control of malaria.
Keywords: Plasmodium falciparum, PfCDPK4, PfCDPK1, Pyrazolopyrimidine, Exflagellation
1. Introduction
Malaria is a major health concern for much of the tropics and subtropics [1-4]. Malaria infections are transmitted to humans by the bite of female Anopheles mosquitoes. Four Plasmodium species, P. falciparum, P. vivax, P. malariae, and P. ovale are responsible for the majority of human infections. Recently, a fifth species, P. knowlesi, which was previously thought to infect only non-human primates, has been found to be adapted to spread from human-to-mosquito-to-human in Malaysia [5, 6]. Although insecticide-treated bed nets, vector control, and effective chemotherapies have resulted in reduced mortality, an estimated 1,238,000 people still die of malaria every year [7]. While efforts are underway to effectively control Anopheles mosquito populations, this task remains a major challenge. In order to control and eradicate malaria, it will be necessary to develop new methods of reducing parasite transmission, especially in light of increasing mosquito resistance to insecticide-treated bed nets [8].
Currently available therapeutics mainly target the erythrocytic (asexual) stage of malaria, but not gametocytes (sexual forms) [9]. As circulating gametocytes maintain the capacity to infect mosquitoes for weeks after therapy, the lack of transmission-blocking activity of these therapies hinders malaria control [10, 11]. While primaquine and artemisinin combination therapy (ACT) possess anti-gametocyte activity, these drugs are not ideal for preventing malaria transmission to mosquitoes. Artemisinin only kills immature gametocytes, while primaquine only kills mature gametocytes [12]. Extended primaquine treatment can effectively eradicate gametocytes from the blood, but results in hemolysis for patients with glucose-6-phosphate dehydrogenase deficiency and many others suffer from GI intolerance during primaquine therapy [10, 13]. For these reasons, a number of new transmission blocking strategies have been explored [13-15].
The life cycle stages of malaria parasites alternate between human and mosquito hosts (Figure 1) [15]. Female Anopheles mosquitoes transmit sporozoites to humans, where asexual reproduction produces merozoites. A subset of merozoites develop into gametocytes, which upon the bite of a mosquito can be taken up with blood and allowed to mature in the mosquito midgut. Inhibition of specific calcium-dependent processes can block this maturation and the eventual formation of mature sporozoites in the mosquito salivary glands, which would otherwise be transmitted when the mosquito next bites a human (Figure 1).
Figure 1.

Diagram of the Malaria parasite lifecycle. Asexual mammalian-host stages are shown in white and lifecycle stages that occur in the mosquito are shown in light prurple. A subset of merozoites differentiate into male and female gametocytes, which are ingested by female mosquitos upon biting an infected human. In the mosquito gut, male gametocytes divide into flagellated microgametes, which escape red blood cells (exflagellation) and swim to the macrogamete, resulting in fertilization. The resultant motile zygote forms an ookinete, which moves across the mosquito gut to form oocysts. Oocysts rupture to form sporozoites that mature in mosquito salivary glands and can be injected into humans by mosquito bites. In humans, replication via the asexual lifecycle occurs. Inhibitors of PfCDPK4, block transmission by preventing exflagellation.
Precise control over malaria life cycle stages is coordinated through intricate signal transduction pathways that are regulated by a number of secondary messengers. One such secondary messenger is calcium, which is involved in important life cycle events, such as host cell invasion, protein secretion, gliding motility, and exflagellation [16]. In addition to other enzymes, calcium release in Plasmodium activates a family of plant- and apicomplexan-specific kinases called the calcium-dependent protein kinases (CDPKs) [17, 18]. Binding of calcium to this family of kinases releases an auto-inhibitory domain interaction, increasing the catalytic activity of the kinase domain, which results in the phosphorylation of numerous cellular substrates [19]. Plasmodium CDPKs are attractive drug targets because they are unique to plants and apicomplexans. Furthermore, these kinases are involved in a number of important Plasmodium life cycle processes, including various steps involved in transmission.
While all of the functions of Plasmodium CDPKs have not been fully characterized, their roles in several important calcium-regulated processes have been described. P. falciparum CDPK1 (PfCDPK1) has been reported to be involved in the regulation of parasite motility and in controlling zygote development and transmission [20, 21]. P. falciparum CDPK5 (PfCDPK5) has been implicated in the regulation of malaria parasite egress from erythrocytes, and P. berghei CDPK3 (PbCDPK3) is required for ookinete gliding motility and invasion of mosquito midguts [22, 23]. Genetic experiments have demonstrated that P. berghei CDPK4 (PbCDPK4), and presumably the homologous PfCDPK4, is essential for microgamete (male gametocytes) exflagellation and sexual stage development in the mosquito [24]. Furthermore, we have previously reported that potent and selective PfCDPK4 inhibitors are capable of blocking Plasmodium microgamete exflagellation, thus disrupting malaria transmission [25, 26]. Expression in P. falciparum of an exogenous drug-resistant PfCDPK4 mutant led to a 12-fold reduction in sensitivity to a PfCDPK4 inhibitor in exflagellation assays [26]. Therefore, PfCDPK4 has been validated genetically and pharmacologically as a promising drug target for the development of transmission blocking therapies. Here, we report a detailed molecular analysis of the requirements for potent and selective PfCDPK4 inhibition. We show that certain structural features in PfCDPK4’s ATP-binding site can be exploited to obtain highly potent and selective inhibitors. Furthermore, we demonstrate that pyrazolopyrimidine-based inhibitors containing diverse functional groups, which inhibit the catalytic activity of PfCDPK4 in vitro, are effective at blocking the exflagellation of P. falciparum. These studies suggest PfCDPK4 is an attractive target for the development of malaria transmission-blocking therapy, both for its essential role in exflagellation and due to its pharmacological tractability.
2. Results and Discussion
2.1. Molecular Design
We have generated and characterized a number of pyrazolopyrimidine-based inhibitors that selectively target Toxoplasma gondii CDPK1 (TgCDPK1) and Cryptosporidium parvum CDPK1 (CpCDPK1) [27-30]. These kinases contain an enlarged ATP-binding pocket due to the presence of a glycine at a conserved structural feature called the gatekeeper residue. As mammalian kinases contain gatekeeper residues larger than glycine, this unique structural element can be targeted to obtain highly selective ATP-competitive inhibitors. Through iterative rounds of synthesis and testing, we have identified aryl substituents displayed from the C-3 position of the pyrazolo[2,3-d]pyrimidine scaffold that optimally complement the ATP-binding pockets of TgCDPK1 and CpCDPK1 (Tg/CpCDPK1). Many of these inhibitors are highly selective for Tg/CpCDPK1 over mammalian kinases. While PfCDPK4 contains a serine rather than a glycine at the gatekeeper position, the rarity of mammalian kinases with a gatekeeper residue this small raises the possibility that highly selective inhibitors can be developed for this kinase as well. Furthermore, during our efforts to target Tg/CpCDPK1 we identified an additional region of the ATP-binding site that can be exploited by inhibitors that target these kinases [30]. This structural feature, which is located in the ribose pocket of the ATP-binding site, works in concert with the gatekeeper position to allow certain bulky C-3 substituents to be specifically accommodated in the ATP-binding sites of Tg/CpCDPK1. Due to the high degree of sequence homology in the CDPK family, we hypothesized that we could develop inhibitors that exploit a similar pair of interactions in PfCDPK4.
The core scaffold of pyrazolo[2,3-d]pyrimidine inhibitors make similar hydrophobic and hydrogen-bonding contacts as the adenine ring of ATP [31]. Substituents displayed from the N-1 position (R2) project into the solvent-exposed ribose pocket. C-3 substituents (R1) occupy the hydrophobic pocket adjacent to the gatekeeper residue. To determine the optimal substituents that complement the hydrophobic pocket next to the serine gatekeeper of PfCDPK4, we first generated and tested a series of inhibitors that contain either an i-Pr- or t-Bu-group at the R2 position and highly variable aryl substituents at the C-3 position (R1) (Table 1). A second series of inhibitors that contain optimal R1 substituents and diverse alkyl substituents displayed from the N-1 position (R2) were next investigated (Table 2). Finally, a series of inhibitors in which the pyrazolopyrimidine core was replaced with an imidazopyrazine scaffold was also generated and tested (Table 3).
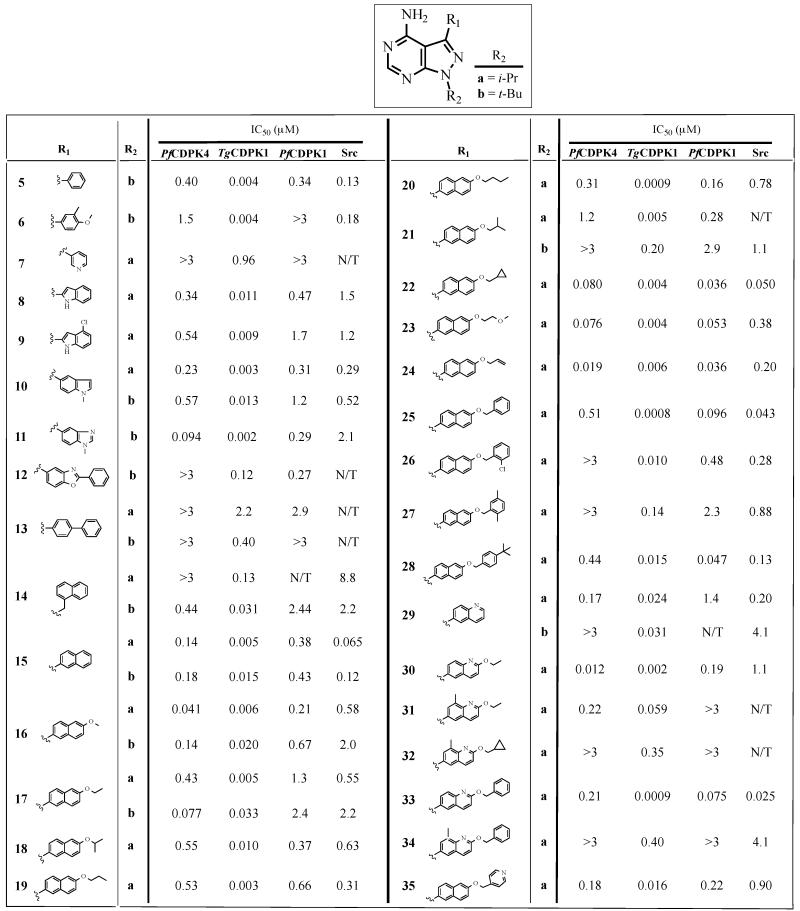
Table 1.
Enzymatic assay (IC50) results for compounds with variable R1 substructures (5-35) across the R2 series a and b. All results are the averages of at least three assays. TgCDPKi IC50 values for a number of compounds have previously been reported [28, 29].
|
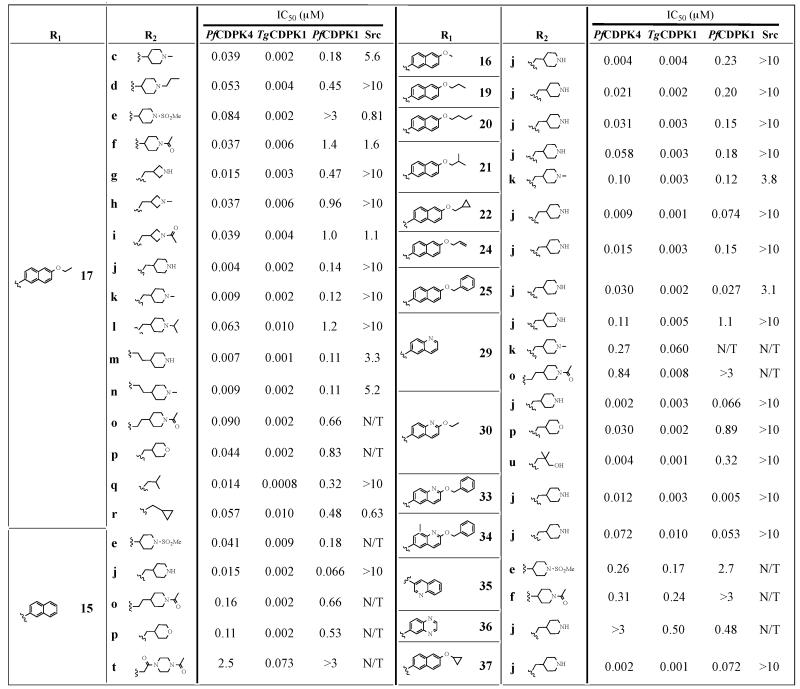
Table 2.
Enzymatic assay (IC50) results for compounds with variable R2 substructures (c-u) across the R1 series containing 2-naphthyl, 6-alkoxy-2-naphthyl or 2-alkoxy-6-quinilino Ri substituents. All results are the averages of at least three assays. TgCDPKi IC50 values for a number of compounds have previously been reported [28, 29].
|
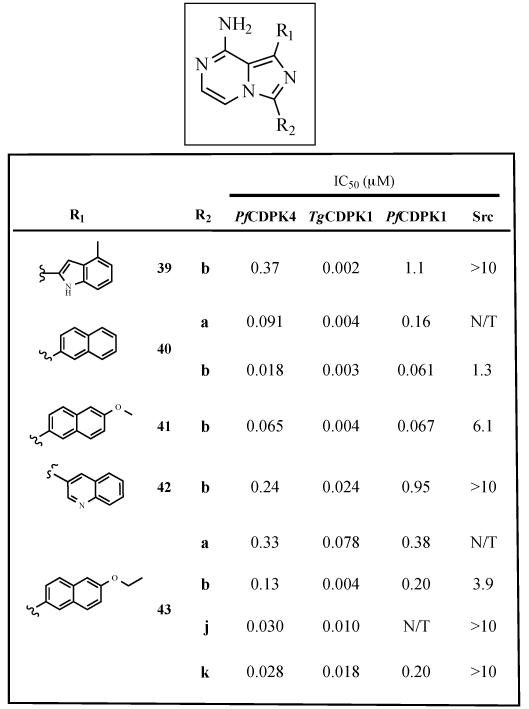
Table 3.
Enzymatic assay (IC50) results for imidazo[1,5-a]pyrazine inhibitors. All results are the averages of at least three assays. N/T=not tested.
|
2.2. Synthesis of PfCDPK4 Inhibitors
Several synthetic strategies were used to generate inhibitors based on a pyrazolo[2,3-d]pyrimidine scaffold. The synthetic route shown in Scheme 1 was used to generate inhibitors with i-Pr- or t-Bu-groups at the R2 position (series a and b). All compounds in Table 1 were generated from halogen-containing intermediates 1 and 2, which were made using previously described synthetic routes [29, 32]. The aryl substituents of compounds 7a-35a and 5b-29b were introduced at the C-3 positions of 1 and 2 via palladium-catalyzed Suzuki-Miyaura coupling reactions with appropriate aryl boronic acids and boronate esters. Boronic acids or esters that were not commercially available were prepared using a palladium-mediated cross coupling between aryl bromides and bis(pinacolato)diboron. Compounds 18a-28a and 21b, which contain extended 6-alkoxy- and 6-benzyloxynaphtyl groups at the C-3 position, were generated by first performing a Suzuki-Miyaura coupling between 6-TBDMS-naphthalene-2-boronic acid and 1 and 2, followed by alkylation of the deprotected naphthols 3 and 4 with alkyl and benzyl halides (Scheme 1). Compounds containing variable groups at the R2 position (Table 2) were generated by alkylating 3-iodo-1H-pyrazolo[3,4-d]pyrimidin-4-amine with alkyl halides, or alkyl mesylates, followed by Suzuki-Miyaura coupling to R1-boronic acids or boronate pinacol esters (Scheme 2). 4-piperidinylmethylene- and 3-azetidinemethylene-containing compounds (15j, 16j,17g, 17j, 19j, 20j, 21j, 22j, 24j, 25j, 29j, 30j, 33j, 34j, 36j, and 37j) were generated by alkylating 3-iodo-1H-pyrazolo[3,4-d]pyrimidin-4-amine with N-Boc-4-piperidinemethanol or N-Boc-3-azetidinemethanol mesylates (Scheme 3). The alkylated intermediates were then deprotected with 50% TFA. After Boc deprotection, alkylation or acetylation was performed to generate analogs containing h, i, k, and l R2 groups.
Scheme 1.

General synthetic procedures for compound series 7a-35a and 5b-29b. i) Na2CO3, Pd(PPh3)4, boronic acid or boronate pinacol ester, H2O/DME, 80 °C; ii) R′-halide, K2CO3, DMF, room temperature or 80 °C.
Scheme 2.
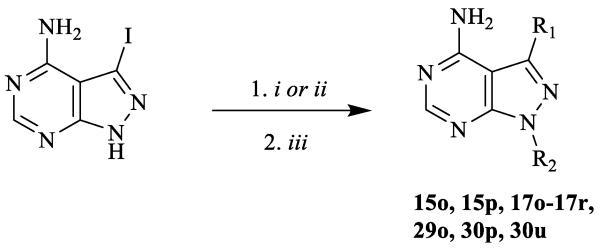
General synthetic procedures for compound with variable R2 groups. i) R2-halide or R2-mesylate, K2CO3, DMF, room temperature; or ii) R2-halide or R2-mesylate, Cs2CO3, DMF, 80 °C; iii) Na2CO3, Pd(PPh3)4, R1-boronic acid or R1-boronate pinacol ester, H2O/DME, 80 °C.
Scheme 3.

General synthetic procedures for compounds with 4-azetidinemethylene and 4-piperidinemethylene substituents. i) N-Boc-4-piperidinemethanol mesylate, Cs2CO3, DMF, 80 °C; ii) N-Boc-3-azetidinemethanol mesylate, Cs2CO3, DMF, 80 °C; iii) Na2CO3, Pd(PPh3)4, boronic acid or boronate pinacol ester, H2O/DME, 80 °C in microwave; iv) TFA/CH2Cl2 (1:1); v) sodium methoxide in MeOH, then 2% AcOH, R″-aldehyde, NaBH3CN; vi) Ac2O, TEA, DMF.
Imidazo[1,5-a]pyrazine-based inhibitors (Table 3) were prepared using the procedure shown in Scheme 4 [33, 34]. R2 substituents were introduced by acylating 3-(chloropyrazin-2-yl)methanamine with appropriate carboxylic acids, followed by cyclization with POCl3. After iodinating the scaffold with NIS and aminolysis in isopropanol, R1 substituents were then introduced at the C-1 position via palladium-mediated Suzuki-Miyaura couplings with commercially available boronic acids. Piperidine-containing compound 43j was generated by removing the CBZ protecting group from intermediate 38 with HCl. Reductive amination of 43j with formaldehyde produced compound 43k. Detailed procedures and characterization data for all compounds are presented in the Supporting Information.
Scheme 4.

General synthetic procedures for imidazo[1,5-a]pyrazine inhibitors. i) R1CO2H, EDCI, HOBt, CH2Cl2; ii) POCl3, DMF (cat.), then TEA, CH2Cl2; iii) NIS, DMF iv) NH4OH, MeOH v) Na2CO3, Pd(PPh3)4, boronic acid or boronate pinacol ester, H2O/DME, 80 °C; vi) HCl (conc.); vii) CH2O, NaBH3CN.
2.3. In vitro activity assays
All of the inhibitors generated were tested for their ability to inhibit recombinant PfCDPK4 in vitro. The catalytic activity of PfCDPK4 was determined using a luminescence-based assay that measures the consumption of ATP in the presence of a peptide substrate [27]. PfCDPK4 efficiently phosphorylates Syntide 2, which is a substrate for mammalian CAMKII. All assays were performed at PfCDPK4’s Km for ATP (10 μM) and in the presence of 2 mM CaCl2. Many of the compounds were also tested against TgCDPK1, PfCDPK1, and the mammalian tyrosine kinase Src. The ATP-binding sites of TgCDPK1 and PfCDPK1 are very similar to that of PfCDPK4 in almost all of the positions predicted to interact with pyrazolopyrimidine-based inhibitors, except, most notably, the gatekeeper residue (Figure 2). TgCDPK1’s glycine gatekeeper residue is smaller than PfCDPK4’s serine, while PfCDPK1’s threonine gatekeeper residue is larger. By comparing the relative sensitivities of these three CDPKs to analogs with variable R1 substituents, the relative contribution of the gatekeeper residue to inhibitor potency can be assessed (Figure 2). The tyrosine kinase Src was selected as a mammalian “off-target” counter-screen because it contains one of the smallest gatekeeper residues, threonine, present in humans, and was one of the original kinase targets for which pyrazolo[2,3-d]pyrimidine inhibitors were developed [35, 36].
Figure 2.

(A) Structure of a pyrazolopyrimidine-based inhibitor (17j) bound to the ATP-binding site of TgCDPK1. The six residues that contribute the largest surface area to the inhibitor binding site are show in blue. The gatekeeper residue is shown in purple. Residue numbering refers to that of TgCDPK1. (B) Conservation of residues making up the CDPK active site. The residues shown are those within 5 Å of bound inhibitor 17j as observed in complex with TgCDPK1 (PDB 3s×9). The black vertical bars show the relative contributions of atoms in each reside to the surface area of the binding site in TgCDPK1. Residues that are shaded grey are not conserved between TgCDPK1, PfCDPK1, and PfCDPK4. The gatekeeper residue (Gly128 in TgCDPK1 and Ser147 in PfCDPK4) is boxed.
2.4. In vitro inhibition of recombinant PfCDPK1 and PfCDPK4
We first explored pyrazolopyrimidine inhibitors with large R1 substructures (Table 1), such as substituted phenyls, indazoles, indoles, naphthyls, and quinolones, because these moieties are directed towards the gatekeeper residue and are likely to impart selectivity for PfCDPK4 over mammalian kinases with bulkier residues at this position. Consistent with the hydrophobic pocket adjacent to the serine gatekeeper residue of PfCDPK4 being fairly permissive, 26 out of 38 analogs in Table 1 have an IC50 of less than 1 μM for this enzyme. However, some R1 substituents, such as 3-pyridyl (7a), biphenyl (13a), and naphthylmethyl (14a), are not tolerated and show no appreciable enzyme inhibition at the highest concentration tested (3 μM). In general, inhibitors containing either 2-naphthyl or 6-quinilino substituents possess the greatest activity against PfCDPK4, with 6-allyloxy-2-naphthyl (24) and 2-ethoxy-6-quinilino (30) substituents appearing to complement the ATP-binding site of this kinase most optimally; 24a and 30a demonstrate an IC50 of less than 20 nM. The extended hydrophobic surface of these substituents appear to make optimal interactions with the binding pocket next to the gatekeeper residue of PfCDPK4, much like these same substituents are able to make extensive contact with the highly similar ATP-binding site of TgCDPK1 [27].
Despite the ability of PfCDPK4’s ATP-binding site to favorably interact with a number of inhibitors, all of the analogs tested in Table 1 are more potent against glycine gatekeeper-containing TgCDPK1, with 63% of the compounds in Table 1 displaying a >20-fold lower IC50 for TgCDPK1 than PfCDPK4. Thus, the enlarged ATP-binding pocket of TgCDPK1 is better able to form favorable interactions with bulky C-3 substituents than the more constricted active site of PfCDPK4. Interestingly, the slightly larger threonine gatekeeper residue of PfCDPK1 does not alter inhibitor efficacy in the majority of cases. Of the inhibitors in Table 1 that were tested against both kinases, 67% differ in IC50 by less than 3-fold. However, several analogs that contain smaller 6-alkoxy-2-naphthyl and 2-alkoxy-6-quinilino C-3 substituents (16a, 17b, 30a, and 31a) are >5-fold more potent against PfCDPK4. In contrast, inhibitors that contain larger 6-alkoxy- and 6-benzyloxynaphtol groups (21a, 25a, 26a, 28a, and 33a) are >3-fold more potent against PfCDPK1 than PfCDPK4 (Table 1). Therefore, even small differences in the gatekeeper residue can result in varying sensitivities to inhibitors with large R1 substituents.
Next, analogs that contain variable R2 groups and 6-alkoxy-2-naphthyl or 2-alkoxy-6-quinilino substituents at the C-3 position (R1) were tested (Table 2). The most comprehensive series of these analogs contain a 6-ethoxy-2-naphthyl R1 group and variable R2 substituents. Like TgCDPK1, R2 groups that contain functionalities that are connected to the N-1 position of the pyrazolopyrimidine scaffold through a methylene linkage are favored by PfCDPK4 [28]. For example, 17k, which contains an N-methylpiperidine attached to the core scaffold through a methylene linkage, is ~4-fold more potent than an analog (17c) that contains no spacer. Similarly, 17q, which contains an isobutyl group at the R2 position, is more than 30-fold more potent than its i-Pr analog 17a. Overall, the most favorable R2 substituent is a 4-piperidinylmethylene group (series j, Table 2), which results in a >100-fold increase in potency against PfCDPK4 in the context of several R1 substituents. The two most potent PfCDPK4 inhibitors identified in this study (compounds 30j and 37j; IC50=2 nM) have a 4-piperidinylmethylene group at the R2 position and either a 2-ethoxyquinoline (30) or 6-cyclopropyloxynaphthyl (37) group at the R1 position. While other R2 groups do not confer similar increases in potency as the piperidinylmethylene (j) group, several substituents increase inhibitor potency by over 10-fold compared to their corresponding i-Pr R2 derivatives.
Much like the inhibitors shown in Table 1, almost all of the analogs in Table 2 are more potent against TgCDPK1 than PfCDPK4. However, the fold differences in IC50 for most inhibitors between these two kinases are much smaller, with several compounds (for example, 15j, 16j, and 29j) essentially being equipotent. Thus, the ability of the R2 position to interact favorably with the R1 position to confer selectivity is even more pronounced in PfCDPK4 than TgCDPK1. The ability of optimal R2 groups to confer increased potency seems to hold for threonine gatekeeper-containing PfCDPK1 as well, although to a lesser degree. The presence of a 4-piperidinylmethylene (j) group increases the potency of inhibitors that contain a 2-ethoxynaphthyl group (17) by ~10-fold relative to the i-Pr-containing analog. Several other R2 groups that contain methylene linkages also increase potency by 10-fold or more. However, favorable R2 groups will not allow 6-alkoxy-2-naphthyl and 2-alkoxy-6-quinilino C-3 substituents to be accommodated in CDPKs with every gatekeeper residue. A PfCDPK4 mutant with a methionine gatekeeper residue (PfCDPK4 S147M) is not sensitive to any of the compounds tested against it (15j, 17j, 17q, 30j, 30u and 37j; Table 4).
Table 4.
Enzymatic assay (IC50) results for PfCDPK4 wt, a methionine gatekeeper mutant of PfCDPK4 (Ser147Met), and the human tyrosine kinase Abl. P. falciparum gametocyte exflagellation inhibition (EC50) and growth inhibition (GI50) of human cell lines. All results are the averages of at least three assays. N/T=not tested.
|
PfCDPK4 IC50 (MM) |
P. falciparum exflagellation inhibition EC50 (MM) |
Abl IC50 (MM) |
S147M PfCDPK4 IC50 (MM) |
HEPG2 GI50 (MM) |
CRL8155 GI50 (mM) |
|
|---|---|---|---|---|---|---|
| 15j | 0.015 | 0.048 | > 10 | > 3 | N/T | > 30 |
| 17j | 0.004 | <0.040 | > 10 | > 3 | > 30 | > 30 |
| 17q | 0.014 | <0.040 | 2.7 | > 3 | > 30 | > 30 |
| 30j | 0.002 | <0.040 | > 10 | > 3 | > 30 | > 30 |
| 30u | 0.004 | <0.040 | > 10 | > 3 | > 30 | > 30 |
| 37j | 0.002 | <0.040 | > 10 | > 3 | > 30 | > 30 |
Beyond varying the R1 and R2 substituents of our PfCDPK4-directed compounds, we also evaluated the contribution of the scaffold to overall inhibitor selectivity and potency. To do this, a series of derivatives that contain optimal R1 and R2 groups displayed from an imidazopyrazine core were generated (Table 3). The imidazo[1,5-a]pyrazine scaffold makes almost the exact same hydrophobic and hydrogen-bonding contacts as the pyrazolopyrimidine scaffold, and projects its R1 and R2 substituents into similar regions of the ATP-binding pockets of kinases. A number of potent and selective inhibitors of IGF1R based on the imidazopyrazine core have previously been reported [33, 34]. As expected, similar combinations of R1 and R2 substituents generally confer similar inhibitory activity against PfCDPK4, independent of the scaffold. For example, the pyrazolopyrimidine-based (17a, Table 1) and imidazopyrazine-based (43a, Table 3) inhibitors that contain a 6-ethoxynapthyl (17) R1 and i-Pr (a) R2 groups have almost the same IC50s against PfCDPK4. The main exceptions to this trend are inhibitors that contain 4-piperidinemethylene (j) R2 groups. Pyrazolopyrimidine-based inhibitors that contain this R2 substituent are generally more potent than their corresponding imidazopyrazine analogs. This is most likely due to a slight variation in the relative presentation of the R1 and R2 substituents from both scaffolds, which we have found to be important for the selectivity and potency of inhibitors that contain bulkier R2 groups [28].
2.5. Pyrazolopyrimidine Inhibitors with Specific R1 and R2 Substituent Combinations are Selective for PfCDPK4 Over Src
While selectivity between threonine gatekeeper-containing-PfCDPK1 and serine gatekeeper-containing-PfCDPK4 provides information on how larger gatekeeper residues affect inhibitor potency in the context of almost identical ATP-binding sites, the main therapeutic liabilities of transmission-blocking kinase inhibitors are most likely off-target human kinases. To obtain a sense of target selectivity, a number of potent PfCDPK4 inhibitors were tested against the mammalian tyrosine kinase Src. Prior studies have shown that some pyrazolopyrimidine-based inhibitors are able to potently inhibit Src kinase [36, 37]. As the Src kinase gatekeeper position contains a threonine, which is one of the smallest gatekeeper residues in the human kinome, this enzyme was selected as a filter for off-target inhibition. A previously reported radioactive kinase assay was used to determine IC50 values for Src kinase [27].
The IC50s of inhibitors that contain i-Pr (a) and t-Bu (b) R2 groups are shown in the last column of Table 1. This inhibitor series clearly shows that it is challenging to obtain selectivity for PfCDPK4 over Src based on the interaction of the R1 substituent with the gatekeeper residue alone. Most aryl R1 substituents provide near equipotent inhibition of PfCDPK4 and Src, while inhibitors containing 6-benzyloxynaphthyl groups are selective for Src. However, a few R1 groups, such as 1-methyl-5-benzimidazole (11), 6-methoxynaphthyl (16), 6-ethoxynaphthyl (17), and 2-ethoxyquinoline (30) confer greater than 10-fold selectivity for PfCDPK4 over Src. Similar to our previous observations with TgCDPK1, inhibitors that contain R2 groups that are connected to the N-1 position of the pyrazolopyrimidine scaffold through a methylene linkage are significantly more selective for PfCDPK4 than their i-Pr- and t-Bu-containing analogs [29, 30]. For example, replacing the t-Bu R2 group of inhibitor 17b with a 4-azetidinemethylene (17g) or 4-piperidinemethylene (17j) group increases the selectivity for PfCDPK4 over Src from ~30-fold to greater than 1300- and 5,000-fold, respectively. This trend holds, regardless of the substituent displayed from the R1 position. It should be noted that the observed increase in PfCDPK4 selectivity is not only from increased potency against this enzyme, but also reduced affinity for Src. Therefore, like TgCDPK1, the magnitude of selective inhibition of PfCDPK4 over Src results from the interplay between the R1 and R2 substructures [30]. The ability of these analogs to potently inhibit PfCDPK4 with minimal potency against Src points to the likelihood that with an optimal combination of R1 and R2 groups it should be possible to selectively target this kinase over potential mammalian off-target kinases. Consistent with this notion, inhibitors that are highly selective for PfCDPK4 over Src show minimal inhibition of the human tyrosine kinase Abl (Table 4). Abl contains a threonine at the gatekeeper position and is also a likely off-target liability due to its sensitivity to pyrazolopyrimidine-based inhibitors [35].
2.6. Cellular Activity of PfCDPK4 Inhibitors
As an initial indicator of potential host cell toxicity of our transmission blocking compounds, we determined whether a number of potent PfCDPK4 inhibitors block the growth of human liver (HepG2) and lymphocyte (CRL-8155) cell lines (Table 4). Assays were performed with a previously reported procedure [29]. All of the compounds tested show minimal growth inhibition at a concentration of 30 μM, which is consistent with the absence of off-target toxicity for this series of inhibitors.
We have previously demonstrated that when nanomolar concentrations of 17j and 17k are present in human blood containing P. falciparum gametocytes, microgamete exflagellation and infection of A. stephensi is prevented [25, 26] In addition, a higher concentration of these drugs leads to the total absence of infective sporozoites in the dissected salivary glands of A. stephensi, with a lower dose still resulting in a significant decrease in observed sporozoites [25]. Importantly, intraperitoneal administration of 17j to mice that are infected with P. berghei expressing PfCDPK4 suppresses exflagellation in blood samples for up to 14 hours post-injection [25]. The strong correlation between the ability of compounds to inhibit PfCDPK4 in vitro and to block exflagellation strongly suggests that this effect is mediated through this kinase. Furthermore, P. falciparum expressing an exogenous drug-resistant PfCDPK4 mutant [substitution of the serine gatekeeper residue with methionine] show decreased sensitivity to compound 17j [26]. While several of our previously reported compounds show promising transmission blocking activity, we wanted to further demonstrate that potent PfCDPK4 inhibitors with diverse functionalities have the potential to act as transmission blocking agents. To do this 17q, 30u, and 37j were tested for their abilities to prevent P. falciparum exflagellation. All three of these inhibitors demonstrate an EC50 of less than 40 nM for parasite exflagellation. The efficacy of potent PfCDPK4 inhibitors to block exflagellation further strengthens our hypothesis that our compounds are eliciting their effects through this enzyme. Moreover, the cellular efficacy of inhibitors with diverse functional groups allows greater flexibility in the optimization of the PK/ADME properties of transmission blocking compounds.
3. Conclusions
In the present study, we have profiled a number of pyrazolopyrimidine- and imidazopyrazine-based ATP-competitive inhibitors against a CDPK, PfCDPK4, that is involved in parasite exflagellation. We found that it was possible to exploit similar interactions as our previous efforts to target Tg/CpCDPK1, and that despite PfCDPK4 possessing a larger serine gatekeeper residue, a number of these inhibitors possess low nanomolar potencies against this enzyme while retaining specificity against potential off-target human kinases. This is accomplished by exploiting the slightly enlarged pocket next to the serine gatekeeper residue of PfCDPK4, in combination with an additional interaction in the ribose-binding pocket. The observed selectivity and potency of our PfCDPK4 inhibitors carries over into cellular assays, where compounds potently inhibit P. falciparum exflagellation, but not the growth of human cell lines. Several compounds exhibit a large therapeutic window between inhibition of parasite exflagellation and human cell growth (e.g., >100-1000×). These inhibitors are undergoing additional testing to evaluate pharmacological properties such as solubility, pharmacokinetics, pharmacodynamics, and metabolism. Ideally, a compound would be given at the time of malaria therapy and would remain in the bloodstream for 3-4 weeks, the time it takes for gametocytes to be eliminated from humans after artemesinin combination therapy. Retention in the bloodstream for 3-4 weeks is necessary because it appears the effect on transmission is reversible, and the compound must be ingested with gametocytes to be effective [25]. Dosing at the time of antimalarial therapy is ideal, as it will be difficult, operationally, to ask people in endemic areas to take multiple doses at defined intervals to prevent transmission when they feel well. Only a few of our compounds have been tested to date in animal models, but they are cleared too quickly for 3-4 week persistence after co-administration with antimalarial therapy. Thus, future studies will focus on decreasing clearance of the compounds, while retaining favorable oral bioavailability, efficacy, and non-toxic parameters. In addition, strategies for obtaining sustained drug release are also being explored. The results obtained here will direct these future studies to evaluate lead candidates in parasitic transmission models, in order to select a few compounds to undergo final pre-clinical drug development testing as malaria transmission-blocking agents.
4. EXPERIMENTAL PROCEDURES
4.1. General synthetic methods
Unless otherwise stated, all chemicals were purchased from commercial suppliers and used without further purification. Reaction progress was monitored by thin-layer chromatography on silica gel 60 F254 coated glass plates (EM Sciences). Chromatography was performed using an IntelliFlash 280 automated flash chromatography system, eluting on pre-packed Varian SuperFlash silica gel columns with hexanes/EtOAc or CH2Cl2/MeOH gradient solvent systems. For preparatory HPLC purification, samples were chromatographically separated using a Varian Dynamax Microsorb 100-5 C18 column (250 mm × 21.4 mm), eluting with H2O/CH3CN or H2O/MeOH gradient solvent systems (+0.05% TFA). The purity of all final compounds was determined by two analytical RP-HPLC methods, using an Agilent ZORBAX SB-C18 (2.1 mm × 150 mm) or Varian Microsorb-MV 100-5 C18 column (4.6 mm × 150 mm), and eluting with either H2O/CH3CN or H2O/MeOH gradient solvent systems (0.05% TFA) run over 30 min. Products were detected by UV at =254 nm, with all final compounds displaying >95% purity. NMR spectra were recorded on Bruker 300 or 500 MHz spectrometers at ambient temperature. Chemical shifts are reported in parts per million (δ) and coupling constants in Hz. 1H-NMR spectra were referenced d6 to the residual solvent peaks as internal standards (7.26 ppm for CDCl3, 2.50 ppm for -DMSO, and 3.34 ppm for CD3OD). Mass spectra were recorded with a Bruker Esquire Liquid Chromatograph - Ion Trap Mass Spectrometer. Inhibitors were synthesized through several different routes, as represented in Schemes 1-3. Compound characterization data is presented in the Supporting Information.
4.2. Spectral data of compounds
4.2.1. 1-(4-(2-(4-amino-3-(naphthalen-2-yl)-1H-pyrazolo[3,4-d]pyrimidin-1-yl)ethyl)piperidin-1-yl)ethanone (15o)
1H NMR (301 MHz, d6-DMSO): δ ppm 8.29 (s, 1H), 8.20 (s, 1H), 8.11-7.48 (m, 3H), 7.83 (dd, J=8.5, 1.6 Hz, 1H), 7.63-7.56 (m, 2H), 4.42 (t, J=6.8 Hz, 2H), 3.40 (m, 2H), 3.25 (m, 2H), 2.57 (m, 2H), 1.97 (s, 3H), 1.80 (m, 2H), 1.42 (m, 1H), 1.22 (m, 2H); MS (ESI): 415.4 m/z [MH+], C24H26N6O requires 415.2; HPLC-1=99.0% pure, HPLC-2=98.4% pure.
4.2.2. 1-(naphthalen-2-yl)-3-((tetrahydro-2H-pyran-4-yl)methyl)-1H-pyrazolo[3,4-d]pyrimidin-4-amine (15p)
1H NMR (300 MHz, d6-DMSO): δ ppm 8.46 (s, 1H), 8.20 (s, 1H), 8.09 (d, J=8.5 Hz, 1H), 8.02-7.94 (m, 2H), 7.80 (d, J=8.5 Hz, 1H), 7.63-7.59 (m, 1H), 7.58-7.52 (m, 1H), 4.44 (d, J=7.0 Hz, 2H), 4.02-3.92 (m, 2H), 3.47-3.35 (m, 2H), 2.37 (m, 1H), 1.65-1.53 (m, 2H), 1.53-1.40 (m, 2H); MS (ESI): 360.2 m/z [MH+], C21H21N5O requires 360.1; HPLC-1=96.0% pure, HPLC-2=97.0% pure.
4.2.3. 1-(naphthalen-2-yl)-3-((acetylpiperazine-4-yl)ethanone)-1H-pyrazolo[3,4-d]pyrimidin-4-amine (15t)
1H NMR (301 MHz, d6-DMSO): δ ppm 8.26 (s, 1H), 8.21 (s, 2H), 7.99-8.04 (m, 3H), 7.82 (dd, J=8.5, 1.6 Hz, 1H), 7.60 (m, 3H), 5.43 (d, J=4.6 Hz, 2H), 3.71-3.54 (m, 4H), 3.54-3.40 (m, 4H), 2.08 (s, 3H); MS (ESI): 430.4 m/z [MH+], C23H23N7O2 requires 430.2; HPLC-1=97.8% pure, HPLC-2=97.6% pure.
4.2.4. 1-(azetidin-3-ylmethyl)-3-(6-ethoxynaphthalen-2-yl)-1H-pyrazolo[3,4-d]pyrimidin-4-amine (17g)
1H NMR (300 MHz, CD3OD): δ ppm 8.52 (s, 1H), 8.17 (s, 1H), 8.00 (d, J=8.3 Hz, 1H) 7.92 (d, J=8.9 Hz, 1H), 7.79 (d, J=8.5 Hz, 1H), 7.37 (s, 1H), 7.27 (d, J=8.7 Hz, 1H), 4.84 (d,J=6.4 Hz, 2H), 4.23 (m, 6H), 3.63 (m, 1H), 1.50 (t, J=6.2 Hz, 3H); MS (ESI): 375.1 m/z [MH+], C21H22N6O requires 375.4; HPLC-1=96.1% pure, HPLC-2=97.0% pure.
4.2.5. 3-(6-ethoxynaphthalen-2-yl)-1-((1-methylazetidin-3-yl)methyl)-1H-pyrazolo[3,4-d]pyrimidin-4-amine (17h)
1H NMR (300 MHz, CD3OD): δ ppm 8.50 (s, 1H), 8.15 (s, 1H), 8.00 (d, J=8.5 Hz, 1H,) 7.92 (d, J=9.1 Hz, 1H), 7.77 (d, J=8.2 Hz, 1H), 7.37 (d, J=2.4 Hz, 1H), 7.27 (dd, J=9.1, 2.4 Hz, 1H), 4.84 (q, J=6.6 Hz, 2H), 4.45 (q, J=6.2 Hz, 2H), 4.20 (m, 4H), 3.55 (m, 1H), 2.96 (s, 3H), 1.50 (t, J=7.0 Hz, 3H); MS (ESI): 389.1 m/z [MH+], C22H24N6O requires 389.2; HPLC-1=97.0% pure, HPLC-2=97.5% pure.
4.2.6. 1-(3-((4-amino-3-(6-ethoxynaphthalen-2-yl)-1H-pyrazolo[3,4-d]pyrimidin-1-yl)methyl)azetidin-1-yl)ethanone (17i)
1H NMR (300 MHz, CDCl3): δ ppm 8.38 (s, 1H), 8.06 (s, 1H), 7.92-7.70 (m, 3H) 7.25 (m, 2H), 5.70 (br s, 2H), 4.68 (d, J=7.2 Hz, 2H), 4.18-3.97 (m, 6H), 3.29 (m, 1H), 1.86 (s, 3H), 1.51 (t, J=7.0 Hz, 3H); MS (ESI): 417.4 m/z [MH+], C23H24N6O2 requires 417.4; HPLC-1=95.6% pure, HPLC-2=95.3% pure.
4.2.7. 3-(6-ethoxynaphthalen-2-yl)-1-((1-isopropylpiperidin-4-yl)methyl)-1H-pyrazolo[3,4-d]pyrimidin-4-amine (17l)
1H NMR (300 MHz, CD3OD): δ ppm 8.46 (s, 1H), 8.13 (s, 1H), 7.97 (d, J=7.8 Hz, 1H) 7.89 (d, J=8.7 Hz, 1H), 7.77 (d, J=8.2 Hz, 1H), 7.34 (s, 1H), 7.23 (d, J=8.5 Hz, 1H), 4.51 (d,J=6.2 Hz, 2H), 4.20 (q, J=6.6 Hz, 2H), 3.48 (m, 2H), 3.05 (m, 3H,), 2.44 (m, 1H), 2.01 (m, 2H), 1.76 (m, 2H), 1.48 (t, J=6.8 Hz, 3H), 1.34 (d, J=6.0 Hz, 6H); MS (ESI): 445.4 m/z [MH+], C26H32N6O requires 445.1; HPLC-1=95.0% pure, HPLC-2=97.5% pure.
4.2.8. 1-(4-(2-(4-amino-3-(6-ethoxynaphthalen-2-yl)-1H-pyrazolo[3,4-d]pyrimidin-1-yl)ethyl)piperidin-1-yl)ethanone (17o)
1H NMR (300 MHz, CD3OD): δ ppm 8.34 (s, 1H), 7.97 (s, 1H), 7.88-7.73 (m, 2H), 7.62 (d, J=8.5 Hz, 1H), 7.20 (s, 1H), 7.11 (d, J=8.7 Hz, 1H), 4.41 (m, 3H), 4.09 (q, J=6.8 Hz, 2H,), 3.83 (m, 1H), 3.01 (m, 1H), 2.58 (m, 1H), 2.07 (s, 3H), 1.93-1.71 (m, 4H), 1.54-1.32 (m, 4H), 1.25-0.97 (m, 2H); MS (ESI): 459.2 m/z [MH+], C26H30N6O2 requires 459.2; HPLC-1=99.0% pure, HPLC-2=95.2% pure.
4.2.9. 3-(6-ethoxynaphthalen-2-yl)-1-((tetrahydro-2H-pyran-4-yl)methyl)-1H-pyrazolo[3,4-d]pyrimidin-4-amine (17p)
1H NMR (300 MHz, d6-DMSO): δ ppm 8.28 (s, 1H), 8.12 (s, 1H), 7.96 (d, J=8.7 Hz, 2H), 7.75 (d, J=8.7 Hz, 1H), 7.41 (s, 1H), 7.23 (d, J=9.6 Hz, 1H), 4.28 (d, J=7.1 Hz, 2H), 4.20 (q, J=6.8 Hz, 2H), 3.84 (m, 2H), 3.26 (m, 2H), 2.24 (m, 1H), 1.53-1.39 (m, 5H), 1.30 (m, 2H); MS (ESI): 404.3 m/z [MH+], C23H25N5O2 requires 404.1; HPLC-1=98.0% pure, HPLC-2=97.0% pure.
4.2.10. 3-(6-ethoxynaphthalen-2-yl)-1-isobutyl-1H-pyrazolo[3,4-d]pyrimidin-4-amine (17q)
1H NMR (300 MHz, CD3OD): δ ppm 8.29 (s, 1H), 8.12 (s, 1H), 7.98 (d, J=8.2 Hz, 1H), 7.92 (d, J=9.1 Hz, 1H), 7.77 (dd, J=8.7, 2.0 Hz, 1H), 7.35 (d, J=2.4 Hz, 1H), 7.27-7.22 (dd, J=9.1, 2.9 Hz, 1H), 4.29-4.18 (m, 4H), 2.40 (m, 1H), 1.50 (t, J=6.8 Hz, 3H), 0.99 (d, J=6.6 Hz, 6H); MS (ESI): 362.2 m/z [MH+], C21H23N5O requires 362.2; HPLC-1=96.3% pure, HPLC-2=97.2% pure.
4.2.11. 1-(cyclopropylmethyl)-3-(6-ethoxynaphthalen-2-yl)-1H-pyrazolo[3,4-d]pyrimidin-4-amine (17r)
1H NMR (300 MHz, CD3OD): δ ppm 8.43 (s, 1H), 8.11 (s, 1H), 7.96 (d, J=8.5 Hz, 1H), 7.88 (d, J=9.1 Hz, 1H), 7.75 (d, J=8.5 Hz, 1H), 7.32 (d, J=2.0 Hz, 2H) 4.35 (d, J=7.2 Hz, 2H), 4.20 (q, J=6.8 Hz, 2H), 1.48 (m, 4H), 0.62 (m, 2H), 0.53 (m, 2H); MS (ESI): 360.2 m/z [MH+], C21H21N5O requires 360.2; HPLC-1=95.1% pure, HPLC-2=96.6% pure.
4.2.12. 3-(6-(cyclopropylmethoxy)naphthalen-2-yl)-1-isopropyl-1H-pyrazolo[3,4-d]pyrimidin-4-amine (22a)
1H NMR (300 MHz, CD3OD): δ ppm 8.38 (s, 1H), 8.07 (s, 1H), 7.98-7.85 (m, 2H), 7.73 (d, J=8.9 Hz, 1H), 7.29 (d, J=8.7 Hz, 2H), 5.26, (m, 1H), 3.99 (d, J=6.8 Hz, 2H), 1.67 (d, J=6.6 Hz, 6H), 1.37 (m, 1H), 0.70 (q, J=6.0 Hz, 2H), 0.44 (q, J=4.7 Hz, 2H,); MS (ESI): 374.3 m/z [MH+], C22H23N5O requires 374.1; HPLC-1=95.1% pure, HPLC-2=95.2% pure.
4.2.13. 3-(6-(cyclopropylmethoxy)naphthalen-2-yl)-1-(piperidin-4-ylmethyl)-1H-pyrazolo[3,4-d]pyrimidin-4-amine (22j)
1H NMR (300 MHz, CD3OD): δ ppm 8.46 (s, 1H), 8.11 (s, 1H), 7.92 (m, 2H), 7.73 (s, 1H), 7.29 (s, 2H), 4.51 (d, J=6.5 Hz, 2H), 3.99 (d, J=6.8 Hz, 2H), 3.46 (m, 2H), 2.99 (m, 2H), 2.45 (m, 1H), 1.93 (m, 2H), 1.78 (m, 2H), 1.34 (m, 1H), 0.72 (m, 2H), 0.46 (m, 2H); MS (ESI) 429.4 m/z [MH+], C25H28N6O requires 429.2; HPLC-1=99.2% pure, HPLC-2=98.4% pure.
4.2.14. 1-isopropyl-3-(6-(2-methoxyethoxy)naphthalen-2-yl)-1H-pyrazolo[3,4-d]pyrimidin-4-amine (23a)
1H NMR (300 MHz, CD3OD): δ ppm 8.42 (s, 1H), 8.11 (s, 1H), 7.85-8.04 (m, 2H), 7.77 (d, J=8.2 Hz, 1H), 7.33 (s, 2H), 5.28 (m, 1H), 4.31, (t, J=4.3 Hz, 2H), 3.88 (t, J=4.5 Hz, 2H), 3.50 (s, 3H), 1.65 (d, J=6.4 Hz, 6H); MS (ESI): 378.2 m/z [MH+], C21H23N5O2 requires 378.1; HPLC-1=95.0% pure, HPLC-2=95.4% pure.
4.2.15. 1-((1-methylpiperidin-4-yl)methyl)-3-(quinolin-6-yl)-1H-pyrazolo[3,4-d]pyrimidin-4-amine (29k)
1H NMR (300 MHz, CD3OD): δ ppm 9.10 (d, J=3.5 Hz, 1H), 8.82 (d, J=8.2 Hz, 1H), 8.44 (m, 2H), 8.30 (m, 2H), 7.87 (dd, J=8.2, 4.6 Hz, 1H), 4.51 (d, J=4.6 Hz, 2H), 3.54 (m, 2H), 2.97 (t, J=12.0 Hz, 2H), 2.84 (s, 3H), 2.40 (m, 1H), 1.98 (m, 2H), 1.69 (m, 2H); MS (ESI) 374.1 m/z [MH+], C21H24N7 requires 374.2; HPLC-1=97.0% pure, HPLC-2=98.5% pure.
4.2.16. 1-(4-(2-(4-amino-3-(quinolin-6-yl)-1H-pyrazolo[3,4-d]pyrimidin-1-yl)ethyl)piperidin-1-yl)ethanone (29o)
1H NMR (301 MHz, d6-DMSO): δ ppm 8.96 (d, J=2.4 Hz, 1H), 8.50 (d, J=8.2 Hz, 1H), 8.27 (m, 2H), 8.16 (d, J=8.2 Hz, 1H), 8.06 (d, J=8.5 Hz, 1H), 7.61 (dd, J=8.5, 4.1 Hz, 1H), 4.45 (t, J=6.5 Hz, 2H), 4.32-3.74 (m, 2H), 2.92 (m, 1H), 1.97 (s, 3H), 1.84 (m, 2H), 1.47 (m, 4H), 1.24-0.95 (m, 2H); MS (ESI): 416.4 m/z [MH+], C23H25N7O requires 416.1; HPLC-1=98.6% pure, HPLC-2=99.0% pure.
4.2.17. 3-(2-ethoxyquinolin-6-yl)-1-isopropyl-1H-pyrazolo[3,4-d]pyrimidin-4-amine (30a)
1H NMR (300 MHz, CD3OD): δ ppm 8.68 (d, J=8.9 Hz, 1H), 8.47 (s, 1H), 8.33 (s, 1H), 8.26-8.01 (m, 2H), 7.64 (m, 1H), 5.31 (m, 1H), 4.70 (q, J=7.5 Hz, 2H), 1.64 (d, J=6.2 Hz, 6H), 1.55 (t, J=7.0 Hz, 3H); MS (ESI): 349.2 m/z [MH+], C19H20N6O requires 349.1; HPLC-1=98.0% pure, HPLC-2=95.0% pure.
4.2.18. 3-(2-ethoxyquinolin-6-yl)-1-((tetrahydro-2H-pyran-4-yl)methyl)-1H-pyrazolo[3,4-d]pyrimidin-4-amine (30p)
1H NMR (300 MHz, CD3OD): δ ppm 8.45 (s, 1H), 8.35-8.01 (m, 2H), 7.98 (s, 1H), 7.67-7.47 (m, 1H), 7.07 (s, 1H), 4.55 (q, J=6.8 Hz, 2H), 4.41 (m, 2H), 3.95 (d, J=6.6 Hz, 2H), 3.40 (m, 2H), 2.35 (m, 1H), 1.58 (m, 2H), 1.51-1.41 (m, 5H); MS (ESI): 405.2 m/z [MH+], C22H24N6O2 requires 405.1; HPLC-1=98.0% pure, HPLC-2=97.0% pure.
4.2.19. 3-(4-amino-3-(2-ethoxyquinolin-6-yl)-1H-pyrazolo[3,4-d]pyrimidin-1-yl)-2,2-dimethylpropan-1-ol (30u)
1H NMR (300 MHz, CD3OD): δ ppm 8.31 (s, 1H), 8.25 (d, J=9.1 Hz, 1H), 8.14 (s, 1H), 7.99 (s, 2H), 7.03 (d, J=9.1 Hz, 1H ), 4.57 (q, J=6.9 Hz, 2H), 4.35 (s, 4H), 1.49 (t, J=7.3 Hz, 3H), 0.88 (s, 6H); MS (ESI): 393.2 m/z [MH+]; C21H24N6O2 requires 393.5; HPLC-1=95.2% pure, HPLC-2=98.0% pure.
4.2.20. 3-(2-ethoxy-8-methylquinolin-6-yl)-1-isopropyl-1H-pyrazolo[3,4-d]pyrimidin-4-amine (31a)
1H NMR (300 MHz, CD3OD): δ ppm 8.26 (s, 1H), 8.20 (m, 1H), 7.93 (s, 1H), 7.83 (m, 1H), 6.98 (d, J=8.9 Hz, 1H), 5.15 (m, 1H), 4.59 (q, J=7.0 Hz, 2H), 1.60 (d, J=6.6 Hz, 6H), 2.80 (s, 3H), 1.49 (t, J=6.6 Hz, 3H); MS (ESI): 363.1 m/z [MH+], C20H22N6O requires 363.1; HPLC-1=95.0% pure, HPLC-2=96.0% pure.
4.2.21. 3-(2-(cyclopropylmethoxy)-8-methylquinolin-6-yl)-1-isopropyl-1H-pyrazolo[3,4-d]pyrimidin-4-amine (32a)
1H NMR (300 MHz, CD3OD): δ ppm 8.44 (s, 1H), 8.06 (d, J=8.9 Hz, 1H,), 7.87 (s, 1H) 7.77 (s, 1H), 6.71 (d, J=8.9Hz, 1H,), 5.27(m, 1H), 3.45 (m, 2H), 2.63 (s, 3H), 1.70-1.60 (t, J=6.6Hz, 6H), 1.26-1.11 (m, 2H), 0.89 (m, 1H), 0.70-0.42 (m, 2H); MS (ESI): 389.6 m/z [MH+], C22H24N6O requires 389.2; HPLC-1=95.0% pure, HPLC-2=95.0% pure.
4.2.22. 3-(2-(benzyloxy)quinolin-6-yl)-1-isopropyl-1H-pyrazolo[3,4-d]pyrimidin-4-amine (33a)
1H NMR (300 MHz, CD3OD) δ 8.42 (s, 1H), 8.27 (d, J=8.9 Hz, 1H), 8.15 (d, J=1.5 Hz, 2H), 8.04-7.95 (m, 2H), 7.54-7.49 (m, 2H) 7.41-7.31 (m, 2H), 7.10 (d, J=8.9 Hz, 1H), 5.57 (s, 2H), 5.28 (m, 1H), 1.63 (d, J=6.6 Hz, 6H); MS (ESI): 411.1 m/z [MH+], C24H22N6O requires 411.1; HPLC-1=95.0% pure, HPLC-2=99.0% pure.
4.2.23. 3-(2-(benzyloxy)quinolin-6-yl)-1-(piperidin-4-ylmethyl)-1H-pyrazolo[3,4-d]pyrimidin-4-amine (33j)
1H NMR (300 MHz, CD3OD): δ ppm 8.26 (s, 1H), 8.21 (d, J=8.9 Hz, 1H), 8.10 (s, 1H), 7.97 (s, 2H), 7.51 (d, J=7.2 Hz, 2H), 7.41-7.29 (m, 3H), 7.06 (d, J=8.7 Hz, 1H), 5.55 (s, 2H), 4.30 (d, J=6.8 Hz, 2H), 3.03 (m, 2H), 2.55 (m, 2H), 2.19 (m, 1H), 1.60 (m, 2H), 1.35 (m, 2H); MS (ESI): 466.1 m/z [MH+], C27H27N7O requires 466.2; HPLC-1=95.0% pure, HPLC-2=98.2% pure.
4.2.24. 3-(2-(benzyloxy)-8-methylquinolin-6-yl)-1-isopropyl-1H-pyrazolo[3,4-d]pyrimidin-4-amine (34a)
1H NMR (300 MHz, CDCl3): δ ppm 8.25 (s, 1H), 8.06 (d, J=8.9 Hz, 1H), 7.84 (s, 1H), 7.75 (s, 1H), 7.57 (d, J=8.0 Hz, 2H), 7.46-7.33 (m, 3H), 7.08 (d, J=8.8 Hz, 1H), 5.62 (s, 2H), 5.28–5.15 (m, 1H), 2.81 (s, 3H), 1.66 (d, J=6.7 Hz, 6H); MS (ESI): 425.2 m/z [MH+], C25H24N6O requires 425.2; HPLC-1=99.0% pure, HPLC-2=95.0% pure.
4.2.25. 3-(2-(benzyloxy)-8-methylquinolin-6-yl)-1-(piperidin-4-ylmethyl)-1H-pyrazolo[3,4-d]pyrimidin-4-amine (34j)
1H NMR (300 MHz, CD3OD): δ ppm 8.27 (s, 1H), 8.20 (d, J=8.7 Hz, 1H), 7.94 (s, 1H), 7.82 (s, 1H), 7.54 (d, J=7.0 Hz, 2H), 7.35 (m, 3H), 7.06 (d, J=8.9 Hz, 1H), 5.60 (s, 2H), 4.31 (d, J=7.2 Hz, 2H), 3.05 (m, 2H), 2.77 (s, 3H), 2.57 (m, 2H), 2.20 (m, 1H), 1.63 (m, 2H), 1.35 (m, 2H); MS (ESI): 480.1 m/z [MH+], C28H29N7O requires 480.2; HPLC-1=95.5% pure, HPLC-2=95.0% pure.
4.2.26. 1-(1-(methylsulfonyl)piperidin-4-yl)-3-(quinolin-3-yl)-1H-pyrazolo[3,4-d]pyrimidin-4-amine (35e)
1H NMR (300 MHz, d6-DMSO): δ ppm 9.26 (d, J=2.0 Hz, 1H), 8.81 (s,1H), 8.51 (s, 1H), 8.20 (dd, J=7.4, 2.3 Hz, 1H), 7.94 (m, 2H), 7.78 (m, 1H), 4.99 (m, 1H), 3.75 (m, 2H), 3.07 (m, 2H), 2.96 (s, 3H), 2.26 (m, 2H), 2.14 (m, 2H); MS (ESI): 424.3 m/z [MH+]; C20H21N7O2S requires 424.2; HPLC-1=97.8% pure, HPLC-2=98.0% pure.
4.2.27. 1-(4-(4-amino-3-(quinolin-3-yl)-1H-pyrazolo[3,4-d]pyrimidin-1-yl)piperidin-1-yl)ethanone (35f)
1H NMR (300 MHz, d6-DMSO): δ ppm 9.17 (d, J=2.1 Hz, 1H), 8.59 (d, J=2.1 Hz, 1H), 8.30 (s, 1H), 8.11 (m, 2H), 7.83 (t, J=8.5 Hz, 1H), 7.69 (t, J=7.9 Hz, 1H), 5.04 (m, 1H), 4.55 (m, 1H), 4.00 (m, 1H), 3.40-3.33(m, 2H), 2.80 (m,1H), 2.20 (m, 1H), 2.07 (s, 3H), 1.99 (m, 2H); MS (ESI): 388.4 m/z [MH+], C21H21N7O requires 388.2; HPLC-1=99.5% pure, HPLC-2=99.2% pure.
4.2.28. 3-(6-cyclopropoxynaphthalen-2-yl)-1-(piperidin-4-ylmethyl)-1H-pyrazolo[3,4-d]pyrimidin-4-amine (37j)
1H NMR (300 MHz, CD3OD): δ ppm 8.47 (s, 1H), 8.15 (s, 1H), 8.01 (d, J=8.5 Hz, 1H), 7.91 (d, J=9.1 Hz, 1H), 7.78 (d, J=8.5 Hz, 1H), 7.62 (d, J=2.0 Hz, 1H), 7.25 (dd, J=8.9, 1.8 Hz, 1H), 4.50 (d, J=6.0 Hz, 2H), 3.96 (m, 1H), 3.54 (m, 2H), 3.00 (m, 2H), 2.40 (m, 1H), 2.00 (m, 2H), 1.71 (m, 2H), 0.90 (m, 2H), 0.78 (m, 2H); MS (ESI): 415.1 m/z [MH+]; C24H26N6O requires 415.2; HPLC-1=95.5% pure, HPLC-2=97.2% pure.
4.2.29. 1-(6-ethoxynaphthalen-2-yl)-3-(piperidin-4-ylmethyl)imidazo[1,5-a]pyrazin-8-amine (43j)
1H-NMR (500 MHz, CD3OD): δ ppm 8.08 (s, 1H), 7.97 (d, J=8.3 Hz, 1H), 7.87 (d, J=8.7 Hz, 1H), 7.80 (d, J=5.5 Hz, 1H), 7.70 (d, J=7.6 Hz, 1H), 7.34 (s, 1H), 7.24 (d, J=8.1 Hz, 1H), 7.02 (d, J=5.5 Hz, 1H ), 4.20 (q, J=6.8 Hz, 2H), 3.41 (d, J=10.0 Hz, 2H), 3.09 (m, 2H), 3.00 (m, 2H), 2.31 (m, 1H), 2.01 (m, 2H), 1.60 (m, 2H), 1.49 (t, J=6.8 Hz, 3H); MS (ESI): 402.1 m/z [MH+], C24H27N5O requires 402.2; HPLC-1=97.0% pure, HPLC-2=96.0% pure.
4.2.30. 1-(6-ethoxynaphthalen-2-yl)-3-((1-methylpiperidin-4-yl)methyl)imidazo[1,5-a]pyrazin-8-amine (43k)
1H-NMR (500 MHz, CD3OD): δ ppm 8.09 (s, 1H), 7.98 (d, J=9.1 Hz, 1H), 7.89 (d, J=9.1 Hz, 1H), 7.82 (d, J=5.8 Hz, 1H), 7.72 (d, J=8.9 Hz, 1H), 7.35 (s, 1H), 7.25 (d, J=8.9 Hz, 1H), 7.03 (d, J=5.8 Hz, 1H), 4.21 (q, J=6.6 Hz, 2H), 3.53 (d, J=10.0 Hz, 2H), 3.11 (m, 2H,), 3.03 (m, 2H), 2.85 (s, 3H), 2.32 (m, 1H), 2.07 (m, 2H), 1.76-1.56 (m, 2H), 1.48 (t, J=6.6 Hz, 3H); MS (ESI): 416.1 m/z [MH+] C25H29N5O requires 416.2; HPLC-1=96.0% pure, HPLC-2=97.0% pure.
4.3. PfCDPK4 activity assays
The wild type and Ser147Met gatekeeper mutants of PfCDPK4 were expressed, purified, and enzymatically characterized as described previously [25, 26]. PfCDPK4 activity assays were performed in assay buffer containing 20 mM HEPES (pH=7.5), 0.1% BSA, 10 mM MgCl2, 1 mM EGTA, 2 mM CaCl2, 10 μM ATP, and 40 μM Syntide-2 peptide substrate (peptide sequence: PLARTLSVAGLPGKK-OH). After incubating for 90 min at 30 °C, the enzymatic reactions were terminated by adding EGTA [final concentration=5 mM]. The amount of ATP remaining was evaluated using the Kinase Glo luciferase assay from Promega. Sample luminescence was determined using a Microbeta 2 plate reader (Perkin Elmer, Waltham, MA). Data were converted to percent inhibition and IC50 values were calculated with non-linear regression analysis using GraphPad Prism. All IC50 values shown are the average of assays that were performed in triplicate or quadruplicate.
4.4. Human kinase enzymatic assays
Compounds were tested for Src kinase inhibition using either the truncated catalytic kinase domain (Src-KD) or a construct that also contains Src’s regulatory SH2 and SH3 domains (Src-3D). No significant difference in IC50 values was observed between the two constructs, and values are therefore reported together as Src kinase IC50. Several compounds were further tested against Abl tyrosine kinase. For both Src and Abl, compounds were tested in 3-fold dilution series starting at an initial concentration of 10 μM according to previously reported procedures. Data were converted to percent inhibition and IC50 values were calculated with non-linear regression analysis using GraphPad Prism. All IC50 values shown are the average of assays that were performed in triplicate or quadruplicate. Assay buffers, enzyme concentrations, ATP concentrations, substrate peptide sequences and concentrations, and enzymatic reaction times are listed in the assay-specific details presented below.
Src assay buffer: recombinant Src-KD or Src-3D (1-2 nM), 33.5 mM HEPES, pH=7.5, 6.7 mM MgCl2, 1.7 mM EGTA, 67 mM NaCl, 2 mM Na3VO4, 0.08 mg/ml BSA, γ32P ATP (0.2 μCi/well). Enzymatic reaction time: 30 min for Src-KD or 60 min for Src-3D. Substrate peptide sequence and concentration: Ac-EIYGEFKKK-OH (100 μM).
Abl assay buffer: recombinant Abl (1-2 nM), 33.5 mM HEPES, pH=7.5, 6.7 mM MgCl2, 1.7 mM EGTA, 67 mM NaCl, 2 mM Na3VO4, 0.08 mg/ml BSA, γ32P ATP (0.2 μCi/well). Enzymatic reaction time: 30 min for Abl-KD and 60 min for Abl-3D. Substrate peptide sequence and concentration: Ac-EAIYAAPFAKKK-OH (100 μM).
4.5. Human cell growth inhibition assays
PfCDPK4-selective compounds were tested for the growth inhibition of two human cell lines: HepG2 (liver) and CRL-8155 (lymphocytic) cells. Cells were grown in either DMEM/F12 (HepG2) or RPMI-1640 (CRL-8155) growth media supplemented with 10% heat inactivated fetal calf serum and 2 mM L-glutamine. HepG2 growth medium additionally contained 25 mM HEPES. CRL-8155 growth medium additionally contained 10 mM HEPES, 1 mM sodium pyruvate, 4.5 g/L glucose and 1.5 g/L sodium bicarbonate. Cells were grown in the presence of varying concentration of inhibitor (highest concentration=30 μM; 3-fold dilution) for 48 or 72 hours at 37°C and 5% CO2 in 96-well flat-bottom plates (Corning). Growth was quantified using Alamar Blue as a developing reagent and detecting sample absorbance at λ=570 nm (600 nm reference wavelength). Percent growth inhibition by test compounds were calculated based on cultures incubated with DMSO negative and tipifarnib (R115777) positive controls (0% and 100% growth inhibition, respectively). All assays were performed in triplicate.
4.6. P. falciparum exflagellation assays
Cultures of wild-type P. falciparum that had been started at 0.5% were grown for 15 days in RPMI 1640 supplemented with 50 mM hypoxanthine and 10% A+ human serum, with daily media changes. Exflagellation was monitored beginning on day 14. After 15 days, the cultures were divided into flasks and varying concentrations of inhibitors or DMSO were added [25]. A wet mount slide was used for induction and monitoring of exflagellation. Parasites were monitored for exflagellation from 10 minutes to 25 minutes after exflagellation was induced. Exflagellation centers per 10,000 erythrocytes were counted.
Supplementary Material
Highlights.
Potent pyrazolopyrimidine-based inhibitors of PfCDPK4 have been identified
High selectivity for PfCDPK4 over human kinases achieved
Inhibitors of PfCDPK4 are also able to block P. falciparum exflagellation
The ATP-binding sites of several parasite kinases have been profiled
Molecular determinants of selective PfCDPK4 inhibition characterized
Acknowledgement
The authors would like to thank Stephen Nakazawa Hewitt and DNAillustrated.com for designing and depicting the Plasmodium life cycle figure. The authors also acknowledge the helpful support of Lynn K. Barrett. Research reported in this publication was supported by the National Institute of Allergy and Infectious Diseases and the National Institute of General Medical Sciences of the National Institutes of Health under award numbers R01AI089441 and R01GM086858. K.R.K. was supported by a training scholarship from the University of Washington Plein Endowment for Geriatric Pharmacy Research. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Abbreviations List
- ACT
artemisinin combination therapy
- ATP
adenosine triphosphate
- Boc
tert-butyloxycarbonyl
- CAMKII
Ca2+/calmodulin-dependent protein kinase II
- CDPK1
calcium-dependent protein kinase 1
- CpCDPK1
Cryptosporidium parvum CDPK1
- DIAD
diisopropyl azodicarboxylate
- DME
dimethoxyethane
- EC50
half maximal effective concentration
- GI50
half maximal growth inhibition
- IC50
half maximal inhibitory concentration
- IGF1R
insulin-like growth factor 1 receptor
- PfCDPK1
Plasmodium falciparum CDPK1
- PfCDPK4
Plasmodium falciparum CDPK4
- TBDMS
tert-butyldimethylsilyl
- TgCDPK1
Toxoplasma gondii CDPK1
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Synthesis, characterization data for all intermediate compounds and 1H NMR spectra for new compounds were given in supporting information. This material is available free of charge via the Internet at http://
REFERENCES
- [1].W.H. Organization . World Malaria Report 2010. Geneva: 2010. [Google Scholar]
- [2].Carter R, Mendis KN. Evolutionary and historical aspects of the burden of malaria. Clin Microbiol Rev. 15(2002):564–594. doi: 10.1128/CMR.15.4.564-594.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Snow RW, Guerra CA, Noor AM, Myint HY, Hay SI. The global distribution of clinical episodes of Plasmodium falciparum malaria. Nature. 2005;434:214–217. doi: 10.1038/nature03342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Greenwood BM, Fidock DA, Kyle DE, Kappe SH, Alonso PL, Collins FH, Duffy PE. Malaria: progress, perils, and prospects for eradication. J Clin Invest. 2008;118:1266–1276. doi: 10.1172/JCI33996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Jongwutiwes S, Putaporntip C, Iwasaki T, Sata T, Kanbara H. Naturally acquired Plasmodium knowlesi malaria in human, Thailand, Emerg. Infect. Dis. 2004;10:2211–2213. doi: 10.3201/eid1012.040293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Cox-Singh J, Davis TME, Lee KS, Shamsul SSG, Matusop A, Ratnam S, Rahman HA, Conway DJ, Singh B. Plasmodium knowlesi malaria in humans is widely distributed and potentially life threatening. Clin. Infect. Dis. 2008;46:165–171. doi: 10.1086/524888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Murray CJL, Rosenfeld LC, Lim SS, Andrews KG, Foreman KJ, Haring D, Fullman N, Naghavi M, Lozano R, Lopez AD. Global malaria mortality between 1980 and 2010: a systematic analysis. Lancet. 2012;379:413–431. doi: 10.1016/S0140-6736(12)60034-8. [DOI] [PubMed] [Google Scholar]
- [8].Trape JF, Tall A, Diagne N, Ndiath O, Ly AB, Faye J, Dieye-Ba F, Roucher C, Bouganali C, Badiane A, Sarr FD, Mazenot C, Toure-Balde A, Raoult D, Druilhe P, Mercereau-Puijalon O, Rogier C, Sokhna C. Malaria morbidity and pyrethroid resistance after the introduction of insecticide-treated bednets and artemisinin-based combination therapies: a longitudinal study. Lancet Infect. Dis. 2011;11:925–932. doi: 10.1016/S1473-3099(11)70194-3. [DOI] [PubMed] [Google Scholar]
- [9].Schneider P, Bousema T, Omar S, Gouagna L, Sawa P, Schallig H, Sauerwein R. (Sub)microscopic Plasmodium falciparum gametocytaemia in Kenyan children after treatment with sulphadoxine-pyrimethamine monotherapy or in combination with artesunate. Int. J. Parasitol. 2006;36:403–408. doi: 10.1016/j.ijpara.2006.01.002. [DOI] [PubMed] [Google Scholar]
- [10].Bousema T, Drakeley C. Epidemiology and Infectivity of Plasmodium falciparum and Plasmodium vivax Gametocytes in Relation to Malaria Control and Elimination. Clin. Microbiol. Rev. 2011;24:377. doi: 10.1128/CMR.00051-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Bousema T, Okell L, Shekalaghe S, Griffin JT, Omar S, Sawa P, Sutherland C, Sauerwein R, Ghani AC, Drakeley C. Revisiting the circulation time of Plasmodium falciparum gametocytes: molecular detection methods to estimate the duration of gametocyte carriage and the effect of gametocytocidal drugs. Malaria J. 2010;9 doi: 10.1186/1475-2875-9-136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Wilairatana P, Krudsood S, Tangpukdee N. Appropriate time for primaquine treatment to reduce Plasmodium falciparum transmission in hypoendemic areas. Korean J. Parasitol. 2010;48:179–182. doi: 10.3347/kjp.2010.48.2.179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Eastman RT, Pattaradilokrat S, Raj DK, Dixit S, Deng BB, Miura K, Yuan J, Tanaka TQ, Johnson RL, Jiang HY, Huang RL, Williamson KC, Lambert LE, Long C, Austin CP, Wu YM, Su XZ. A Class of Tricyclic Compounds Blocking Malaria Parasite Oocyst Development and Transmission. Antimicrob. Agents CH. 2013;57:425–435. doi: 10.1128/AAC.00920-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Alonso PL, Brown G, Arevalo-Herrera M, Binka F, Chitnis C, Collins F, Doumbo OK, Greenwood B, Hall BF, Levine MM, Mendis K, Newman RD, Plowe CV, Rodriguez MH, Sinden R, Slutsker L, Tanner M. A Research Agenda to Underpin Malaria Eradication. PLoS Med. 2011;8 doi: 10.1371/journal.pmed.1000406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Delves MJ. Plasmodium cell biology should inform strategies used in the development of antimalarial transmission-blocking drugs. Future Med. Chem. 2012;4:2251–2263. doi: 10.4155/fmc.12.182. [DOI] [PubMed] [Google Scholar]
- [16].Nagamune K, Sibley LD. Comparative genomic and phylogenetic analyses of calcium ATPases and calcium-regulated proteins in the apicomplexa. Mol. Biol. Evol. 2006;23:1613–1627. doi: 10.1093/molbev/msl026. [DOI] [PubMed] [Google Scholar]
- [17].Doerig C, Billker O, Haystead T, Sharma P, Tobin AB, Waters NC. Protein kinases of malaria parasites: an update. Trends Parasitol. 2008;24:570–577. doi: 10.1016/j.pt.2008.08.007. [DOI] [PubMed] [Google Scholar]
- [18].Billker O, Lourido S, Sibley LD. Calcium-Dependent Signaling and Kinases in Apicomplexan Parasites. Cell Host Microbe. 2009;5:612–622. doi: 10.1016/j.chom.2009.05.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Wernimont AK, Amani M, Qiu W, Pizarro JC, Artz JD, Lin YH, Lew J, Hutchinson A, Hui R. Structures of parasitic CDPK domains point to a common mechanism of activation. Proteins. 2011;79:803–820. doi: 10.1002/prot.22919. [DOI] [PubMed] [Google Scholar]
- [20].Kato N, Sakata T, Breton G, Le Roch KG, Nagle A, Andersen C, Bursulaya B, Henson K, Johnson J, Kumar KA, Marr F, Mason D, McNamara C, Plouffe D, Ramachandran V, Spooner M, Tuntland T, Zhou Y, Peters EC, Chatterjee A, Schultz PG, Ward GE, Gray N, Harper J, Winzeler EA. Gene expression signatures and small-molecule compounds link a protein kinase to Plasmodium falciparum motility. Nat. Chem. Biol. 2008;4:347–356. doi: 10.1038/nchembio.87. [DOI] [PubMed] [Google Scholar]
- [21].Sebastian S, Brochet M, Collins MO, Schwach F, Jones ML, Goulding D, Rayner JC, Choudhary JS, Billker O. A Plasmodium Calcium-Dependent Protein Kinase Controls Zygote Development and Transmission by Translationally Activating Repressed mRNAs. Cell Host Microbe. 2012;12:9–19. doi: 10.1016/j.chom.2012.05.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Dvorin JD, Martyn DC, Patel SD, Grimley JS, Collins CR, Hopp CS, Bright AT, Westenberger S, Winzeler E, Blackman MJ, Baker DA, Wandless TJ, Duraisingh MT. A Plant-Like Kinase in Plasmodium falciparum Regulates Parasite Egress from Erythrocytes. Science. 2010;328:910–912. doi: 10.1126/science.1188191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Siden-Kiamos I, Ecker A, Nyback S, Louis C, Sinden RE, Billker O. Plasmodium berghei calcium-dependent protein kinase 3 is required for ookinete gliding motility and mosquito midgut invasion. Mol. Microbiol. 2006;60:1355–1363. doi: 10.1111/j.1365-2958.2006.05189.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Billker O, Dechamps S, Tewari R, Wenig G, Franke-Fayard B, Brinkmann V. Calcium and a calcium-dependent protein kinase regulate gamete formation and mosquito transmission in a malaria parasite. Cell. 2004;117:503–514. doi: 10.1016/s0092-8674(04)00449-0. [DOI] [PubMed] [Google Scholar]
- [25].Ojo KK, Pfander C, Mueller NR, Burstroem C, Larson ET, Bryan CM, Fox AM, Reid MC, Johnson SM, Murphy RC, Kennedy M, Mann H, Leibly DJ, Hewitt SN, Verlinde CL, Kappe S, Merritt EA, Maly DJ, Billker O, Van Voorhis WC. Transmission of malaria to mosquitoes blocked by bumped kinase inhibitors. J. Clin. Invest. 2012;122:2301–2305. doi: 10.1172/JCI61822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Ojo, R. T. KKE, Vidadala RSR, Zhang Z, Rivas KL, Choi R, Lutz JD, Reid MC, Fox AMW, Hulverson MA, Kennedy M, Isoherranen N, Kim L, Comess KM, Kempf DJ, Verlinde CLMJ, Su X, Kappe S, Maly DJ, Fan E, Van Voorhis WC. Specific inhibitor of PfCDPK4 blocks malaria transmission: Chemical-genetic validation. J. Infect. Dis. 2013 doi: 10.1093/infdis/jit522. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Ojo KK, Larson ET, Keyloun KR, Castaneda LJ, DeRocher AE, Inampudi KK, Kim JE, Arakaki TL, Murphy RC, Zhang L, Napuli AJ, Maly DJ, Verlinde CLMJ, Buckner FS, Parsons M, Hol WGJ, Merritt EA, Van Voorhis WC. Toxoplasma gondii calcium-dependent protein kinase 1 is a target for selective kinase inhibitors. Nat. Struct. Mol. Biol. 2010;17:602–U102. doi: 10.1038/nsmb.1818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Murphy RC, Ojo KK, Larson ET, Castellanos-Gonzalez A, Perera BG, Keyloun KR, Kim JE, Bhandari JG, Muller NR, Verlinde CL, White AC, Jr., Merritt EA, Van Voorhis WC, Maly DJ. Discovery of Potent and Selective Inhibitors of Calcium-Dependent Protein Kinase 1 (CDPK1) from C. parvum and T. gondii. ACS Med. Chem. Lett. 2010;1:331–335. doi: 10.1021/ml100096t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Johnson SM, Murphy RC, Geiger JA, DeRocher AE, Zhang ZS, Ojo KK, Larson ET, Perera BGK, Dale EJ, He PQ, Reid MC, Fox AMW, Mueller NR, Merritt EA, Fan EK, Parsons M, Van Voorhis WC, Maly DJ. Development of Toxoplasma gondii Calcium-Dependent Protein Kinase 1 (TgCDPK1) Inhibitors with Potent Anti-Toxoplasma Activity. J. Med. Chem. 2012;55:2416–2426. doi: 10.1021/jm201713h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Larson ET, Ojo KK, Murphy RC, Johnson SM, Zhang ZS, Kim JE, Leibly DJ, Fox AMW, Reid MC, Dale EJ, Perera BGK, Kim J, Hewitt SN, Hol WGJ, Verlinde CLMJ, Fan EK, Van Voorhis WC, Maly DJ, Merritt EA. Multiple Determinants for Selective Inhibition of Apicomplexan Calcium-Dependent Protein Kinase CDPK1. J. Med. Chem. 2012;55:2803–2810. doi: 10.1021/jm201725v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Liu Y, Bishop A, Witucki L, Kraybill B, Shimizu E, Tsien J, Ubersax J, Blethrow J, Morgan DO, Shokat KM. Structural basis for selective inhibition of Src family kinases by PP1. Chem. Biol. 1999;6:671–678. doi: 10.1016/s1074-5521(99)80118-5. [DOI] [PubMed] [Google Scholar]
- [32].Bulawa, M. CED, Elbaum D. Preparation of pyrazolopyrimidinamines as modulators of protein trafficking. 2009 WO2009062118A. [Google Scholar]
- [33].Mulvihill MJ, Ji QS, Werner D, Beck P, Cesario C, Cooke A, Cox M, Crew A, Dong HQ, Feng LX, Foreman KW, Mak G, Nigro A, O’Connor M, Saroglou L, Stolz KM, Sujka I, Volk B, Weng QH, Wilkes R. 1,3-Disubstituted-imidazo[1,5-a]pyrazines as insulinlike growth-factor-I receptor (IGF-IR) inhibitors. Bioorg. Med. Chem. Lett. 2007;17:1091–1097. doi: 10.1016/j.bmcl.2006.11.016. [DOI] [PubMed] [Google Scholar]
- [34].Mulvihill MJ, Ji QS, Coate HR, Cooke A, Dong HQ, Feng LX, Foreman K, Rosenfeld-Franklin M, Honda A, Mak G, Mulvihill KM, Nigro AI, O’Connor M, Pirrit C, Steinig AG, Siu K, Stolz KM, Sun YC, Tavares PAR, Yao Y, Gibson NW. Novel 2-phenylquinolin-7-yl-derived imidazo[1,5-a]pyrazines as potent insulin-like growth factor-I receptor (IGF-IR) inhibitors. Bioorg. Med. Chem. 2008;16:1359–1375. doi: 10.1016/j.bmc.2007.10.061. [DOI] [PubMed] [Google Scholar]
- [35].Bain J, Plater L, Elliott M, Shpiro N, Hastie CJ, McLauchlan H, Klevernic I, Arthur JS, Alessi DR, Cohen P. The selectivity of protein kinase inhibitors: a further update. Biochem. J. 2007;408:297–315. doi: 10.1042/BJ20070797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Hanke JH, Gardner JP, Dow RL, Changelian PS, Brissette WH, Weringer EJ, Pollok K, Connelly PA. Discovery of a novel, potent, and Src family-selective tyrosine kinase inhibitor - Study of Lck- and FynT-dependent T cell activation. J. Biol. Chem. 1996;271:695–701. doi: 10.1074/jbc.271.2.695. [DOI] [PubMed] [Google Scholar]
- [37].Brandvold KR, Steffey ME, Fox CC, Soellner MB. Development of a Highly Selective c-Src Kinase Inhibitor. ACS Chem. Biol. 2012;7:1393–1398. doi: 10.1021/cb300172e. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.