

Abstract
The increased concentration of glutamate in synaptic vesicles, mediated by the vesicular glutamate transporter (VGLUT), is an initial vital step in glutamate synaptic transmission. Evidence indicates that aberrant overexpression of VGLUT is involved in certain pathophysiologies of the central nervous system. VGLUT is subject to inhibition by various types of agents. The most potent VGLUT-specific inhibitor currently known is Trypan Blue, which is highly charged, hence membrane-impermeable. We have sought a potent, VGLUT-specific agent amenable to easy modification to a membrane-permeable analog. We provide evidence that Brilliant Yellow exhibits potent, VGLUT-specific inhibition, with a Ki value of 12 nM. Based upon structure-activity relationship studies and molecular modeling, we have defined the potent inhibitory pharmacophore of VGLUT Brilliant Yellow. This study provides new insight into development of a membrane-permeable agent to lead to specific blockade, with high potency, of accumulation of glutamate into synaptic vesicles in neurons.
Keywords: Synaptic vesicles, glutamate uptake, inhibitor, Brilliant Yellow, pharmacophore
Introduction
Glutamate synaptic transmission underlies the major neural communication system in the central nervous system (CNS). As such, glutamate transmission is involved in various CNS functions, and dysfunction of this synaptic transmission is implicated in a variety of CNS pathophysiologies. Loading of glutamate into synaptic vesicles is the initial pivotal step in glutamate transmission. This ensures sufficient concentration of glutamate in the synaptic vesicle prior to its release to the synaptic cleft. Released glutamate thus concentrated signals activation of its various receptors. Vesicular glutamate loading is achieved by an ATP-dependent, glutamate-specific vesicular uptake system, consisting of v-type proton-pump ATPase and a vesicular glutamate transporter (VGLUT) (1-8). Bellocchio et al. and Takamori et al. have demonstrated that a brain-specific isoform of the Na+-dependent phosphate transporter functions as VGLUT (9, 10), and three isoforms (VGLUT1-3) are now known (11, 12). They have different brain regional, cellular, and subcellular distributions. However, they exhibit no difference in biochemical uptake function. Overexpression of VGLUT1 has been shown to enhance glutamate synaptic transmission (13).
VGLUT1 is overexpressed in the hippocampus of temporal lobe epilepsy patients (14). It is also overexpressed in a seizure-prone animal model as well as in an animal model with pilocarpin-induced seizure (15, 16). VGLUT2 is increased in the cortex of absence epilepsy animals (17). ATP-dependent vesicular glutamate uptake had been shown to be elevated in the cerebrum of a genetic animal model of human complex partial seizures (18). VGLUT1 has also been shown to be overexpressed in the amygdala of major depressive disorder and bipolar disorder patients (19). These lines of evidence suggest that excess vesicular glutamate accumulation in distinct brain regions is involved in various types of brain pathophysiology. Despite the potential role of VGLUT in CNS pathophysiology, no drugs have hitherto been developed targeting VGLUT.
Several classes of VGLUT inhibitors have been reported, however. They include (a) glutamate analogs such as trans-1-amino-cyclopentane-1,3-dicarboxylate (trans-ACPD) and trans-1-amino-cyclohexane-1,3-dicarboxylate (trans-ACHD) (20-22); (b) quinoline and quinoxaline analogs such as 7-chloro-kynurenate, xanthurenate, and 2-quinoxaline carboxylate (23, 24); (c) certain azo dyes bearing disulfonic acid-substituted naphthylamine groups and a biphenyl linker, such as Trypan Blue (25, 26) and Evans Blue (27); (d) an inhibitory protein factor referred to as IPF (28); (e) polyhalogenated fluorescein such as Rose Bengal and its analogs (29, 30); and (f) tetrapeptide inhibitors such as D-Gln- L-Ile- D-Glu- L-Try (31).
Compounds belonging to groups (a), (b), and (f) are specific to VGLUT, but have relatively low potencies compared to some of the most potent inhibitors; in addition, they all lack cell-membrane permeability. Trypan Blue, IPF, and Rose Bengal are the most potent inhibitors known (IC50 = 20-50 nM). However, Trypan Blue, though specific to VGLUT, does not penetrate the cellular membrane. The endogenous protein inhibitor IPF, though highly potent, is neither specific to VGLUT nor membrane-permeable. The most potent inhibitor Rose Bengal proved membrane-permeable, leading to reduced glutamate release by inhibiting vesicular glutamate accumulation, but was not highly specific to VGLUT. Thus, there is great need to develop a potent, membrane-permeable VGLUT-specific inhibitor. The purpose of this study was to seek a potent, small molecule VGLUT-specific inhibitor amenable to transformation to a membrane-permeable agent. We report here the transporter specificity and pharmacophore of such an inhibitor.
Materials and Methods
Animals
Rat brain was used to prepare synaptic vesicles and synaptosomes, except where otherwise indicated. Rats were sacrificed by decapitation without drug treatment. All animal procedures were approved by the University of Michigan Committee on Use and Care of Animals, in accordance with the National Institutes of Health Guide for the Use and Care of Laboratory Animals.
Reagents and Chemicals
L-[3,4-3H]Glutamic acid (50 Ci/mmol) and [2,3-3H(N]] γ-aminobutyric acid (40 Ci/mmol) were purchased from PerkinElmer, and potassium glutamate, γ-aminobutyric acid, ATP, and 4,4’-dinitrostilbene-2,2’-disulfonate, IR-1048, and IR-1061 from Sigma-Aldrich. 4,4’-Dibenzamidostilbene-2,2’-disulfonate and 4,4’-diaminostilbene-2,2’-disulfonate were obtained from Molecular Probes and Acros Organics, respectively.
Assay for Vesicular Glutamate Uptake
Since there was no significant difference in inhibitor dose-response curve of vesicular glutamate between crude vesicles and purified vesicles (30), crude synaptic vesicles prepared from rat cerebrum or calf cerebral cortex, as described previously (32), were used for this study. Vesicular glutamate uptake was assayed by a modification of Kish and Ueda (32). The final incubation mixture (100 μl) contained: synaptic vesicles (50 μg protein), 20 mM K-Hepes (pH 7.4), 4 mM MgSO4, 4 mM KCl, 2 mM aspartate, 0.25 M sucrose, various concentrations of test agent, ± 2 mM ATP (neutralized with Tris), and 50 μM [3H]glutamate (0.5 Ci/mmol). Pre-incubation was carried out at 30° C for 10 min without ATP and [3H]glutamate, followed by addition of a mixture of these agents (20 μl). The final mixture was incubated for an additional 10 min, followed by filtration and thorough washing with 0.15 M KCl.
Assay for kinetics of vesicular glutamate uptake with Brilliant Yellow
The assay conditions were similar to the one described above, except that (a) K-Hepes was replaced with Tris-Hepes, (b) pre-incubation was carried out for 30 min in the presence of Brilliant Yellow, and (c) [H3]glutamate uptake time was 1.5 min.
Assay for Synaptosomal Glutamate Uptake
Synaptosomes were prepared from rat brain cerebrum, as described by Krueger et al. (33). Synaptosomes (100 μg protein) were incubated at 37° C for 10 min in a Krebs solution (200 μl) containing 20 mM Hepes-Tris (pH 7.4), 150 mM NaCl or choline chloride, 6.2 mM KCl, 1.2 mM Na2PO4, 1.2 mM MgSO4, 10 mM glucose, and I μM [3H]glutamate (5 Ci/mmol), in the absence or presence of 50 μM test agent. Uptake in the presence of NaCl minus that in the presence of choline chloride was presented as Na+-dependent synaptosomal uptake (34).
Preparation of IPF
IPF was partially purified through hydroxyapatite column chromatography (HTP) as described (28), and used for this study.
Statistical Analysis
Data in tables are presented as mean ± SEM, except where otherwise indicated.
Computational Analysis
PCMODEL V 9.0 (Serena Software, Bloomington, IN, USA) was used to calculate intra-molecular atomic distances for some of the agents tested. In this program, molecular mechanics calculations are performed with the MM3 force field (35). This is capable not only of geometric optimization but also of conformational searches of flexible, small-to medium-sized organic and inorganic molecules. The complementary software Gaussian performs detailed electronic structure calculations based on the density functional theory. Since quantum methods, used in the latter, include all electrons, the performance is slower, and hence this program is applicable to much smaller systems than can be treated by force field methods. PCMODEL has been used in a number of studies (e.g., refs. 36-40). PCMODEL-computed bond distances and angles, as well as the global minimal energy conformation, of organic molecules agree approximately with those determined by X-ray diffraction in the crystal form (36, 37).
Results
Brilliant Yellow is a potent inhibitor of VGLUT
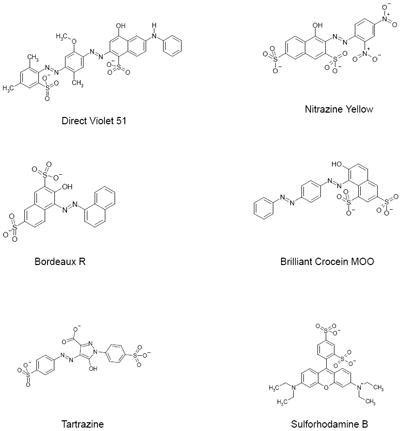
We have tested 200 compounds bearing the sulfonic acid functional group, as well as those devoid of this group. We found Brilliant Yellow to be the most potent, followed by Direct Violet 51, as shown in Fig. 1 and Table 1. The IC50 of Brilliant Yellow was determined to be 44 ± 6 nM. Some of the other sulfonic acid-bearing compounds which showed lower potency than Direct Violet 51 are Brilliant Blue G, Nitrosulfonazo III, Direct Yellow 27, and Cibacron Brilliant Red 3B-A (IC50 = 0.67 - 1.4 μM), followed by Nitrazine Yellow, Brilliant Black BN, Bordeau R, Brilliant Crocein MOO, Bordeau R, and Ponceau S (IC50 = 4.4 - 26 μM). Some examples of the rather inactive inhibitors are tartrazine and sulforhodamine.
Fig.1.
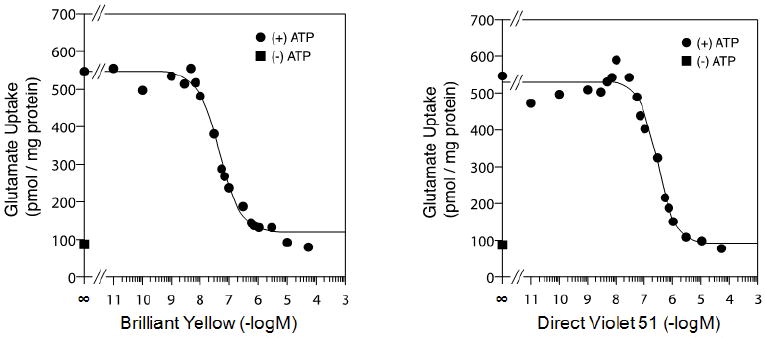
Brilliant Yellow and Direct Violet 51 are potent inhibitors of vesicular glutamate uptake. Rat brain synaptic vesicles were pre-incubated in the presence of various concentrations of Brilliant Yellow (left) or Direct Violet 51 (right) in a mixture described in Materials and Methods, followed by addition of a mixture of [3H]glutamate ± ATP, and an additional 10-min incubation.
Table 1.
IC50 of some of the vesicular glutamate uptake inhibitors tested
| Test Compounds | IC50 μM |
|---|---|
| Brilliant Yellow | 0.044 ± 0.007* |
| Direct Yellow 51 | 0.148 ± 0.031* |
| Nitrazine Yellow | 4.4 |
| Bordeau R | 6.5 |
| Brilliant Crocein MOO | 7.7 |
| Tartrazine | > 50 |
| Sulforhodamine | > 50 |
Synaptic vesicles were preincubated in the presence of various concentrations of test compounds, followed by addition of a mixture of [3H]glutamate ± ATP, with an additional 10-min incubation, as described in Fig. 1.
mean ± SEM (n=4)
The potency of Brilliant Yellow is equivalent to that of Trypan Blue, the most potent VGLUT-specific inhibitor hitherto reported (IC50 = 49 nM) (25, 26). Both compounds have diazo groups and sulfonic acid substitutions. However, Brilliant Yellow differs from Trypan Blue in several respects. The former is a significantly smaller molecule than the latter (MW 578.55 vs. MW 868.81). Brilliant Yellow has a stilbene moiety, but lacks the naphthalenyl and amino groups, both present in Trypan Blue. Brilliant Yellow’s sulfonic acid substitution occurs in the benzene ring of the stilbene moiety, as opposed to the naphthalene ring seen in Trypan Blue. Brilliant Yellow has two sulfonic acid groups, as opposed to four with Trypan Blue; this makes the former less charged than the latter. Perhaps the most important difference for our purpose between these diazo compounds is that Brilliant Yellow is free of the amino group and has a hydroxyl group in each of the molecule’s terminal positions, as opposed to the presence of the hydrogen-bonded amino groups in the Trypan Blue molecule. This renders Brilliant Yellow amenable to easy modification.
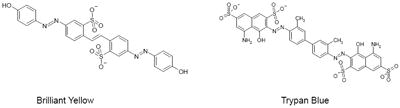
Direct Violet 51, though not as potent as Brilliant Yellow, was also found to be a quite strong inhibitor of vesicular glutamate uptake with IC50 of 148 ± 31 nM. Unlike Brilliant Yellow, it lacks the stilbene moiety, but has a moiety containing sulfonic acid group-substituted naphthol linked to the azenyl group. In this respect, Direct Violet 51 is similar to Trypan Blue, but has only one naphthol group, and only one (rather than two) sulfonic acid group is substituted per naphthol group, in contrast to Trypan Blue.
Brilliant Yellow does not affect vesicular GABA uptake or synaptosomal glutamate uptake
In order to determine how specific Brilliant Yellow inhibition is to VGLUT, we have examined the effect of this agent on GABA uptake into synaptic vesicles as well as glutamate uptake into synaptosomes. As shown in Fig. 2 and Table 2, Brilliant Yellow affected neither vesicular GABA uptake nor synaptosomal glutamate uptake at concentrations up to 50 μM. These data suggest Brilliant Yellow acts as a specific inhibitor for VGLUT. Consistent with this, kinetic experiments, shown in Fig. 3, indicate that Brilliant Yellow competes with glutamate for VGLUT, suggesting that Brilliant Yellow binds at or near the glutamate binding site. The Ki value of Brilliant Yellow was determined to be 12.5 nM (n=3), using 0, 40, 60, and 80 nM Brilliant Yellow.
Fig. 2.
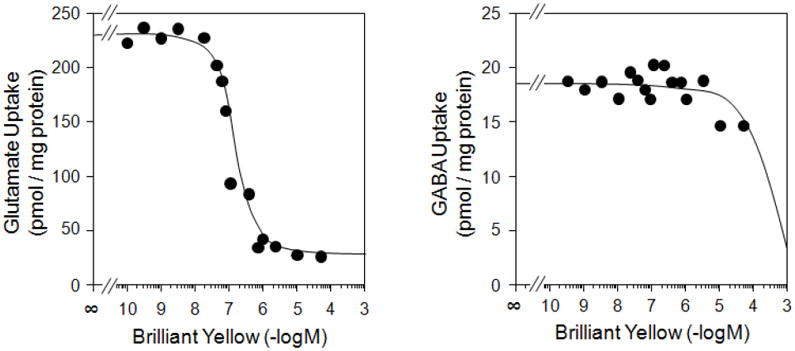
Brilliant Yellow inhibits vesicular glutamate uptake without affecting GABA uptake. Bovine brain synaptic vesicles were pre-incubated in the presence of various concentrations of Brilliant Yellow, and further incubated with 2 mM ATP and 50 μM [3H]glutamate (left) or [3H]GABA (right), as described in Materials and Methods.
Table 2.
Inability of Brilliant Yellow, Direct Violet 51, and XAS 0919 to affect synaptosomal glutamate uptake
| Test compounds | Concentration μM | Glutamate uptake dpm | %Control |
|---|---|---|---|
| None | 247,286 | 100 | |
| Brilliant Yellow | 50 | 279,032 | 112 |
| Direct Violet 51 | 50 | 258,020 | 104 |
Synaptosomes were incubated with [3H]glutamate in the absence or presence of 50 μM test agent in a Krebs-Ringer solution, as described in Materials and Methods. Na+-dependent [3H]glutamate uptake into synaptosomes is presented.
Fig. 3.
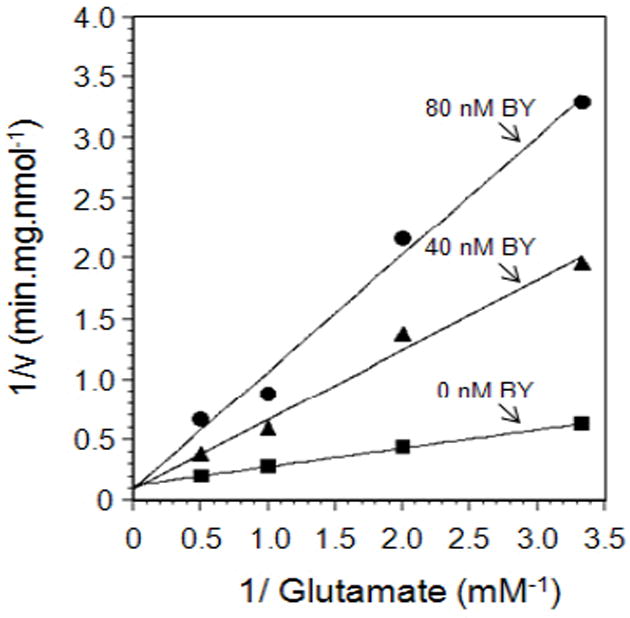
Brilliant Yellow is a competitive inhibitor with glutamate for VGLUT. Synaptic vesicles were pre-incubated with 0, 40, and 80 nM Brilliant Yellow, followed by addition of a mixture of ATP and various concentrations of [3H]glutamate, with an additional 1.5- min incubation, as described in Materials and Methods. This represents a mean of three experimental data. v, initial rate of vesicular glutamate uptake; BY, Brilliant Yellow.
Pharmacophore of Brilliant Yellow
In an effort to determine the inhibitory pharmacophore, we have tested various Brilliant Yellow analogs and related compounds. This includes (a) chrysophenine, the dimethyl ester of Brilliant Yellow; (b) 4,4’-dinitrostilbene-2,2’-disulfonate; (c) 4,4’-dibenzamidostilbene-2,2’-disulfonate; (d) 4,4’-diacetamidostilbene-2,2’-disulfonate; (e) 4,4’-diphenylsulfonamidostilbene-2,2’-disulfonate; (f) 4,4’-diaminostilbene-2,2’-disulfonate [in (b-f), the p-hydroxyphenyl-azenyl moiety is replaced by the nitro group, the benzoylamino group, the acetylamino group, the phenylsufonylamino group, and the amino group, respectively]; (g) 4,4’-di-p-hydroxybenzylidene-aminostilbene-2,2’-disulfonate (in which one of the nitrogen atoms of the azenyl group, the one linked to the p-hydroxyphenyl group, is replaced by the carbon atom); and (h) 4,4-dinitrostilbene-2,2’-disulfonic acid diphenyl ester.
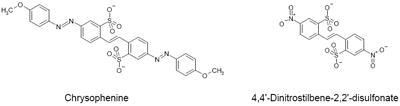
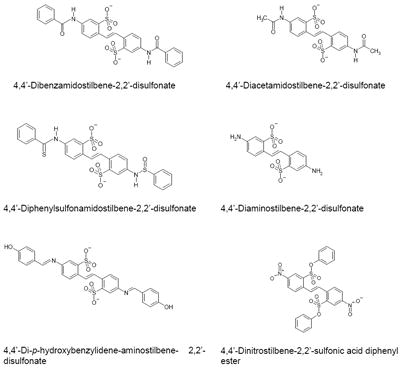
As shown in Table 3, chrysophenine is somewhat, but not drastically, less potent than Brilliant Yellow. This indicates that the free hydroxyl group linked to the phenyl group is not critical in causing inhibition. However, it is important in optimizing the inhibitory effect. Potency of 4,4’-dinitrostilbene-2,2’-disulfonate is substantially reduced compared to Brilliant Yellow, although it can still act as an inhibitor of vesicular glutamate uptake. This indicates that the (p-hydroxyphenyl)azenyl moiety is crucial in producing highly potent inhibition. Replacement of the nitro group with the amino group resulted in further marked reduction in potency, as seen in 4,4’-diaminostilbene-2,2’-disulfonate. Replacement of one of the amino group’s hydrogen atoms with the benzoyl, but not acetyl, group restored inhibitory potency to that of 4,4’-dinitrostilbene-2,2’-disulfonate, as seen in 4,4’-dibenzamidostilbene-2,2’-disulfonate (Table 3, Fig. 4). This indicates that the phenyl group is an important element of the inhibitory pharmacophore. Of great interest, simply replacing one Brilliant Yellow azenyl group nitrogen atom with the carbon atom eliminated its potent inhibitory activity, as evident in 4,4’-di-p-hydroxybenzylidene-aminostilbene-2,2’-disulfonate. This shows the critical importance of the azenyl group in generating Brilliant Yellow’s high potency. Consistent with this, replacement of an azenyl group’s nitrogen atom with the sufonyl group abolishes inhibitory activity, as seen in 4,4’-diphenylsulfonamidostilbene-2,2’-disulfonate. Finally, esterification of the sulphonic acid group with phenol also abolished Brilliant Yellow’s highly potent inhibitory activity, as seen in 4,4’-dinitrostilbene-2,2’-sulfonic acid diphenyl ester. This indicates that the negatively charged sulfonate group is also a critical element of Brilliant Yellow’s pharmacophore. These results altogether indicate that the sulfonate, azenyl, phenyl and hydroxyl groups constitute the highly potent pharmacophore of Brilliant Yellow.
Table 3.
Effect of functional group alterations of Brilliant Yellow and 4,4’-dinitrostilbene-2,2’-disulfonate on vesicular glutamate uptake inhibition by Brilliant Yellow
| Test compounds | IC50 μM |
|---|---|
| Brilliant Yellow | 0.044 ± 0.006a |
| Chrysophenine | 0.266 ± 0.070b |
| 4,4’-Dinitrostilbene-2,2’-disulfonate | 1.69 ± 0.11c |
| 4,4’-Dibenzamidostilbene-2,2’-disulfonate | 2.0d |
| 4,4-Diacetamidostilbene-2,2’-disulfonate | > 50 |
| 4,4-Diphenylsulfonamidostilbene-2,2’-disulfonate | > 50 |
| 4,4-Diaminostilbene-2,2’-disulfonate | > 50 |
| 4,4-Di-p-hydroxybenzylidene-aminostilbene-2,2’-disulfonate | > 50 |
| 4,4-Dinitrostilbene-2,2’-sulfonic acid diphenyl ester | > 50 |
Fig. 4.
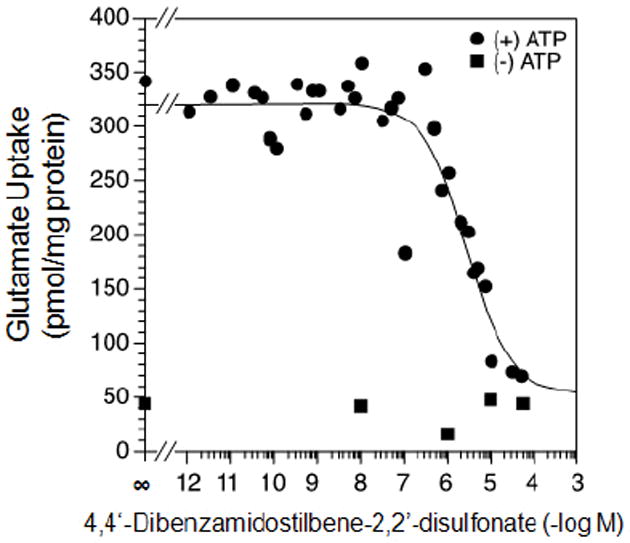
Dose-response curve of 4,4’-dibenzamidostilbene-2,2’-disulfonate for vesicular glutamate uptake. Rat brain synaptic vesicles were incubated with [3H]glutamate ± ATP in the presence of various concentrations of 4,4’-dibenzamidostilbene-2,2’-disulfonate, as described in Fig. 1.
Inhibition by Brilliant Yellow is prevented by IR-1048
Since an agent which would relieve Brilliant Yellow inhibition could be useful in the study of Brilliant Yellow-induced biological phenomena, we have sought its de-inhibitor. We have found that IR-1048 alleviates vesicular glutamate uptake inhibition by Brilliant Yellow in a dose-dependent manner, as shown in Fig. 5. Brilliant Yellow inhibited vesicular glutamate uptake 80% at 0.3 μM. This inhibition was almost completely abolished in the presence of 10 μM IR-1048. IR-1048 alone had no effect on vesicular glutamate uptake in the absence of Brilliant Yellow. The IR-1048 concentration required to cause 50% de-inhibition was 0.8-1.0 μM under these conditions. IR-1061, a somewhat similar compound to IR-1048, did not act as a de-inhibitor of Brilliant Yellow (data not shown). Thus, IR-1048 could serve as quite a potent, selective antagonist of the vesicular glutamate inhibitor Brilliant Yellow. The mechanism of de-inhibition is not known. Whether IR-1048 competes with Brilliant Yellow or binds to an allosteric site remains to be determined.
Fig. 5.
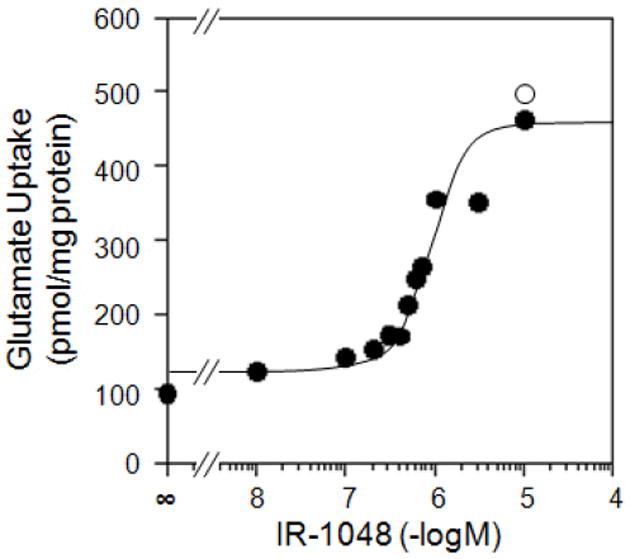
IR-1048 reverses inhibition by Brilliant Yellow. Synaptic vesicles were incubated with [3H]glutamate, ATP, and various concentrations of IR-1048 in the absence (open circle) or presence (closed circle) of 0.3 μM Brilliant Yellow, as described in Fig. 1.
Among potent inhibitors of vesicular glutamate uptake is an inhibitory protein factor referred to as IPF (28, 41-42). In contrast to Brilliant Yellow-induced inhibition, IPF-induced inhibition was not prevented by IR-1048 (Fig. 6). It may be pointed out that the former inhibition is specific to VGLUT, whereas the latter is not; IPF affects vesicular GABA and monoamine uptake as well (28, 42). Thus, IR-1048 distinguishes these two types of VGLUT inhibition.
Fig. 6.
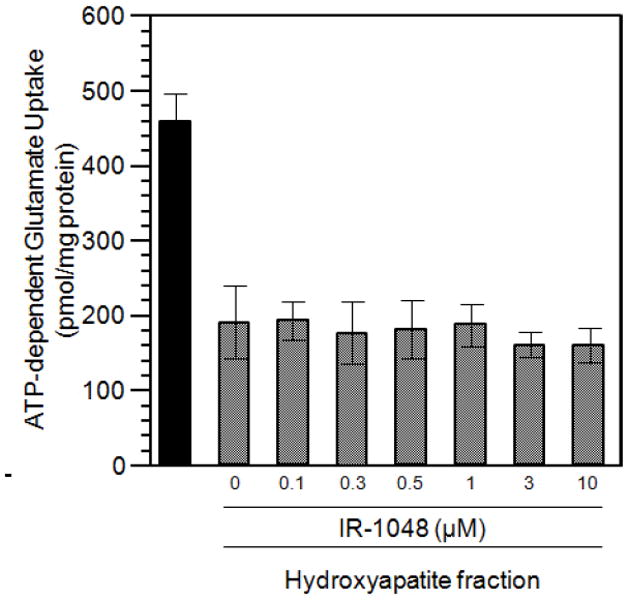
IR-1048 does not reverse inhibition by IPF. Synaptic vesicles were incubated with [3H]glutamate, ATP, and various concentration of IR-1048 in the absence or presence of partially purified IPF (hydroxyapatite chromatography fraction 0.24 mg/ml), as described in Fig. 1.
Discussion
We have shown that Brilliant Yellow is a potent VGLUT-specific inhibitor, as illustrated in Fig. 7. Its potency is similar to that of Trypan Blue. These diazo compounds are the most potent VGLUT-specific inhibitors currently known. These agents have azenyl and sulfonate groups in common. However, Brilliant Yellow is distinct from Trypan Blue in several respects, as mentioned in Results. The most important difference for our purpose is that Brilliant Yellow is free of amino groups but bears a hydroxyl group in each terminal position, whereas Trypan Blue has amino groups as well as hydroxyl groups, which are hydrogen-bonded to the amino groups. This would render Brilliant Yellow amenable to easy modification.
Fig. 7.
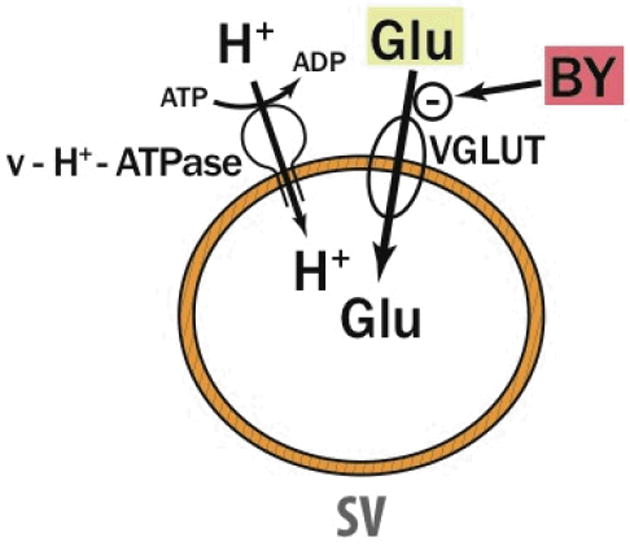
Specific action of Brilliant Yellow. Brilliant Yellow (BY) specifically inhibits VLGUT with high potency. Glu, glutamate; SV, synaptic vesicle.
Transporter specificity of Brilliant Yellow is quite remarkable. Affinity difference between VGLUT and the vesicular GABA transporter, as well as the plasma membrane glutamate transporter, is greater than three orders of magnitude. Thus, it is unlikely that Brilliant Yellow, at low concentrations sufficient to inhibit VGLUT, affects ATPase, membrane potential formation, chloride influx, or pH gradient formation, all of which are required for vesicular GABA transport. We have provided evidence that Brilliant Yellow is a competitive inhibitor with respect to glutamate, with a Ki value of 12.5 nM. This suggests that Brilliant Yellow interacts with VGLUT at a site close to the glutamate-binding site, not at an allosteric site. The Ki value of Trypan Blue has been reported to be about 50 nM (25). Thus, Brilliant Yellow appears to have higher affinity for VGLUT than does Trypan Blue, rendering Brilliant Yellow the most potent, VGLUT-specific inhibitor at present.
Reserpine, tetrabenazine, and ketanserin are well-known potent, specific inhibitors of vesicular monoamine transporters (VMAT) (43-51). VMAT2, the isoform primarily expressed in monoaminergic neurons of the central and peripheral sympathetic nervous systems, is more sensitive to these inhibitors than is VMAT1, which is preferentially expressed in peripheral endocrine cells such as chromaffin cells (50, 51). The high affinity binding of [11C]tetrabenazine and [11C]dihydrotetrabenazine to VMAT2 has been used for non-invasive imaging of VMAT2 in the human brain (52, 53). The inhibitory action of tetrabenazine on VMAT2 in the brain is also considered to underlie its therapeutic effect on Huntington’s chorea (54, 55). Vesamicol, a neuromuscular blocking agent, has been shown to attenuate cholinergic transmission by acting as a potent, allosteric vesicular acetylcholine transporter (VAChT)-specific inhibitor (56, 57). A number of radioligands have been developed based upon the vesamicol scaffold, aimed at brain VAChT imaging (58). None of these VMAT-or VAChT-specific agents or their analogs resemble Brilliant Yellow or Trypan Blue in structure. Hence, it is unlikely that Brilliant Yellow affects vesicular amine transporter activity. However, this, as well as VGLUT isoform specificity, remains to be investigated.
Data presented in Table 3 suggest that the sulfonate, azenyl, and phenyl groups are all essential for high potency of Brilliant Yellow. The question is how they should be coordinated. Molecular modeling indicates that Brilliant Yellow and Trypan Blue, though structurally significantly different, have in common the O- (negatively charged sulfonate oxygen) - N (phenyl group-linked azenyl nitrogen) distance. This is consistent with the azenyl group being an essential element of the pharmacophore of Brilliant Yellow.
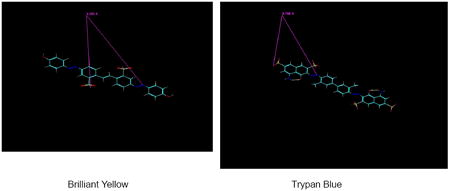
The distance between the negatively charged oxygen of Brilliant Yellow’s left side sulfonate and the azenyl group nitrogen atom linked to the right side hydroxyphenyl group is 9.324 ± 0.008 A (in the frozen sate; n = 6). This distance is close to the distance [9.799 ± 0.007 (in the frozen sate; n = 6)] between the negatively charged oxygen of Trypan Blue’s external sulfonate group (meta-positioned relative to the amino group of the naphtholamine moiety) and the azenyl group nitrogen linked to the methylphenyl group in the same half of the molecule. This similarity supports the notion that azenyl group nitrogen and negatively charged sulfonate oxygen are involved in binding of Brilliant Yellow and Trypan Blue to VGLUT. It is feasible that the former interacts with VGLUT’s Tyr195 residue, forming the hydrogen bond with the phenolic proton, and that the latter interacts with its His128 residue. These interactions could block access of glutamate to the binding site of VGLUT. Juge et al. (59) have demonstrated that His128 and Arg184, as well as Glu191 (which is close to Tyr195), are essential for VGLUT’s glutamate transport function, suggesting that these amino acid residues constitute its glutamate binding site.
Another common aspect of Brilliant Yellow and Trypan Blue is their structural symmetry, enabling their endowment with two identical pharmacophores per molecule. This may contribute to their potency.
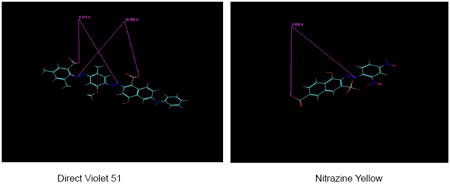
Direct Violet 51, the second potent inhibitor in Table 1, also appears to bear two pharmacophores per molecule. The distance between O- of the sulfonate group linked to the dimethylphenyl group and the naphthol group-linked N of the azenyl group is 8.274 ± 0.002 A (n=5), somewhat close to the O-–N distance of Brilliant Yellow mentioned above (9.324 A). The distance between O- of the sulfonate group linked to the naphthol group and the dimethylphenyl group-linked N of the azenyl group is 10.354 ± 0 A (n=5), close to the O-–N distance of Trypan Blue mentioned above (9.799 A). The additional hydrophobic phenyl group fused with the hydroxyphenyl group may also contribute to the high potency of Direct Violet 51. It is feasible that this extra phenyl group interacts with Tyr195 and/or the hydrophobic region of Pro196.
Nitrazine Yellow is similar in structure to one-half of the Trypan Blue molecule. It is not a symmetric molecule, but the distance between the external naphthol-linked sulfonate O- and the phenyl group-linked azenyl group N, 9.607 ± 0.011 A (n=8), is close to the Trypan Blue O--N distance. However, the potency of Nitrazine Yellow is substantially lower than that of Trypan Blue. This is most likely due to its lack of dual pharmacophores, in contrast to Trypan Blue.
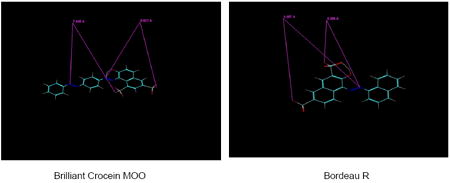
Brilliant Crocein MOO, like Nitrazine Yellow, lacks symmetry. The distance between the external sulfonate O- and the central azenyl N linked to the phenyl group is 8.819 ± 0.007 A (n=5). This is 0.5 A shorter than the Brilliant Yellow O--N distance. The Nitrazine Yellow O--N distance is only about 0.3 A longer than the Brilliant Yellow O--N distance. These similar, rather small deviations from the optimal O--N distance (i.e., that of Brilliant Yellow) could account for both Brilliant Crocein MOO and Nitrazine Yellow exhibiting decent VGLUT inhibition with similar potency. Brilliant Crocein MOO’s potency is similar to that reported by Roseth et al. (25). The other O--N distance of Brilliant Crocein MOO (7.845 A) is significantly shorter than optimal, and hence barely contributes to potency.
The O--N distance of Brilliant Crocein MOO (8.819 A) is similar to that of Bordeau R (8.857 ± 0 A, n=3), the distance between the O- of the sulfonate group (not adjacent to the hydroxyl group) linked to one naphthalane group and the azenyl N linked to the other naphthalane group. These almost identical distances would also account for very similar potencies of Brilliant Crocein MOO and Bordeau R (7.7 vs. 6.5 μM IC50).
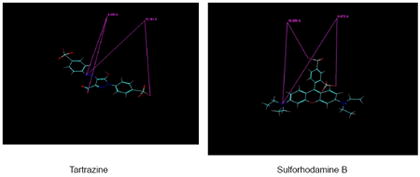
The even lower potency of Tartrazine could be explained by its much longer O--N distance compared to that of Brilliant Yellow or Trypan Blue. The distance between the right side sulfonate O- and the azenyl N linked to the phenyl group is 12.191 ± 0 A (n=3). The distance between the carboxyl group O- and the azenyl N linked to the phenyl group is 4.430 ± 0 A (n=3); this is much shorter than the O--N distance of Brilliant Yellow or Trypan Blue. The distance between the O- of the left terminal sulfonate group and the azenyl N linked to the pyrazol moiety, rather than to the phenyl group, is not taken into account.
Another example of poor sulfonate-containing VGLUT inhibitors is sulforhodamine. The distance between the ortho-sulfonate group O- and the uncharged N of the dimethyl group is 8.721 ± 0.129 A (n=5). The distance between the para-sulfonate group O- and the uncharged N of the dimethylamino group is 10.291 ± 0.194 A (n=5). The former is similar to the O--N distance of Brilliant Crocein MOO (8.816 A), and fairly close to that of Brilliant Yellow. The latter is also fairly close to the O--N distance of Trypan Blue. However, this nitrogen of sulforhodamine is neither hydrogen bond-forming nor linked to the phenyl group. This would account for the poor sulforhodamine affinity for VGLUT. This is consistent with hydrogen bond-forming nitrogen and phenyl group requirements for the potent pharmacophore of Brilliant Yellow.
The fact that Brilliant Yellow can be easily modified raises the possibility that a hydrophobic derivative could be synthesized. Such an agent could provide a useful tool in cellular experiments, investigating the role of VGLUT in synaptic transmission. It could also serve as a prototype compound for a novel anti-epileptic drug lead.
In conclusion, the pharmacophore of Brilliant Yellow is defined by the distance between the negatively charged oxygen in one half of the molecule and the hydrogen bond-forming nitrogen linked to the hydroxyphenyl group in the other half, together with its molecular symmetry, as shown below.
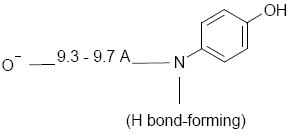
Other molecules containing two of these components per molecule are also likely to exhibit potent, specific VGLUT inhibition.
Acknowledgments
This work was supported by a National Institutes of Health Javits Neuroscience Investigator Award NS 26884 (T.U.) and a grant from Taisho Pharmaceutical, Tokyo (T.U.). We thank Taisho Pharmaceutical for the gift of some of the 4,4’-dinitrostilbene-2,2’-disulfonate analogs. We also thank Dr. Minor J. Coon for reading the manuscript, as well as Mary Sisson for technical assistance on kinetic experiments.
Abbreviations
- VGLUT
vesicular glutamate transporter
- GABA
γ-aminobutyric acid
- VMAT
vesicular monoamine transporter
- VAChT
vesicular acetylcholine transporter
References
- 1.Naito S, Ueda T. Adenosine triphosphate-dependent uptake of glutamate into protein I associated synaptic vesicles. J Biol Chem. 1983;258:696–699. [PubMed] [Google Scholar]
- 2.Naito S, Ueda T. Characterization of glutamate uptake into synaptic vesicles. J Neurochem. 1985;44:99–109. doi: 10.1111/j.1471-4159.1985.tb07118.x. [DOI] [PubMed] [Google Scholar]
- 3.Maycox PR, Deckwerth T, Hell JW, Jahn R. Glutamate uptake by brain synaptic vesicles. Energy dependence of transport and functional reconstitution in proteoliposomes. J Biol Chem. 1988;263:15423–15428. [PubMed] [Google Scholar]
- 4.Fykse EM, Christensen H, Fonnum F. Comparison of the properties of γ-aminobutyric acid and L-glutamate uptake into synaptic vesicles isolated from rat brain. J Neurochem. 1989;52:946–951. doi: 10.1111/j.1471-4159.1989.tb02546.x. [DOI] [PubMed] [Google Scholar]
- 5.Tabb JS, Ueda T. Phylogenetic studies on the synaptic vesicle glutamate transport system. J Neurosci. 1991;11:1822–1828. doi: 10.1523/JNEUROSCI.11-06-01822.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Tabb JS, Kish PE, Van Dyke R, Ueda T. Glutamate transport into synaptic vesicles. Roles of membrane potential, pH gradient, and intravesicular pH. J Biol Chem. 1992;267:15412–15418. [PubMed] [Google Scholar]
- 7.Wolosker H, de Souza DO, de Meis L. Regulation of glutamate transport into synaptic vesicles by chloride and proton gradient. J Biol Chem. 1996;271:11726–11731. doi: 10.1074/jbc.271.20.11726. [DOI] [PubMed] [Google Scholar]
- 8.Özkan ED, Ueda T. Glutamate transport and storage in synaptic vesicles. Jpn J Pharmacol. 1998;77:1–10. doi: 10.1254/jjp.77.1. [DOI] [PubMed] [Google Scholar]
- 9.Bellocchio EE, Reimer RJ, Fremeau RTJ, Edwards RH. Uptake of glutamate into synaptic vesicles by an inorganic phosphate transporter. Science. 2000;289:957–960. doi: 10.1126/science.289.5481.957. [DOI] [PubMed] [Google Scholar]
- 10.Takamori S, Rhee JS, Rosenmund C, Jahn R. Identification of a vesicular glutamate transporter that defines a glutamatergic phenotype in neurons. Nature. 2000;407:189–194. doi: 10.1038/35025070. [DOI] [PubMed] [Google Scholar]
- 11.Fremeau RT, Jr, Voglmaier S, Seal RP, Edwards RH. VGLUTs define subsets of excitatory neurons and suggest novel roles for glutamate. Trends Neurosci. 2004;27:98–103. doi: 10.1016/j.tins.2003.11.005. [DOI] [PubMed] [Google Scholar]
- 12.Erickson JD, De Gois S, Varoqui H, Schafer MKH, Weihe E. Activity-dependent regulation of vesicular glutamate and GABA transporters: A means to scale quantal size. Neurochem Int. 2006;48:643–649. doi: 10.1016/j.neuint.2005.12.029. [DOI] [PubMed] [Google Scholar]
- 13.Wilson NR, Kang J, Hueske EV, Leung T, Varoqui H, Murnick JG, Erickson JD, Liu G. Presynaptic regulation of quantal size by VGLUT1. J Neurosci. 2005;25:6221–6234. doi: 10.1523/JNEUROSCI.3003-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Van der Hel W, et al. Hippocampal distribution of vesicular glutamate transporter 1 in patients with temporal lobe epilepsy. Epilepsia. 2009;50:1717–1728. doi: 10.1111/j.1528-1167.2009.02054.x. [DOI] [PubMed] [Google Scholar]
- 15.Kang T-C, Kim D-S, Kwak S-E, Kim J-E, Kim DW, Kang JH, Won MH, Kwon O-S, Choi S-Y. Valproic acid reduces enhanced vesicular glutamate transporter immunoreactivities in the dentate gyrus of the seizure prone gerbil. Neuropharmacol. 2005;49:912–921. doi: 10.1016/j.neuropharm.2005.08.007. [DOI] [PubMed] [Google Scholar]
- 16.Kim J-E, et al. Anti-glutamatergic efect of rizuzole: comparison with valoproic acid. Neuroscience. 2007;147:136–145. doi: 10.1016/j.neuroscience.2007.04.018. [DOI] [PubMed] [Google Scholar]
- 17.Touret M, Parrot S, Denoroy L, Belin M-F, Didier-Bazes M. Glutamatergic alterations in the cortex of genetic absence epilepsy rats. BMC Neurosci. 2007;8:69–76. doi: 10.1186/1471-2202-8-69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lewis SM, Lee FS, Todorova M, Seyfried TN, Ueda T. Synaptic vesicle glutamate uptake in epileptic (EL) mice. Neurochem Int. 1997;31:581–585. doi: 10.1016/s0197-0186(97)00014-4. [DOI] [PubMed] [Google Scholar]
- 19.Varea E, Guirado R, Gilabert-Juan J, Marti U, Castillo-Gomez E, Blasco-Ibanez JM, Crespo C, Nacher J. Expression of PSA-NCAM and synaptic proteins in the amygdala of psychiatric disorder patients. J Psychiatr Res. 2012;46:189–197. doi: 10.1016/j.jpsychires.2011.10.011. [DOI] [PubMed] [Google Scholar]
- 20.Winter HC, Ueda T. Glutamate uptake system in the presynapric vesicle: Glutamic acid analogs as inhibitors and alternative substrates. Neurochem Res. 1993;18:79–85. doi: 10.1007/BF00966925. [DOI] [PubMed] [Google Scholar]
- 21.Winter HC, Ueda T. The glutamate uptake system in presynaptic vesicles: Further characterization of structural requirements for inhibitors and substrates. Neurochem Res. 2008;33:223–231. doi: 10.1007/s11064-007-9493-8. [DOI] [PubMed] [Google Scholar]
- 22.Moriyama Y, Yamamoto A. Vesicular L-glutamate transporter in microvesicles from bovine pineal glands. Driving force, mechanism of chloride anion activation, and substrate specificity. J Biol Chem. 1995;270:22314–22320. doi: 10.1074/jbc.270.38.22314. [DOI] [PubMed] [Google Scholar]
- 23.Bartlett RD, Esslinger CS, Thompson CM, Bridges RJ. Substituted quinolines as inhibitors of L-glutamate transport into synaptic vesicles. Neuropharmacology. 1998;37:839–846. doi: 10.1016/s0028-3908(98)00080-x. [DOI] [PubMed] [Google Scholar]
- 24.Thompson CM, Davis E, Carrigan CN, Cox HD, Bridges RJ, Gerdes JM. Inhibitors of the glutamate vesicular transporter (VGLUT) Curr Med Chem. 2005;12:2041–2056. doi: 10.2174/0929867054637635. [DOI] [PubMed] [Google Scholar]
- 25.Roseth S, Fykse EM, Fonnum F. Uptake of Lglutamate into synaptic vesicles: Competitive inhibition by dyes with biphenyl and amino and sulphonic acid substituted naphthyl groups. Biochem Pharmacol. 1998;56:1243–1249. doi: 10.1016/s0006-2952(98)00200-7. [DOI] [PubMed] [Google Scholar]
- 26.Fonnum F, Fykse EM, Roseth S. Uptake of glutamate into synaptic vesicles. Prog Brain Res. 1998;116:87–101. doi: 10.1016/s0079-6123(08)60432-x. [DOI] [PubMed] [Google Scholar]
- 27.Roseth S, Fykse EM, Fonnum F. Uptake of L-glutamate into rat brain synaptic vesicles: effect of inhibitors that bind specifically to the glutamate transporter. J Neurochem. 1995;65:96–103. doi: 10.1046/j.1471-4159.1995.65010096.x. [DOI] [PubMed] [Google Scholar]
- 28.Özkan ED, Lee FS, Ueda T. A protein factor that inhibits ATP-dependent glutamate and γ-aminobutyric acid accumulation into synaptic vesicles: purification and initial characterization. Proc Natl Acad Sci USA. 1997;94:4137–4142. doi: 10.1073/pnas.94.8.4137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ogita K, Hirata K, Bole DG, Yoshida S, Tamura Y, Leckenby AM, Ueda T. Inhibition of vesicular glutamate storage and exocytotic release by Rose Bengal. J Neurochem. 2001;77:34–42. doi: 10.1046/j.1471-4159.2001.t01-1-00200.x. [DOI] [PubMed] [Google Scholar]
- 30.Bole DG, Ueda T. Inhibition of vesicular uptake by Rose Bengal-related compounds: structure activity relationship. Neurochem Res. 2005;30:363–369. doi: 10.1007/s11064-005-2610-7. [DOI] [PubMed] [Google Scholar]
- 31.Patel SA, Nagy JO, Bolstad ED, Gerdes JM, Thompson CM. Tetrapeptide inhibitors of the glutamate vesicular transporter (VGLUT) Bioorg Med Chem Lett. 2007;17:5125–5128. doi: 10.1016/j.bmcl.2007.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kish PE, Ueda T. Glutamate accumulation into synaptic vesicles. Meth Enzymol. 1989;174:9–25. doi: 10.1016/0076-6879(89)74005-2. [DOI] [PubMed] [Google Scholar]
- 33.Krueger BK, Forn J, Greengard P. Depolarization-induced phosphorylation of specific proteins, mediated by calcium ion flux, in rat brain synaptosomes. J Biol Chem. 1977;252:2764–2773. [PubMed] [Google Scholar]
- 34.Winter HC, Ueda T. Glutamate uptake system in the presynapric vesicle: Glutamic acid analogs as inhibitors and alternative substrates. Neurochem Res. 1993;18:79–85. doi: 10.1007/BF00966925. [DOI] [PubMed] [Google Scholar]
- 35.Allinger NL, Yeh YH, Lii J-H. Molecular mechanics. The MM3 force field for hydrocarbons. J Am Chem Soc. 1989;111:8551–8556. [Google Scholar]
- 36.Sun G, Fronczek FR, Gandour RD. (R)-2-hydroxy-3-iodo-2-methylpropyl 4-nitrobenzenesulfonate. Acta Crystallogr. 1993;C49:1507–1509. doi: 10.1107/s0108270193000344. [DOI] [PubMed] [Google Scholar]
- 37.Sun G, Fronczek FR, Gandour RD. Methyl (2S,6S:2R,6R)-6-(2-cyanoethyl)-4,6-dimethyl-2-morpholineacetate. Acta Crystallogr. 1995;C51:980–982. doi: 10.1107/s0108270194003604. [DOI] [PubMed] [Google Scholar]
- 38.Karunakaran V, Ramamurthy P, Josephrajan T, Ramakrishnan VT. Solvent effects and photophysical studies of a new class of acridine(1,8)dione dyes. Spectrochim Acta A Mol Biomol Spectrosc. 2002;58:1443–1451. doi: 10.1016/s1386-1425(01)00599-6. [DOI] [PubMed] [Google Scholar]
- 39.Hay BP, Oliferenko AA, Uddin J, Zhang CG, Firman TK. Search for improved host architectures: Application of de novo structure-based design and high-throughput screening methods to identify optimal building blocks for multidentate ethers. J Am Chem. 2005;127:17043–17053. doi: 10.1021/ja055169x. [DOI] [PubMed] [Google Scholar]
- 40.Vukovic S, Hay BP. De novo structure-based design of bis-amidoxime uranophiles. Inorg Chem. 2013;52:7805–7810. doi: 10.1021/ic401089u. [DOI] [PubMed] [Google Scholar]
- 41.Lobur AT, Kish PE, Ueda T. Synaptic vesicular glutamate uptake: modulation by a synaptosomal cytosolic factor. J Neurochem. 1990;54:1614–1618. doi: 10.1111/j.1471-4159.1990.tb01212.x. [DOI] [PubMed] [Google Scholar]
- 42.Tamura Y, Özkan ED, Bole DG, Ueda T. IPF, a vesicular uptake inhibitory protein factor, can reduce the Ca2+-dependent, evoked release of glutamate, GABA and serotonin. J Neurochem. 2001;76:1153–1164. doi: 10.1046/j.1471-4159.2001.00120.x. [DOI] [PubMed] [Google Scholar]
- 43.Kirschner N. Uptake of catecholamines by a particulate fraction of the adrenal medulla. J Biol Chem. 1962;237:2311–2317. [PubMed] [Google Scholar]
- 44.Johnson RG., Jr Accumulation of biological amines into chromaffin granules: a model for hormone and neurotransmitter transport. Physiol Rev. 1988;68:232–307. doi: 10.1152/physrev.1988.68.1.232. [DOI] [PubMed] [Google Scholar]
- 45.Sherman D, Jaudon P, Henry JP. Characterization of the monoamine carrier of chromaffin granule membrane by binding of [2-3H]dihydrotetrabenazine. Proc Natl Acad Sci USA. 1983;80:584–588. doi: 10.1073/pnas.80.2.584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Stern-Bach Y, Greenberg-Ofrath N, Flechner I, Schuldiner S. Identification and purification of a functional amine transporter from bovine chromaffin granules. J Biol Chem. 1990;265:3961–3966. [PubMed] [Google Scholar]
- 47.Liu Y, Peter D, Roghani A, Schuldiner S, Prive GG, Eisenberg D, Brecha N, Edwards RH. A cDNA that suppresses MPP+ toxicity encodes a vesicular amine transporter. Cell. 1992;70:539–551. doi: 10.1016/0092-8674(92)90425-c. [DOI] [PubMed] [Google Scholar]
- 48.Erickson JD, Eiden LE, Hoffman B. Expression cloning of a reserpine-sensitive monoamine transporter. Proc Natl Acad Sci USA. 1992;89:10993–10997. doi: 10.1073/pnas.89.22.10993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Schuldiner S, Shirvan A, Linial M. Vesicular neurotransmitter transporters: From bacteria to humans. Physiol Rev. 1996;75:369–392. doi: 10.1152/physrev.1995.75.2.369. [DOI] [PubMed] [Google Scholar]
- 50.Erickson JD, Schafer MKH, Bonner TI, Eiden LE, Weihe E. Distinct pharmacological properties and distribution in neurons and endocrine cells of two isoforms of the human vesicular monoamine transporter. Proc Natl Acad Sci USA. 1996;93:5166–5171. doi: 10.1073/pnas.93.10.5166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Wimalasena K. Vesicular monoamine transporters: Structure-function, pharmacology, and medicinal chemistry. Med Res Rev. 2011;31:483–519. doi: 10.1002/med.20187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kilbourn MR, DaSilva N, Frey KA, Koeppe RA, Kuhl DE. In vivo imaging of vesicular monoamine transporters in human brain using [11C]tetrabenazine and positron emission tomography. J Neurochem. 1993;60:2315–2318. doi: 10.1111/j.1471-4159.1993.tb03521.x. [DOI] [PubMed] [Google Scholar]
- 53.Frey KA, Koeppe RA, Kilbourn MR, Vander Borght TM, Albin RL, Gilman S, Kuhl DE. Presynaptic monoaminergic vesicles in Parkinson’s disease and normal aging. Ann Neurol. 1996;40:873–884. doi: 10.1002/ana.410400609. [DOI] [PubMed] [Google Scholar]
- 54.Ondo WG, Tintner R, Thomas M, Jankovic J. Tetrabenazine treatment for Huntington’s disease associated chorea. Clin Neuropharmacol. 2002;25:300–302. doi: 10.1097/00002826-200211000-00003. [DOI] [PubMed] [Google Scholar]
- 55.Marshall FJ, Walker F, Frank S, Oakes D, Plumb S, Factor SA, Fahn S, Hunt VP, Jankovic J, Shinaman A, Shoulson I. Tetrabenazine as antichorea therapy in Huntington’s disease: A randomized controlled trial. Neurology. 2006;66:366–372. doi: 10.1212/01.wnl.0000198586.85250.13. [DOI] [PubMed] [Google Scholar]
- 56.Parsons SM, Prior C, Marshall IG. Acetylcholine transport, storage, and release. Int Rev Neurobiol. 1993;35:279–390. doi: 10.1016/s0074-7742(08)60572-3. [DOI] [PubMed] [Google Scholar]
- 57.Bahr BA, Clarkson ED, Rogers GA, Noremberg K, Prsons SM. A kinetic and allosteric model for the acetylcholine transporter-vesamicol receptor in synaptic vesicles. Biochemistry. 1992;31:5752–5762. doi: 10.1021/bi00140a010. [DOI] [PubMed] [Google Scholar]
- 58.Giboureau N, Som IM, Boucher-Arnold A, Guilloteau D, Kassiou M. PET radioligands for the vesicular acetylcholine transporter (VAChT) Curr Top Med Chem. 2010;15:1569–1583. doi: 10.2174/156802610793176846. [DOI] [PubMed] [Google Scholar]
- 59.Juge N, Yoshida Y, Yatsushiro S, Omote H, Moriyama Y. Vesicular glutamate transporter contains two independent transport machineries. J Biol Chem. 2006;281:39499–39506. doi: 10.1074/jbc.M607670200. [DOI] [PubMed] [Google Scholar]