

Abstract
Enantiopure mono-cycloplatinated-[8]helicene and bis-cycloplatinated-[6]helicene derivatives were prepared through column chromatography combined with crystallization of diastereomeric complexes using a chiral ancillary sulfoxide ligand. The UV-visible spectra, circular dichroism, molar rotations, and (circularly polarized) luminescence activity of these new helical complexes have been examined in detail and analysed with the help of first-principles quantum-chemical calculations.
Introduction
Helicenes are among the rare classes of molecules displaying exceptionally strong chiroptical properties (optical rotation, circular dichroism into the visible region), thanks to their inherent helical chirality combined with extended π-conjugation.1 Structural and electronic features provide helicenes with additional properties that are essential for the development of chiral molecular materials and devices.2 For instance, helicenes have been reported displaying circularly polarized luminescence (CPL),3a–d redox3e–g or optical switching,3h,i electrical conductivity,3j,k non-linear optical (NLO) activity,3l to name a few. The conception of new chiral materials and devices is closely related to efficient and innovative synthetic and enantiomer-resolution methods allowing not only structural diversity but also gram-scale synthesis. We have recently demonstrated the potential of the organometallic approach to prepare the first metallahelicenes bearing a metal ion incorporated into the helical backbone.4 We have shown that using a 3rd row transition-metal ion, this strategy confers multifunctionality on the helicene, by combining the chiroptical properties with room temperature phosphorescence and redox activity.
Apart from HPLC and column chromatographic separations,1,5a,b the few known preparations of enantiopure helicene derivatives are essentially based on asymmetric synthesis using either a chiral catalyst,5c,d kinetic resolution,5e enantio-5f or diastereoselective syntheses.5g,h Although the parent carbo[n]helicenes have been known to form conglomerates,6a resolution of helicene enantiomers by crystallization methods is rarely used.6b–e
In this paper, we describe a straightforward access to the unprecedented mono-platina[8]helicene (3a) and bis-platina[6]helicene (3b) (Scheme 1). These new complexes were obtained in enantiopure forms through simple crystallization of diastereomeric complexes {(M*,RS)-2a1,2 and (M*,RS,RS)-2b1,2, respectively} bearing a chiral ancillary sulfoxide ligand, which was then replaced by the achiral acetylacetonate (acac) ligand. The chiroptical properties (circular dichroism and molar rotations), and the photophysical properties (UV-visible absorption, non-polarized and circularly-polarized luminescence) of the complexes are examined and analyzed with the help of quantum-chemical calculations in the context of parameters including the size of the helicene, the helicity, and the role of the metal ions within the helicene.
Scheme 1.

General synthesis of enantiopure Pt-[8]helicene and Pt2-[6]helicene derivatives M-3a and P-3b from (±)-1a and 1b respectively. i) (SS,SS)-PtCl2(p-tolyl-MeSO)2, toluene, Na2CO3, Ar, reflux, overnight, 60%; ii) column chromatography and/or crystallization; iii) pentane-2,4-dione, toluene, Na2CO3, Ar, reflux, 2 h. X-ray crystallographic structures of 3a and 3b (one enantiomer shown) and chemical structure of 3c.4a,b
B Results and discussion
B.1. Synthesis and solid state structure
Phenylpyridine ligands are known to form stable platinacycles bearing a variety of ancillary ligands, including monodentate sulfoxides.7 In a few examples, the sulfoxide ligand is a chiral one (usually R- or S-methyl-p-tolyl-sulfoxide) and therefore leads to enantiopure chiral cycloplatinated complexes.8 Using this methodology, the diastereomeric mixture of monoplatinated [8]helicenes (M,RS)-2a1 and (P,RS)-2a2 were prepared in one step by orthoplatination of racemic 2-pyridyl-[6]helicene4d 1a using (SS,SS)-PtCl2(p-tolyl-MeSO)2 (trans/cis mixture) as the enantiopure platinum source (ee>97%), in refluxing toluene and under basic conditions (Na2CO3). This cycloplatination reaction increases the size of the helicene from a hexa- to an octahelicene derivative. The presence of two (M,RS)-2a1and (P,RS)-2a2 diastereomers is ascertained by 1H and 13C NMR spectra which show the presence of two sets of signals in a 1:1 ratio. Compared to the non-metallated ligand 1a, complexes 2a1,2 display well-resolved 1H NMR spectra in which 195Pt-1H couplings constants are observed (2a1: 34 Hz for H6’ and 52 Hz for H3, see ESI†).
Gratifyingly, crystallization by slow diffusion of pentane vapour into a CH2Cl2 solution yielded single crystals of the pure diastereomer (M,RS)-2a1. The X-ray crystallographic structure (P21 space group, Figure 1) clearly depicts (i) the eight ortho-fused cycles with the platinacycle incorporated within the helical backbone with a helicity (dihedral angle between terminal rings) of 11.42°, a value which is very low due to intramolecular π–π stacking (centroid-to-centroid distances of 3.465 Å between the pyridyl ring and the 7th benzene ring); (ii) the sulfoxide and chloride disposed trans to the N and C atoms respectively, due to the strong trans influence of the latter; and (iii) the M and the RS configurations only. Note that the sulfoxide ligand is coordinated through the S atom and that the p-tolyl group is directed away from the helicene. Another important feature is the columnar packing of the molecules along the x axis arising from intermolecular π–π stacking between adjacent helicene moieties (vide supra, Figure 1). Such a supramolecular arrangement may be the driving force for the efficient diastereoselective crystallization process.
Figure 1.

X-ray crystallographic structures of diastereomerically pure Pt-[8]helicene derivatives (M,RS)-2a1 and (P,RS)-2a2. Columnar packing of (M,RS)-2a1 along the x axis (CPK model representation).
This encouraging result prompted us to conduct the separation of (M,RS)-2a1 and (P,RS)-2a2 diastereomers on a preparative scale. This was achieved by initial column chromatography (using heptane/ethyl acetate 3:1 as the eluent) to obtain partially resolved samples, which subsequently crystallized out of solution as enantiomerically and diastereomerically pure (M,RS)-2a1 and (P,RS)-2a2 in 33 and 18% yield respectively. Single crystals of (P,RS)-2a2 suitable for X-ray diffraction analysis were grown from the heptane/ethyl acetate solution. The X-ray crystallographic structure of (P,RS)-2a2 (Figure 1) displayed the same features as for (M,RS)-2a1 except that (i) the helicene has its configuration inverted and is directed towards the p-tolyl substituent, and (ii) no supramolecular organization is observed.
In a final step, the chloride ligand and the p-tolyl methyl sulfoxide used as a chiral auxiliary agent were replaced by a bidentate acetylacetonate ligand, giving rise to the enantiomeric pair M- and P-3a (Scheme 1) in 95% yield. Note that no racemization was observed during this step since the molecules are stable in refluxing toluene. The X-ray structure of 3a obtained from a racemic sample is displayed in Scheme 1. It shows that the Pt(II) complex 3a is a structural analogue of a carbo[8]helicene with a small helicity (15.04°) due to the strong intramolecular π–π interaction.
Since we have demonstrated that diastereomeric resolution by column chromatography coupled with crystallization is a convenient way for producing enantiopure helicene derivatives, enabling the preparation of enantiopure samples on a scale of several hundred milligrams (see ESI), a straightforward synthesis of a [6]-helicene incorporating two platinum centres was performed by taking advantage of the fact that cycloplatination increases the helicene size by two fused rings. Indeed, the first bis-cycloplatinated [6]helicene derivative was prepared in enantiopure form in only three steps through the double cycloplatination of a 1,8-bisarylnaphthalene scaffold.9 The proligand 1,8-bis(2-pyridyl)naphthalene 1b was prepared by a Stille coupling between 1,8-diiodonaphthalene and 2-(trimethylstannyl)-pyridine, according to a known procedure9c,e (see ESI). 1,8-Bis-aryl-naphthalene derivatives are known to display cis-trans isomerism, due to steric hindrance of the aryl groups that are forced into close proximity by the naphthalene platform. While the cis-form is centrosymmetric, the trans-form is C2-symmetric and exists as two enantiomers.9
Proligand 1b crystallizes in a non-centrosymmetric space group (P21), where the two pyridyl groups are transoid and twisted with a dihedral angle of 35.44° (Figure 2), with weak π–π interaction observed (centroid-centroid distance = 3.683 Å). While molecule 1b adopts a helical shape in the solid state, it is conformationally labile in solution and is overall achiral. The double orthometallation of 1b was then accomplished by reaction with 2 equiv of (RS,RS)-PtCl2(p-tolyl-MeSO)2 (trans/cis mixture) as an enantiopure platinum source in refluxing toluene in the presence of a base (Na2CO3). The 1H NMR and 13C NMR spectra of the product of this reaction revealed the presence of a mixture of two bis-cyclometallated compounds (2b1 and 2b2) in the ratio 1.5:1 respectively, based on integration of the H6-py proton (see ESI) in the 9–10 ppm region of the 1H NMR spectrum.
Figure 2.

X-ray crystallographic structures of ligand 1b and the pure diastereomeric complex (P,RS,RS)-2b1. Columnar packing of (P,RS,RS)-2b1 is observed along the z axis (Pt2-[6]helicene shown in CPK model representation).
Slow crystallization by diffusing pentane vapour over a CHCl3 solution yielded single crystals that were identified as the pure diastereomer (P,RS,RS)-2b2 by X-ray crystallography. Indeed, the solid state structure (P212121 space group) shows that the bis-Pt(II) complex 2b2 adopts a helical topology (Figure 2), with a helicity angle between the two terminal pyridyl groups of 58.8°, in very good agreement with mono-platina[6]helicenes and carbo[6]helicene derivatives.4 In this unprecedented structure, two almost planar platinacycles (dihedral angles between phenyl and pyridyl rings < 19°) are incorporated within the helical backbone (with the two Cl trans to the metallated carbon atoms and two S-sulfoxides trans to the N atoms). A supramolecular columnar arrangement is also present in the solid state due to intermolecular π–π stacking between adjacent helicene moieties (Figure 2). Clearly, bis-metallahelicene derivatives can be straightforwardly produced by double orthometallation of a bis-substituted naphthalene platform. Furthermore, given the 1.5:1 diastereomeric ratio of (M,RS,RS)-2b1 and (P,RS,RS)-2b2, the C–H activation that occurs during the cycloplatination appears to be diastereoselective.4d,10 Despite the moderate efficiency, such diastereoselectivity is very rare in cycloplatination reactions since high ee values are difficult to achieve when high-temperature reactions are required (refluxing toluene here).
Based on the successful isolation of enantiomerically and diastereomerically pure single crystals of (P,RS,RS)-2b2, the preparative resolution of 2b1,2 was studied by taking advantage of their different solubilities. This was accomplished by simply using the appropriate quantity of heptane/CHCl3 (3:1) as the crystallization solvent: (P,RS,RS)-2b2 precipitated in 34% yield, whereas the mother liquors contained mainly (M,RS,RS)-2b1, which was finally obtained in pure form by column chromatography (SiO2, EtOAc as the eluent) followed by crystallization in heptane/CHCl3 (3:1) (33% yield). The mirror-image experiment starting from (SS,SS)-PtCl2(p-tolyl-MeSO)2 yielded pure (M,SS,SS)-2b1 and (P,SS,SS)-2b2. In a final step, the chloride and sulfoxide ligands were replaced by acac, yielding enantiomeric pairs M- and P-3b (Scheme 1). The X-ray structure shows that 3b has the same helicity (51.24°) as the former monoplatina[6]helicene analogue 3c (52.3°),4a but a more open skeleton and therefore is more prone to racemization. This was experimentally verified on complexes 2b1 and 2b2 that revealed 30% loss of diastereomeric purity after refluxing in toluene for 72 hours, while complex 3b displays a long half-life (~19 days at 100°C, see ESI).
B.2. Photophysical properties
Having to hand the different platinahelicene derivatives, with either different lengths (Pt-[6]-vs.Pt-[8]-helicene) or different numbers of Pt(II) atoms (Pt-[6]- vs. Pt2-[6]-helicene), their absorption and photophysical properties were measured and compared. The UV-visible absorption data of compounds 1a, 2a1,2 and 3a are summarized in Table 1 and the spectra are shown in Figure 3.
Table 1.
Photophysical data of ligand 1a and of the PtII-helicene complexes 2a1, 3a,b, together with that of the parent Pt-[6]helicene 3c for comparison.
| 1a | 2a1 | 3a | 3b | 3c(f) | |
|---|---|---|---|---|---|
| Absorption λmax/nm (ε/M−1cm−1) (a) | 232 (43400), 265 (40800), 310 (21600), 339 (15600), 399sh (1280), 422 (760) | 233 (66100), 263 (40900), 282 (37100), 320 (24100), 378 (8700), 466 (4340) | 260 (49900), 281 (45400), 318sh (34300), 369 (15000), 467 (5320) | 255 (42000), 290 (34200), 389 (21400), 433 (11200), 471 (6640) | 255 (61300), 294 (34000), 316 (33500), 346 (3350), 435sh (7110), 452 (7800) |
| Emission λmax/nm (298K) (a) | 430, 453 | 677 | 648 | 633, 673 | 644 |
| Φf× 102 (298 K)(b) | 3.3 | 4.1 | 5.6 | 13 | 10 |
| τlum/ns (298 K) deg. [aer.](c) | 5.9 | 24000 [780] | 16500 [630] | 18700 [970] | 21000 |
| kQO2/108 M−1s−1 (d) | 5.6 | 6.9 | 4.4 | ||
| Emission λmax/nm (77 K) (e) | 420, 446, 474, 504 (fluo) 541, 589, 639 (phos) |
641, 677 | 598, 644 | 608, 664, 727 | 592, 638, 691(sh) |
| τlum (77 K) | 8.9 ns (fluo) 1.1 s (phos) |
61 μs | 32 μs | 28 μs | 45 μs |
In CH2Cl2 at 298 K.
Measured using [Ru(bpy)3]Cl2 as the standard.
Values in air-equilibrated solution in parentheses.
Bimolecular rate constant for quenching by O2.
In diethyl ether/isopentane/ethanol (2:2:1 v/v).
Data from references 4a,b.
Figure 3.

UV-vis and CD spectra of (P,SS)-2a1, (M,SS)-2a2, P-3a, (P,SS,SS)-2b1, (P,RS,RS)-2b1, and P-3b in solution in dichloromethane at room temperature, compared to the starting organic ligands P-1a and 1b.
The electronic absorption spectrum of 1a displays an intense high-energy band (ε > 3 × 104 M−1 cm−1) around 290 nm, with longer-wavelength shoulders and a tail out to 420 nm (Figure 3). This spectrum is typical of a [6]helicene derivative. The electronic absorption spectra of Pt-[8]helicene complexes 2a1,2 and 3a display several intense absorption bands between 250 and 380 nm and two weaker (ε ~ 300–1000 M−1 cm−1) lower-energy broad bands at 380 and 480 nm arising from interactions between the metal and the π-ligand. The absorption at these lowest energy bands in 2a1,2 and 3a are bathochromically shifted by ca. 50 nm compared to 1a (vide infra). This red-shift is consistent with an elongation of the π-conjugated system from a [6]helicene to an [8]helicene via the ring closure that accompanies orthometallation (Scheme 1). UV-vis and CD spectra have been calculated by time-dependent DFT (TDDFT) at the BHLYP/SV(P) level (c.f. ESI),11 see Figure 4, Tables 2 and 3. An excitation analysis in terms of MO-pair contributions of [6]helicene ligand 1a shows that the main low-energy transitions are of π-π* type, with sizable dipolar oscillator strength values (e.g. excitation no. 3, Table 2, Figure 4). In the case of 2a1 and 3a, the excitations at lower energy are assigned predominantly as HOMO-LUMO (P-2a1, 84%; P-3a, 81%) (excitations no. 1, Table 2, Figure 4) and may be considered as a π-π* transition localized in the N^C ligand system with some participation of the platinum 5d orbitals. The calculations therefore show that the orbitals of the metal centre efficiently interact with those of the helicoidal π-system.
Figure 4.

Upper panels: comparison of the calculated UV-vis (top) and CD spectra (bottom) of P-1a, P-3a, (P,SS)-2a1 and (M,SS)-2a2 (left) and P-1b, P-3b, (P,SS,SS)-2b1 and (P,RS,RS)-2b2 systems (right). Color and line schemes correspond to those in Figure 3. No spectral shifts were applied. Numbers correspond to excitations that were analyzed in detail. Lower panels: isosurface plots of the HOMO and LUMO of series 1a, 2a1, and 3a and of series 1b, 2b1, and 3b are shown. Further details are provided in the ESI.
Table 2.
Selected excitations and occupied (occ) – unoccupied (unocc) MO pair contributions (greater than 10%) of P-1a, (P,SS)-2a1, and P-3a.
| Excitation | E/eV | f | R/10−40cgs | occ no. | unocc no. | % |
|---|---|---|---|---|---|---|
| 1a | ||||||
| #3 | 3.82 | 0.167 | 789.76 | 113 | 115 | 33.1 |
| 114 | 116 | 30.4 | ||||
| 112 | 115 | 20.8 | ||||
|
| ||||||
| 2a1 | ||||||
| #1 | 3.14 | 0.088 | 69.58 | 172 | 173 | 83.6 |
| #3 | 3.52 | 0.017 | 67.04 | 172 | 174 | 56.3 |
| #4 | 3.62 | 0.173 | 697.14 | 171 | 173 | 49.0 |
| 172 | 174 | 16.7 | ||||
| 172 | 175 | 10.1 | ||||
|
| ||||||
| 3a | ||||||
| #1 | 3.16 | 0.085 | 45.01 | 149 | 150 | 80.7 |
| #2 | 3.46 | 0.029 | 89.37 | 149 | 151 | 70.6 |
| 148 | 150 | 10.0 | ||||
| #3 | 3.48 | 0.021 | 58.90 | 149 | 152 | 41.3 |
| 147 | 150 | 16.0 | ||||
| 148 | 151 | 15.0 | ||||
| 148 | 150 | 11.8 | ||||
| #4 | 3.66 | 0.162 | 649.16 | 148 | 150 | 58.3 |
Table 3.
Selected excitations and occupied (occ) – unoccupied (unocc) MO pair contributions (greater than 10%) of 1b, (P,SS,SS)-2b1, and P-3b.
| Excitation | E/eV | f | R/10−40cgs | occ no. | unocc no. | % |
|---|---|---|---|---|---|---|
| 1b | ||||||
| #1 | 4.07 | 0.328 | 32.04 | 74 | 75 | 94.7 |
|
| ||||||
| 2b1 | ||||||
| #1 | 3.11 | 0.139 | 129.61 | 190 | 191 | 93.6 |
| #2 | 3.38 | 0.031 | −46.58 | 190 | 192 | 58.9 |
| 189 | 191 | 30.9 | ||||
| #3 | 3.71 | 0.419 | 132.57 | 189 | 191 | 45.6 |
| 190 | 192 | 33.3 | ||||
| #5a | 3.94 | 0.086 | −59.32 | - | - | - |
| #10 | 4.36 | 0.023 | −56.21 | 190 | 193 | 52.6 |
| 189 | 192 | 29.7 | ||||
| #12 | 4.56 | 0.108 | 179.44 | 187 | 191 | 36.7 |
| 190 | 196 | 21.3 | ||||
|
| ||||||
| 3b | ||||||
| #1 | 3.18 | 0.145 | 126.70 | 144 | 145 | 92.7 |
| #2 | 3.34 | 0.029 | −70.28 | 144 | 146 | 57.1 |
| 143 | 145 | 33.7 | ||||
| #3 | 3.72 | 0.493 | 122.44 | 143 | 145 | 52.3 |
| 144 | 146 | 35.1 | ||||
| #8 | 4.29 | 0.007 | 80.65 | 144 | 150 | 43.6 |
| 140 | 145 | 16.3 | ||||
| #10 | 4.46 | 0.013 | −45.43 | 142 | 145 | 39.5 |
| 141 | 146 | 14.4 | ||||
| #11 | 4.56 | 0.039 | 245.20 | 142 | 148 | 11.7 |
No contributions exceeded 10%.
Similarly, whilst the starting proligand 1b displays two bands of moderate intensity at 233 nm (ε ~ 34 × 103 M−1 cm−1) and 303 nm (ε ~ 11 × 103 M−1 cm−1), the electronic absorption spectra of its Pt2-[6]helicene complexes 2b1,2 and 3b show much more intense absorption between 250 nm and 350 nm (Figure 3), and three lower-energy, broad bands between 380 and 500 nm originating from the interaction between the two metallic ions and the π-ligand (vide infra). This strong modification and red-shift of the UV-vis spectra show the efficiency of the double orthometallation ring closure in creating elongated helical π-conjugated [6]helicene frameworks. Furthermore, metal-to-ligand charge transfers are more efficient when two metal centres are involved within the π-ligand. For example, comparing 3b with the mononuclear Pt-[6]helicene 3c (Table 1), it is apparent that the absorption bands in the visible region are not only shifted to lower energy in the dinuclear compounds, but that their molar absorptivity is also increased (e.g. ε values at 434 ± 1 nm are 11200 and 7110 M−1cm−1 for the di- and mononuclear compounds respectively). A similar red-shift has been observed recently when the bridging unit is a heterocycle, for example, in diplatinum complexes of diphenylpyrazines and 2,3-diphenylpyrimidines.12 Typically, in cycloplatinated complexes of arylpyridine ligands, electrochemical studies and first-principles theory indicate that the HOMO is predominantly localised on the metallated ring and the metal ion, whereas the LUMO resides primarily on the pyridyl rings.13 In organometallic helicenes 2b1,2 and 3b, the metal atom functions not only to construct the helicoidal framework by double orthometallation (Scheme 1) but also to markedly impact the electronic properties of the π-conjugated system. Indeed, the excitations at lower energy are assigned to HOMO-LUMO (P-2b1, 94%; P-3b, 93%) (see excitations no. 1, Figure 4 and Table 3). This excitation can be considered as a π-π* transition localized in the π-ligand with non-negligible involvement of metal orbitals. The same type of excitation can be also found in the case of the mono-platinum complex 3c but with much weaker intensity.4a,b Accordingly, higher dipolar strengths of this transition in (2b/3b) may be attributed to the presence of two Pt centres built into the N^C π-system and modifying the electronic properties of the helicene platform compared to 3c.
To summarize: A direct comparison of the UV-vis spectra of 1a and 3a-c (Figure 5) along with an analysis of the calculated spectra highlights the differences and common features shared by these four helicene systems: i) additional low-energy HOMO-LUMO transitions present in cycloplatinated helicenes 3a–c with non-negligible involvement of Pt orbitals that do not exist in organic helicene 1a; ii) similar UV-vis spectra of Pt-[8]helicene 3a, Pt2-[6]helicene 3b, and Pt-[6]helicene 3c reflecting an extended π-conjugation involving one or two Pt atoms.
Figure 5.

Comparison of the experimental (left) and calculated (right) UV-vis (top) and CD spectra (bottom) of P-1a (black line), P-3a (blue line), P-3b (red line), and P-3c (green line). No spectral shifts were applied in the calculated spectra. Numbers correspond to excitations that were analyzed in detail.
Interactions between the metal and the π-ligands are not only responsible for the intense, low-energy absorbance in the UV-vis spectra but also for dramatic differences in the emission properties of these new platinahelicenes compared to the metal-free proligands. Proligand 1a exhibits the typical emission behaviour of organic π-conjugated systems.4b In dichloromethane solution at room temperature, it displays structured blue fluorescence around 430 nm (Φ = 3.3%, τ = 5.9 ns; see Table 1 and Figure 6). At 77K, the fluorescence is only slightly blue-shifted but is then accompanied by a very long-lived green phosphorescence around 540 nm emanating from the triplet state (τ = 1.1 s), with similar vibrational structure to the fluorescence (Δν ~ 1300 cm−1) (see ESI). As usual for such organic molecules, the phosphorescence is not observable at room temperature, the radiative rate constant is too small to compete with non-radiative decay processes.
Figure 6.

CPL (upper curves within each panel) and total luminescence (lower curves within each panel) spectra of M-1a (top left red), P-1a (top left black), M-3a (top right red), P-3a (top right black), M-3b (bottom left red), P-3b (bottom left black), M-3c (bottom right red), P-3b (bottom right black) in degassed dichloromethane solution (1 mM) at 295 K, upon excitation at 371–382, 452, 459–469 nm, and 452 nm respectively.
When the platinum(II) ion is incorporated into the π-conjugated system, the emission properties change drastically. The platinahelicenes show no fluorescence but instead display strong room-temperature phosphorescence. As found in simpler complexes of arylpyridines, efficient spin-orbit coupling of the metal ion valence orbitals promotes very rapid intersystem crossing to the triplet state, eliminating the fluorescence from the S1 state that was seen for the proligand, whilst facilitating the subsequent T1 → S0 phosphorescence transition. Phosphorescence can then successfully compete with triplet non-radiative decay pathways even at room temperature. Indeed, platinahelicenes 2a1 and 3a are efficient deep-red phosphors, with emission maxima of 677 and 648 nm respectively, and quantum yields of around 5% in deoxygenated solution at room temperature (Figure 6 and Table 1). The luminescence lifetimes of around 20 μs are, however, somewhat longer than those found for complexes of simpler arylpyridine ligands such as phenylpyridine,13 likely because the metal ion has a proportionately smaller influence in the excited state extended over a larger π–system.
The presence of the second platinum centre is seen to slightly stabilise the triplet excited state of the diplatinum complex 3b compared to the mononuclear Pt-[6]helicene 3c (λmax = 608 and 592 nm respectively at 77K), although an apparently larger stabilising effect of the solvent in the latter case leads to very similar spectra at room temperature. The phosphorescence quantum yield Φ of the dinuclear complex is slightly higher, though since the difference is close to the uncertainty on the measurement of Φ, it is not possible to make any definitive conclusions as to whether the presence of a second Pt ion leads to more efficient spin-orbit coupling pathways.12 The phosphorescence quantum yield of Pt-[8]helicene 3a, on the other hand, is lower than the analogous Pt-[6]helicene 3c and the lifetime somewhat shorter. This might reflect an increased scope for non-radiative decay in the more extended structure of the [8]helicene, though again the differences are too small to draw firm conclusions.
As seen in Table 4 below, quantum-chemical calculations of luminescence properties for 1a, and 3a,b,c support the experiment assignment: the calculated fluorescence and phosphorescence energies (2.9 eV and ~1.7 eV, respectively) of [6]helicene ligand 1a correspond well to the experimental low-temperature results. Similarly, in the case of Pt complexes 3a,b,c the calculated emission energies S1 → S0 (~2.9 eV) were found to be very different from the measured luminescence values, while the energies of T1 → S0 phosphorescence transitions (~ 1.9 eV) agree fairly well with the experimental data. A rapid intersystem crossing from the excited singlet to the triplet state responsible for elimination of the fluorescence processes in these complexes may be facilitated by a high structural similarity between S1 and T1 as the calculations showed (see ESI, Figures S35–38).
Table 4.
Experimental and calculated photophysical data of ligand 1a and of the mono- and bis-cycloplatinated helicenes 3a,b,c. Energies in eV.
| System | P-1a | P-3a | P-3b | P-3c |
|---|---|---|---|---|
| Experiment | ||||
| Emission 298 K a | 2.88, 2.74 | 1.91 | 1.96, 1.84 | 1.93 |
| Emission 77 K a | 2.95, 2.78, 2.62, 2.46 (fluo) 2.29, 2.10, 1.94 (phos) |
2.07, 1.93 | 2.04, 1.87, 1.71 | 2.09, 1.94, 1.79 |
|
| ||||
| BHLYP/SV(P) | ||||
| S1{TDDFT}b | 2.93 (0.0477/390.92) | 2.88 (0.1044/55.84) | 2.95 (0.1399/106.02) | 2.92 (0.1524/6.75) |
| T1{TDDFT}c | 1.63 | 1.90 | 1.92 | 1.89 |
| T1{DFT}d | 1.76 | 1.89 | 1.93 | 1.89 |
| S0//T1{DFT} - T1//T1{DFT}e | 1.70 | 1.81 | 1.93 | 1.80 |
|
| ||||
| CASSCF | ||||
| T1{DFT}f | 1.68(0.00/0.00) | - | - | 2.11(1.31·10−6/9.19·10−5) |
|
| ||||
| CASPT2 | ||||
| T1{DFT}g | 2.42(0.00/0.00) | - | - | 2.05(1.35·10−5/1.57·10−3) |
Experimental data.
TDDFT S0-S1 energy difference at TDDFT BHLYP/SV(P) optimized S1 geometry. In parentheses oscillator strength, in au/rotatory strength values, in 10−40 cgs, are listed. To convert to integrated CPL intensity, in cgs units of power output per molecule, the following conversion can be used: ΔI = 1.60041320·1037·Rcgs·Ecgs4.
TDDFT S0-T1 energy difference at TDDFT BHLYP/SV(P) optimized T1 geometry.
TDDFT S0-T1 energy difference at DFT BP/SV(P) optimized triplet configuration.
S0-T1 energy gap calculated as difference between S0//T1{DFT} and T1//T1{DFT}.
B.3. Chiroptical properties (circular dichroism, specific and molar rotations)
The electronic circular dichroism (ECD) spectra and molar rotations (MRs) of enantiopure ligand 1a, PtII-[8]helicene derivatives 2a1,2, 3a and PtII2-[6]helicenes 2b1,2, 3b also revealed the role of the platinum centres (Table 5, Figure 3). The two M and P enantiomers of ligand 1a with respective ee’s higher than 98% were obtained through HPLC separation over a chiral stationary phase (see ESI). Ligand P-1a displays the classical behaviour of a [6]helicene derivative, with a strong negative band (Δε = −84 M−1cm−1) at 262 nm accompanied by a smaller negative band (Δε = −24 M−1cm−1) at 295 nm, a strong positive band (Δε = +112 M−1cm−1) at 340 nm and a smaller one (Δε = +61 M−1cm−1) at 361 nm. Complex (P,SS)-2a1 displays ECD active bands that are 30–40 nm red-shifted and stronger intensity than for 1a ((P,SS)-2a1: Δε = +100, −91, −62, +131 M−1cm−1 at 245, 285, 325 and 374 nm respectively) together with low-energy ECD-active bands above 430 nm ((P,SS)-2a1: Δε = +10 M−1cm−1 at 462 nm). Complex (M,SS)-2a2 affords nearly a mirror-image ECD spectrum of (P,SS)-2a1 even though this is not strictly an enantiomer pair. The ECD spectra of enantiomeric complexes P- and M-3a (see Figure 3 and ESI) show a similar shape to (P,SS)-2a1 and (M,SS)-2a2.
Table 5.
Experimental and calculated optical rotations for the [6]helicene ligand 1a and mono- and bis-cycloplatinated helicenes 2a,b1,2 and 3a,b. a)
| BHLYP/SV(P)
|
Expt. (23°C)
|
||
|---|---|---|---|
| [α]D | [ϕ]D | [ϕ]D | |
| P-1a | 3767 | 16406 | 8290 |
| (M,RS)-2a1 | −2930 | −24008 | −19920 |
| (P,RS)-2a2 | 2337 | 19147 | 19210 |
| P-3a | 3197 | 23298 | 22500 |
| (P,SS,SS)-2b1 | 933.4 | 9800 | 11720 |
| (P,RS,RS)-2b2 | 413.1 | 4337 | 9616 |
| P-3b | 1185 | 10297 | 8950 |
| P-3cb) | 1588 | 9982 | 8170 |
Specific rotations and molar rotations are given in the usual units of degree/(dm g/cm−3) and degree cm2/dmol, respectively.
Data from references 4a,b.
The corresponding TDDFT calculated CD envelopes, in general, agree will with the experimental. The TDDFT electronic excitation energies, and the resulting band peaks in the simulated spectra, are somewhat blue-shifted with respect to experiment (Figures 4 and S29, ESI). As seen in Figure 4, important experimentally observed trends are correctly reproduced by the calculations. There is an almost uniform red-shift of the 2a1,2 and 3a spectra compared to 1a, and the CD spectra of (P,SS)-2a1 and (M,SS)-2a2 are nearly mirror images with respect to reflection on the abscissa. The ordering of intensities between 1a, 2a, and 3a does not match the experiment. A strong overestimation of the main positive CD band in 1a is in line with a too large magnitude of the calculated OR (vide infra). The 3a excitation revealing the strongest rotatory strength, no. 4 (E = 3.66 eV, 339 nm), involves one dominant contribution (58%), from π to π* orbitals in the helicene ligand system. In terms of individual MO pairs, the transition is assigned as the HOMO-1 to LUMO (MO148 to MO150, Figures 8). Because the excitation involves orbitals centered in different parts of the ligand π-system, one can consider it as having some degree of charge transfer (CT) character. Such a CT character may be responsible for a noticeable red-shift of this excitation as compared to the corresponding excitation of 1a, (no. 3, E = 3.82 eV, 324 nm, Table 2), which involves orbitals that are more uniformly spread over the helicene π-system (Figure 7). Two other CD-intense excitations of 3a are close in energy (no. 3, 3.48 eV, 356 nm, and no. 2, 3.46 eV, 358 nm) and also exhibit a (partial) π-to-π* CT character within the helicene ligand. In both excitations the dominant contributions, 41% HOMO to LUMO+2 (MO149 to MO152) for no. 3 and 71% HOMO to LUMO+1 (MO149 to MO151) for no. 2, respectively, involve an orbital that has some visible Pt 5d contribution. The HOMO (MO149, Figure 4) for 3a is similar to the HOMO (MO114) of 1a but with some Pt character. Even though there is no obvious π-conjugation between the metal and the pyridyl substituent (the Pt d lone pair orbital is oriented perpendicular to the pyridyl π-orbitals), the involvement of the metal orbital correlates with a significant increase in the rotatory strength of the 3a transitions in comparison with the corresponding 1a ligand. The lowest-energy 3a excitation no. 1 calculated at E = 3.16 eV (393 nm), is assigned almost purely as HOMO to LUMO (81%). Similar to excitations no. 3 and no. 2, it is assigned as a π-to-π* transition localized in the 2-pyridyl-helicene ligand system with some participation of platinum 5d orbitals. Excitations that are similar in terms of energy and rotatory strength to those of 3a are also present in the low-energy part of the CD spectrum of 2a1 (compare Figure 4, Table 2, and Figure S34 in ESI).
Figure 8.
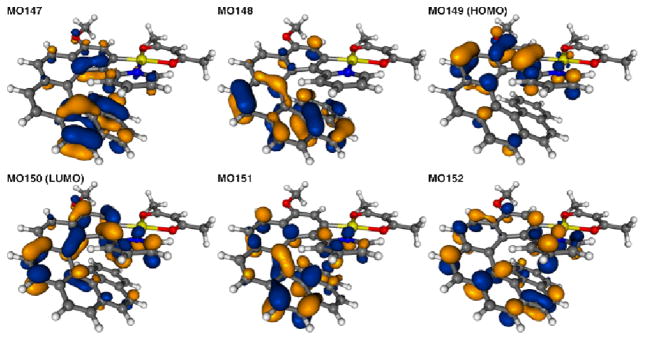
Isosurfaces(0.04 au) of MOs involved in selected excitations of 3a.
Figure 7.

Isosurfaces (0.04 au) of MOs involved in selected excitations of 1a.
The red-shift and higher intensity of ECD-active bands for the Pt complexes is also reflected in the strong increase of the molar rotations when going from the [6]helicene ligand to the Pt-[8]helicene complexes (P-1a: +8290, (P,SS)-2a1: +19210, P-3a: +22500 (±5%) (CH2Cl2, C 10−4 M)). The calculated MR values agree reasonably well with the experiment (see Table 5). Although the theoretical data do not reproduce the full magnitude of the differences between the experimental optical rotations, the trend of increased optical rotation upon formation of the platinum helicenes is correctly predicted by the calculations. In line with the spatial extension of the π-conjugated systems, the MRs of both a-type Pt-[8]helicene complexes are about twice the magnitude of the parent Pt-[6]helicene 3c derivative which itself has a MR close to the pyridyl-[6]helicene ligand 1a. A strong over-estimation relative to the measured value (16406 vs. 8290 degree cm2/dmol) was calculated for the 2-pyridyl-[6]helicene ligand 1a. As shown in references 15, pristine and substituted helicenes can reveal a pronounced dependence of the optical rotations on their molecular structure. Inclusion of van der Waals (dispersive) forces in the geometry optimizations may be needed to improve the calculated chiroptical properties of 1a (vide supra). The MR for the 2a1 complex is also overestimated by the calculations (−24008 vs. −19920 degree cm2/dmol). The optical rotations of both 2a1 and 2a2 are likely to be somewhat dependent on the spatial orientation of the chiral sulfoxide ligand (in particular, the p-tolyl substituent) relative to the helicene π-system, due to excitonic coupling effects. Such effects have been identified and analysed previously for related metal-helicene complexes, as described in references 16. See also the discussion of the b-series (vide infra).
In the b-series of complexes, (P,SS,SS)-2b1 and (P,RS,RS)-2b2 display almost identical experimental CD spectra except in the 250–280 nm region (Figure 3). Their overall shape is surprisingly different from organic [6]helicene derivatives, since the strongest bands are found at 302, 350 and 371 nm with respectively Δε = +47, 24 and 27 M−1cm−1 only, together with several positive or negative bands between 330 and 430 nm of even lower intensities. The CD spectrum of P-3b shows an overall similar behaviour, with a set of one rather strong negative band (Δε = −85 M−1cm−1) at 243 nm, one strong positive band (Δε = +107 M−1cm−1) at 293 nm, and four moderately intense positive bands (Δε = +14, 22, 8, 21 M−1cm−1 at 342, 374, 443, 468 respectively). As Figures 4 and S30–32 (ESI) show, the calculated spectral envelopes of complexes 2b1,2 and 3b agree generally quite well with experiment, provided that a red-shift is applied to the calculated spectra. In accordance with experiment, complexes 2b1 and 3b display positive CD bands of significant intensity in the low-energy part of the spectra (excitation no. 1, E = 3.11 eV (399 nm) and 3.18 eV (390 nm) for 2b1 and 3b, respectively) that are of almost pure HOMO-to-LUMO character (2b1: MO190 to MO191: 94%, Figure 9, and 3b: MO144 to MO145: 93%, Figure 10) correspond to π-to-π* transition localized in the pyridyl-based helicene moiety with slight involvement of platinum orbitals. The next excitation, no. 2, calculated at E = 3.38 eV, 366 nm (2b1) and 3.34 eV, 371 nm (3b) has a negative rotatory strength and consequently suppresses positive band intensity at the low-energy spectral region for both compounds. For both 2b1 and 3b this excitation is a π-to-π* transition localized in the pyridyl-based helicene moiety with participation of Pt 5d orbitals (HOMO to LUMO+1: ~ 58% and HOMO-1 to LUMO: ~ 33%, see Figures 9, 10). Unlike HOMO-1 for 2,3a which are localized predominantly on the carbohelical fragment, the corresponding orbitals of 2,3b show a noticeable contribution from lone pairs of both platinum centres which become a part of the naphthalene π-system, along with non-negligible participation of the acetylacetonate (acac) ligand moieties. Excitations no. 3 (3.71 eV, 334 nm for 2b1 and 3.72 eV, 333 nm for 3b) have qualitatively the same assignment as no. 2, with two main contributions, HOMO to LUMO+1 and HOMO-1 to LUMO but more weight on the latter (46% for 2b1 and 52% for 3b versus 33% and 35%, respectively). The HOMO to LUMO+1 orbital pair indicates a degree of CT character in the excitation as it involves orbitals centered in different parts of the ligand π-system.
Figure 9.

Isosurfaces (0.04 au) of MOs involved in selected excitations of 2b1.
Figure 10.

Isosurfaces (0.04 au) of MOs involved in selected excitations of 3b.
Despite some unique features of the 2,3b CD spectra, the experimental molar rotation values are typical for [6]helicene derivatives (complexes (P,SS,SS)-2b1: ,(P,RS,RS)-2b2: +9616, P-3b: +8950 (±5%) (in CH2Cl2 solution at 10−4 M)) and of same order of magnitude as the MR of P-3c (Table 5). We attribute this to the similar sizes of the helical π-systems. Satisfactory agreement of calculated ORs with the experiment is found for all the systems except 2b2 for which a striking underestimation occurs (4337 vs. 9616 degree cm2/dmol, see Table 5). Due to this discrepancy, a series of additional calculations were carried out to study possible influences of density functional, basis set as well as an effect of structural parameters on the calculated MR (see ESI, Tables S1 and S2, and Figures S31–S33). As indicated above, the orientation of the sulfoxide ligand has a strong effect on the calculated optical rotation values which we tentatively attribute to excitonic effects similar to those described in references 16.
A comparison of the CD spectra of the ligand 1a and complexes 3a-c as depicted in Figure 5 highlights their common and different general features: i) the CD spectra of the organic 2-pyridyl-[6]helicene 1a and organometallic Pt-[6]helicene 3c display similar overall shape; ii) Pt-[8]helicene 3a displays a stronger and more red-shifted CD spectrum compared to Pt-[6]helicene 3c, a well-known tendency for helicenes that can be attributed to an enlargement of the π-electron system; iii) Pt2-[6]helicene 3b displays a significantly different CD spectrum from 3a,c and 1a. The TDDFT results indicate that this is due to a cancellation of CD-active transitions with opposite sign rotatory strengths and similar energies.
B.4. Circularly polarized luminescence (CPL)
We have resorted to CPL, the emission analogue of ECD, to further investigate the influence of the helicity in the compounds of interest on the chiroptical properties. The circularly polarized luminescence (ΔI) and total luminescence (I) spectra measured for P-/M-1a, P-/M-3a, P-/M-3b, and P-/M-3c in degassed dichloromethane solutions at 295 K are shown in Figure 6, respectively. The degree of circularly polarized luminescence is given by the luminescence dissymmetry ratio, glum(λ) = 2ΔI/I = 2(IL − IR)/(IL + IR), where IL and IR refer, respectively, to the intensity of left and right circularly polarized emissions.17 The solid lines in the CPL plot are presented to show the luminescence spectral line shape.
As usual for most chiral organic chromophores and transition metal complexes, the |glum| values obtained are small: −0.0008/+0.0008, −0.0050/+0.0040, −0.0005/+0.0005 and −0.011/+0.013 for M-1a/P-1a, M-3a/P-3a, M-3b/P-3b, and M-3c/P-3c respectively, as determined at the maximum emission wavelengths. Although these glum values are very small (a value equal to ~0.004 corresponding to light that is only 0.4% circularly polarized), opposite CPL signals were measured for the four sets of compounds having opposite P and M helical configurations. These results confirm that the organic and organometallic helicene solutions in dichloromethane exhibit an active CPL signal and also that the emitted light is polarized in opposite directions for the two enantiomeric forms for each set of these helicenes. Overall, the experimental |glum| values are similar to the ones obtained for organic helicenes,3a,18 except for Pt-[6]helicene which displays a higher |glum| value of the order 10−2, the biggest value obtained for molecular helicenes.3b,d It must be pointed out that the CPL activity is dependent on the structural properties of the compounds of interest: of special importance is that the addition of the Pt fragment in only three steps resulted in the formation of organometallic helicenes that possessed a CPL activity more efficient than their organic counterparts (|0.01| vs. |0.0008|), even if Pt is not a stereogenic centre in these platinahelicene derivatives (a square planar geometry around the Pt centre is observed). Notably, compared to former CPL active helicenes that are fluorescent organic systems, these are phosphorescent. As already mentioned, phosphorescence of these helicene derivatives comes from the presence of the heavy platinum atom which has a large spin-orbit coupling constant. Interestingly, although the platinum centre is non-chiral in itself, it provides helicenes with circularly polarized phosphorescence. This can be explained by the fact that the platinum orbitals are conjugated within the π-system of the helical ligand. The quantum-chemical calculations discussed above confirm participation of Pt 5d orbitals in the MOs that contribute to the electronic transitions. Additional preliminary calculations of spectral emission properties were carried out for 1a and 3c (Table 4) with complete active space self-consistent field (CASSCF) and complete active space second-order perturbation theory (CASPT2) including spin-orbit coupling effects (see ESI).14 A full account of calculated emission spectra will be given in a follow-up publication. The dependence of the wave-function based results on the size of the active spaces needs to be investigated in more detail. However, the preliminary calculations confirm that the CD of the S0–S1 excitation of P-1a and the CPL of the S1–S0 emission have the same sign. For P-3c, calculations at the optimized structure of T1 at different levels of theory including spin-orbit coupling predict nonzero positive rotatory strengths for the T1-S0 emission, which is also consistent with the sign of the CPL in the experiment. The smaller CPL activity of the bis-platinated [6]helicene 3b compared to its mono-platinated helicene[6] analogue 3c (|0.0005| vs. |0.01|) seen experimentally is probably a consequence of two factors: (i) weaker ECD active bands and stronger UV-vis bands involving the platinum centres, (ii) a possibility of racemization of 3b under irradiation by the xenon lamp used for CPL measurements. This is in line with the fact that the CPL response is influenced by the chiral arrangement surrounding the luminescent centre.17a,b
C Conclusions
We have developed an efficient crystallization process combined with chromatography of diastereomeric cycloplatinated complexes bearing a chiral sulfoxide ligand to obtain unprecedented enantiopure mono- and bis-platinahelicenes bearing respectively an [8] and a [6]helicenic backbone. The easy access to these enantiopure cycloplatinated helicenes enabled us to examine the influence of different structural features on the UV-visible spectra, circular dichroism, molar rotations, luminescence and CPL activity. It was shown that, although the platinum centre is not a stereogenic element, these platinahelicenes displayed circularly polarized phosphorescence, with highest |glum| values of the order of 10−2.
Supplementary Material
Acknowledgments
We thank the Ministère de l’Education Nationale, de la Recherche et de la Technologie, the Centre National de la Recherche Scientifique (CNRS) and the ANR (12-BS07-0004-METALHEL-01) and the LIA Rennes-Durham for financial support. The theoretical component of this work has received financial support from the National Science Foundation (CHE 0952253, CHE 1265833 to J.A.) and the Foundation for Polish Science Homing Plus programme co-financed by the European Regional Development Fund under the Operational Programme Innovative Economy (to M. S.) M. S. is also grateful for financial support from the Ministry of Science and Higher Education in Poland (‘Outstanding Young Scientist’ scholarship to M. S. and J. A. acknowledge the Centre for Computational Research (CCR) at the University at Buffalo for providing computational resources. G.M. thanks the National Institute of Health, Minority Biomedical Research Support (1 SC3 GM089589-03 and 3 S06 GM008192-27S1) and the Henry Dreyfus Teacher-Scholar Award for financial support, whilst T. D. J. and K. K. D. thank the NIH (RISE Grant 5R25GM071381 and MARC Grant 2T34GM008253-26, respectively) for research fellowships.
Footnotes
Electronic Supplementary Information (ESI) available: experimental procedures, fluorescence and phosphorescence emission and excitation spectra, X-ray data, theoretical calculations details. See DOI: 10.1039/b000000x/
Contributor Information
Jochen Autschbach, Email: jochena@buffalo.edu.
Jeanne Crassous, Email: jeanne.crassous@univ-rennes1.fr.
References
- 1.Selected reviews, see: Shen Y, Chen C-F. Chem Rev. 2012;112:1463. doi: 10.1021/cr200087r.and references therein. Gingras M. Chem Soc Rev. 2013;42:1051. doi: 10.1039/c2cs35134j.Stará IG, Starý I. In: Science of Synthesis. Siegel JS, Tobe Y, editors. Vol. 45. Thieme; Stuttgart: 2010. pp. 885–953.Rajca A, Miyasaka M. In: Functional Organic Materials. Müller TJJ, Bunz UHF, editors. Wiley-VCH; Weinheim: 2007. pp. 543–577.Urbano A. Angew Chem Int Ed. 2003;42:3986. doi: 10.1002/anie.200301667.Katz TJ. Angew Chem Int Ed. 2000;39:1921. doi: 10.1002/1521-3773(20000602)39:11<1921::aid-anie1921>3.0.co;2-f.Martin RH. Angew Chem Int Ed. 1974;13:649.
- 2.(a) Amabilino D. Chirality at the Nanoscale, Nanoparticles, Surfaces, Materials and more. Wiley-VCH; 2009. [Google Scholar]; (b) Feringa BL, Browne WR. Molecular Switches. Wiley-VCH; 2001. [Google Scholar]
- 3.Selected examples: Field JE, Muller G, Riehl JP, Venkataraman D. J Am Chem Soc. 2003;125:11808. doi: 10.1021/ja035626e.Sawada Y, Furumi S, Takai A, Takeuchi M, Noguchi K, Tanaka K. J Am Chem Soc. 2012;134:4080. doi: 10.1021/ja300278e.Phillips KES, Katz TJ, Jockusch S, Lovinger AJ, Turro NJ. J Am Chem Soc. 2001;123:11899. doi: 10.1021/ja011706b.Kaseyama T, Furumi S, Zhang X, Tanaka K, Takeuchi M. Angew Chem, Int Ed. 2011;50:3684. doi: 10.1002/anie.201007849.Anger E, Srebro M, Vanthuyne N, Toupet L, Rigaut S, Roussel C, Autschbach J, Crassous J, Réau R. J Am Chem Soc. 2012;134:15628. doi: 10.1021/ja304424t.Nishida J, Suzuki T, Ohkita M, Tsuji T. Angew Chem Int Ed. 2001;40:3251. doi: 10.1002/1521-3773(20010903)40:17<3251::AID-ANIE3251>3.0.CO;2-P.Biet T, Fihey A, Cauchy T, Vanthuyne N, Roussel C, Crassous J, Avarvari N. Chem Eur J. 2013;19:13160. doi: 10.1002/chem.201301095.Chen CT, Chen CH, Ong TG. J Am Chem Soc. 2013;135:5294. doi: 10.1021/ja401214t.Wigglesworth TJ, Sud D, Norsten TB, Lekhi VS, Branda NR. J Am Chem Soc. 2005;127:7272. doi: 10.1021/ja050190j.Hatakeyama T, Hashimoto S, Oba T, Nakamura M. J Am Chem Soc. 2012;134:19600. doi: 10.1021/ja310372f.Yang Y, da Costa RC, Fuchter MJ, Campbell AJ. Nature Photonics. 2013;7:634.Verbiest T, Van Elshocht S, Kauranen M, Hellemans L, Snauwaert J, Nuckolls C, Katz TJ, Persoons A. Science. 1998;282:913. doi: 10.1126/science.282.5390.913.
- 4.(a) Norel L, Rudolph M, Vanthuyne N, Williams JAG, Lescop C, Roussel C, Autschbach J, Crassous J, Réau R. Angew Chem Int Ed. 2010;49:99. doi: 10.1002/anie.200905099. [DOI] [PubMed] [Google Scholar]; (b) Anger E, Rudolph M, Norel L, Zrig S, Shen C, Vanthuyne N, Toupet L, Williams JAG, Roussel C, Autschbach J, Crassous J, Réau R. Chem Eur J. 2011;17:14178. doi: 10.1002/chem.201101866. [DOI] [PubMed] [Google Scholar]; (c) Anger E, Rudolph M, Shen C, Vanthuyne N, Toupet L, Roussel C, Autschbach J, Crassous J, Réau R. J Am Chem Soc. 2011;133:3800. doi: 10.1021/ja200129y. [DOI] [PubMed] [Google Scholar]; (d) Shen C, Anger E, Srebro M, Vanthuyne N, Toupet L, Roussel C, Autschbach J, Réau R, Crassous J. Chem Eur J. 2013;19:16722. doi: 10.1002/chem.201302479. [DOI] [PubMed] [Google Scholar]; (e) Crespo O, Eguillor B, Esteruelas MA, Fernandez I, Garcia-Raboso J, Gomez-Gallego M, Martin-Ortiz M, Olivan M, Sierra MA. Chem Comm. 2012;48:5328. doi: 10.1039/c2cc30356f. [DOI] [PubMed] [Google Scholar]
- 5.Selected examples: Gingras M. Chem Soc Rev. 2013;42:1007. doi: 10.1039/c2cs35111k.Laleu B, Mobian P, Herse C, Laursen BW, Hopfgartner G, Bernardinelli G, Lacour J. Angew Chem Int Ed. 2005;44:1879. doi: 10.1002/anie.200462321.Heller B, Hapke M, Fischer C, Andronova A, Stary I, Stara IG. J Organomet Chem. 2013;723:98.and references therein. Sawada Y, Furumi S, Takai A, Takeuchi M, Noguchi K, Tanaka K. J Am Chem Soc. 2012;134:4080. doi: 10.1021/ja300278e.Latorre A, Urbano A, Carreno CM. Chem Comm. 2011;47:8103. doi: 10.1039/c1cc12921j.Weimar M, Correa da Costa R, Lee F-H, Fuchter MJ. Org Lett. 2013;15:1706. doi: 10.1021/ol400493x.Yavari K, Moussa S, Ben Hassine B, Retailleau P, Voituriez A, Marinetti A. Angew Chem Int Ed. 2012;51:6748. doi: 10.1002/anie.201202024.Zadny J, Jancarik A, Andronova A, Samal M, Chocholousova JV, Vacek J, Pohl R, Saman D, Cisarova I, Stara IG, Stary I. Angew Chem Int Ed. 2012;51:5857. doi: 10.1002/anie.201108307.
- 6.Jacques J, Collet A, Wilen SH. Enantiomers, Racemates, and Resolutions. 2. Krieger; Malabar, Florida: 1994. See Vavra J, Severa L, Cisarova I, Klepetarova B, Saman D, Koval D, Kasicka V, Teply F. J Org Chem. 2013;78:1329. doi: 10.1021/jo301615k.and references therein. Vavra J, Severa L, Svec P, Cisarova I, Koval D, Sazelova P, Kasicka V, Teply F. Eur J Org Chem. 2012:489. doi: 10.1021/jo301615k.Paruch K, Katz TJ, Incarvito C, Lam KC, Rhatigan B, Rheingold AL. J Org Chem. 2000;65:7602. doi: 10.1021/jo001055m.Newman MS, Lednicer D. J Am Chem Soc. 1956;78:4765.
- 7.(a) Kobayashi M, Masaoka S, Sakai K. Acta Cryst. 2008;E64:1325. [Google Scholar]; (b) Santoro A, Whitwood AC, Williams JAG, Kozhevnikov VN, Bruce DW. Chem Mater. 2009;21:3871. [Google Scholar]; (c) Santoro A, Wegrzyn M, Whitwood AC, Donnio B, Bruce DW. J Am Chem Soc. 2010;132:10689. doi: 10.1021/ja105469a. [DOI] [PubMed] [Google Scholar]
- 8.(a) Ryabov AD, Panyashkina IM, Polyakov VA, Howard JAK, Kuz’mina LG, Datt MS, Sacht C. Organometallics. 1998;17:3615. [Google Scholar]; (b) Suenkel K, Branzan R, Weigand S. Inorg Chim Acta. 2013;399:193. [Google Scholar]; (c) Djukic JP, Hijazi A, Flack HD, Bernardinelli G. Chem Soc Rev. 2008;37:406. doi: 10.1039/b618557f. [DOI] [PubMed] [Google Scholar]
- 9.(a) Pieters G, Gaucher A, Prim D, Marrot J. Chem Comm. 2009:4827. doi: 10.1039/b905670j. [DOI] [PubMed] [Google Scholar]; (b) Zoltewicz JA, Maier NM. J Org Chem. 1997;62:3215. doi: 10.1021/jo961825n. [DOI] [PubMed] [Google Scholar]; (c) Mei X, Martin RM, Wolf C. J Org Chem. 2006;71:2854. doi: 10.1021/jo0600353. [DOI] [PubMed] [Google Scholar]; (d) Vyskocil S, Meca L, Tislerova I, Cisarova I, Polasek M, Harutyunyan SR, Belokon YN, Stead RMJ, Farrugia L, Lockhart SC, Mitchell WL, Kocovsky P. Chem Eur J. 2002;8:4633. doi: 10.1002/1521-3765(20021018)8:20<4633::AID-CHEM4633>3.0.CO;2-N. [DOI] [PubMed] [Google Scholar]; (e) Wolf C, Mei X. J Am Chem Soc. 2003;125:10651. doi: 10.1021/ja0358145. [DOI] [PubMed] [Google Scholar]
- 10.Example of diastereoselective cycloplatination reaction, see: Huang H, Peters R. Angew Chem Int Ed. 2009;48:604. doi: 10.1002/anie.200804944.Examples of chiral platinated complexes: Anderson C, Crespo M, Morris J, Tanski JM. J Organomet Chem. 2006;691:5635.Plutino MR, MonsuScolaro L, Albinati A, Romeo R. J Am Chem Soc. 2004;126:6470. doi: 10.1021/ja030486u.Günay ME, Hughes DL, Richards CJ. Organometallics. 2011;30:3901.Lopez C, Caubet A, Perez S, Solans X, Font-Bardia M. Chem Comm. 2004:540. doi: 10.1039/b315157c.
- 11.(a) Valiev M, Bylaska E, Govind N, Kowalski K, Straatsma T, Dam HV, Wang D, Nieplocha J, Apra E, Windus T, de Jong W. Comput Phys Commun. 2010;181:1477. [Google Scholar]; (b) Hirata S, Zhan CG, Aprà E, Windus TL, Dixon DA. J Phys Chem A. 2003;107:10154. [Google Scholar]; (c) Autschbach J. Chem Phys Chem. 2011;12:3224. doi: 10.1002/cphc.201100225. [DOI] [PubMed] [Google Scholar]
- 12.(a) Kozhevnikov VN, Durrant MC, Williams JAG. Inorg Chem. 2011;50:6304. doi: 10.1021/ic200706e. [DOI] [PubMed] [Google Scholar]; (b) Culham S, Lanoë PH, Whittle VL, Durrant MC, Williams JAG, Kozhevnikov VN. Inorg Chem. 2013;52:10992. doi: 10.1021/ic401131x. [DOI] [PubMed] [Google Scholar]
- 13.(a) Tam AYY, Tsang DP-K, Chan MY, Zhu NY, Yam VWW. Chem Commun. 2011;47:3383. doi: 10.1039/c0cc05538g. [DOI] [PubMed] [Google Scholar]; (b) Strassert CA, Chien C-H, Galvez Lopez MD, Kourkoulos D, Hertel D, Meerholz K, De Cola L. Angew Chem Int Ed. 2011;50:946. doi: 10.1002/anie.201003818. [DOI] [PubMed] [Google Scholar]; (c) Mroz W, Botta C, Giovanella U, Rossi E, Colombo A, Dragonetti C, Roberto D, Ugo R, Valore A, Williams JAG. J Mater Chem. 2011;21:8653. [Google Scholar]; (d) Rossi E, Murphy L, Brothwood PL, Colombo A, Dragonetti C, Roberto D, Ugo R, Cocchi M, Williams JAG. J Mater Chem. 2011;21:15501. [Google Scholar]; (e) Hui SCF, Chow PK, Tong GSM, Lai SL, Cheng G, Kwok CC, Low KH, Ko MY, Che CM. Chem Eur J. 2013;19:69. doi: 10.1002/chem.201203687. [DOI] [PubMed] [Google Scholar]; (f) Zhang J, Zhao FC, Zhu XJ, Wong WK, Ma DG, Wong WY. J Mater Chem. 2012;22:16448. [Google Scholar]; (g) Hang XC, Fleetham T, Turner E, Brooks J, Li J. Angew Chem Int Ed. 2013;52:6753. doi: 10.1002/anie.201302541. [DOI] [PubMed] [Google Scholar]; (h) Turner E, Bakken N, Li J. Inorg Chem. 2013;52:7344. doi: 10.1021/ic302490c. [DOI] [PubMed] [Google Scholar]
- 14.Aquilante F, De Vico L, Ferré N, Ghigo G, Malmqvist P-Å, Neogrády P, Pedersen TB, Pitoňák M, Reiher M, Roos BO, Serrano-Andrés L, Urban M, Veryazov V, Lindh R. J Comput Chem. 2010;31:224. doi: 10.1002/jcc.21318. [DOI] [PubMed] [Google Scholar]
- 15.(a) Srebro M, Govind N, de Jong WA, Autschbach J. J Phys Chem A. 2011;115:10930. doi: 10.1021/jp2055409. [DOI] [PubMed] [Google Scholar]; (b) El Sayed Moussa M, Srebro M, Anger E, Vanthuyne N, Roussel C, Lescop C, Autschbach J, Crassous J. Chirality. 2013;25:455. doi: 10.1002/chir.22201. [DOI] [PubMed] [Google Scholar]
- 16.(a) Graule S, Rudolph M, Vanthuyne N, Autschbach J, Roussel C, Crassous J, Réau R. J Am Chem Soc. 2009;131:3183. doi: 10.1021/ja809396f. [DOI] [PubMed] [Google Scholar]; (b) Graule S, Rudolph M, Shen W, Lescop C, Williams JAG, Autschbach J, Crassous J, Réau R. Chem Eur J. 2010;16:5976. doi: 10.1002/chem.200903234. [DOI] [PubMed] [Google Scholar]
- 17.Riehl JP, Muller G. Circularly Polarized Luminescence Spectroscopy and Emission-Detected Circular Dichroism. In: Berova N, Polavarapu PL, Nakanishi K, Woody RW, editors. Comprehensive Chiroptical Spectroscopy. 1. Vol. 1. John Wiley & Sons, Inc; Hoboken, New Jersey: 2012. pp. 65–90. Instrumentation, Methodologies, and Theoretical Simulations, Chapter 3.Solntsev KM, Gould EA, Pan G, Muller G, Bommireddy S, Huppert D, Tolbert LM. Isr J Chem. 2009;49:227. doi: 10.1560/IJC.49.2.227.Riehl JP, Muller G. Circularly Polarized Luminescence Spectroscopy from Lanthanide Systems; In: Gschneidner KA Jr, Bünzli J-CG, Pecharsky VK, editors. In Handbook on the Physics and Chemistry of Rare Earths. Vol. 34. North-Holland Publishing Company; Amsterdam: 2005. pp. 289–357. Chapter 220., and references therein Tohgha U, Deol KK, Porter AG, Bartko SG, Choi JK, Leonard BM, Varga K, Kubelka J, Muller G, Balaz M. ACS Nano.
- 18.For different examples of CPL active helicenes see ref. 3a-d and Tang Y, Cook TA, Cohen AE. J Phys Chem A. 2009;111:6213. doi: 10.1021/jp903598t.Goto K, Yamaguchi R, Hiroto S, Ueno H, Kawai T, Shinokubo H. Angew Chem Int Ed. 2012;51:10333. doi: 10.1002/anie.201204863.Cyphersmith A, Surampudi S, Casey MJ, Jankowski K, Venkataraman D, Barnes MD. J Phys Chem A. 2012;116:5349. doi: 10.1021/jp300352n.Yang Y, da Costa RC, Smilgies DM, Campbell AJ, Fuchter MJ. Adv Mater. 2013;15:2624. doi: 10.1002/adma.201204961.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.