

Abstract
The morphogenesis of viruses belonging to the genus Enterovirus in the family Picornaviridae is still poorly understood despite decades-long investigations. However, we recently provided evidence that 2CATPase gives specificity to poliovirus encapsidation through an interaction with capsid protein VP3. The polypeptide 2CATPase is a highly conserved non-structural protein of enteroviruses with important roles in RNA replication, encapsidation and uncoating. We have identified a site (K279/R280) near the C terminus of the polypeptide that is required for morphogenesis. The aim of the current project was to search for additional functional sites near the C terminus of the 2CATPase polypeptide, with particular interest in those that are required for encapsidation. We selected for analysis a cysteine-rich site of the polypeptide and constructed four mutants in which cysteines or a histidine was changed to an alanine. The RNA transcripts were transfected into HeLa cells yielding two lethal, one temperature-sensitive and one quasi-infectious mutants. All four mutants exhibited normal protein translation in vitro and three of them possessed severe RNA replication defects. The quasi-infectious mutant (C286A) yielded variants with a pseudo-reversion at the original site (A286D), but some also contained one additional mutation: A138V or M293V. The temperature-sensitive mutant (C272A/H273A) exhibited an encapsidation and possibly also an uncoating defect at 37 °C. Variants of this mutant revealed suppressor mutations at three different sites in the 2CATPase polypeptide: A138V, M293V and K295R. We concluded that the cysteine-rich site near the C terminus of 2CATPase is involved in encapsidation, possibly through an interaction with an upstream segment located between boxes A and B of the nucleotide-binding domain.
Introduction
The family Picornaviridae contains a large number of human and animal pathogens, the prototype of which is poliovirus (PV), a member of the genus Enterovirus. The last step in the life cycle of picornaviruses is encapsidation of the newly made viral RNA, which takes place in the cytoplasm of the infected cell. Although the basic steps in particle assembly are known, few details are available about the mechanism and the factors that regulate this process. Encapsidation is difficult to study because this process is tightly linked to protein translation and RNA replication. So far, only a single non-structural protein, 2CATPase, has been identified that has an essential role in morphogenesis (Liu et al., 2010; Vance et al., 1997; Wang et al., 2012). The aim of this study was to identify domains in the 2CATPase protein that are involved in this complex process.
The plus-strand genome (7.5 kb) of PV contains a long 5′ non-translated region (NTR), a single ORF, a short 3′ NTR and a poly(A) tail. It encodes a polyprotein with one structural (P1) and two non-structural (P2, P3) domains (Fig. 1a), which are processed into precursor and mature viral proteins by 2Apro and 3Cpro/3CDpro (Wimmer et al., 1993). 2CATPase is a highly conserved and multifunctional membrane protein of picornaviruses (Fig. 1b). This 329 aa polypeptide contains an NTP-binding motif (Fig. 1b, boxes A, B and C) and exhibits ATPase activity in vitro (Mirzayan & Wimmer, 1994; Pfister & Wimmer, 1999; Rodríguez & Carrasco, 1993). Guanidine hydrochloride (GnHCl), a potent inhibitor of RNA replication, inhibits the ATPase activity of purified 2CATPase protein (Pfister & Wimmer, 1999). Mutants resistant to or dependent on GnHCl map to the 2CATPase polypeptide (Baltera & Tershak, 1989; Pincus & Wimmer, 1986). Genetic studies have identified numerous functions for the protein including particle uncoating, host-cell membrane alterations, viral RNA binding, viral RNA replication and encapsidation (Aldabe & Carrasco, 1995; Banerjee et al., 1997; Barton & Flanegan, 1997; Cho et al., 1994; Li & Baltimore, 1988, 1990; Liu et al., 2010; Paul et al., 1994; Rodríguez & Carrasco, 1995; Teterina et al., 1992, 1997; Vance et al., 1997; Verlinden et al., 2000). The protein was shown to oligomerize and to interact with capsid protein VP3 and with non-structural proteins 2B and 2BC, 3A and 3AB, and 3Cpro (Adams et al., 2009; Banerjee et al., 2004; Cuconati et al., 1998; Liu et al., 2010; Yin et al., 2007). The N-terminal domain of the protein contains the oligomerization and RNA-binding domains and an amphipathic helix, which anchors the protein to membranes (Adams et al., 2009; Echeverri et al., 1998; Paul et al., 1994; Rodríguez & Carrasco, 1995). The central and C-terminal segments of the protein possess serpin (serine protease inhibitor) motifs (Banerjee et al., 2004). Near the C terminus of the protein, there is another amphipathic helix and a cysteine-rich site (CRS), which binds zinc (Pfister et al., 2000; Teterina et al., 1997). Finally, the PV 2CATPase polypeptide binds specifically to an RNA structure near the 3′ end of PV minus-strand RNA (Banerjee et al., 1997). At the RNA level, a small hairpin, cre(2C), in the 2CATPase coding sequence, is required for the protein-primed initiation of RNA replication (Goodfellow et al., 2000; Paul et al., 2000).
Fig. 1.

Genome structure of PV, the functional domains of 2CATPase and location of mutants in the cysteine-rich site (CRS). (a) The PV RNA contains a long 5′ NTR, a single ORF, a short 3′ NTR and a poly(A) tail. The polyprotein is processed into one structural (P1) and two non-structural domains (P2 and P3). Protein 2CATPase is located in the P2 domain. (b) Functional domains of PV 2CATPase and locations of known mutations involved in encapsidation (N252S, K279A/R280A) or uncoating (M293V, K295R) are shown. The locations of the new mutants are also illustrated. The primary GnHCl-resistant (N179G) and -dependent (M187L) mutations are also shown. (c) Conservation of cysteine/histidine residues in the CRSs (aa 269–286) of enterovirus 2CATPase polypeptides.
Both genetic and drug inhibition studies have indicated the involvement of 2CATPase in encapsidation. Hydantoin, a drug that inhibits PV morphogenesis, yielded resistant mutants located near the centre or N terminus of the 2CATPase polypeptide (Vance et al., 1997; A. V. Paul, J. Mugavero, N. Dhiman, and E. Wimmer unpublished data). A cold-sensitive mutant with an insertion in 2CATPase at S255 yielded suppressor mutations M293V and K295R, whose phenotype was interpreted as a defect in uncoating. This observation suggests that 2CATPase might possess some role in determining virion structure (Li & Baltimore, 1988, 1990). Most importantly, a chimaera of PV, containing the capsid of CAV20, was found to be defective in encapsidation. This chimaera yielded suppressor mutations either in capsid protein VP3 or in 2CATPase (Liu et al., 2010). From these studies, we concluded that the specificity of encapsidation is provided by an interaction between 2CATPase and capsid proteins. Our recent mutational analyses of the 2CATPase polypeptide strongly supported this conclusion. It also led to the identification of a C-terminal site (K279/R280) that is involved in both RNA replication and encapsidation (Wang et al., 2012).
Previous studies with other viruses have indicated the importance of zinc-binding domains of proteins in morphogenesis. For example, the cysteine/histidine boxes of the human immunodeficiency virus type 1 nucleocapsid have been shown to play an important role in encapsidation (Schwartz et al., 1997). In addition, the cysteine-rich region of hepatitis B virus is involved in the encapsidation of the pre-genomic RNA (Kim et al., 2009). The cysteine-rich domain of PV was shown previously by us to have an essential function in RNA replication and viral growth (Pfister et al., 2000). However, at that time, the role of this domain in encapsidation was not examined. This domain is conserved among enteroviruses, and in PV it consists of the following motif: CX2CHX7CCX3C (Pfister et al., 2000) (Fig. 1c). It should be noted that the thiol groups of cysteines in 2CATPase occur in the reduced form in infected cells (Pfister et al., 2000). The aim of this study was to re-examine the cysteine-rich region of PV 2CATPase in particular with respect to a possible function in encapsidation. We produced four mutants in which a histidine or cysteines were changed to alanines (Table 1, Fig. 1b, c). Of these, two mutants possessed lethal growth phenotypes, one was temperature sensitive (ts) and one was quasi-infectious (qi). The mutant with the ts growth phenotype was found to be defective at 37 °C, specifically in morphogenesis. The delayed growth kinetics of this virus at 37 °C suggested the possibility of an additional uncoating defect. Variants with suppressor mutations either were in a spacer between boxes A and B of the NTP-binding domain or contained the same suppressor mutations (M293V, K295R) that were reported previously for an uncoating mutant (Li & Baltimore, 1988, 1990). We concluded that the CRS in PV 2CATPase is involved in morphogenesis, possibly through an interaction with a spacer region between boxes A and B of the NTP-binding domain.
Table 1. List of the CRS mutants of 2CATPase, the corresponding amino acid and nucleotide changes, and their growth phenotypes.
| Mutants | Wild-type codon(s) | Alanine codon(s)* | Growth phenotype | Time of full CPE at indicated temperature† | ||
| 33 °C | 37 °C | 39.5 °C | ||||
| CRS1 (C269A) | TGT | gcT | Lethal | – | – | – |
| CRS2 (C272A/H273A) | TGTCAC | gcTgcC | ts | Tf | Tf | Passage 1 |
| CRS3 (C281A/C282A) | TGCTGT | gcCgcT | Lethal | – | – | – |
| CRS4 (C286A) | TGT | gcT | qi | Passage 1 | Passage 1 | Passage 1 |
Lower-case letters indicate the nucleotide changes.
–, No CPE, even after four blind passages; Tf, full CPE at transfection; passage 1, full CPE at passage 1.
Results
Studying the role of 2CATPase in encapsidation is difficult because the protein is essential for RNA replication, a step in the life cycle of the virus prior to particle assembly. Thus, a lack of RNA replication results in a complete loss of encapsidation. Conditional-lethal mutants, such as ts or qi mutants, have been used frequently in the past to distinguish between defects in RNA replication and encapsidation. In addition, ts and qi mutants are useful because they are prone to produce suppressor mutations that identify interacting partners of the protein examined. Our aim was to search for ts or qi mutants by changing the cysteines or a histidine to alanines in the zinc-binding domain of 2CATPase. A C272S/H273Q mutant at this site was previously shown to be a ts mutant in terms of growth (Pfister et al., 2000).
CRS 2CATPase mutants exhibit normal translation and polyprotein processing
We constructed four mutants containing one or two histidine or cysteine changes to alanines (Fig. 1c, Table 1). To rule out the possibility that the mutants were defective in translation or protein processing we translated in vitro in HeLa cell-free extracts (Molla et al., 1991) RNA transcripts of the four mutants. After incubation for 8 h at 34 °C, samples were analysed by SDS-PAGE. As shown in Fig. 2, all four mutant transcript RNAs exhibited the same translation and polyprotein processing profiles as the wt, and no aberrant migration patterns of the 2C-related polypeptides, due to the amino acid substitutions, could be detected.
Fig. 2.
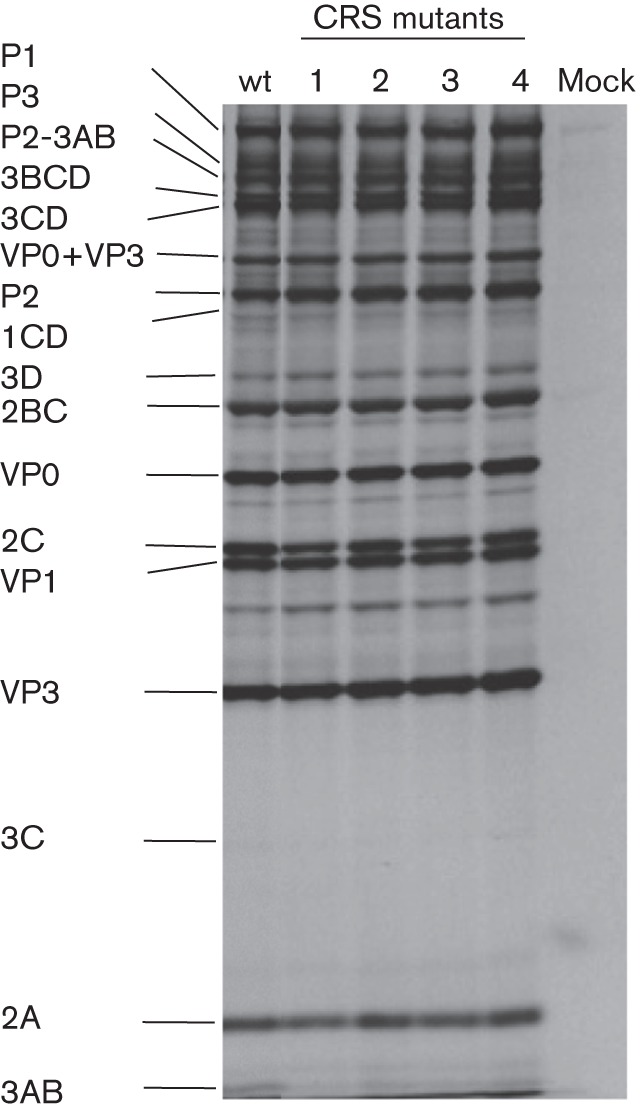
In vitro translation and polyprotein processing. RNA transcripts of the wt and mutant constructs were translated in HeLa cell-free extracts at 34 °C for 8 h (see Methods). The viral proteins were analysed by SDS-PAGE. The positions of the viral precursor and mature proteins are indicated on the left of the figure.
Growth phenotypes of the four CRS 2CATPase mutants
To compare the growth properties of the four CRS mutants with those of the wt, transcript RNAs were transfected into HeLa R19 cells and incubated at 33, 37 and 39.5 °C for up to 72 h or until a full cytopathic effect (CPE) developed. Lysates of mutants producing no full CPE upon transfection were subjected to up to four blind passages at the same temperatures. Two lethal (CRS1 and CRS3), one ts (CRS2) and one qi (CRS4) mutant were obtained (Table 1). The ‘lethal’ mutants (CRS1 and CRS3) that yielded no CPE after transfection at all three temperatures were passaged at the same temperature four times but still exhibited no sign of CPE.
The growth phenotypes of the resulting viruses, derived from mutant clones CRS2 and CRS4, were examined by plaque assay (Fig. 3). Mutant CRS2 produced full CPE upon transfection at 33 °C, yielding virus with smaller plaques and slightly lower titres than the wt virus at the same temperature (Table 1, Fig. 3a). This CRS2 virus, derived from RNA transfection at 33 °C, exhibited tiny plaques at 37 °C and minute plaques at 39.5 °C (Fig. 3b). The viral titre at 39.5 °C was 3 logs lower than that of the wt, indicating a severe ts growth phenotype (Fig. 3a). When RNA transcripts of mutant CRS2 were transfected at 37 °C, full CPE developed upon transfection (Table 1) but it was delayed (72 h) compared with that of the wt virus (48 h) (data not shown). Transfection of CRS2 mutant RNA at 39.5 °C yielded full CPE only after the first passage (Table 1).
Fig. 3.
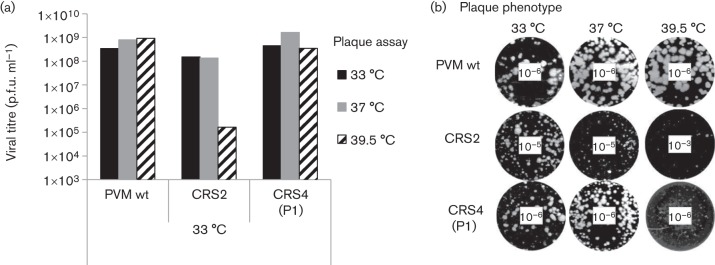
Growth phenotypes of a ts (CRS2) and a qi (CRS4) mutant. Cell lysates from 33 °C cultures were collected at full CPE. Mutant CRS2 was harvested from transfections and mutant CRS4 after the first passage on fresh HeLa cells. (a) The lysates were titrated at three different temperatures (33, 37 and 39.5 °C) by plaque assay (see Methods). (b) Pictures of plaque assays were taken at different virus dilutions, as indicated in the centre of the well. The plaques of variant CRS4 obtained after the first passage (P1) are shown.
The growth properties of the ts CRS2 mutant were analysed in more detail with one-step growth curves at 33 and 37 °C (Fig. 4). HeLa cells were infected at an m.o.i. of 1 with wt or CRS2 viruses and the virus yield (p.f.u. ml−1) was determined at various times post-infection (p.i.). The results indicated that, at 33 °C, the virus yield was close to the wt PV type 1 Mahoney (PVM) at early time points but was reduced about 10-fold later in infection. Interestingly, at 37 °C, virus production was much reduced at early times in infection and the final yield of progeny was only moderately reduced relative to the wt virus at 8–24 h p.i. Whether the delayed growth of the mutant at 37 °C was related to an abnormal uncoating step, possibly related to a defective encapsidation step in the previous growth cycle (see below), remains to be determined.
Fig. 4.
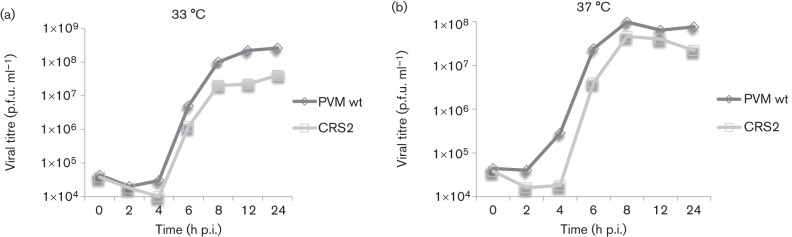
One-step growth curves of wt PV type 1 Mahoney (PVM) and CRS2 at 33 and 37 °C in HeLa cells. HeLa R19 cells were infected with wt or CRS2 virus at an m.o.i. of 1. Cultures of infected cells were harvested a 0, 2, 4, 6, 8, 12 and 24 h post-infection (p.i.) at 33 °C (a) and 37 °C (b), respectively. Viral titres were determined by plaque assay (see Methods).
Transfection with mutant CRS4 transcript RNAs resulted in full CPE only after the first passage at all three temperatures tested (33, 37 and 39.5 °C; Table 1). The viral progeny of mutant CRS4, isolated at 33 °C at passage 1, was also subjected to plaque assay at all three temperatures. It yielded somewhat smaller plaques at 33 and 37 °C but minute plaques at 39.5 °C when compared with the wt virus (Fig. 3b). The viral titres of CRS4 variants at all three different temperatures were comparable to that of the wt (Fig. 3a).
RNA replication and encapsidation phenotypes of CRS 2CATPase mutants
To characterize our mutants further for possible defects in RNA replication and/or encapsidation we used a Renilla luciferase (R-Luc) reporter virus (R-PPP) in which the R-Luc gene was fused to the N terminus of the PV polyprotein (PPP indicates domains P1, P2 and P3) (Liu et al., 2010). After translation, the R-Luc polypeptide was cleaved from the polyprotein by 3CDpro (Fig. 5a). The advantage of using this reporter virus over conventional reporter replicons, in which P1 is replaced by the luciferase gene, is that it can distinguish between defects in RNA replication and morphogenesis. An R-Luc-containing reporter virus that is unable to encapsidate itself will exhibit normal RNA levels as shown by a wt-like R-Luc signal after transfection. However, it will not generate infectious virus and, as a consequence, will not produce an R-Luc signal after passage to fresh HeLa cells. It should be noted, however, that the assay with the R-Luc virus cannot directly distinguish between encapsidation or uncoating, two closely linked processes. A defect in the encapsidation of the progeny virus might cause an abnormal capsid structure that can interfere with normal uncoating in the next round of infection.
Fig. 5.

RNA replication and encapsidation phenotypes of the four CRS mutants. (a) The genome structure of the R-PPP virus is illustrated. R-Luc was fused to the N terminus of the polyprotein and cleaved off during translation at a 3CDpro cleavage site. (b) RNA transfections of wt and mutant RNAs were carried out in the absence (−) and presence (+) of GnHCl. Lysates were passaged onto fresh HeLa cells, in the absence and presence of the drug, and R-Luc activity was determined (see Methods). R-Luc activity produced by wt and mutant reporter viruses (CRS1, CRS3 and CRS4) after transfection (indicating replication) and after first passage (P1; indicating encapsidation) at 33 °C are shown. For the sake of simplicity, a ratio of R-Luc activity in the absence and presence of GnHCl is plotted on the figure. (c) Transfection and passage of the wt and ts CRS2 mutant were carried out at all three temperatures (33, 37 and 39.5 °C) and R-Luc activity was determined. Mock, no transcript RNA was added. (d) Immunofluorescence imaging of PVM wt and CRS2 virus-infected HeLa cells at 33 and 37 °C. HeLa cells were infected with wt or CRS2 viruses at an m.o.i. of 1 at 33 or at 37 °C. The infected cell cultures were incubated for 4 h (37 °C) or 5 h (33 °C). The infected cells were probed with primary antibody A12 (a gift from N. Altan-Bonnet, Laboratory of Host-Pathogen Dynamics, National Institutes of Health, Bethesda, Maryland, USA), which recognizes the mature virus, followed by Alex Fluor 488-conjugated secondary antibody (green). VP3 localization in the same cell was with a polyclonal antibody to VP3 followed by Alex Fluor 555-conjugated secondary antibody (red). The blue immunofluorescence is the DAPI staining of the nucleus. Bars, 5 µm.
RNA transcripts of the wt or mutant R-Luc reporter viruses were transfected into HeLa cells at different temperatures in the absence and presence of GnHCl, a potent inhibitor of RNA replication (Pincus & Wimmer, 1986). R-Luc activity was measured at 16 h post-transfection. In the presence of the drug, the R-Luc signal measures translation, whilst in its absence it indicates the level of RNA replication. Subsequently, cell lysates from transfections carried out in the absence of GnHCl were subjected to fresh HeLa cells to test for encapsidation. These cells were then again incubated with or without GnHCl, followed by an assay for R-Luc activity at 8 h p.i. The level of encapsidation was estimated from the R-Luc activity in lysates of cells incubated without the drug during the passage (Liu et al., 2010).
Using this assay, we determined that both lethal mutants (CRS1 and CRS3) and the qi mutant (CRS4) were defective in RNA replication at 33 (Fig. 5b), 37 and 39.5 °C (data not shown). As expected, the defect in RNA replication with these mutants resulted in a lack of encapsidation (Fig. 5b). The ts mutant (CRS2) exhibited normal RNA replication and encapsidation at 33 °C, and it was defective in both processes at 39.5 °C (Fig. 5c). Interestingly, at 37 °C, CRS2 was nearly normal in RNA replication but produced no R-Luc signal after passaging, indicating a specific defect in encapsidation and/or uncoating (Fig. 5c).
Immunofluorescence imaging of wt- and CRS2-infected HeLa cells at 33 and 37 °C
We compared the amounts of VP3 polypeptide and mature virus produced by wt PVM and the CRS2 mutants at 33 and 37 °C using immunofluorescence imaging. HeLa cells were infected with wt or CRS2 virus at an m.o.i. of 1 at 33 and 37 °C, and were incubated at the same temperature for 5 or 4 h, respectively. Infected cells were probed with A12 antibodies, which recognize mature virus (N. Altan-Bonnett, unpublished results) and polyclonal antibodies to the VP3 capsid protein (Fig. 5d). The results indicated that the amount of VP3 capsid protein produced by the mutant was similar to that produced by the wt virus at both temperatures. However, there was a strong reduction in the amount of mature virus produced in CRS2-infected cells relative to the wt, which was particularly evident at 37 °C but could also be observed at 33 °C. These results strongly suggested that the deficiency in mature virus production was the result of a defect in encapsidation and confirmed the results obtained with the CRS2 R-Luc virus (Fig. 5c).
Characterization of suppressor variants derived from mutants CRS2 and CRS4
To enhance the number of variants from CRS2 or CRS4, we used a strategy of growing, plaquing and propagating the mutants from the plaques at combinations of low and high temperatures (33 and 39.5 °C) (Wang et al., 2012). This resulted in the identification of a pseudo-revertant and/or numerous second-site suppressor mutants following reverse transcription (RT)-PCR of the total RNA and whole genome sequencing (Figs 6 and 7).
Fig. 6.

Growth phenotypes of pseudo-revertant and suppressor variants of CRS2 and CRS4. (a) Growth phenotypes of suppressor variants of mutant CRS2 are shown. Viral titres were measured by plaque assay (see Methods) at three different temperatures. The genotypes and the selection temperatures and phenotypes of the three suppressor variants are summarized in Fig. 7. (b) Growth phenotypes of the three suppressor variants of mutant CRS4 are shown. Plaques were picked and amplified at the temperatures indicated in Fig. 7 and the lysates were titrated by plaque assay at 33, 37 and 39.5 °C. Plaque pictures were taken from different dilutions, as indicated for each well.
Fig. 7.

Mutations in PV 2CATPase known to affect encapsidation or uncoating. The genomic structure of PV is shown at the top of the figure. The locations of the pseudo-revertant and suppressor variants of mutants CRS2 and CRS4 and of previously published 2CATPase mutant variants 11b (Wang et al., 2012), 2C-31 R1 and 2C-31 R3 (Li & Baltimore, 1990) are indicated. The temperatures at which the transfection (Tf) and first passage (P1) and the plaque purifications were carried out are listed on the right.
When mutant CRS2 was transfected at 33 °C but plaque purified and amplified at 39.5 °C, we detected two different variants (Figs 6a and 7). Both contained the original mutations (C272A/H273A) but acquired a suppressor mutation at either A138V (CRS2a) or M293V (CRS2b) (Fig. 7). As expected, only CRS2 genomes (C272A/H273A) but no revertants or suppressors of mutant CRS2 were detected in samples transfected, plaque purified and amplified at the permissive temperature (33 °C). When CRS2 RNA was transfected, plaque purified and grown at 39.5 °C, a suppressor mutation, K295R, was observed (Fig. 7), whilst the original two mutations were retained (CRS2c). All three suppressor mutations rescued the ts phenotype of CRS2 and the variants exhibited wt-like growth phenotypes at all three temperatures (Fig. 6a). The plaque sizes of the variants were smaller than those of the wt at all three temperatures tested, 33, 37 and 39.5 °C (Fig. 6b).
We carried out the same selection process at two different temperatures (33 and 39.5 °C) to enhance the number of variants of the qi mutant CRS4 (Fig. 7). RNA transcripts of CRS4 were transfected into HeLa cells at both temperatures and, after the development of full CPE at passage 1, the resulting viruses were plaque purified and isolated. As mutant CRS4 is a qi mutant, the progeny isolated from cell lysates were always found to be genetic variants of the original mutant. When the CRS4 variants were propagated at 33 °C, they lost the original C286A mutation and acquired a pseudo-reversion at the same position A286D (CRS4a, Fig. 7). It should be noted that the A→D change required only a single nucleotide change, whilst a reversion back to C would have involved two nucleotide changes. It appears that a D at this position in the 2CATPase polypeptide is fully functional at 33 °C. However, when the CRS4 variants were passaged at 39.5 °C, in addition to the above noted pseudo-reversion (A286D), an additional suppressor mutation was required to regain function: A138V (CRS4b) or M293V (CRS4c) (Fig. 7). The plaque phenotypes and viral titres of the CRS4 variants were also determined (Fig. 6b). Variant CRS4a (A286D), which was selected at 33 °C, yielded a wt-like titre but minute plaques at 39.5 °C. The growth phenotypes of CRS4b and CRS4c were comparable to that of the wt virus (Fig. 6b).
Summary of the C-terminal 2CATPase sites involved in encapsidation
The locations of suppressor mutations, in variants derived from CRS2 and CRS4, are summarized in Fig. 7. We also included the locations of suppressor variants of mutant 11, K279A/R280A, from our previous alanine-scanning study (Wang et al., 2012) and of cold-sensitive mutant 2C-31 R1/R3 from the study by Li & Baltimore (1990). It should be noted that residues K279/R280 in the parental virus of mutant 11 are involved in encapsidation, whilst the cold-sensitive mutant has an uncoating defect, most likely the consequence of an encapsidation defect. Thus, we concluded that a C-terminal domain of 2CATPase, which includes the CRSs, is involved in encapsidation, probably through an interaction with an amino acid sequence contained between boxes A and B of the NTP-binding domain (aa 136–172) (Fig. 1b).
Discussion
Morphogenesis is the last step of the viral life cycle in which the progeny viral RNA is enclosed in a protective coat that ensures a proper uncoating process during the next round of infection of susceptible host cells. We believe that encapsidation and uncoating are related processes, as improper encapsidation of the viral RNA will lead to a defect in uncoating. Previous studies have already identified two sites in a domain near the C terminus of 2CATPase that are important either for morphogenesis or perhaps for uncoating (Li & Baltimore, 1990). Recently, using chimaeras of PV and coxsackie A virus 20, residue N252 of 2CATPase was identified as an interacting partner of E180 of the capsid protein VP3, providing specificity to the encapsidation process (Liu et al., 2010). In addition, clustered charge-to-alanine mutagenesis, which yielded suppressor mutations either in 2CATPase or in both 2CATPase and capsid proteins VP1 or VP3, has identified residues K279/R280 of 2CATPase as important for encapsidation (Wang et al., 2012).
The aim of the current study was to identify and characterize additional sites near the C terminus of the 2CATPase polypeptide that are involved in morphogenesis. We were particularly interested in generating conditional-lethal or qi mutants, which in the past have yielded useful information on protein–protein interactions in morphogenesis (Wang et al., 2012). Previous studies from our laboratory have indicated that a CRS near the C terminus, which binds Zn2+, is required for viral growth and RNA replication (Pfister et al., 2000). The availability of a reporter virus that can distinguish between defects in RNA replication and encapsidation/uncoating (Liu et al., 2010) has made it possible to examine the possibility that one or more of the cysteine/histidine residues are also important for encapsidation. Therefore, we constructed and analysed the properties of four mutants in the cysteine-rich domain.
All of the mutants (CRS1–4) exhibited normal protein translation and polyprotein processing profiles, but they were defective in RNA replication, at least to some extent, indicating the importance of the CRS in this process. As normal RNA replication is a prerequisite of encapsidation, we cannot rule out the possibility that some or all of the other cysteine/histidine residues in the CRS are also involved in encapsidation. Interestingly, our studies with a reporter virus indicated a specific defect of the ts mutant (CRS2) in encapsidation at 37 °C, whilst the delayed growth kinetics of this mutant, observed by imaging, suggested an early defect in the virus life cycle. We are currently investigating the possibility that the defect in encapsidation of CRS2 at 37 °C leads to improper uncoating during the next round of infection.
We subjected the original CRS2 and CRS4 mutants to different selection pressures with a combination of low and high temperatures during transfection and passage, and also during plaque purification and amplification, a method that led to a variety of interesting suppressor mutants and a pseudo-revertant in our previous study (Wang et al., 2012). The CRS2 mutant yielded suppressor variants (M293V, K295R) either near the original mutated site (C272/H273) or in a spacer region (A138V) between boxes A and B of the nucleotide-binding site (Figs 1b and 7). Curiously, the rescue of the CRS2 alanine substitutions at nearby sites could occur either by the replacement of a less hydrophobic residue with a more hydrophobic residue (M293V) or by the replacement of one strongly basic residue with another (K295R). The observation that the CRS2 mutation can be rescued by the same suppressor mutations (M293V, K295R) as the uncoating defect, previously analysed by Li & Baltimore (1990), supports a linkage between the encapsidation and uncoating processes. The quasi-infectious CRS4 (C286A) mutant generated a pseudo-revertant (A286D), either alone or in combination with other suppressor mutations (A138V or M293V). Both CRS2 and CRS4 yielded a suppressor mutation A138V, close to the site (E148K) of the suppressor mutant of alanine-scanning mutant 11 (K279A/R280A) (Wang et al., 2012). These results support our proposal that there is a functional interaction between the C-terminal domain of the 2CATPase polypeptide and a spacer region between boxes A and B of the NTP-binding domains. Whether this interaction occurs intramolecularly or between different 2CATPase protein molecules remains to be determined. It should be noted that cysteine-rich zinc-binding domains in proteins frequently mediate protein–protein interactions, particularly those that contribute to the formation and architecture of large molecular scaffolds (Borden, 2000; Kentsis & Borden, 2000). Previous studies have indicated that purified MBP-tagged 2CATPase formed oligomers that appeared by electron microscopy as ring-like structures composed of five to eight protomers, and that these oligomers are essential for function (Adams et al., 2009). It is not yet known whether the zinc-binding domain of 2CATPase has a role in the formation of such functional oligomers.
The CRSs of enterovirus 2CATPase proteins are highly conserved both in the amino acids they contain and in the spacing of residues within the sites (Fig. 1c) (Pfister et al., 2000). The CX2CHX7CCX3C pattern resembles a zinc-binding motif with four cysteine-containing locations (Coleman, 1992; Pfister et al., 2000). Within the motif, the two single C residues are fully conserved in all enteroviruses examined, including the more distantly related rhinoviruses. Of the adjacent CC residues, the first C is fully conserved and the second is highly conserved. The CH residues on the other hand are highly divergent, and in rhinoviruses they appear to be nearly completely absent. Interestingly, the growth phenotypes of the four CRS mutants correlated very well with the extent of conservation of amino acid residues within the motif. The two non-viable mutants, CRS1 and CRS3, and the qi mutant CRS4, had mutations in the fully/highly conserved single Cs or CC of the motif. The ts mutants, CRS2, however, was mutated at the variable CH site.
Many unanswered questions remain about the role of 2CATPase in morphogenesis. However, the information obtained from these and our previous results (Liu et al., 2010; Wang et al., 2012) clearly demonstrate the usefulness of genetic studies with conditional-lethal and qi mutants for the identification of important residues and protein–protein interactions involved in encapsidation.
Methods
Cells.
HeLa R19 cells were maintained in Dulbecco’s minimal essential medium (DMEM; Life Technology) supplemented with 10 % v/v bovine calf serum (BCS), 100 mg streptomycin ml−1, and 100 U penicillin. Transfection and passages of HeLa R19 cells were carried out with supplementation of 2 % (v/v) BCS.
Plasmids.
pT7PVM contains a full-length infectious cDNA of PVM. The pGEM-T vector was obtained from Promega. pR-PPP is an infectious R-Luc reporter virus construct in which R-Luc (311 aa) is expressed as an N-terminal fusion to the PV polyprotein (Liu et al., 2010).
T-vector-based site-directed mutagenesis.
Site-directed mutagenesis was used to obtain the desired mutations. In each CRS mutation, cysteine and histidine, if present, in the four-conserved-cysteine-rich zinc-binding site of PV 2CATPase protein (Pfister et al., 2000) were replaced with alanine by changing the corresponding codons. First, four pairs of oligonucleotide primers were designed containing the alanine mutations and introduced into the pGEM-T vector containing the wt 2CATPase nucleotide sequence (Wang et al., 2012), using a Stratagene QuikChange Site-directed Mutagenesis kit according to the instruction manual. The mutated sites and the corresponding codon changes are summarized in Table 1 (left three columns). After being confirmed by sequencing analysis, the designed 2CATPase mutations were then subcloned from the pGEM-T vector into pT7PVM or the R-Luc reporter virus R-PPP using restriction sites XhoI and HpaI.
In vitro RNA transcription.
Wt and mutant plasmids of pT7PVM were linearized at a unique EcoRI restriction site and used as templates for in vitro RNA transcription using T7 RNA polymerase.
In vitro translation.
HeLa cell S10 cytoplasmic extracts were prepared using HeLa S3 cells. As described previously (Molla et al., 1991), in vitro RNA translations were performed with these cytoplasmic extracts and the newly made RNA transcripts were incubated at 34 °C for 8 h. The viral proteins were labelled with a mixture of [35S]methionine/cysteine and separated by SDS-PAGE (12.5 % acrylamide). The bands were visualized by autoradiography.
Transfection.
RNA transcripts (3–10 µg) were transfected into 35 mm diameter HeLa R19 cell monolayers by the DEAE-dextran method as described previously (van der Werf et al., 1986) and incubated at 33, 37 and 39.5 °C. At 3 days post-transfection or at the time of full CPE, viruses, if any, were harvested. Full CPE was defined as the point where 90–95 % of the cells displayed CPE. Lysates of cells transfected with mutants lacking CPE were inoculated into fresh 35 mm diameter HeLa R19 cell monolayers for up to four blind passages (Wang et al., 2012). To assess the viral titres (p.f.u. ml−1) and plaque phenotypes of the viable CRS mutants, any samples displaying full CPE at 33 °C were then subjected to plaque assays at 33, 37 and 39.5 °C. The identity of the viruses was confirmed or determined by plaque purification, full-length RT-PCR and sequencing analysis.
Plaque assays.
Plaque assays were performed on HeLa R19 monolayers using 0.6 % (w/v) tragacanth gum. After 72 h incubation at 33 °C or 48 h at 37 or 39.5 °C, the viral plaques were visualized by 1 % (w/v) crystal violet staining (Liu et al., 2010).
RT-PCR and sequencing analysis of viral RNAs isolated from purified plaques.
Single plaques were picked from the plaque assay plates before staining, and amplified by one passage at the same temperature in fresh 35 mm diameter HeLa R19 monolayers. Total RNA was extracted from 200 µl lysates with 1 ml Trizol reagent (Invitrogen) and reverse transcribed into cDNA using SuperScript III Reverse Transcriptase (Invitrogen). The PCR products, generated by the Expand Long Template PCR System (Roche), were purified and sequenced.
Luciferase assays.
Dishes (35 mm diameter) of HeLa R19 monolayer cells were transfected with 5 µg R-PPP RNA transcripts (linearized with PvuI for pR-PPP) and incubated at 33, 37 or 39.5 °C in standard tissue culture medium (DMEM) with 2 % (v/v) BCS and in the absence or presence of 2 mM GnHCl. Luciferase activity was determined in the lysates of cells harvested at 16 h post-transfection. Cell lysates (20 µl) were mixed with 20 µl R-Luc assay reagent (Promega), and R-Luc activity was measured in an OPTOCOMP I luminometer (MGM Instruments). Cell lysates (250 µl) from transfections were passaged once in HeLa R19 cells in the absence or presence of 2 mM GnHCl. Luciferase activity was determined in the lysates of cells harvested at 8 h p.i. The R-Luc activity ratio (−GnHCl/+GnHCl) was calculated as: luciferase activity without GnHCl divided by luciferase activity with GnHCl in either transfection or infection.
Immunofluorescence cell imaging.
HeLa cells were infected with wt or CRS2 virus at an m.o.i. of 1 and incubated at 37 °C for 4 h or at 33 °C for 5 h. The infected cells were probed for mature virus with A12 primary antibody, which recognizes the mature virus, followed by Alexa Fluor 488-conjugated secondary antibody. The localization of VP3, a capsid protein, was determined in the same cell using VP3 polyclonal antibodies and Alexa Fluor 555-conjugated secondary antibody.
Acknowledgements
This work was supported by a grant from the NIH NIAID (5R37AI15122). We thank Nihal Altan-Bonnet for mAb A12 and Steffen Mueller for the plasmid of the R-Luc reporter virus.
References
- Adams P., Kandiah E., Effantin G., Steven A. C., Ehrenfeld E. (2009). Poliovirus 2C protein forms homo-oligomeric structures required for ATPase activity. J Biol Chem 284, 22012–22021 10.1074/jbc.M109.031807 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aldabe R., Carrasco L. (1995). Induction of membrane proliferation by poliovirus proteins 2C and 2BC. Biochem Biophys Res Commun 206, 64–76 10.1006/bbrc.1995.1010 [DOI] [PubMed] [Google Scholar]
- Baltera R. F., Jr, Tershak D. R. (1989). Guanidine-resistant mutants of poliovirus have distinct mutations in peptide 2C. J Virol 63, 4441–4444 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banerjee R., Echeverri A., Dasgupta A. (1997). Poliovirus-encoded 2C polypeptide specifically binds to the 3′-terminal sequences of viral negative-strand RNA. J Virol 71, 9570–9578 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banerjee R., Weidman M. K., Echeverri A., Kundu P., Dasgupta A. (2004). Regulation of poliovirus 3C protease by the 2C polypeptide. J Virol 78, 9243–9256 10.1128/JVI.78.17.9243-9256.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barton D. J., Flanegan J. B. (1997). Synchronous replication of poliovirus RNA: initiation of negative-strand RNA synthesis requires the guanidine-inhibited activity of protein 2C. J Virol 71, 8482–8489 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borden K. L. B. (2000). RING domains: master builders of molecular scaffolds? J Mol Biol 295, 1103–1112 10.1006/jmbi.1999.3429 [DOI] [PubMed] [Google Scholar]
- Cho M. W., Teterina N., Egger D., Bienz K., Ehrenfeld E. (1994). Membrane rearrangement and vesicle induction by recombinant poliovirus 2C and 2BC in human cells. Virology 202, 129–145 10.1006/viro.1994.1329 [DOI] [PubMed] [Google Scholar]
- Coleman J. E. (1992). Zinc proteins: enzymes, storage proteins, transcription factors, and replication proteins. Annu Rev Biochem 61, 897–946 10.1146/annurev.bi.61.070192.004341 [DOI] [PubMed] [Google Scholar]
- Cuconati A., Xiang W., Lahser F., Pfister T., Wimmer E. (1998). A protein linkage map of the P2 nonstructural proteins of poliovirus. J Virol 72, 1297–1307 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Echeverri A., Banerjee R., Dasgupta A. (1998). Amino-terminal region of poliovirus 2C protein is sufficient for membrane binding. Virus Res 54, 217–223 10.1016/S0168-1702(98)00016-1 [DOI] [PubMed] [Google Scholar]
- Goodfellow I., Chaudhry Y., Richardson A., Meredith J., Almond J. W., Barclay W., Evans D. J. (2000). Identification of a cis-acting replication element within the poliovirus coding region. J Virol 74, 4590–4600 10.1128/JVI.74.10.4590-4600.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kentsis A., Borden K. L. (2000). Construction of macromolecular assemblages in eukaryotic processes and their role in human disease: linking RINGs together. Curr Protein Pept Sci 1, 49–73 10.2174/1389203003381478 [DOI] [PubMed] [Google Scholar]
- Kim S., Lee J., Ryu W. S. (2009). Four conserved cysteine residues of the hepatitis B virus polymerase are critical for RNA pregenome encapsidation. J Virol 83, 8032–8040 10.1128/JVI.00332-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J. P., Baltimore D. (1988). Isolation of poliovirus 2C mutants defective in viral RNA synthesis. J Virol 62, 4016–4021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J. P., Baltimore D. (1990). An intragenic revertant of a poliovirus 2C mutant has an uncoating defect. J Virol 64, 1102–1107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y., Wang C., Mueller S., Paul A. V., Wimmer E., Jiang P. (2010). Direct interaction between two viral proteins, the nonstructural protein 2C and the capsid protein VP3, is required for enterovirus morphogenesis. PLoS Pathog 6, e1001066 10.1371/journal.ppat.1001066 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mirzayan C., Wimmer E. (1994). Biochemical studies on poliovirus polypeptide 2C: evidence for ATPase activity. Virology 199, 176–187 10.1006/viro.1994.1110 [DOI] [PubMed] [Google Scholar]
- Molla A., Paul A. V., Wimmer E. (1991). Cell-free, de novo synthesis of poliovirus. Science 254, 1647–1651 10.1126/science.1661029 [DOI] [PubMed] [Google Scholar]
- Paul A. V., Molla A., Wimmer E. (1994). Studies of a putative amphipathic helix in the N-terminus of poliovirus protein 2C. Virology 199, 188–199 10.1006/viro.1994.1111 [DOI] [PubMed] [Google Scholar]
- Paul A. V., Rieder E., Kim D. W., van Boom J. H., Wimmer E. (2000). Identification of an RNA hairpin in poliovirus RNA that serves as the primary template in the in vitro uridylylation of VPg. J Virol 74, 10359–10370 10.1128/JVI.74.22.10359-10370.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pfister T., Wimmer E. (1999). Characterization of the nucleoside triphosphatase activity of poliovirus protein 2C reveals a mechanism by which guanidine inhibits poliovirus replication. J Biol Chem 274, 6992–7001 10.1074/jbc.274.11.6992 [DOI] [PubMed] [Google Scholar]
- Pfister T., Jones K. W., Wimmer E. (2000). A cysteine-rich motif in poliovirus protein 2CATPase is involved in RNA replication and binds zinc in vitro. J Virol 74, 334–343 10.1128/JVI.74.1.334-343.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pincus S. E., Wimmer E. (1986). Production of guanidine-resistant and -dependent poliovirus mutants from cloned cDNA: mutations in polypeptide 2C are directly responsible for altered guanidine sensitivity. J Virol 60, 793–796 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodríguez P. L., Carrasco L. (1993). Poliovirus protein 2C has ATPase and GTPase activities. J Biol Chem 268, 8105–8110 [PubMed] [Google Scholar]
- Rodríguez P. L., Carrasco L. (1995). Poliovirus protein 2C contains two regions involved in RNA binding activity. J Biol Chem 270, 10105–10112 10.1074/jbc.270.17.10105 [DOI] [PubMed] [Google Scholar]
- Schwartz M. D., Fiore D., Panganiban A. T. (1997). Distinct functions and requirements for the Cys-His boxes of the human immunodeficiency virus type 1 nucleocapsid protein during RNA encapsidation and replication. J Virol 71, 9295–9305 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teterina N. L., Kean K. M., Gorbalenya A. E., Agol V. I., Girard M. (1992). Analysis of the functional significance of amino acid residues in the putative NTP-binding pattern of the poliovirus 2C protein. J Gen Virol 73, 1977–1986 10.1099/0022-1317-73-8-1977 [DOI] [PubMed] [Google Scholar]
- Teterina N. L., Gorbalenya A. E., Egger D., Bienz K., Ehrenfeld E. (1997). Poliovirus 2C protein determinants of membrane binding and rearrangements in mammalian cells. J Virol 71, 8962–8972 [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Werf S., Bradley J., Wimmer E., Studier F. W., Dunn J. J. (1986). Synthesis of infectious poliovirus RNA by purified T7 RNA polymerase. Proc Natl Acad Sci U S A 83, 2330–2334 10.1073/pnas.83.8.2330 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vance L. M., Moscufo N., Chow M., Heinz B. A. (1997). Poliovirus 2C region functions during encapsidation of viral RNA. J Virol 71, 8759–8765 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verlinden Y., Cuconati A., Wimmer E., Rombaut B. (2000). Cell-free synthesis of poliovirus: 14S subunits are the key intermediates in the encapsidation of poliovirus RNA. J Gen Virol 81, 2751–2754 [DOI] [PubMed] [Google Scholar]
- Wang C., Jiang P., Sand C., Paul A. V., Wimmer E. (2012). Alanine scanning of poliovirus 2CATPase reveals new genetic evidence that capsid protein/2CATPase interactions are essential for morphogenesis. J Virol 86, 9964–9975 10.1128/JVI.00914-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wimmer E., Hellen C. U., Cao X. (1993). Genetics of poliovirus. Annu Rev Genet 27, 353–436 10.1146/annurev.ge.27.120193.002033 [DOI] [PubMed] [Google Scholar]
- Yin J., Liu Y., Wimmer E., Paul A. V. (2007). Complete protein linkage map between the P2 and P3 non-structural proteins of poliovirus. J Gen Virol 88, 2259–2267 10.1099/vir.0.82795-0 [DOI] [PubMed] [Google Scholar]