

Abstract
This study was aimed at investigating the antitumor activity of novel 2-oxindole derivatives against a well-characterized human nonsmall cell lung cancer (NSCLC) cell line. Test compounds produced an antiproliferative activity in the low micromolar/submicromolar range of concentrations and significantly induced typical apoptotic morphology with cell shrinkage, nuclear condensation and fragmentation, and rupture of cells into debris in a relatively low percentage of A549 cells. Cell cycle arrest occurred at the G1/S phase (1a and 2), and Akt phosphorylation was significantly inhibited at Thr308 and Ser473. The most active compound (1a) has an IC50 6-fold lower than the Akt inhibitor, perifosine. These data suggest that the new compounds may be cytostatic and may have maximum clinical effects in NSCLC patients who do not respond to EGFR inhibitors. These findings prompt us to further explore the oxindole structure as leading scaffold to design new molecules with potent antitumor activity against NSCLC.
Keywords: Antiproliferative activity, nonsmall cell lung cancer, cell cycle, PDK1/Akt inhibitors, oxindole
Lung cancer is one of the most common cancers worldwide and about 85% of lung cancers are nonsmall cell lung cancer (NSCLC). Patients with NSCLC have a poor prognosis and limited treatment options including conventional chemotherapy and targeted agents that inhibit epidermal growth factor receptor (EGFR) activation. However, these drugs are not curative because of both primary and acquired treatment resistance1 highlighting the need to identify new molecules with potent antitumor activity against NSCLC cells.
The role of the PI3K/Akt/mTOR pathway in the development of the neoplastic phenotype has been extensively reviewed.2 Noteworthy, activation of Akt was found in 51% of NSCLC patient samples and in 74% of NSCLC cell lines.3 Persistent activity of this pathway was associated with increased resistance to multiple chemotherapeutic agents for gastric4 and ovarian5 cancer and in the lack of sensitivity of NSCLC cell lines to EGFR inhibitors.6
After stimulation with growth factors and cytokines, Akt is recruited from the cytosol to the plasma membrane and is phosphorylated at two key regulatory sites, Thr308 (in the activation domain) and Ser473 (in the carboxy-terminal hydrophobic motif) by 3-phosphoinositide-dependent protein kinase-1 (PDK1) and PDK2, respectively.7,8 Once activated, Akt stimulates cell growth and survival by phosphorylating numerous downstream substrates including caspase-9,9 glycogen synthase kinase-3 (GSK3), BCL antagonist of cell death (BAD),10 and forkhead box transcription factors (FOXO).11
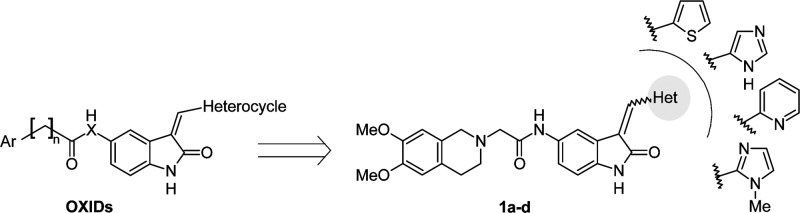
Compounds with an indole/oxindole moiety are promising pharmacophore endowed of interesting biological and pharmacological properties as anticancer agents.12 Recently, new compounds designed to target the activity of the serine/threonine kinases PDK1 and/or Akt were described.13,14 Moreover, the therapeutic potential of tetrahydroisoquinoline (THI) derivatives characterized by antineoplastic,15,16 multidrug resistance reversal17,18 and kinases inhibitor19 activities have encouraged us to design and synthesize a new class of novel chemotherapeutic agents active on EGFR-resistant NSCLC cell line, by anchoring the THI-nucleus to the oxindole scaffold. While providing further insights into the structure–activity relationship, the introduction of different nuclei could also be the starting point for the development of a new class of cytotoxic agents. On this basis, we designed and synthesized a series of 2-oxindole-derivatives (OXIDs, 1a–d) characterized by the presence of different heterocycles (i.e., thiophene, imidazole, N-methylimidazole, and pyridine) in 3-position and 3,4-dimethoxy-THI group anchored in 5-position through an amidomethyl chain (Figure 1). The structure of compounds 1a–d matches well with the common structure of recently patented PDK1/Akt inhibitors.20,21
Figure 1.
General structures of new oxindole-derivatives (OXIDs).
In the current study, we report findings clearly demonstrating the potent antiproliferative activity of test compounds associated to inhibition of Akt phosphorylation and cell cycle arrest at the G1/S phase in human NSCLC cells.
The desired compounds 1a–d were synthesized according to the procedure reported in Scheme 1. The 5-amino-2-oxindole 3 was obtained by catalytic hydrogenation of the commercial 5-nitro-1,3-dihydro-2H-indol-2-one. The subsequent reaction of 3 with 2-chloroacetyl chloride afforded the 2-chloro-N-(2-oxo-2,3-dihydro-1H-indol-5-yl)acetamide 4, which reacted with 6,7-dimethoxy-1,2,3,4-tetrahydroisoquinoline hydrochloride in the presence of K2CO3 affording derivative 2. Final compounds 1a–d were obtained by reaction of 2 with 2-thiophencarboxaldehyde, 1-methyl-5-imidazolecarboxaldehyde, 1H-imidazole-4-carbaldehyde, and 2-pyridinecarboxaldehyde, respectively, in the presence of a catalytic amount of pirrolidine. The configuration of final products 1a–d was assigned by comparison of their 1H NMR spectra with those reported in literature for analogue structures.22
Scheme 1. Synthesis of OXIDs Derivatives 1a–d and 2.

The in vitro antitumor activity of compounds was evaluated against A549 cells that have been reported to be gefitinib-resistant due to the maintenance of an EGFR-independent activity of the PI3K/Akt pathway.23 All tested compounds produced a concentration and time-dependent cell growth inhibition with IC50 mean values in the low micromolar/submicromolar range (Figure 2).
Figure 2.

Concentration- and time-dependent effect on A549 cell proliferation by test compounds and the Akt inhibitor, perifosine. Data were expressed as mean values ± SD (n = 3).
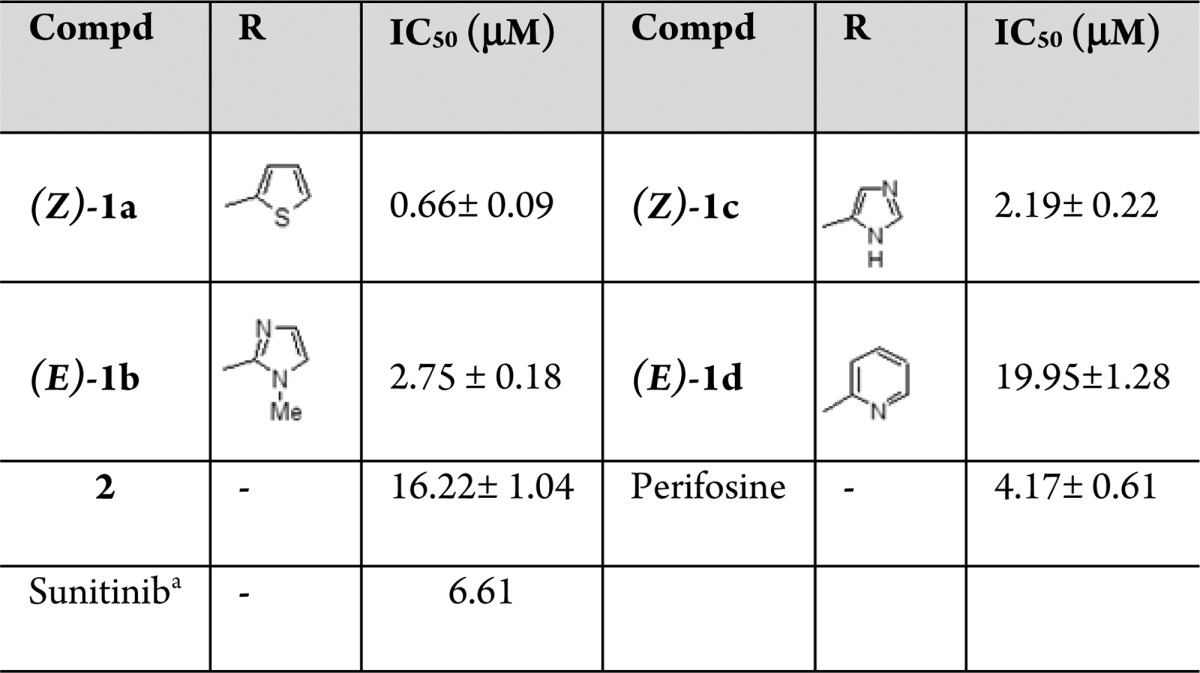
Compound 1a–c showed 2- to 6-fold higher antitumor activity than the Akt inhibitor perifosine, and 2.5- to 10-fold higher than sunitinib (a 2-oxindole-based anticancer drug), while compounds 1d and 2 showed IC50 mean values of 19.95 ± 1.28 and 16.22 ± 1.04 μM, respectively (Table 1).
Table 1. IC50 Values for OXIDs Compounds 1a–d and 2, Sunitinib, and the Akt Inhibitor Perifosine on A549 Cells.
See ref (12).
The ability to escape apoptosis through the phosphorylation of the above downstream substrates is a critical characteristic of cancer cells that allow their uncontrolled growth and invasive behavior. In the current study, the ability of tested compounds to induce apoptosis on A549 cell line was assessed by microscopy that remains the gold standard for measurement of nuclear fragmentation.24 Upon exposure to OXIDs derivatives at their corresponding IC50 values, A549 cells presented typical apoptotic morphology with cell shrinkage, nuclear fragmentation, and cellular rupture into debris. The apoptotic index, determined by morphological metrics, was significantly higher in cells treated with test compounds than controls with values ranging from 12% (2) to 24% (1a). The apoptotic index for the Akt inhibitor, perifosine, was 32% (Figure 3). In addition to play a crucial role for cell survival, Akt signaling pathway modulates cell cycle progression.25
Figure 3.
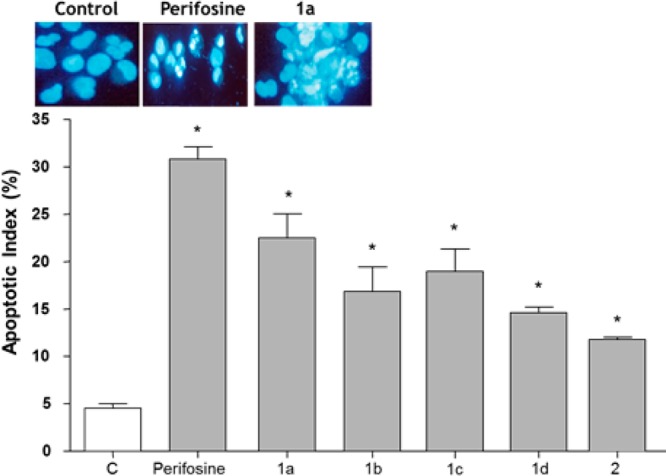
Percentage of A549 cells with damaged DNA after exposure to test compounds and the Akt inhibitor perifosine. Upper panels, morphological appearance of control and treated cells. Data were expressed as mean values ± SD (n = 3). *P < 0.05, as compared with control.
Therefore, cell-cycle distribution in A549 cells exposed to OXIDs derivatives was assessed by flow cytometry, and results were expressed as percentage variation on cells population in ΔG1, ΔS, and ΔG2 phases (Figure 4).
Figure 4.
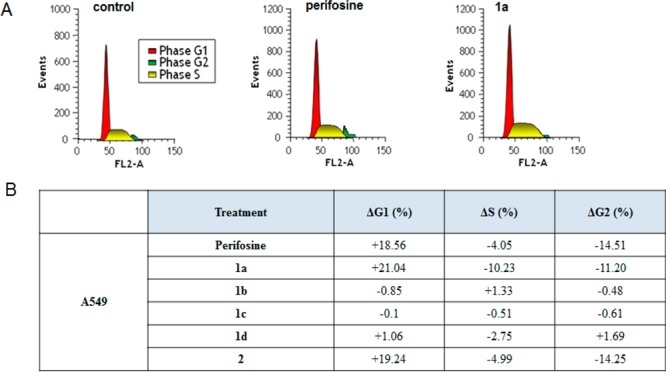
(A) Effect of compound 1a and perifosine on the distribution of events in the A549 cell cycle. (B) Percentage of cells in the G1, S, and G2/M phases of the cell cycle in the presence or absence of test compounds and the Akt inhibitor perifosine. Data were expressed as mean values ± SD (n = 3). *P < 0.05, as compared with control.
Treatment with compounds 1a and 2 caused a significant increase in the proportion of cells in the G1 phase of the cell cycle (21.04 and 19.24%, respectively) associated to a percentage decrease of S-phase and G2/M-phase cells, as a result of the inhibition of DNA synthesis.
Similar cell cycle changes were observed when A549 was treated with perifosine, while compounds 1b–d did not have any effect (Figure 4). Therefore, the OXID derivatives 1a and 2 could affect cell survival and cell cycle progression, a pharmacological profile similar to that observed for the Akt inhibitor, perifosine.
To provide further insight into the molecular mechanism of compounds’ action, we used a specific and sensitive immunoenzymatic method to assess Akt activation in treated and untreated cells. Specifically, we measured the phosphorylation of the threonine and serine residues at position 308 and 473 of the aminoacid sequence of the enzyme, respectively. OXIDs at their IC50s for 72 h could significantly decrease the phosphorylation of Akt by about 20–40%, with the maximal effect observed for the compound 1a on the pThr308 site. It is worth mentioning that pThr308 and pSer473 are two target sites of the PDK1 and mTORC2 action, respectively.26 In our experimental setting, compounds 1a,c and 2 and the Akt inhibitor, perifosine, showed a similar ability to inhibit both regulatory sites, whereas compounds 1b and 1d appeared to have some selectivity toward pThr308 (see Supporting Information).
In order to confirm selectivity of the compounds, we evaluated the phosphorylation status of several important proteins of the PDK1/Akt cascade by Western Blot analysis (Figure 5). While the OXIDs 1a and 1b did not affect EGFR (Tyr992) and Her2 (Tyr1221/1222) phosphorylation, they inhibited phosphorylation of Akt (Thr308) and FoxO1 (Thr24)/FoxO3a (Thr32), two downstream effectors of the PDK1/Akt pathway. These findings suggest that tested compounds are selective inhibitors of PDK1/Akt survival pathway in NSCLC cells.
Figure 5.

Effect of 1a and 1b on protein phosphorylation of different targets involved in the PDK1/Akt pathway. Data are representative of 3 independent experiments (see Supporting Information).
We also performed cytotoxicity experiments on MDA-MB-231, a cell line reported to grow independently from PDK1/Akt pathway.27,28 Our findings clearly demonstrated that compounds 1a and 1b at 1 and 10 μM for 72 h do not affect MDA-MB-231 cell proliferation (see Supporting Information, Figure S1), supporting the selectivity of the compounds on the PDK1/Akt pathway.
Finally, taking into account that A549 cells carry wild-type EGFR gene coding sequences and are resistant to gefitinib, we tested the most active compound 1a against the human NSCLC cell line HCC827 that carries mutant EGFR and is highly sensitive to gefitinib.29,30 Compound 1a inhibited HCC827 cell proliferation with an IC50 lower than that obtained in A549 cells (0.66 ± 0.09 and 0.045 ± 0.09 μM, respectively). However, since HCC827 cells are about 1700 times more sensitive than A549 cells to gefitinib,27 these findings seem to confirm that EGFR inhibition did not substantially account for the antiproliferative effect of compounds tested in the current study.
Our findings clearly demonstrated that OXID derivatives, in the low micromolar/submicromolar range of concentrations, could exert a substantial antitumor activity against human NSCLC cells. This activity appears to be clinically relevant, particularly in NSCLC patients previously treated with chemotherapeutic drugs who do not respond to EGFR inhibitors.
In addition, our data suggest that the 2-oxindole nucleus may be considered as central core to develop new ligands able to disrupt the PDK/Akt pathway. The substituent in 3 position on the oxindole nucleus can affect antitumor activity of test compounds. Indeed, the presence of the thiophene ring characterized the most active compound (1a), while the OXID derivatives with methyl imidazole (1b) and imidazole (1c) show a reduced activity. The detrimental effects of a substituent in 3 position is also highlighted by the loss of activity for the oxindole, which lacks the substituent (2).
Future perspectives include investigation of the most promising compounds in combination with drugs currently used in NSCLC patients, both in vitro and in vivo.
Glossary
ABBREVIATIONS
- PI3K
phosphoinositide-3-kinase
- PDK1
3-phosphoinositide-dependent kinase-1
- Akt
protein kinase B
- OXIDs
2-oxindole derivatives
- NSCLC
nonsmall cell lung cancer
- EGFR
epidermal growth factor receptor
- CAC
chloroacetilchloride
- mTORC2
mammalian target of rapamycin complex 2
Supporting Information Available
Synthetic procedures, analytical data, procedures for pharmacological activities, and ELISA on Akt. This material is available free of charge via the Internet at http://pubs.acs.org.
Author Contributions
∥ These authors (G.N. and S.S.) contributed equally to this work. All authors have given approval to the final version of the manuscript.
The authors declare no competing financial interest.
Supplementary Material
References
- Gadgeel S. M.; Wozniak A. Preclinical rationale for PI3K/Akt/mTOR pathway inhibitors as therapy for epidermal growth factor receptor inhibitor-resistant non-small-cell lung cancer. Clin. Lung Cancer 2013, 14, 322–332. [DOI] [PubMed] [Google Scholar]
- Laplante M.; Sabatini D. M. mTOR signaling in growth control and disease. Cell 2012, 149, 274–293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balsara B. R.; Pei J.; Mitsuuchi Y.; Page R.; Klein-Szanto A.; Wang H.; Unger M.; Testa J. R. Frequent activation of AKT in non-small cell lung carcinomas and preneoplastic bronchial lesions. Carcinogenesis 2004, 25, 2053–2059. [DOI] [PubMed] [Google Scholar]
- Oki E.; Baba H.; Tokunaga E.; Nakamura T.; Ueda N.; Futatsugi M.; Mashino K.; Yamamoto M.; Ikebe M.; Kakeji Y.; Maehara Y. Akt phosphorylation associates with LOH of PTEN and leads to chemoresistance for gastric cancer. Int. J. Cancer 2005, 117, 376–380. [DOI] [PubMed] [Google Scholar]
- Carden C. P.; Stewart A.; Thavasu P.; Kipps E.; Pope L.; Crespo M.; Miranda S.; Attard G.; Garrett M. D.; Clarke P. A.; Workman P.; de Bono J. S.; Gore M.; Kaye S. B.; Banerji U. The association of PI3 kinase signaling and chemoresistance in advanced ovarian cancer. Mol. Cancer Ther. 2012, 11, 1609–1617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janmaat M. L.; Kruyt F. A. E.; Rodriguez J. A.; Giaccone G. Response to epidermal growth factor receptor inhibitors in non- small cell lung cancer cells: limited antiproliferative effects and absence of apoptosis associated with persistent activity of extracellular signal-regulated kinase or Akt kinase pathways. Clin. Cancer Res. 2003, 9, 2316–2326. [PubMed] [Google Scholar]
- Altomare D. A.; Testa J. R. Perturbations of the AKT signaling pathway in human cancer. Oncogene 2005, 24, 7455–7464. [DOI] [PubMed] [Google Scholar]
- Cheng J. Q.; Lindsley C. W.; Cheng G. Z.; Yang H.; Nicosia S. V. The Akt/PKB pathway: molecular target for cancer drug discovery. Oncogene 2005, 24, 7482–7492. [DOI] [PubMed] [Google Scholar]
- Cardone M. H.; Roy N.; Stennicke H. R.; Salvesen G. S.; Franke T. F.; Stanbridge E.; Frisch S.; Reed J. C. Regulation of cell death protease caspase-9 by phosphorylation. Science 1998, 282, 1318–1321. [DOI] [PubMed] [Google Scholar]
- Datta S. R.; Dudek H.; Tao X.; Masters S.; Fu H.; Gotoh Y.; Greenberg M. E. Akt phosphorylation of BAD couples survival signals to the cell-intrinsic death machinery. Cell 1997, 91, 231–241. [DOI] [PubMed] [Google Scholar]
- Brunet A.; Bonni A.; Zigmond M. J.; Lin M. Z.; Juo P.; Hu L. S.; Anderson M. J.; Arden K. C.; Blenis J.; Greenberg M. E. Akt promotes cell survival by phosphorylating and inhibiting a Forkhead transcription factor. Cell 1999, 96, 857–868. [DOI] [PubMed] [Google Scholar]
- Lv K.; Wang L. L.; Zhou X. B.; Liu M. L.; Liu H. Y.; Zheng Z. B.; Li S. Synthesis and in vitro antitumor activity of 1-(3-dimethylamino)propyl indolin-2-one derivatives. Med. Chem. Res. 2013, 2, 1723–1729. [Google Scholar]
- Islam I.; Bryant J.; Chou Y. L.; Kochanny M. J.; Lee W.; Phillips G. B.; Yu H.; Adler M.; Whitlow M.; Ho E.; Lentz D.; Polokoff M. A.; Subramanyam B.; Wu J. M.; Zhu D.; Feldman R. I.; Arnaiz D. O. Indolinone based phosphoinositide-dependent kinase-1 (PDK1) inhibitors. Part 1: design, synthesis and biological activity. Bioorg. Med. Chem. Lett. 2007, 17, 3814–3818. [DOI] [PubMed] [Google Scholar]
- Islam I.; Brown G.; Bryant J.; Hrvatin P.; Kochanny M. J.; Phillips G. B.; Yuan S.; Adler M.; Whitlow M.; Lentz D.; Polokoff M. A.; Wu J.; Shen J.; Walters J.; Ho E.; Subramanyam B.; Zhu D.; Feldman R. I.; Arnaiz D. O. Indolinone based phosphoinositide-dependent kinase-1 (PDK1) inhibitors. Part 2: optimization of BX-517. Bioorg. Med. Chem. Lett. 2007, 17, 3819–3825. [DOI] [PubMed] [Google Scholar]
- Mohler M. L.; Kang G. S.; Hong S. S.; Patil R.; Kirichenko O. V.; Li W.; Rakov I. M.; Geisert E.; Miller D. D. Discovery of antiglioma activity of biaryl 1,2,3,4-tetrahydroisoquinoline derivatives and conformationally flexible analogues. J. Med. Chem. 2006, 49, 5845–5848. [DOI] [PubMed] [Google Scholar]
- Lin H. R.; Safo M. K.; Abraham D. J. Identification of a series of tetrahydroisoquinoline derivatives as potential therapeutic agents for breast cancer. Bioorg. Med. Chem. Lett. 2007, 17, 2581–2589. [DOI] [PubMed] [Google Scholar]
- Li Y.; Zhang H. B.; Huang W. L.; Li Y. M. Design and synthesis of tetrahydroisoquinoline derivatives as potential multidrug resistance reversal agents in cancer. Bioorg. Med. Chem. Lett. 2008, 18, 3652–3655. [DOI] [PubMed] [Google Scholar]
- Colabufo N. A.; Berardi F.; Perrone R.; Rapposelli S.; Digiacomo M.; Vanni M.; Balsamo A. 2-[(3-Methoxyphenylethyl)phenoxy]-based ABCB1 inhibitors: effect of different basic side-chains on their biological properties. J. Med. Chem. 2008, 51, 7602–7613. [DOI] [PubMed] [Google Scholar]
- Choquette D.; Teffera Y.; Polverino A.; Harmange J. C. Discovery of novel 1,2,3,4-tetrahydroisoquinolines and 3,4-dihydroisoquinoline-1(2H)-ones as potent and selective inhibitors of KDR: synthesis, SAR, and pharmacokinetic properties. Bioorg. Med. Chem. Lett. 2008, 18, 4054–4058. [DOI] [PubMed] [Google Scholar]
- Arnaiz D.; Bryant J.; Chou Y. L.; Feldman R.; Hrvatin P.; Islam I.; Kochanny M.; Lee W.; Polokoff M.; Yu H.; Yuan S.. Indolinone Derivatives and Their Use in Treating Disease-States Such As Cancer. US 07105563, 2006.
- Tang F.; Shen H.; Jin Q.; Ding L.; Yang J.; Yin X.; Lu S.. 3-Pyrrole-cyclohexylene-2-dihydro-indolinone Derivatives and Uses Thereof. WO2008067756, 2008.
- Sun L.; Tran N.; Tang F.; App H.; Hirth P.; McMahon G.; Tang C. Synthesis and biological evaluations of 3-substituted indolin-2-ones: A novel class of tyrosine kinase inhibitors that exhibit selectivity toward particular receptor tyrosine kinases. J. Med. Chem. 1998, 41, 2588–2603. [DOI] [PubMed] [Google Scholar]
- Zou Z. Q.; Zhang X. H.; Wang F.; Shen Q. J.; Xu J.; Zhang L. N.; Xing W. H.; Zhuo R. J.; Li D. A. Novel dual PI3Kalpha/mTOR inhibitor PI-103 with high antitumor activity in non-small cell lung cancer cells. Int. J. Mol. Med. 2009, 24, 97–101. [DOI] [PubMed] [Google Scholar]
- Henery S.; George T.; Hall B.; Basiji D.; Ortyn W.; Morrissey P. Quantitative image based apoptotic index measurement using multispectral imaging flow cytometry: a comparison with standard photometric methods. Apoptosis 2008, 13, 1054–1063. [DOI] [PubMed] [Google Scholar]
- Sun H.; Lesche R.; Li D. M.; Liliental J.; Zhang H.; Gao J.; Gavrilova N.; Mueller B.; Liu X.; Wu H. PTEN modulates cell cycle progression and cell survival by regulating phosphatidylinositol 3,4,5,-trisphosphate and Akt/protein kinase B signaling pathway. Proc. Natl. Acad. Sci. U.S.A. 1999, 96, 6199–6204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alessi D. R.; James S. R.; Downes C. P.; Holmes A. B.; Gaffney P. R.; Reese C. B.; Cohen P. Characterization of a 3-phosphoinositide-dependent protein kinase which phosphorylates and activates protein kinase Balpha. Curr. Biol. 1997, 7, 261–269. [DOI] [PubMed] [Google Scholar]
- Nagashima K.; Shumway S. D.; Sathyanarayanan S.; Chen A. H.; et al. Genetic and pharmacological inhibition of PDK1 in cancer cells: characterization of a selective allosteric kinase inhibitor. J. Biol. Chem. 2011, 286, 6433–6448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raimondi C.; Chikh A.; Wheeler A. P.; Maffucci T.; Falasca M. A novel regulatory mechanism links PLCγ1 to PDK1. J. Cell. Sci. 2012, 125, 3153–3163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amann J.; Kalyankrishna S.; Massion P. P.; Ohm J. E.; Girard L.; Shigematsu H.; Peyton M.; Juroske D.; Huang Y.; Stuart S. J.; Kim Y. H.; Pollack J. R.; Yanagisawa K.; Gazdar A.; Minna J. D.; Kurie J. M.; Carbone D. P. Aberrant epidermal growth factor receptor signaling and enhanced sensitivity to EGFR inhibitors in lung cancer. Cancer Res. 2005, 65, 226–235. [PubMed] [Google Scholar]
- Mukohara T.; Engelman J. A.; Hanna N. H.; Yeap B. Y.; Kobayashi S.; Lindeman N.; Halmos B.; Pearlberg J.; Tsuchihashi Z.; Cantley L. C.; Tenen D. G.; Johnson B. E.; Jänne P. A. Differential effects of gefitinib and cetuximab on non-small-cell lung cancers bearing epidermal growth factor receptor mutations. J. Natl. Cancer Inst. 2005, 97, 1185–1194. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.