

Abstract
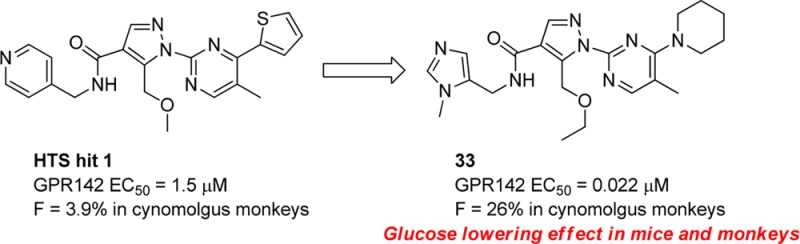
GPR142 is a G protein-coupled receptor that is predominantly expressed in pancreatic β-cells. GPR142 agonists stimulate insulin secretion in the presence of high glucose concentration, so that they could be novel insulin secretagogues with reduced or no risk of hypoglycemia. We report here the optimization of HTS hit compound 1 toward a proof of concept compound 33, which showed potent glucose lowering effects during an oral glucose tolerance test in mice and monkeys.
Keywords: GPR142, agonist, insulin secretagogue, diabetes, glucose lowering
Type 2 diabetes is characterized by high blood glucose resulting from reduced insulin production from pancreatic β-cell or insulin resistance or both of them.1 Uncontrolled hyperglycemia increases the risk of cardiovascular complications such as coronary heart disease, stroke, nephropathy, neuropathy, and retinopathy in patients with diabetes.2 Thus, effective glycemic control is important to prevent chronic diabetic complications. Currently, insulin secretagogues such as sulfonylureas and meglitinide are widely used for patients with a moderate degree of β-cell dysfunction.3 They trigger insulin release independently of blood glucose level. As a result, some of them could increase the risk of hypoglycemia.4 Therefore, novel glucose dependent insulin secretagogues, such as DPP-IV inhibitors,5 GLP-1 analogues,5 GPR40 agonists,6 and GPR119 agonists,7 are highly attractive alternatives for the treatment of diabetes.
GPR142 was identified as an orphan G protein-coupled receptor (GPCR). GPR142 is predominantly expressed in pancreatic β-cells and coupled with the Gq signaling pathway, and its endogenous ligands are aromatic amino acids with tryptophan, which is the most potent ligand in inositol phosphate accumulation assay (IP assay).8 Tryptophan stimulates insulin secretion from isolated mice pancreatic islets only under high glucose conditions and oral administration of tryptophan improves glucose tolerance in mice. These findings indicate that GPR142 agonists could be novel insulin secretagogues with reduced or no risk of hypoglycemia.
High throughput screening (HTS) campaigns identified several hit compounds including compound 1 (Figure 1) as well as phenylalanine derivatives9,10 already reported. Compound 1 showed modest agonistic activity against human GPR142 (IP assay, EC50 = 1.5 μM). Compound 1 potentiated insulin secretion in rat primary islets at high glucose conditions, but not at low glucose conditions (see Supporting Information). This data showed the glucose-dependent insulin secretion of GPR142 agonist. Subcutaneous injection of compound 1 improved glucose tolerance in mice. However, poor pharmacokinetic (PK) profile of compound 1 was inadequate for further proof of concept studies in vivo. High in vivo clearance (1.3 L/h/kg) and very poor oral bioavailability (F = 3.9%) of compound 1 in monkey are presumably due to metabolic instability (Table 4). Moreover, compound 1 inhibited CYP3A4 (IC50 = 2.9 μM), which can cause unfavorable drug–drug interaction.
Figure 1.

Structure of GPR142 agonist from HTS.
Table 4. Monkey PK Parameters of 33.
Compounds were dosed iv at 0.5 mg/kg.
Compounds were dosed po at 1 mg/kg.
Not applicable.
We describe here the optimization of a HTS hit compound 1 to provide a potent orally bioavailable GPR142 agonist and its pharmacological effects in mice and monkeys.
First, we investigated the structure–activity relationship (SAR) of the pyrazole side chain (Table 1). Synthesis of compounds 7a–f started from Suzuki coupling between 2,4-dichloro-5-methylpyrimidine (2) and thiopheneboronic acid to give 3 (Scheme 1).11 Compound 3 was heated with NH2NH2 in pyridine to give 4. Keto-ester 9 was conveniently prepared by heating 8 with N,N-dimethylforamide dimethyl acetal. Condensation of 4 and 9 under acidic conditions resulted in methyl ester 5, which was hydrolyzed to give acid 6. Finally, coupling of 6 with 4-(aminomethyl)pyridine using HBTU gave compounds 7a–f. n-Propyl (7b) and n-butyl (7c) derivatives were found to improve agonistic activities, but their poor metabolic stability was not improved. The size of alkyl side chains seems to be important for GPR142 agonism.
Table 1. SAR of Pyrazole Side Chains.
| cmpd | R1 | hGPR142 EC50 (μM)a,b | MLMc (% remaining) |
|---|---|---|---|
| 1 | MOM | 1.5 | 3 |
| 7a | Et | 1.0 | NT |
| 7b | nPr | 0.25 | 1 |
| 7c | nBu | 0.0070 | NT |
| 7d | nPent | 1.0 | NT |
| 7e | secBu | 1.2 | NT |
| 7f | CH2OEt | 0.030 | NT |
Assay protocols are provided in the Supporting Information.
Assay results are the average of three replicates; standard deviation was ±20%.
% Remaining after incubation for 15 min in mouse liver microsomes. NT = not tested.
Scheme 1. General Synthesis of Compound.

Reagents and conditions: (a) 2-thiopheneboronic acid, Pd(PPh3)4, K2CO3, DME, reflux; (b) NH2NH2, pyridine; (c) 9, HOAc, EtOAc; (d) LiOH, THF/H2O; (e) 4-(aminomethyl)pyridine, HBTU, iPr2NEt, DMF; (f) N,N-dimethylforamide dimethyl acetal, heat.
Next, we explored the 4-pyridyl headgroup in the compound 7b or 7c to improve metabolic stability (Table 2). Compounds 10–22 were synthesized with the same method in Scheme 1. Dimethyl substitution (10) at benzyl position was tolerated in terms of agonistic activity but did not improve metabolic stability. Substitution on the amide nitrogen (11) resulted in significant loss of GPR142 potency. Replacement of pyridine ring with 4-fluorophenyl ring (12) gave an inactive analogue. 5-Substituted imidazole derivative (13) showed very potent agonistic activity and modest metabolic stability. In contrast, 4-substitued imidazole derivative (14) lost its GPR142 agonistic activity. The orientation of a nitrogen lone pair is likely to be crucial to GPR142 agonism. Methyl substitution at 4-position (15) or 2-position (16) in the imidazole ring of 13 was tolerated in agonistic activity. However, metabolic stability of both compounds was significantly decreased. A conformationally restricted analogue (17), which might resist metabolism, resulted in the loss of agonistic activity and no improvement of metabolic stability. Introduction of carboxylic acid in the headgroup (18) was tolerated and improved metabolic stability; however, its membrane permeability was too low to achieve oral bioavailability. Pyrazole derivative (19) was a modest agonist, which had poor metabolic stability. Isoxazole (20), thiazole (21), and indole (22) derivatives were less potent agonists than imidazole derivatives. By this moment, we selected the imidazole derivative 13 for further optimization.
Table 2. Exploration of 4-Pyridyl Head Group.
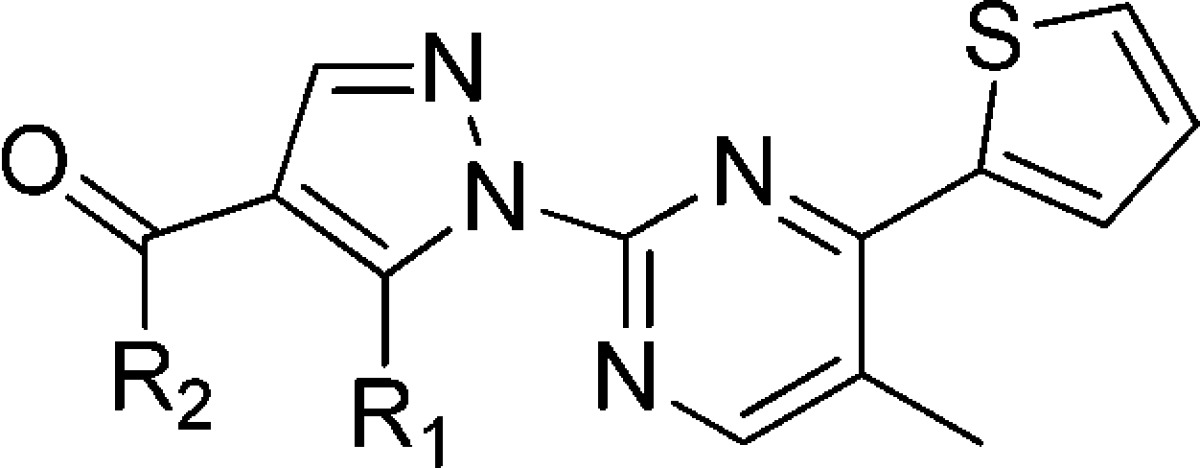
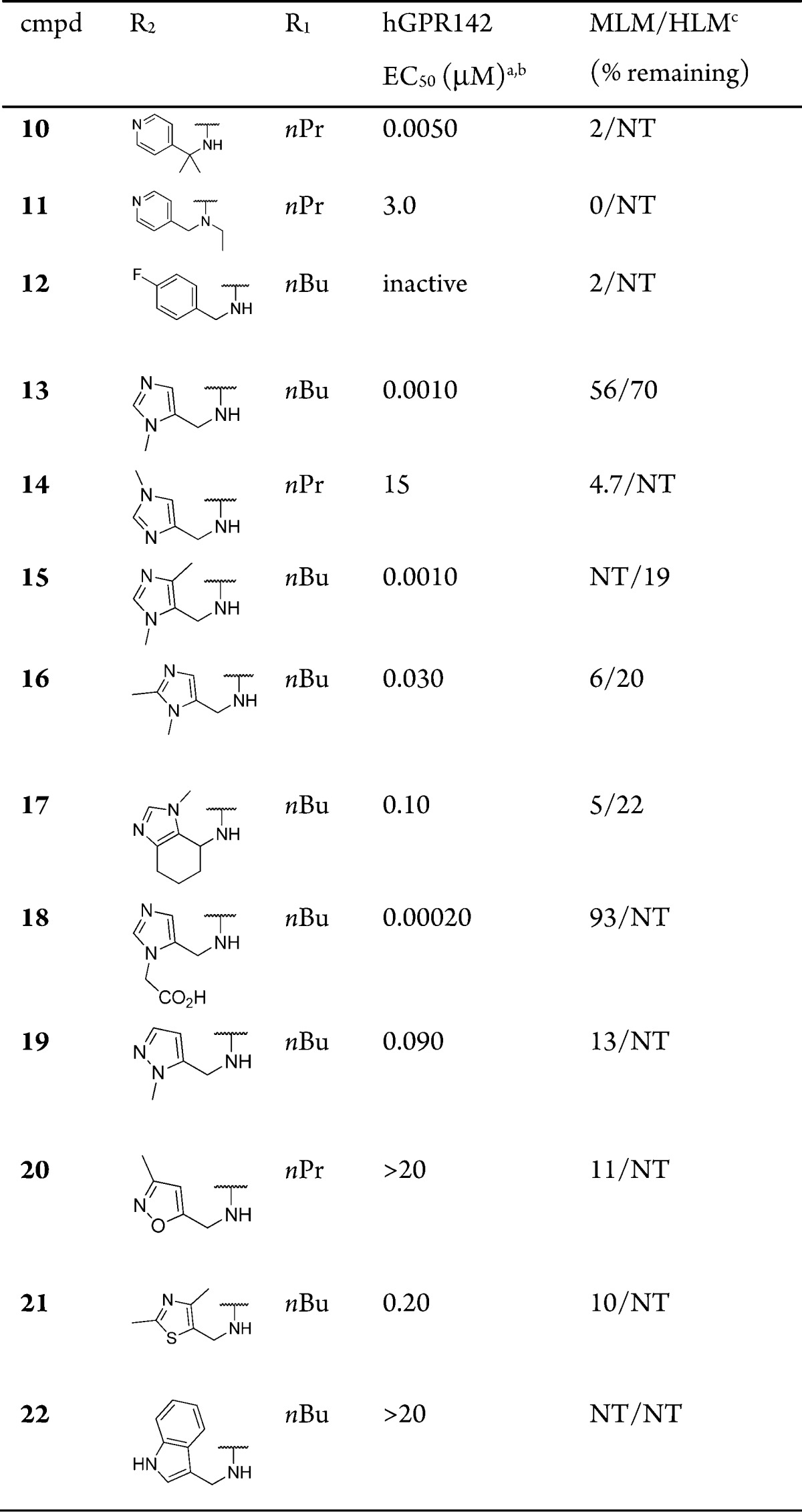
Assay protocols are provided in the Supporting Information.
Assay results are the average of three replicates; standard deviation was ±20%.
% Remaining after incubation for 15 min in mouse liver microsomes or human liver microsomes. NT = not tested.
Introduction of the ethoxymethyl side chain instead of the butyl side chain gave compound 23, which showed comparable GPR142 agonistic activity and metabolic stability to 13; moreover, its hydrophobicity (LogD = 2.2) was in an optimal range (Table 3).12 Thiophene is known to give reactive metabolite in vivo and have potential of idiosyncratic toxicity,13 in that we investigated substitution of thiophene ring particularly with a nonaromatic saturated group. To facilitate the exploration of replacements for the thiophene, an efficient synthesis was developed to allow the introduction of this moiety at the last step, as shown in Scheme 2. Compound 24 was obtained by treatment of 2 with the sodium salt of 2-(trimethylsilyl)ethanol in THF according to Lluis et al.14 A two-step sequence to install the pyrazole (as described in Scheme 1) gave compound 25, which was treated with TBAF in MeCN to liberate the hydroxypyrimidine. The methyl ester was then hydrolyzed to the acid 26. The acid 26 was heated at reflux with POCl3 to provide 4-chloro pyrimidine acid chloride, which was isolated as an acid 27 after workup in ice water. Coupling with the methyl imidazole amine gave 28, a key precursor for the preparation of 29–33. Ether and amine derivatives were made by direct nucleophilic aromatic displacement with the appropriate sodium alkoxide or with an excess of the amine in DMF. Compound 29 having ethoxy moiety was found to be a modest GPR142 agonist. A series of alkyl amine analogues (30–33) showed good GPR142 agonistic activity as well as good metabolic stability. Cyclobutylamine (30) and azetidine (31) derivatives had poor membrane permeability. Pyrrolidine (32) and piperidine (33) derivatives had good membrane permeability with increasing hydrophobicity. Unfortunately, CYP inhibition of a series of compounds described here was still not appropriate for further development (CYP3A4 IC50 = 1.0 μM for 33). We assessed PK profiles of 33 in monkeys. Compound 33 demonstrated good oral bioavailability (F = 26%), which was consistent with its good metabolic stability (Table 4).
Table 3. Substitution of Thiophene Ring.
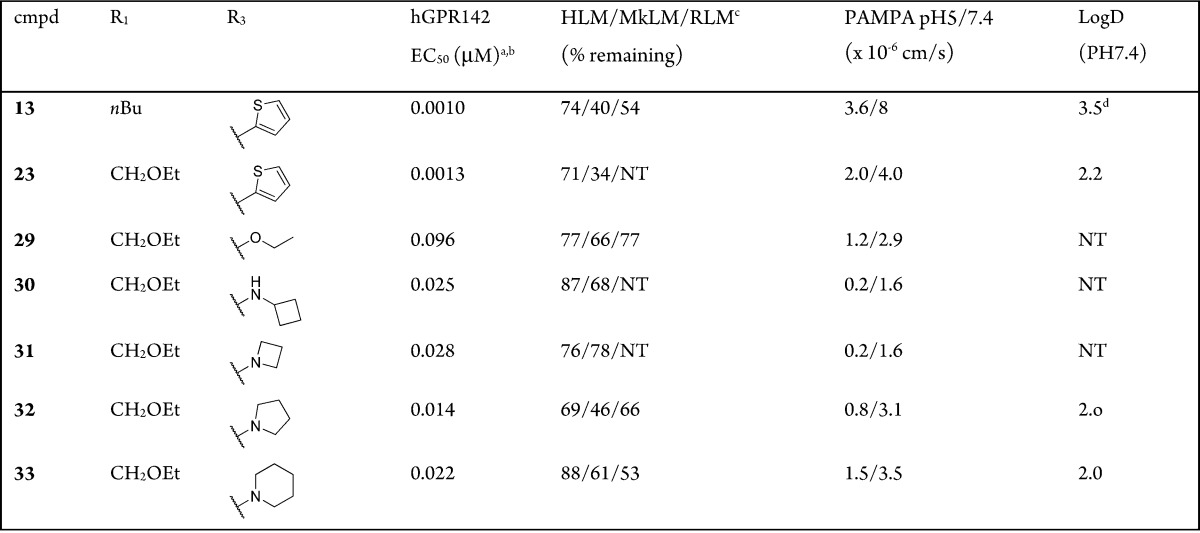
Assay protocols are provided in the Supporting Information.
Assay results are the average of three replicates; standard deviation was ±20%.
% Remaining after incubation for 30 min in human, monkey, and rat liver microsomes.
Calculated by ACD. NT = not tested.
Scheme 2. Synthesis of 4-Substituted Analogues to Replace Thiophene.

Reagents and conditions: (a) 2-(trimethylsilyl)ethanol, NaH, THF; (b) NH2NH2, pyridine; (c) 9, HOAc, EtOAc; (d) 1. TBAF, THF, MeCN; 2. LiOH, THF/H2O; (e) POCl3, 110 °C, quench with H2O; (f) 1-methyl-5-(aminomethyl)-1H-imidazole, HBTU, iPr2NEt, DMF; (g) alkoxide or amine, DMF.
Therefore, we selected 33 as a proof of concept compound for in vivo studies in mice (Figure 2) and monkeys (Figure 3). C57BL/6J mice were dosed orally with compound 33. After that, mice were challenged with an oral glucose, and blood samples were collected for the assay of blood glucose levels. Compound 33 (1–30 mg/kg) decreased blood glucose concentration in C57BL/6J mice in a dose dependent manner. The maximum glucose lowering effect of 33 is similar to that of sitagliptin,15 a marketed DPP-IV inhibitor. Because plasma insulin level of C57BL/6J mice was so low that it was difficult to detect the increase, we confirmed the plasma insulin increase based on GPR142 activation by using ob/ob mice, the diabetic animal model that strongly secreted insulin. Obese ob/ob mice were dosed orally with compound 33, and blood samples were collected 20 min after dosing for insulin measurement. Compound 33 (1–10 mg/kg) increased plasma insulin concentration in a dose dependent manner.
Figure 2.
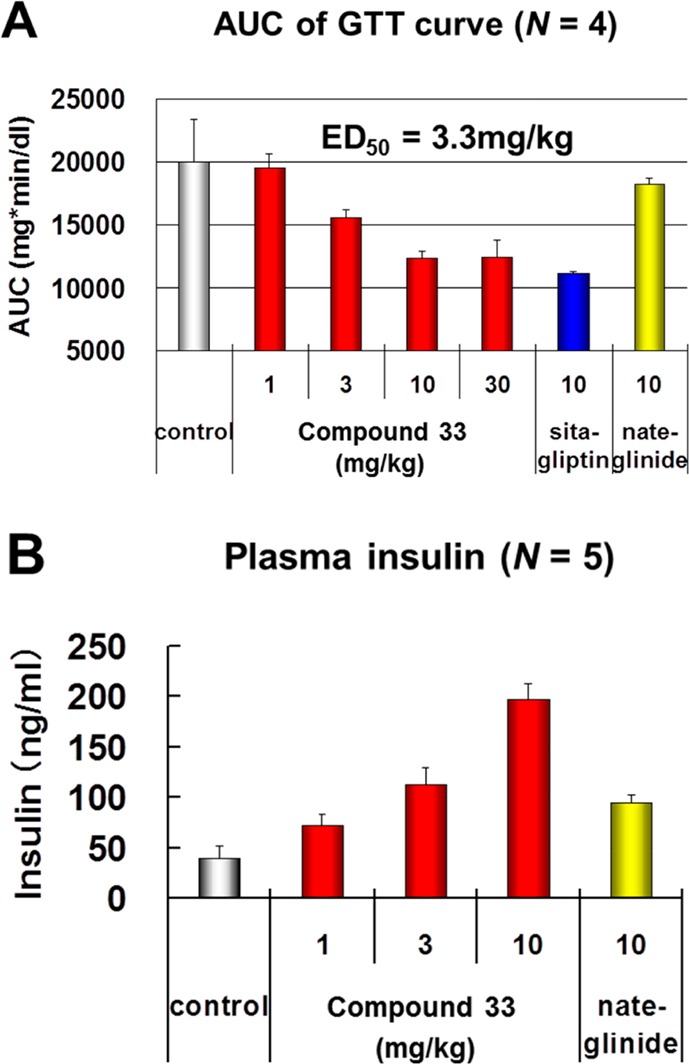
In vivo efficacy of 33 in mice. (A) Oral glucose tolerance test (C57BL/6J mouse). (B) Plasma insulin level at 15 min after bolus dosing (ob/ob mouse).
Figure 3.
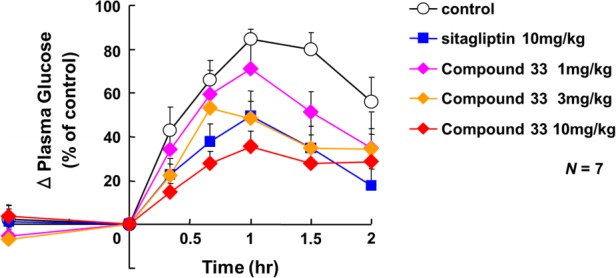
In vivo efficacy of 33 in monkeys with impaired glucose tolerance.
Compound 33 was evaluated for its ability to improve glycemic control in cynomolgus monkeys (Figure 3). Single oral dosing of 33 (1–10 mg/kg) reduced the blood glucose level in a dose dependent manner during an oral glucose tolerance test. Furthermore, at a dose of 10 mg/kg, compound 33 tended to show greater efficacy than sitagliptin.15
In conclusion, we described the optimization leading from HTS hit compound 1 to the orally bioavailable compound 33, a structurally novel potent GPR142 agonist. The antidiabetic effect that compound 33 demonstrated in mice and monkeys strongly suggests that GPR142 agonists have great potential for the treatment of type II diabetes. Detailed pharmacological evaluation and further optimization of this series will be reported elsewhere.
Glossary
Abbreviations
- HTS
high throughput screening
- CYP
cytochrome P450
- HBTU
o-benzotriazole-N,N,N′,N′-tetramethyl-uronium-hexafluoro-phosphate
- MLM
mouse liver microsome
- HLM
human liver microsome
- MkLM
monkey liver microsome
- RLM
rat liver microsome
Supporting Information Available
Experimental procedures and analytical data. This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest.
Supplementary Material
References
- Defronzo R. A. From the triumvirate to the ominous octet: a new paradigm for the treatment of type 2 diabetes. Diabetes 2009, 58, 773–795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nazimek-Siewniak B.; Moczulski D.; Grzeszczak W. J. Risk of macrovascular and microvascular complications in type 2 diabetes: results of longitudinal study design. Diabetes Complications 2002, 16, 271. [DOI] [PubMed] [Google Scholar]
- Randell M. The role of sulfonylureas in the management of type 2 diabetes mellitus. Drugs 2004, 64, 1339–1358. [DOI] [PubMed] [Google Scholar]
- Burge M. R.; Sood V.; Sobhy T. A.; Rassam A. G.; Schade D. S. Sulfonylurea-induced hypoglycemia in type 2 diabetes mellitus: a review. Diabetes, Obes. Metab. 1999, 1, 199–206. [DOI] [PubMed] [Google Scholar]
- Drucker D. J.; Nauck M. A. The incretin system: glucagon-like peptide-1 receptor agonists and dipeptidyl peptidase-4 inhibitors in type 2 diabetes. Lancet 2006, 368, 1696–1706. [DOI] [PubMed] [Google Scholar]
- Bharate S. B.; Nemmani K. V.; Vishwakarma R. A. Progress in the discovery and development of small-molecule modulators of G-protein-coupled receptor 40 (GPR40/FFA1/FFAR1): An emerging target for type 2 diabetes. Expert Opin. Ther. Pat. 2009, 19, 237–294. [DOI] [PubMed] [Google Scholar]
- Ohishi T.; Yoshida S. The therapeutic potential of GPR119 agonists for type 2 diabetes. Expert Opin. Invest. Drugs 2012, 21, 237–294. [DOI] [PubMed] [Google Scholar]
- Xiong Y.; Motani A.; Reagan J.; Gao X.; Yang H.; Ma J.; Schwandner R.; Zhang Y.; Liu Q.; Miao L.; Luo J.; Tian H.; Chen J.-L.; Murakoshi M.; Nara F.; Yeh W.-C.; Cao Z. Manuscript in preparation.
- Lizarzabura M.; Turcotte S.; Du X.; Duquette J.; Fu A.; Houze J.; Li L.; Liu J.; Oda K.; Okuyama R.; Yu M.; Reagan J.; Medina J. C. Discovery and optimization of a novel series of GPR142 agonists for the treatment of type 2 diabetes mellitus. Bioorg. Med. Chem. Lett. 2012, 22, 5942–5947. [DOI] [PubMed] [Google Scholar]
- Du X.; Kim Y.-J.; Lai S.; Chen X.; Lizarzaburu M.; Turcotte S.; Fu Z.; Liu Q.; Zhang Y.; Motani A.; Oda K.; Okuyama R.; Nara F.; Murakoshi M.; Fu A.; Reagan J. D.; Fan P.; Xiong Y.; Shen W.; Li L.; Houze J.; Medina J. C. Phenylalanine derivatives as GPR142 agonists for the treatment of Type II diabetes. Bioorg. Med. Chem. Lett. 2012, 22, 6218–6223. [DOI] [PubMed] [Google Scholar]
- Ogawa Y.; Okuyama R.; Shibuya S.; Toda N.; Cao Z.; Fu Z.; Hao X.; Kim Y.; Li L.; Lively S.; Lizarzaburu M.; Tian H.; Yu M.. WO 2008/045484.
- Waring M. J. Lipophilicity in drug discovery. Expert Opin. Drug Discovery 2010, 5, 235–248. [DOI] [PubMed] [Google Scholar]
- Hu Y.; Yang S.; Shilliday B.; Heyde B. R.; Mandrell K. M.; Robins R. H.; Xie J.; Reding M. T.; Lai Y.; Thompson D. C. Novel metabolic bioactivation mechanism for a series of anti-inflammatory agents (2,5-diaminothiophene derivatives) mediated by cytochrome P450 enzymes. Drug Metab. Dispos. 2010, 38, 1522–1531. [DOI] [PubMed] [Google Scholar]
- Lluís J.; Matheu M.; Castillón S. Stereoselective synthesis of 2′,3′-dideoxy-nucleosides via intramolecular glycosylation of phenyl 1-seleno-glycosides. Synthesis of 2′,3′-dideoxythymidine. Tetrahedron Lett. 1998, 39, 1807–1810. [Google Scholar]
- Kim D.; Wang L.; Beconi M.; Eiermann G.; Fisher M.; He H.; Hickey G.; Kowalchick J.; Leiting B.; Lyons K.; Marsilio F.; McCann M.; Patel R.; Petrov A.; Scapin G.; Patel S.; Roy R.; Wu J.; Wyvratt M.; Zhang B.; Zhu L.; Thornberry N.; Weber A. (2R)-4-oxo-4-[3-(Trifluoromethyl)-5,6-dihydro[1,2,4]triazolo[4,3-a]pyrazin-7(8H)-yl]-1-(2,4,5-trifluorophenyl)butan-2-amine: a potent, orally active dipeptidyl peptidase IV inhibitor for the treatment of type 2 diabetes. J. Med. Chem. 2005, 48, 141–151. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.