

Abstract
Human β-N-acetyl-d-hexosaminidase has gained much attention due to its roles in several pathological processes and been considered as potential targets for disease therapy. A novel and efficient skeleton, which was an unsymmetrical dyad containing naphthalimide and methoxyphenyl moieties with an alkylamine spacer linkage as a noncarbohydrate-based inhibitor, was synthesized, and the activities were valuated against human β-N-acetyl-d-hexosaminidase. The most potent inhibitor exhibits high inhibitory activity with Ki values of 0.63 μM. The straightforward synthetic manners of these unsymmetrical dyads and understanding of the binding model could be advantageous for further structure optimization and development of new therapeutic agents for Hex-related diseases.
Keywords: Naphthalimide, β-N-acetyl-d-hexosaminidase, inhibitors, noncarbohydrates, GM2 gangliosides
Family 20 β-N-acetyl-d-hexosaminidase (EC 3.2.1.52) is a widely distributed enzyme that catalyzes the release of N-acetyl-β-d-glucosamine (GlcNAc) or N-acetyl-β-d-galactosamine (GalNAc) residues from the nonreducing ends of oligosaccharides and sugar components of glycoproteins and glycolipids.1 The human β-N-acetyl-d-hexosaminidase (hHex) has gained much attention due to its roles in several pathological processes and been considered as potential target for disease therapy. It is the predominant glycosaminoglycan-degrading glycosidase in the synovial fluid from patients with osteoarthritis.2 So the inhibition of this enzyme may prevent cartilage matrix degradation and thus help the treatment of osteoarthritis.3 Moreover, the human β-N-acetyl-d-hexosaminidase degrades GM2 gangliosides in the lysosome of neuronal cells.4 Disfunction of this enzyme results in the accumulation of GM2 gangliosides and leads to Tay-Sachs or Sandhoff disease.5 The adult forms of these diseases are caused by the presence of large quantities of unstable but still active mutant enzymes, which could not be transported to lysosome.6 So inhibitors against β-N-acetyl-d-hexosaminidase can be used as pharmacological chaperones to stabilize these mutant enzymes and facilitate the transport of them to increase the enzymatic activity in the lysosome and thus help the treatment of these diseases.7−9
The promising application in biology and medicine has induced the appearance of more specific and potent inhibitors of β-N-acetyl-d-hexosaminidases in recent years.10−14 However, most of these inhibitors (Figure 1) possess carbohydrate scaffolds. Comparing with noncarbohydrate-based structure, it is more challenging to convert these leads of sugars into therapeutic agents because of poor pharmacokinetic properties and complex synthetic chemistry, in particular, the difficulties in further optimization by medicinal chemistry.7,15 Thus, it is of significance to develop novel noncarbohydrate-based inhibitors that could be readily modified or synthesized in a more straightforward manner and is easy to improve selectivity, solubility, and toxicity profiles.
Figure 1.

Potency and selectivity of inhibitors against GH20 β-N-acetyl-d-hexosaminidase.
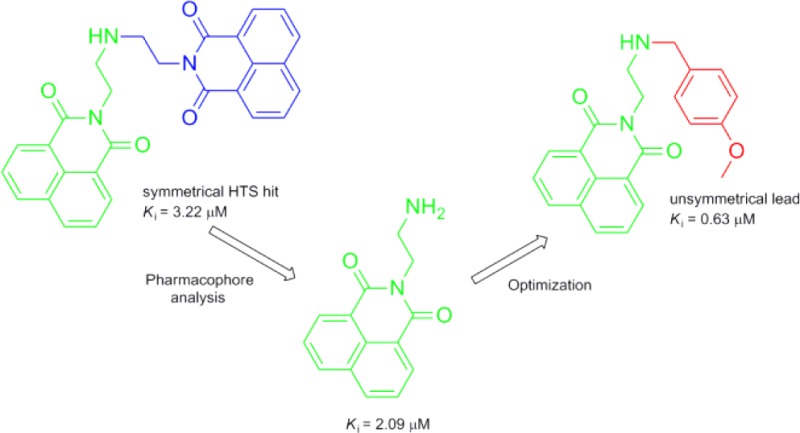
High-throughput screening has been performed to find noncarbohydrate-based inhibitors of human β-N-acetyl-d-hexosaminidase, resulting in four structurally distinct micromolar competitive inhibitors.16,17 Among them, the symmetrical bisnapthalimide (M-31850) is the most potent one against human β-N-acetyl-d-hexosaminidase (IC50 = 6.0 and 3.1 μM for hHex A and hHex B, respectively). Furthermore, the bisnaphthalimide is in a framework that differs significantly from the carbohydrate-based inhibitors of hexosaminidase and supplies a novel scaffold that could be further optimized. In the last two decades, many researchers have made great efforts to promote applications of naphthalimide derivatives in the fields of medicine and biological sensing and imaging.18−20 Most importantly, they are in relatively simple structures whose facile and straightforward syntheses have been established.
We herein report the synthesis and the structure–function relationship of a novel and efficient skeleton, which was an unsymmetrical dyad containing naphthalimide and methoxyphenyl moiety with an alkylamine spacer linkage as a noncarbohydrate-based inhibitor against human β-N-acetyl-d-hexosaminidases A/B.
Our strategy was first to determine the pharmacophore of M-31850, which directly occupies the active site of the human β-N-acetyl-d-hexosaminidase and interacts with the key residues constituting the active pocket. Then various groups were incorporated to this moiety to acquire additional electrostatic and other interactions between the substrate and the enzyme. Since these unsymmetrical dyads exhibit higher inhibitory activity with Ki values of less than 1 μm, they show a promise for further development of drugs.
In the structure–activity relationship of M-31850 proposed by Tropak et al.,16 the naphthalimide group binds to the active site of hHex and the secondary amine of the N-alkylamine linker may form a stabilizing hydrogen bond with a residue around. Besides, the other naphthalimide moiety binds to another hydrophobic patch, making important contributions to its high binding affinity. Though this binding model gives a reasonable explanation for the inhibitory activities of naphthalimide derivatives, there is much unknown in terms of a specificity mechanism, which is important for further drug development.
A series of naphthalimide derivatives or analogous with different N-alkylamine chains 1a–k has been synthesized and assayed their inhibitory activities toward the human β-N-acetyl-D-hexosaminidase A/B (Scheme 1).
Scheme 1. Synthesis of Compounds 1a–k.

n, R1, and R2 are as defined in Table 1. Reagents and conditions: (a) EtOH, reflux, 4 h, 50-100%; (b) MeOH, K2CO3, DMF, 60 °C, 4 h, 62% or NH(CH3)2, DMF, rt, 12 h, 82%; (c) CF3COOH, DCM, rt, 4 h, 100%; (d) AcOH, 100 °C, 1 h, 69%; (e) CF3COOH, DCM, rt, 1 h, 90%; (f) K2CO3, DMF, 50 °C, 8 h, 89%; (g) NH2NH2, MeOH, reflux, 4 h, 80%.
Table 1. Structures and Evaluation Datas of Compounds 1a–k against Human β-N-Acetyl-d-hexosaminidases A/B.
| compd | n | R1 | R2 | Ki (μM)a |
|---|---|---|---|---|
| 1a | 2 | H | NH2 | 2.09 |
| 1b | 3 | H | NH2 | 29.08 |
| 1c | 4 | H | NH2 | 61.74 |
| 1d | 6 | H | NH2 | Nd |
| 1e | 2 | H | OH | 30.87 |
| 1f | 2 | H | N(CH3)2 | 2.81 |
| 1g | 2 | Br | NH2 | Nd |
| 1h | 2 | OCH3 | NH2 | Nd |
| 1i | 2 | N(CH3)2 | NH2 | Nd |
| 1j | Nd | |||
| 1k | Nd | |||
| M-31850b | 3.22 |
Measured with 4MU-β-GlcNAc as substrate and calculated by linear regression of data in Dixon plots. Values with SD were shown in Table S1, Supporting Information. Nd: Not determined (less than 50% inhibition at 50 μM).
For comparison, the Ki of M-31850 was also determined with the same condition.
Compounds 1a–g were obtained by reacting a suspension of the commercially available 1,8-naphthalic anhydrides in ethanol with excess amines followed by recrystallization. 4-Nitro-1,8-naphthalic anhydride was refluxed with tert-butyl (2-aminoethyl)carbamate in ethanol to afford intermediate in an almost quantitative yield. The preparations of 1h–i were finished by the reactions of the intermediate with dimethylamine or methanol followed by deprotection in acidic conditions. Compound 1j was prepared by reaction of phthalic anhydride in acetic acid with Boc-protected ethylenediamine followed by deprotection in the presence of trifluoroacetic acid. The preparation of 1k started with the substitution of 1,8-naphtholactam with 1,2-dibromoethane in basic conditions. Then, the bromine of the intermediate was converted to primary amine by Gabriel synthesis to afford 1m.
To our surprise, the single naphthalimide moiety with the N-alkylamine chain, 1a, is a bit more potent than the symmetrical dyad M-31850. This suggests that the naphthalimide group and the N-alkylamine chain make important contributions, and perhaps, the second naphthalimide group of M-31850 may be not necessary or suitable for binding the hydrophobic aglycon pocket outside the active site.
With the extension of chain length between the terminal N atom and naphthalimide group, the inhibitory activities of 1a–d decrease dramatically. This may indicate that the terminal amine of 1a should be in an appropriate distance away from the naphthalimide group so as to form a hydrogen bond with a key residue in charge of catalysis. The change of terminal amine to alcohol (1e) also affects the activity tremendously. However, the change of the primary amine to tertiary amine as 1f maintains the activity. Thus, we deduce that it may be a good strategy to modify and optimize this scaffold from the terminal amine.
The affinity difference between 1g–k and 1a suggests that the naphthalimide group plays an irreplaceable role for inhibitory activity. Any modification to the naphthalimide group would lead to a decrease or loss in activity, further confirming its importance.
Computation-based molecular modeling of β chain of hHex in complex with 1a explains the above observations (Figure 2). The naphthalimide group binds the hydrophobic pocket composed of residues, W405, W424, and W489, and interacts with the indole ring of the W489 by a π–π stacking force, and the hydrogen atom of terminal amine interacts through a hydrogen bond with the catalytic residue E355, which is for protonation of the glycosidic oxygen atom during catalysis.21 Computational studies also support that, if there is increase in the length of the alkyl chain, the NH group would be within a more appropriate distance to interact with the D452 instead of the catalytic E355 (Figure S2, Supporting Information). The hydrophilic groups at 4-position of naphthalimide leads to a restriction for the interaction between the compound and the active site.
Figure 2.
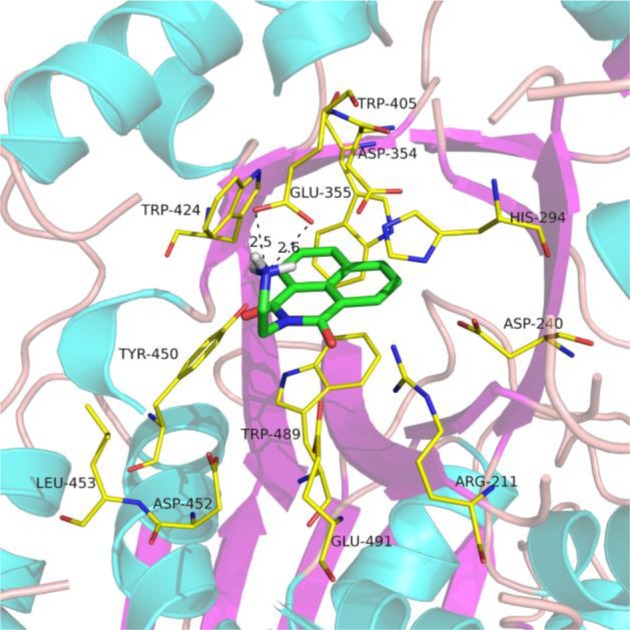
Computational molecular model of 1a against β chain of hHex (PDB code, 3LMY) in the active site as shown in cartoon (hHexB) and stick (1a). Key residues around the active site are shown as lines including carbon (yellow), nitrogen (blue), and oxygen (red) atoms. The carbon atoms of 1a are colored in green. The hydrogen bonds are labeled as a black dash. The unit of hydrogen bonds is Å. The molecular model was created using the software PyMOL.
So compound 1a was selected as an excellent scaffold for developing noncarbohydrate-based inhibitors. To further improve the activity, we proposed to attach other groups to the terminal nitrogen of naphthalimide 1a, which afford unsymmetrical dyads. A series of planar conjugate structures was first introduced to acquire other contact with the hydrophobic aglycon pockets near the active site. After failure trials, we found the anisole analogues, instead of largely hydrophobic structures, could enhance binding affinity. On the basis of studies by Lin et al.,14 the p-methoxybenzyl substituents of GlcNAc-type iminocyclitiol (Figure 1) could be surrounded by a hydrophobic pocket, and thus, the potency and selectivity of inhibitors are improved. To acquire similar electrostatic or other interactions in our case, different methoxybenzyl groups with different linkers were then introduced to obtain compounds 3a–m, 6a–c, and 7a–c, which were also evaluated under the same condition as 1a–m.
Compounds 3a–m were first prepared by corresponding phenols or phenthiols reacting with 1,2-dibromoethane or 1,3- dibromopropane in a solution of sodium hydroxide and tetrabutylammonium bromide in ethanol or a solution of sodium hydroxide in water (Scheme 2). Then 1a was refluxed with the bromide 2a–m and potassium carbonate in acetonitrile for 12 h to afford compounds 3a–m. Compounds 6a–c were synthesized by following steps. Compounds 4a–c were obtained by CuI-catalyzed Ullmann-type coupling reactions of bromobenzene and ethanolamine.22 These alcohols were converted into the corresponding bromides 5a–c using a system of Ph3P, DDQ, and TBAB in high yield. Compounds 5a–c were substituted and refluxed with 1a and potassium carbonate in acetonitrile, giving the compounds 6a–c. Compound 1a was easily transformed into 7a–c by performing a Schiff condensation in methanol in the presence of equivalent different benzaldehydes following the reducing reaction with sodium triacetoxyborohydride.
Scheme 2. Synthesis of Compounds 3a–m, 6a–c, and 7a–c.

Reagents and conditions: (a) NaOH, H2O, 90 °C, 48 h, 68–73% or NaOH, TBAB, EtOH, reflux, 24 h, 32–61%; (b) 1a, K2CO3, MeCN, reflux, 12 h, 44–72%; (c) HO(CH2)2NH2, K2CO3, CuI, l-proline, DMSO, 60 °C, 12 h, 78–90%; (d) TBAB, PPh3, DDQ, DCM, rt, 30 min, 44–82%; (e) 1a, K2CO3, MeCN, reflux, 12 h, 55–70%; (f) 1a, MeOH, rt; (g) NaBH(CH3COO)3, AcOH, DCM, rt, 12 h, 50–65% (for steps f and g). X, n, and R are as defined in Table 2.
Table 2. Structures and Evaluation Datas of Dyads 3a–m, 6a–c and 7a–c against Human β-N-Acetyl-d-hexosaminidases A/B.
| compd | n | X | R | Ki (μM)a |
|---|---|---|---|---|
| 3a | 2 | O | p-OMe | 1.04 |
| 3b | 2 | O | m-OMe | 0.93 |
| 3c | 2 | O | o-OMe | 1.35 |
| 3d | 2 | S | p-OMe | 1.13 |
| 3e | 3 | O | p-OMe | 1.23 |
| 3f | 3 | O | m-OMe | 4.39 |
| 3g | 3 | S | p-OMe | Nd |
| 3h | 2 | O | m,p-(OMe)2 | 0.69 |
| 3i | 2 | O | [1,3]dioxole | 3.00 |
| 3j | 2 | O | p-OCF3 | Nd |
| 3k | 2 | O | p-COOMe | 4.20 |
| 3l | 2 | O | p-Cl | Nd |
| 3m | 2 | O | p-NO2 | Nd |
| 6a | p-OMe | 1.08 | ||
| 6b | m-OMe | Nd | ||
| 6c | o, p-(OMe)2 | 4.63 | ||
| 7a | p-OMe | 0.63 | ||
| 7b | m-OMe | 6.22 | ||
| 7c | m, p-(OMe)2 | 4.99 | ||
| NGTb | 0.27, 0.29 |
Measured with 4MU-β-GlcNAc as substrate and calculated by linear regression of data in Dixon plots. Values with SD were shown in Table S1, Supporting Information. Nd: not determined (less than 50% inhibition at 50 μM).
Ki against hHex A and hHex B, respectively; Values from ref (16).
The introduction of anisole groups results in an increase in the inhibitory activity when comparing these dyads 3a–e, 3h, and 7a with 1a, suggesting the importance of the methoxyphenoxy group attached to the terminal nitrogen of N-alkylamine naphthalimide by an appropriate linker. In contrast, the absence of methoxyl group dramatically decreases the inhibitory activities of 3i–j and 3l–m. By the activity comparison of 3a–h, 6a–c, and 7a–c, it can be drawn that the molecules with a para-methoxyl group of the benzene ring are more potent. This may suggest that the methoxyl group and the NH of the alkylamine linker must be spaced appropriately in order to effectively bind a corresponding residue. The connecting type of the methoxyphenoxy group and the alkylamine linker also affects the activity. The dyads 3a–d containing the oxygen or sulfur atom between benzene ring and alkane chain are more potent than dyads 6a–c containing a nitrogen atom.
Besides determining the Ki value, Dixon plots (Figure 3) also revealed that these active dyads are competitive inhibitors against human β-N-acetyl-d-hexosaminidase. The trendlines drawn for each concentration of substrate intersect in a single point above the x-axis, indicating competitive inhibition. These results supported that these inhibitors are able to compete with the substrate for binding directly to the catalytic pocket and interact with critical residues locating in the active site.
Figure 3.

Representative Dixon plots (A) for inhibition of human β-N-acetyl-d-hexosaminidases A/B by 7a. The trendlines represent two substrate concentrations (■, 0.1 mM; ●, 0.2 mM). The inset B is the partial enlarged view of A.
Molecular docking has also been performed to analyze the binding model of these unsymmetrical dyads. However, the results can not give perfect explanations to the inhibitory activity difference (Figure S3, Supporting Information).
In summary, we have described the molecular design, synthesis, and inhibitory activities against human β-N-acetyl-d-hexosaminidase of a series of unsymmetrical dyads containing naphthalimide and methoxyphenyl moiety with an alkylamine spacer linkage. The enzymatic activity testing indicated that the potent unsymmetrical dyads exhibit better competitive inhibition activity than the HTS hit symmetrical dyad M-31850 when the human β-N-acetyl-d-hexosaminidase is the target. Especially, dyads 3h and 7a have the highest activities with Ki values of 0.69 and 0.63 μM, respectively. Furthermore, these structures are much different from the reported carbohydrate-based inhibitors and supply a novel and efficient skeleton, which is easier and more straightforward to be synthesized and modified. The understanding of the binding model of these inhibitor potentors against the human β-N-acetyl-d-hexosaminidase may be advantageous for further structure optimization and thereby provides some insight into the rational design of new therapeutic agents for Hex-related diseases.
Acknowledgments
The authors thank the technical guidance for molecular docking provided by Dr. Yongliang Yang at Dalian University of Technology.
Glossary
Abbreviations
- Hex
β-N-acetyl-d-hexosaminidase
- hHex
human β-N-acetyl-d-hexosaminidase
- HTS
high-throughput screening
- 4MU-β-GlcNAc
4-methylumbelliferyl N-acetyl-β-d-glucosaminide
- PDB
protein data bank
Supporting Information Available
Experimental procedures for the synthesis, characterization, and enzymatic assay protocols of compounds 1a–k, 3a–m, 6a–c, and 7a–c as well as molecular docking of selected compounds. This material is available free of charge via the Internet at http://pubs.acs.org.
Author Contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.
This work is supported by National Natural Science Foundation of China (21236002, 31070715, and 31101671), National Special Fund for State Key Laboratory of Bioreactor Engineering (ECUST) (Shanghai, China), National Key Project for Basic Research (2010CB126100), the National High Technology Research and Development Program of China (2011AA10A204), the National Key Technology R&D Program (2011BAE06B05), and the Fundamental Research Funds for the Central Universities (DUT11ZD113 and DUT11RC(3)73).
The authors declare no competing financial interest.
Supplementary Material
References
- Wendeler M.; Sandhoff K. Hexosaminidase assays. Glycoconjugate J. 2009, 26, 945–952. [DOI] [PubMed] [Google Scholar]
- Liu J. J.; Shikhman A. R.; Lotz M. K.; Wong C. H. Hexosaminidase inhibitors as new drug candidates for the therapy of osteoarthritis. Chem. Biol. 2001, 8, 701–711. [DOI] [PubMed] [Google Scholar]
- Shikhman A. R.; Brinson D. C.; Lotz M. Profile of glycosaminoglycan-degrading glycosidases and glycoside sulfatases secreted by human articular chondrocytes in homeostasis and inflammation. Arthritis Rheum. 2000, 43, 1307–1314. [DOI] [PubMed] [Google Scholar]
- Werth N.; Schuette C. G.; Wilkening G.; Lemm T.; Sandhoff K. Degradation of membrane-bound ganglioside GM2 by beta-hexosaminidase A: Stimulation by GM2 activator protein and lysosomal lipids. J. Biol. Chem. 2001, 276, 12685–12690. [DOI] [PubMed] [Google Scholar]
- Gilbert F.; Kucherlapati R.; Creagan R. P.; Murnane M. J.; Darlington G. J.; Ruddle F. H. Tay-Sachs’ and Sandhoff’s diseases: the assignment of genes for hexosaminidase A and B to individual human chromosomes. Proc. Natl. Acad. Sci. U.S.A. 1975, 72, 263–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahuran D. J. Biochemical consequences of mutations causing the GM2 gangliosidoses. Biochim. Biophys. Acta, Mol. Basis Dis. 1999, 1455, 105–138. [DOI] [PubMed] [Google Scholar]
- Tropak M. B.; Mahuran D. Lending a helping hand, screening chemical libraries for compounds that enhance beta-hexosaminidase A activity in GM2 gangliosidosis cells. FEBS J 2007, 274194951–4961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tropak M. B.; Reid S. P.; Guiral M.; Withers S. G.; Mahuran D. Pharmacological enhancement of beta-hexosaminidase activity in fibroblasts from adult Tay-Sachs and Sandhoff patients. J. Biol. Chem. 2004, 2791413478–13487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valenzano K. J.; Khanna R.; Powe A. C.; Boyd R.; Lee G.; Flanagan J. J.; Benjamin E. R. Identification and characterization of pharmacological chaperones to correct enzyme deficiencies in lysosomal storage disorders. Assay Drug Dev. Technol. 2011, 9, 213–235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu T.; Yan J.; Yang Q. Comparative biochemistry of GH3, GH20 and GH84 beta-N-acetyl-d-hexosaminidases and recent progress in selective inhibitor discovery. Curr. Drug Targets 2012, 13, 512–525. [DOI] [PubMed] [Google Scholar]
- Ho C. W.; Popat S. D.; Liu T. W.; Tsai K. C.; Ho M. J.; Chen W. H.; Yang A. S.; Lin C. H. Development of GlcNAc-Inspired Iminocyclitiols as potent and selective N-acetyl-beta-hexosaminidase inhibitors. ACS Chem. Biol. 2010, 5, 489–497. [DOI] [PubMed] [Google Scholar]
- Stubbs K. A.; Macauley M. S.; Vocadlo D. J. A selective inhibitor Gal-PUGNAc of human lysosomal beta-hexosaminidases modulates levels of the ganglioside GM2 in neuroblastoma cells. Angew. Chem., Int. Ed. 2009, 48, 1300–1303. [DOI] [PubMed] [Google Scholar]
- Knapp S.; Vocadlo D.; Gao Z. N.; Kirk B.; Lou J. P.; Withers S. G. NAG-thiazoline, an N-acetyl-beta-hexosaminidase inhibitor that implicates acetamido participation. J. Am. Chem. Soc. 1996, 118, 6804–6805. [Google Scholar]
- Horsch M.; Hoesch L.; Vasella A.; Rast D. M. N-Acetylglucosaminono-1,5-lactone oxime and the corresponding (phenylcarbamoyl)oxime. Novel and potent inhibitors of beta-N-acetylglucosaminidase. Eur. J. Biochem. 1991, 197, 815–818. [DOI] [PubMed] [Google Scholar]
- Dorfmueller H. C.; van Aalten D. M. F. Screening-based discovery of drug-like O-GlcNAcase inhibitor scaffolds. FEBS Lett. 2010, 584, 694–700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tropak M. B.; Blanchard J. E.; Withers S. G.; Brown E. D.; Mahuran D. High-throughput screening for human lysosomal beta-N-acetyl hexosaminidase inhibitors acting as pharmacological chaperones. Chem. Biol. 2007, 14, 153–164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maegawa G. H. B.; Tropak M.; Buttner J.; Stockley T.; Kok F.; Clarke J. T. R.; Mahuran D. J. Pyrimethamine as a potential pharmacological chaperone for late-onset forms of GM2 gangliosidosis. J. Biol. Chem. 2007, 282, 9150–9161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duke R. M.; Veale E. B.; Pfeffer F. M.; Kruger P. E.; Gunnlaugsson T. Colorimetric and fluorescent anion sensors: an overview of recent developments in the use of 1,8-naphthalimide-based chemosensors. Chem. Soc. Rev. 2010, 39, 3936–3953. [DOI] [PubMed] [Google Scholar]
- Ingrassia L.; Lefranc F.; Kiss R.; Mijatovic T. Naphthalimides and azonafides as promising anti-cancer agents. Curr. Med. Chem. 2009, 16, 1192–1213. [DOI] [PubMed] [Google Scholar]
- Brana M. F.; Ramos A. Naphthalimides as anti-cancer agents: synthesis and biological activity. Curr. Med. Chem.: Anti-Cancer Agents 2001, 1, 237–255. [DOI] [PubMed] [Google Scholar]
- Lemieux M. J.; Mark B. L.; Cherney M. M.; Withers S. G.; Mahuran D. J.; James M. N. G. Crystallographic structure of human β-hexosaminidase A: interpretation of Tay-Sachs mutations and loss of GM2 ganglioside hydrolysis. J. Mol. Biol. 2006, 359, 913–929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang H.; Cai Q.; Ma D. Amino acid promoted CuI-catalyzed C–N bond formation between aryl halides and amines or N-containing heterocycles. J. Org. Chem. 2005, 70, 5164–5173. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.