

Abstract
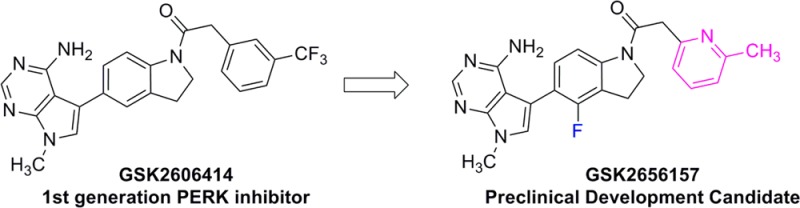
We recently reported the discovery of GSK2606414 (1), a selective first in class inhibitor of protein kinase R (PKR)-like endoplasmic reticulum kinase (PERK), which inhibited PERK activation in cells and demonstrated tumor growth inhibition in a human tumor xenograft in mice. In continuation of our drug discovery program, we applied a strategy to decrease inhibitor lipophilicity as a means to improve physical properties and pharmacokinetics. This report describes our medicinal chemistry optimization culminating in the discovery of the PERK inhibitor GSK2656157 (6), which was selected for advancement to preclinical development.
Keywords: PERK, UPR, kinase, lead optimization, structure−activity relationship, fluorine interaction
Increased endoplasmic reticulum (ER) stress resulting from nutrient deprivation and unfolded protein accumulation is associated with debilitating conditions such as neurodegeneration, heart disease, diabetes, and cancer.1 To maintain ER homeostasis, the unfolded folded protein response (UPR) coordinates an adaptive cellular signaling cascade to alleviate the impact of the stress and enhance cell survival.2 Protein kinase R (PKR)-like ER kinase (PERK) is one of three primary effectors of the UPR.3,4 Once activated, PERK phosphorylates eukaryotic initiation factor 2α (eIF2α) at serine 51, which inhibits the ribosome translation initiation complex and reduces overall protein synthesis.5−7 The reduction in translation reduces the ER burden, providing time for the cell to process or degrade the accumulated unfolded proteins. Although global protein synthesis is decreased, there is also specific increased transcription of certain messages regulated by downstream PERK effectors, such as ATF4, which activate genes that enhance UPR function. For example, in situations of extreme hypoxia or nutrient starvation, UPR activation is associated with increased vascularization. A number of studies using genetic manipulation or siRNA knock down provide evidence that PERK function contributes to transcriptional activation and up-regulation of pro-angiogenic genes.9−14 Therefore, inhibiting PERK in cancer cells may limit their ability to thrive under hypoxia or nutrient deprived conditions and lead to apoptosis or tumor growth inhibition. Recently, we reported the discovery and characterization of GSK2606414 (1), a highly selective, first in class inhibitor of PERK that demonstrated tumor growth inhibition in a human tumor xenograft in mice.15 In this communication, we disclose the medicinal chemistry optimization leading to the discovery of the preclinical development candidate GSK2656157 (6), which was recently reported with extensive biological characterization.16
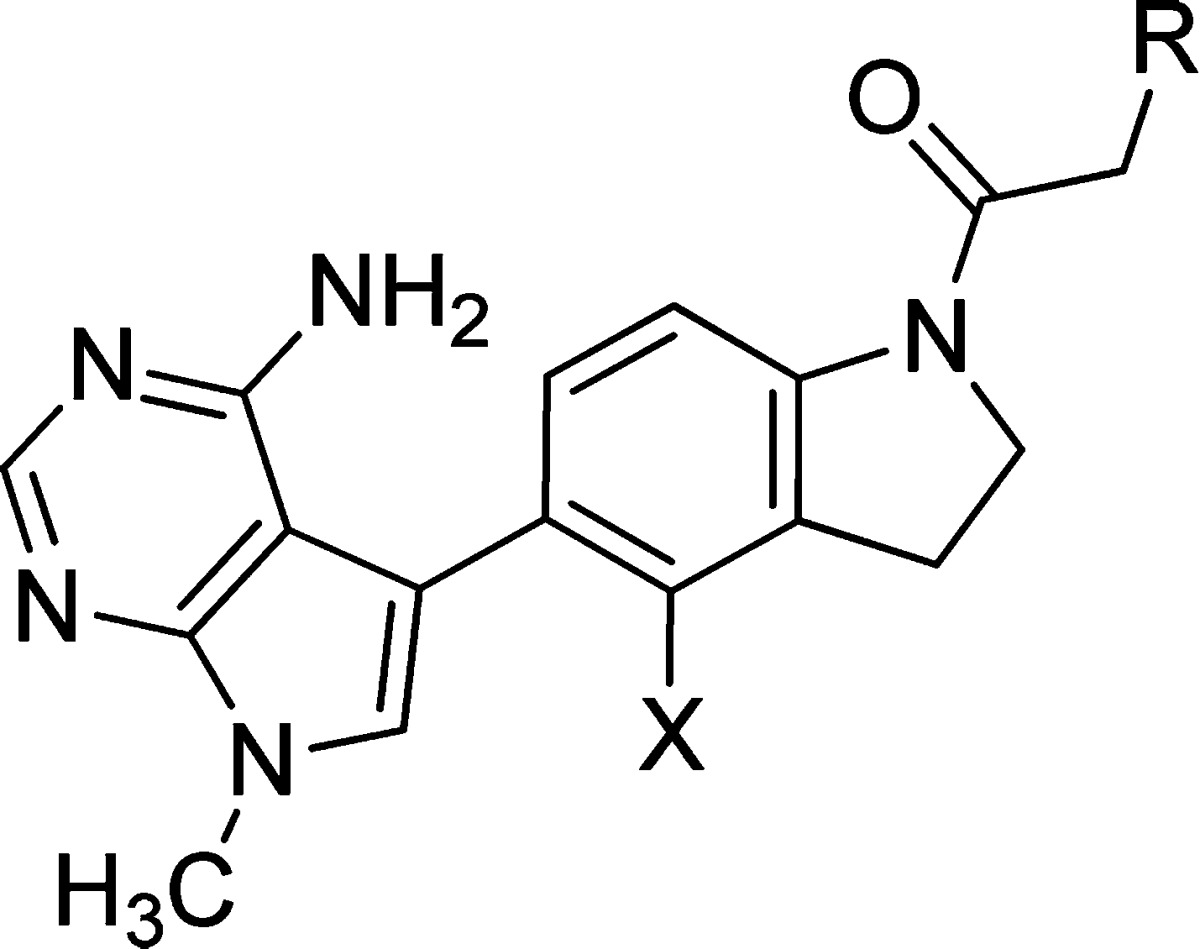
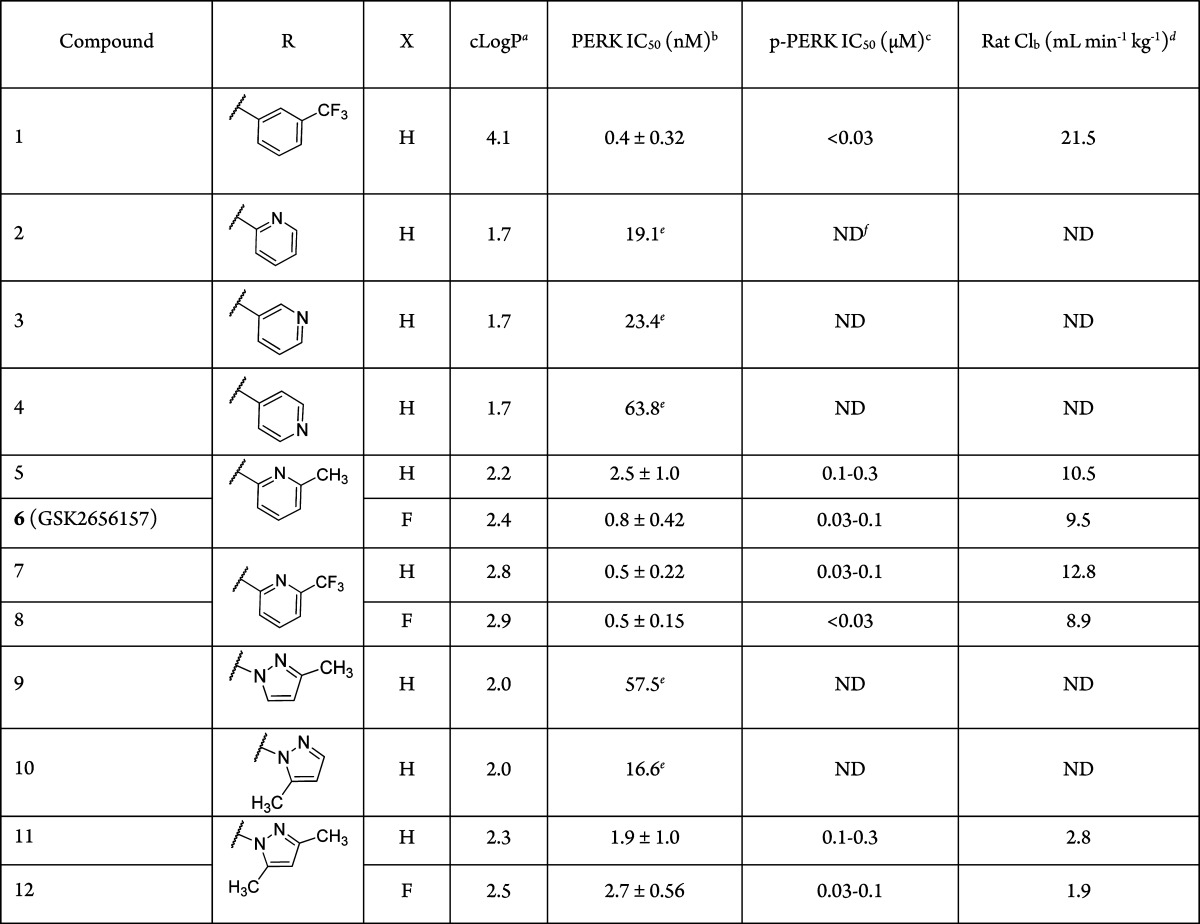
Although compound 1 served as an excellent, orally available tool to elucidate the function of PERK in cells and animals, we sought improvements to the physicochemical properties, metabolism, and pharmacokinetics. Data from in vitro metabolic studies showed that 1 broadly inhibited cytochrome P450s in human liver microsomes, with submicromolar inhibition of CYP2C8 (IC50 = 0.89 μM, Table 2). We suspected that the broad inhibitory activity against cytochrome P450s was related to the overall lipophilicity of the molecules and electronic nature of the arylacetamide based on our previous work relating this part of the molecule to in vivo rat clearance.15 Thus, our optimization strategy to increase polarity focused on modifying the aryl acetamide. We specifically targeted heteroaryl acetamides to minimize molecular weight gain and to evenly distribute polarity throughout the molecules, with an emphasis on the design of analogues with cLogP values <3. In early structure–activity relationship (SAR) investigations, we prepared the pyridyl acetamide isomers 2–4 (Table 1) and observed a significant loss of PERK inhibitory activity compared to the parent phenyl analogue. The 4-pyridyl analogue 4 was the least potent (IC50 = 63.8 nM), consistent with the sensitivity of the aryl acetamide to para-substitution. We selected the 2-pyridyl isomer 2 (IC50 = 19.1 nM) for further investigation, hypothesizing that substitution at the 6-position may improve potency by filling the lipophilic back pocket observed in X-ray structures. Additionally, this substitution could also serve to impede interactions of the pyridine moiety with P450s. The corresponding 6-methylpyridyl analogue 5 (IC50 = 2.5) and trifluoromethylpyridyl analogue 7 (IC50 = 0.5 nM) offered significant (7- to 30-fold) increases in potency. X-ray structural data confirmed the binding mode and the interaction of the pyridyl methyl group with the small pocket adjacent to the pyridine ring (data not shown).
Table 2. Human Liver Microsomal P450 Inhibition Data Reported As IC50 (μM) for Selected PERK Inhibitorsa.
| compound | cLogPb | CYP1A2 phenacetin | CYP2C9 diclofenac | CYP2C19 mephenytoin | CYP2D6 bufuralol | CYP3A4 midazolam | CYP3A4 nifedipine | CYP2C8 paclitaxel |
|---|---|---|---|---|---|---|---|---|
| 1 | 4.1 | 21 | 4.5 | 7.0 | 23c | 13 | 10 | 0.89 |
| 6 | 2.4 | >25 | 24.7 | >25 | >25 | >25 | 19.7 | >25 |
| 8 | 2.9 | >25 | 19.2 | >25 | 23.7 | 9.09 | 7.4 | 5.83 |
| 12 | 2.5 | >25 | >25 | >25 | >25 | >25 | 20.5 | >25 |
CYP450s with molecular probe listed under specific isoform. For assay details, see Supporting Information.
Calculated using BioByte cLogP (the calculated logarithm of the 1-octanol–water partition coefficient of the nonionized molecule; see www.biobyte.com.)
Dextromethorphan was used as the probe in this experiment.
Table 1. PERK Inhibition and Pharmacokinetic Data for Compound 1 and Heteroaryl Acetamide Analogues.
Calculated using BioByte cLogP (the calculated logarithm of the 1-octanol–water partition coefficient of the nonionized molecule; see www.biobyte.com).
Inhibitory activity as measured by cytoplasmic PERK domain phosphorylation of EIF2α. Average values are reported with standard deviation.
IC50 estimated from a Western blot 6-point dose response, except for compounds 1 and 9a (3-point dose response).
Average in vivo blood clearance determined after i.v. infusion in SD rats (n = 2).
Data reported as the average of a single experiment.
Not determined.
During our studies, we also found that 5-membered heteroaryl acetamides were potent PERK inhibitors when appropriately substituted. In a series of pyrazoles, the 3-methyl-pyrazol-1-yl-acetamide 9 (IC50 = 57.5 nM) had decreased activity, yet the 5-methyl-pyrazol-1-yl-acetamide isomer 10 (IC50 = 16.6 nM) preserved a large amount of inhibitory potency. Unlike the parent phenyl acetamides, monosubstitution adjacent to the acetamide linkage in the pyrazole acetamides was favored over the corresponding 3-subtituted analogue. An apparent synergistic effect was observed when both 3- and 5-positions were substituted with methyl groups to give compound 11 (IC50 = 1.9 nM). The increased potency observed with this substitution pattern is speculated to arise from a better overall fit within the volume of the lipophilic acetamide binding pocket.
Compounds 5, 7, and 11 were assayed in A549 cells for their ability to inhibit thapsigargin-induced PERK autophosphorylation (Table 1). Consistent with the biochemical activity of these three analogues, the most potent compound 7 (PERK IC50 = 0.5 nM) demonstrated the most potent inhibition of PERK autophosphorylation in cells (IC50 = 0.03–0.1 μM). Compounds 5 and 11, which were 3 to 4 times weaker than 7 in the PERK biochemical assay, showed similar weaker activity in the pPERK cellular assay (IC50 = 0.1–0.3 μM). Overall, the data indicated that we could in fact greatly decrease the lipophilicity of the PERK inhibitors (cLogP = 2.3–2.8 compared to 4.1 for compound 1) while maintaining biochemical and cellular potency.
To evaluate the impact of structure and physiochemical property changes on the pharmacokinetics of advanced inhibitors, we monitored the in vivo rat blood clearance for the potent and cell active compounds 5, 7, and 11. When compared to 1, each analogue showed improved pharmacokinetic profiles and low blood clearance in rats (Table 1), suggestive of a decrease in metabolic clearance. The rat blood clearance of the pyridyl acetamides 5 and 7 (Clb = 10.5 and 12.8 mL min–1 kg–1, respectively) was about 2-fold lower than that of 1 (Clb = 21.5 mL min–1 kg–1), whereas the pyrazole acetamide 11 exhibited very low rat blood clearance (Clb = 2.8 mL min–1 kg–1). Overall, the data supported our strategy given that analogues with lower cLogP values led to improved blood clearance; however, there was a trade off in PERK biochemical potency resulting in a corresponding decrease in cell activity as measured by PERK autophosphorylation (vide supra).
Fluorination, a well documented and proven strategy to improve compound permeability and pharmacokinetics in drug discovery, was implemented in further optimization of our PERK inhibitors.17 To minimize potential impact on binding affinity, we selected the indoline 4-position for fluorine substitution since it was not likely to have an effect on the biaryl torsion or the critical amide conformation observed in the X-ray structures. During the course of our research we observed a trend in which the majority of 4-fluoroindolines offered small improvements to the PERK activity relative to the indolines. We synthesized 4-fluoroindoline derivatives of the advanced analogues and found that 6-methylpyridyl acetamide 6 had a more than 2-fold improvement in PERK biochemical activity (IC50 = 0.8 nM vs 2.5 nM for 5), whereas the 6-trifluoromethylpyridyl acetamide 8 (IC50 = 0.5 nM vs 0.5 nM for 7) and the 3,5-dimethylpyrazolyl acetamide 12 (IC50 = 2.7 nM vs 1.9 nM for 11) maintained similar activities to their parent compounds. A more pronounced effect of the fluorine substitution was observed when characterizing the intracellular inhibition of PERK. The IC50 for PERK autophosphorylation in A549 cells was uniformly improved 3- to 10-fold for compounds 6, 8, and 12 relative to their parent 4-unsubstituted indolines 5, 7, and 11 (Table 1).
The subtle, general trend of increased biochemical potency was not predicted, but subsequently rationalized from crystal structures of PERK with inhibitors such as 6, which binds in the kinase ATP pocket in an identical fashion to that previously described for 1 (Figure 1a).15 Upon close inspection, the crystal structure revealed that the indoline fluorine atom of 6 is directed toward the methyl groups of valine 606 in the kinase P-loop (Figure 1b). Fluorine can assume diverse roles in protein binding events, most of which are described as polar interactions.18 However, evidence also suggests that it is reasonable for fluorine to induce subtle energetic gains with nonpolar C–H bonds.19 We hypothesize that the fluorine-Val606 interaction is favorable due to the lipophilic nature of the fluorine atom, which is reflected in a general trend of increased biochemical activity we observed for 4-fluoroindoline analogues.
Figure 1.
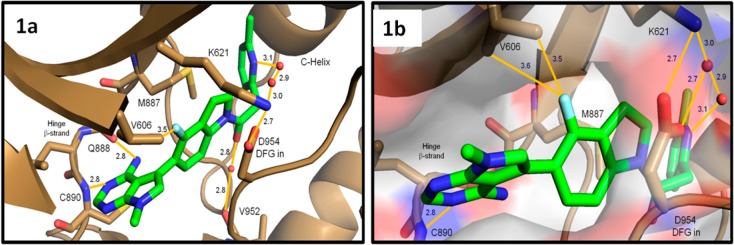
Crystal structure of compound 6 bound to the PERK kinase domain. (a) Close-up view of 6 (green sticks) in the PERK active site (brown cartoon and sticks). The 6-methylpyridyl group occupies the large inner lipophilic pocket, while the fluoroindoline fills a narrow channel between M887 and D954 of the inward facing DFG motif. The amino-pyrimidine binds the hinge β-strand with hydrogen bonds to the backbone amide atoms of Q888 and C890. (b) Close-up view of 6 bound to the PERK active site with surface rendering. Close spatial arrangement between the indoline fluorine atom and the V606 methyl groups may account for some of the improved biochemical activity. Atomic interactions are indicated with orange solid lines and distances reported in Angstroms. The crystal structure of compound 6 bound to the PERK kinase domain is available with the PDB access code 4M7I .
In addition to the increased functional potency in cells, there was also a noticeable modest improvement to in vivo rat blood clearance for each 4-fluoroindoline compound. Concordantly, we characterized cytochrome P450 inhibition in human liver microsome preparations to gauge potential for drug–drug interactions and metabolism associated with these derivatives relative to the tool inhibitor 1 (Table 2). Unlike 1, which broadly inhibited P450s to varying degrees with most potent inhibition observed for CYP2C8 (IC50 = 0.89 μM), the 4-fluoroindoline containing heteroaryl acetamides 6, 8, and 12 generally displayed weaker P450 inhibitory potency. The only exception was 8, which was a slightly more potent inhibitor of the CYP3A4 enzyme than 1. Nevertheless, 8 had an improved overall profile, and compounds 6 and 12 were uniformly less active versus the P450 panel with IC50s for all isoforms >19 μM. The levels of P450 activity of the compounds profiled in Table 2 align with their corresponding lipophilicity indicated by their respective cLogP (Table 1). Taken together, the improvements in cell potency, P450 profile, and rat blood clearance guided our selection of the 4-fluoroindolines 6, 8, and 12 for extensive selectivity screening.
In our initial assessment of selectivity, compounds 6, 8, and 12 were assayed against HRI, PKR, and GCN2, closely related EIF2AK family members that also phosphorylate eIF2α (Table 3). All three compounds showed selectivity for PERK over other members of the EIF2AK family; however, the magnitude of the selectivity varied depending on the identity of the heteroarylacetamide. PERK inhibitor 6 is the least potent inhibitor of other EIF2AK family members and showed 500-fold selectivity over the most sensitive kinase HRI (IC50 = 460 nM). Compound 8 is greater than 700-fold selective over PKR and GCN2, but does have significant activity against HRI (IC50 = 37 nM). The pyrazole acetamide 12 has the poorest selectivity of the three analogues since it potently inhibits both HRI and PKR (IC50s = 37 and 99 nM, respectively) with <50-fold selectivity for PERK. Compounds 6 and 8, the two most selective PERK inhibitors within the EIF2AK family, were profiled against a panel of 300 kinases. In line with the EIF2AK data, 6 continued to demonstrate superior kinase selectivity, inhibiting only 17/300 kinases >80% at 10 μM, whereas compound 8 inhibited 39/300 kinases >80% at the same concentration.
Table 3. Kinase Inhibiton Summary for Compounds 6, 8, and 12.
| compound | EIF2AK1 HRI IC50 (nM)a | HRI/PERK IC50 ratio | EIF2AK2 (PKR) IC50 (nM)a | PKR/PERK IC50 ratio | EIF2AK4 (GCN2) IC50 (nM)b | GCN2/PERK IC50 ratio | X/300 Kinases Inhibited >80% @ 10 μMc |
|---|---|---|---|---|---|---|---|
| 6 | 460 | 511 | 905 | 1,006 | 3,388 | 3,764 | 17 |
| 8 | 37 | 74 | 359 | 718 | 776 | 1,552 | 39 |
| 12 | 61 | 23 | 99 | 37 | ND | ND | ND |
Data from a single 10-point dose response performed by Reaction Biology Corp. (http://www.reactionbiology.com).
Values reported are the average of at least two experiments (see the Experimental Section in the Supporting Information for details).
See Supporting Information for complete kinase profile.
Pharmacokinetic studies were performed in mouse, rat, and dog for compound 6 (Table 4). Data collected from i.v./p.o. crossover studies showed that compound 6 was well absorbed providing good exposure, high oral availability, and low to moderate blood clearance in mouse, rat, and dog. Estimated volume of distribution at steady-state was low in rodents and moderate to high in the dog. Half-lives were less than 2 h in the mouse and rat (t1/2 = 1.25 and 1.4 h, respectively), which influenced the use of twice a day dosing in efficacy studies in mice.
Table 4. Pharmacokinetic Parameters of Compound 6a.
| mousec | ratd | doge | |
|---|---|---|---|
| i.v. dose (mg kg–1) | 2.0 | 2.2 | 2.9 |
| AUC(0–inf) (ng·h mL–1) | 3270 (2817–4085) | 3921.0 (3226.8–4615.2) | 3128.5 (2577.1–3594.8) |
| CLb (mL min–1 kg–1) | 10.5 (8.2–11.8) | 9.5 (8.0–10.9) | 15.5 (13.4–18.2) |
| Vdss (L kg–1) | 0.72 (0.50–0.86) | 0.6 (0.6) | 2.8 (2.8–2.9) |
| t1/2 (h) | 1.25 (1.13–1.36) | 1.4 (1.3–1.5) | 3.1 (2.7–3.6) |
| oral dose (mg kg–1) | 13.4 | 4.3 | 5.2 |
| AUC(0–inf) (ng·h mL–1) | 13378.6 (13110.6–13645.5) | 8015.7 (7174.5–8856.8) | 6210.0 (5274.9–7144.2) |
| oral F(%) | 52b | ∼100 | ∼100 |
Data is reported as a mean, with ranges provided in parentheses.
Bioavailability (F%) was estimated using mean AUC(0–t) values due to the noncrossover study design.
Mouse i.v. (bolus, n = 3), 1% DMSO and 20% Captisol in saline, pH = 4; p.o. (suspension, n = 2), 2% DMSO and 40% PEG 400 in water, pH = 4.0.
Rat i.v. (60 min infusion, n = 2), 1% DMSO and 20% Captisol in saline, pH = 4; oral (solution, n = 2), 1% DMSO and 20% PEG 400 in water, pH = 4.
Dog i.v. (60 min infusion, n = 3), 1% DMSO and 20% Captisol in saline, pH = 6.5; p.o. (solution, n = 3), 1% DMSO and 40% PEG 400 in water, pH = 4.9.
We recently reported the results of extensive biological evaluation of 6 in cell culture and in vivo.16 Chemical stress induced PERK activation was inhibited by 6 in multiple cell lines, with corresponding decreases in eIF2α phosphorylation and downstream transcriptional activation. In efficacy studies, oral treatment with 6 resulted in dose-dependent inhibition of multiple human tumor xenografts growth in mice. The tumor growth inhibition was mechanistically associated with an antiangiogenic affect, which is in agreement with observations reported using genetic means to down-regulate PERK in mouse tumor studies.9,10 In addition, after treatment with 6 for several weeks, we observed a pancreas specific phenotype in mice characteristic of genetic PERK ablation.20−22
Taken together, the data strongly supported that the observed pharmacologic effects upon treatment with 6 were associated with PERK inhibition. The collective biological effects, exquisite kinase selectivity, and pharmacokinetic profile supported the selection of 6 (GSK2656157) for progression to preclinical development.
In summary, our PERK inhibitor-lead optimization was guided by a strategy to decrease analogue lipophilicity while maintaining the potency and exquisite kinase selectivity of tool inhibitor 1. We focused on heteroaryl acetamide analogues to minimize molecular weight gain and designed a series of inhibitors with low to very low in vivo rat blood clearance. Fluorination of the indoline 4-position was important for the recovery of potent biochemical and cell potency, and the optimized 4-fluorindoline analogues 6, 8, and 12 had favorable pharmacokinetics and lower levels of P450 inhibition in human liver microsomes. Expanded profiling established the superior kinase selectivity of 6, which was selected as a preclinical development candidate.
Supporting Information Available
General synthetic scheme and experimental procedures for the synthesis of compounds 2–12, DMPK, and biological assay descriptions, crystallographic methods, and kinase selectivity profile information. This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest.
Supplementary Material
References
- Kim I.; Xu W.; Reed J. C. Cell death and endoplasmic reticulum stress: disease relevance and therapeutic opportunities. Nat. Rev. Drug Discovery 2008, 7, 1013–1030. [DOI] [PubMed] [Google Scholar]
- Walter P.; Ron D. The unfolded protein response: from stress pathway to homeostatic regulation. Science 2011, 34, 1081–1086. [DOI] [PubMed] [Google Scholar]
- Shi Y.; Vattem K. M.; Sood R.; An J.; Liang J.; Stramm L.; Wek R. C. Identification and characterization of pancreatic eukaryotic initiation factor 2 alpha-subunit kinase, PEK, involved in translational control. Mol. Cell. Biol. 1998, 18, 7499–7509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harding H. P.; Zhang Y.; Ron D. Protein translation and folding are coupled by an endoplasmic-reticulum-resident kinase. Nature 1999, 397, 271–274. [DOI] [PubMed] [Google Scholar]
- Sood R.; Porter A. C.; Ma K.; Quilliam L. A.; Wek R. C. Pancreatic eukaryotic initiation factor-2alpha kinase (PEK) homologues in humans, Drosophila melanogaster and Caenorhabditis elegans that mediate translational control in response to endoplasmic reticulum stress. Biochem. J. 2000, 346, 81–93. [PMC free article] [PubMed] [Google Scholar]
- Marciniak S. J.; Garcia-Bonilla L.; Hu J.; Harding H. P.; Ron D. Activation-dependent substrate recruitment by the eukaryotic translation initiation factor 2 kinase PERK. J. Cell Biol. 2006, 172, 201–209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ron D.; Walter P. Signal integration in the endoplasmic reticulum unfolded protein response. Nat. Rev. Mol. Cell Bio 2007, 8, 519–529. [DOI] [PubMed] [Google Scholar]
- Bi M.; Naczki C.; Koritzinsky M.; Fels D.; Blais J.; Hu N.; Harding H.; Novoa I.; Varia M.; Raleigh J.; Scheuner D.; Kaufman R. J.; Bell J.; Ron D.; Wouters B. G.; Koumenis C. ER stress-regulated translation increases tolerance to extreme hypoxia and promotes tumor growth. EMBO J. 2005, 24, 3470–3481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blais J. D.; Addison C. L.; Edge R.; Falls T.; Zhao H.; Wary K.; Koumenis C.; Harding H. P.; Ron D.; Holcik M.; Bell J. C. Perk-dependent translational regulation promotes tumor cell adaptation and angiogenesis in response to hypoxic stress. Mol. Cell. Biol. 2006, 26, 9517–9532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghosh R.; Lipson K. L.; Sargent K. E.; Mercurio A. M.; Hunt J. S.; Ron D.; Urano F. Transcriptional regulation of VEGF-A in the unfolded protein response pathway. PLoS One 2010, 5, e9575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pereira E. R.; Liao N.; Neale G. A.; Hendershot L. M. Transcriptional and post-transcriptional regulation of proangiogenic factors by the unfolded protein response. PloS One 2010, 5, e12521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y.; Alam G. N.; Ning Y.; Visioli F.; Dong Z.; Nör J. E.; Polverini P. J. The unfolded protein response induces the angiogenic switch in human tumor cells through the PERK/ATF4 pathway. Cancer Res. 2012, 72, 5396–5406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Axten J. M.; Medina J. R.; Feng Y.; Shu A.; Romeril S. P.; Grant S. W.; Li W. H. H.; Heerding D. A.; Minthorn E.; Mencken T.; Atkins C.; Liu Q.; Rabindran S.; Kumar R.; Hong X.; Goetz A.; Stanley T.; Taylor J. D.; Sigethy S. D.; Tomberlin G. H.; Hassell A. M.; Kahler K. M.; Shewchuk L. M.; Gampe R. T. Discovery of 7-methyl-5-(1-{[3-(trifluoromethyl)phenyl]acetyl}-2,3-dihydro-1H-indol-5-yl)-7H-pyrrolo[2,3-d]pyrimidin-4-amine (GSK2606414), a potent and selective first-in-class inhibitor of protein kinase R (PKR)-like endoplasmic reticulum kinase (PERK). J. Med. Chem. 2012, 55167193–7207. [DOI] [PubMed] [Google Scholar]
- Atkins C.; Liu Q.; Minthorn E.; Zhang S.; Figueroa D. J.; Moss K.; Stanley T. B.; Sanders B.; Goetz A.; Gaul N.; Choudhry A. E.; Alsaid H.; Jucker B. M.; Axten J. M.; Kumar R. Characterization of a novel PERK kinase inhibitor with anti-tumor and anti-angiogenic activity. Cancer Res. 2013, 73, 1993–2002. [DOI] [PubMed] [Google Scholar]
- Shah P.; Westwell A. D. The role of fluorine in medicinal chemistry. J. Enzyme Inhib. Med. Chem. 2007, 22, 527–540. [DOI] [PubMed] [Google Scholar]
- Müller K.; Faeh C.; Diederich F. Fluorine in pharmaceuticals: looking beyond intuition. Science 2007, 317, 1881–1886. [DOI] [PubMed] [Google Scholar]
- Zhou P.; Zou J.; Tian F.; Shang Z. Fluorine bonding: how does it work in protein–ligand interactions?. J. Chem. Inf. Model. 2009, 49, 2344–2355. [DOI] [PubMed] [Google Scholar]
- Harding H. P.; Zeng H.; Zhang Y.; Jungries R.; Chung P.; Plesken H.; Sabatini D. D.; Ron D. Diabetes mellitus and exocrine pancreatic dysfunction in Perk–/– mice reveals a role for translational control in secretory cell survival. Mol. Cell 2001, 7, 1153–1163. [DOI] [PubMed] [Google Scholar]
- Zhang P.; McGrath B.; Li S.; Frank A.; Zambito F.; Reinert J.; Gannon M.; Ma K.; McNaughton K.; Cavener D. R. Mol. Cell. Biol. 2002, 22, 3864–3874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao Y.; Sartori D. J.; Li C.; Yu Q.; Kushner J. A.; Simon M. C.; Diehl J. A. PERK is required in the adult pancreas and is essential for maintenance of glucose homeostasis. Mol. Cell. Biol. 2012, 32, 5129–5139. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.