

Abstract
Selective inhibitors for the human immunoproteasome LMP7 (β5i) subunit over the constitutive proteasome hold promise for the treatment of autoimmune and inflammatory diseases and hematologic malignancies. Here we report that oxathiazolones inhibit the immunoproteasome β5i with up to 4700-fold selectivity over the constitutive proteasome, are cell permeable, and inhibit proteasomes inside cells.
Keywords: Immunoproteasome, constitutive proteasome, selective inhibitor, autoimmune disease
Cells maintain protein homeostasis in large part through regulated proteolysis by the ubiquitin–proteasome-system (UPS). The UPS plays essential roles in diverse cellular functions, ranging from degradation of cell cycle checkpoint proteins to generation of peptides for antigen presentation in the immune system. UPS functions or malfunctions contribute to the pathogenesis of neoplastic, autoimmune, autoinflammatory, and neurodegenerative disorders.1,2 The impact of bortezomib and carfilzomib in the treatment of certain hematological malignancies demonstrates the therapeutic potential of proteasome inhibitors.
The proteasome is an ATP-dependent, multisubunit, barrel-shaped N-terminal hydrolase present in all eukaryotic cells3 as well as in archaea and a few eubacteria. Inhibition of the mammalian proteasome can induce ER stress and the unfolded protein response, leading to apoptosis, and reduce antigen presentation, damping the immune response.4−6 In addition to the constitutive proteasome (c-20S), the immune system employs a modified proteasome (i-20S), in which the enzymatically active subunits β1, β2, and β5 are replaced by β1i, β2i, and β5i, respectively. The i-20S is constitutively expressed in conventional dendritic cells, plasmacytoid dendritic cells, and lymphocytes and can be induced in other cells by cytokines such as IFN-γ or TNF-α. Compared to the c-20S, the i-20S generates an altered peptide repertoire for antigen presentation,7 although the roles that the i-20S plays in the immune system are debated. The discovery of intermediate proteasomes with mixed β subunits of c-20S and i-20S adds to the complexity of the human proteasome system.8
Bortezomib has been used to treat systemic lupus erythematosus,9 transplant rejection,10 and other immune-related diseases. However, the toxicity of broad-spectrum proteasome inhibitors, which may be acceptable in the treatment of advanced malignancy, may preclude their chronic use for nonmalignant diseases. Several studies demonstrated an improved therapeutic index in mouse models of autoimmune disease by using inhibitors with some selectivity for i-20S over c-20S. For example, the i-20S inhibitor PR-957 showed promise in mouse models of arthritis,11 systemic lupus erthymatosus,12 and inflammatory bowel disease.13 Inhibition of β5i can suppress Th1 and Th17 expansion and enhance regulatory T cell differentiation,13 but exactly how i-20S inhibitors may mitigate autoimmune and inflammatory disease is incompletely understood.
Immunoproteasome inhibitors have also shown therapeutic potential in hematopoietic malignancies. B-lymphoma cell lines expressing a mixed population of c-20S and i-20S are not only susceptible to pan-proteasome inhibitors such as bortezomib and carfilzomib, but also to immunoproteasome-selective inhibitors. For example, PR-924 was found to cause apoptosis in multiple myeloma cell lines and to inhibit grafted multiple myeloma in mice.14
Thus far, several β1i-selective inhibitors and β5i-selective inhibitors have been reported, as illustrated in Figure 1.15 PR-957 belongs to a class of peptide epoxy-ketones that irreversibly inhibit β5i.5 Recently, a specific β5i probe was introduced.16 Another peptide epoxyketone, UK-101, targets β1i,17 as does a peptidyl boronate, ML604440,18 and a peptide aldehyde, IPSI-001.19 Expanding the classes of i-20S-selective inhibitors may increase the chance of success in targeting this enzyme to provide sustained benefit for patients with autoimmune, inflammatory, and malignant diseases. Earlier studies have demonstrated that targeting β5i provided more therapeutic potential than inhibiting the other two active sites of the immunoproteasome in the treatment of autoimmune and inflammatory diseases. Here we describe a new class of β5i-selective inhibitors.
Figure 1.

Structures of proteasome inhibitor bortezomib and representative i-20S β5i or β1i selective inhibitors.
In comparing proteasomes across species, we noticed certain similarities between the Mycobacterium tuberculosis (Mtb) proteasome and the human (hu) immunoproteasome. Both prefer certain P1 amino acids in N-acetyl-tripeptide-AMC substrates and small hydrophobic amino acids in P3. Moreover, the Mtb proteasome and human β5i share a spacious S1 pocket that is larger than that in constitutive proteasomes.20−22 A high throughput screen against the Mtb proteasome led to discovery of a novel class of 1,3,4-oxathiazol-2-ones (Figure 1) that inhibit the Mtb proteasome selectively over hu c-20S.23 Oxathiazolones inhibit the Mtb proteasome via a competitive, irreversible mechanism that results in cyclocarbonylation of the β-OH and α-NH2 of the active site Thr1N of the Mtb proteasome. This is accompanied by a marked conformational change in the loop around the active site. X-ray crystallography revealed that upon cyclocarbonylation of the active site, three hydrogen bonds that stabilize the loop linking the S4 strand and H1 helix of the Mtb20S to the S4 strand are broken and replaced by three different hydrogen bonds linking S4–H1 to S20 and S′7 (Figure 2A,B). Shifting of the loop over the active site was implicated in favoring suicide-substrate inhibition vs hydrolysis of the reaction intermediate.21 Of the 6 pairs of amino acids involved in these hydrogen bonds, only two pairs are conserved in human β2c, one pair in hu β1, β1i, and β2i, and none in hu β5c and β5i (Figure S1, Supporting Information). However, Cys48 in mouse β5i subunit (conserved in hu β5i) may function to stabilize the rearranged conformation, similar to Thr48 in Mtb20S (Figure 2A,C). Thus, shared substrate preferences, S1 pocket architecture, and the partial conservation of Thr48 in Mtb20S and Cys48 in mouse/human β5i suggested that oxathiazolones active against the Mtb proteasome might inhibit hu β5i.
Figure 2.
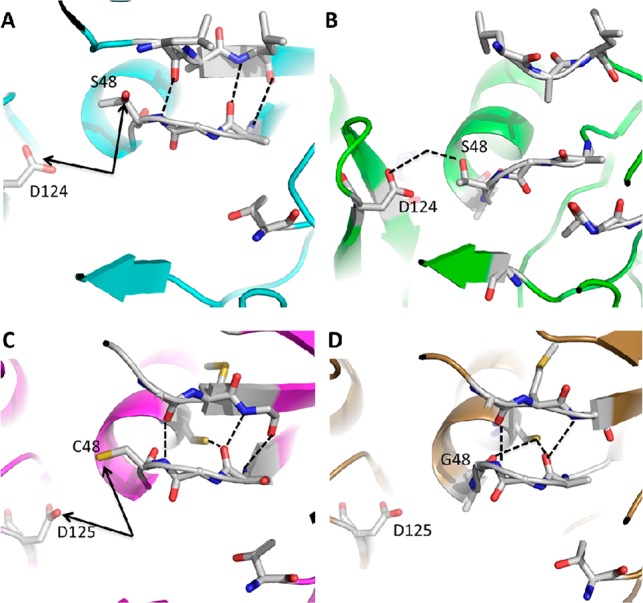
Structure comparison between Mycobacterium tuberculosis proteasome and mouse proteasomes. (A) Mtb 20S: hydrogen bonds that stabilize the S4–H1 loop and S5–H2 loop are shown in black dashed lines. (B) Mtb20S undergoes conformational changes following inhibition by oxathiazolones: four newly formed pairs of hydrogen bonds between three amino acids, three of which are mediated via water. Only the hydrogen bond between S48 and D124 is shown in black dashed line for clarity. (C) Mouse immunoproteasome: three pairs of hydrogen bonds in black dashed lines stabilize the S4–H1 loop and S5–H2 loop. (D) Mouse constitutive proteasome: three pairs of hydrogen bonds in black dashed lines stabilize the S4–H1 loop and S5–H2 loop. Black arrows in panels A and C point to pairs of amino acids (Asp124-Thr48 and Asp125-Cys48) that are partially conserved in Mtb20S and mouse immunoproteasome, which may stabilize the rearranged conformation as shown for Mtb20S in panel B. Images are made from 2FHG, 3H6F, 3UNE, and 3UNH with MacPyMol (DeLano Scientific. Inc.).
To settle this question, we determined the IC50 value for oxathiazolone HT1171 against hu i-20S. Hu i-20S was preincubated with HT1171 at concentrations ranging from 9.8 nM to 100 μM for 15 min prior to addition of substrate suc-LLVY-AMC, resulting in potent inhibition (IC50 = 0.22 μM). We then determined IC50s for 17 oxathiazolones we have synthesized (Table S1, Supporting Information). For their inhibitory activities against β2i and β1i, we measured the percentage of inhibition at 100 μM, and IC50s were determined if the percentage of inhibition was greater than 80%. Since oxathiazolones are irreversible inhibitors, their absolute inhibitory activities are more accurately described by kinact/KI than IC50s. We then determined kinact/KI for oxathiazolones by following the time course of the hydrolysis of suc-LLVY-AMC by hu i-20S in the presence of oxathiazolones at various concentrations. Progress curves indicated that inhibition of hu i-20S by oxathiazolones was time-dependent, similar to the inhibition of Mtb20S (Figure 3). The kobs values of each curve were calculated by fitting to equation I. Plots of kobs against inhibitor concentrations were hyperbolic except for HT1071, HT2210, and HT2109, indicating that the inhibition of hu i-20S by most oxathiazolones tested followed a two-step mechanism: formation of an encounter complex of oxathiazolone and proteasome, followed by an induced conformational change. By fitting the curves to equation II, kinact and KI were estimated for the inhibitors (Table 1). Here kinact denotes the maximal rate of inactivation and KI denotes the concentration at which the rate of inactivation reaches half of the maximum. In contrast to the i-20S results, the plots of kobs for hu c-20S inhibition by oxathiazolones gave linear correlations, indicating a different mechanism of inhibition of c-20S β5c from that of inhibition of i-20S β5i. The values of kinact, KI, and kinact/KI are listed in Table 1. Values of kinact/KI for inhibition of hu c-20S were determined similarly.
Figure 3.

Kinetic analysis of inhibition of hu i-20S and hu c-20S by oxathiazolones. Reaction progress curves of cleavage of Suc-LLVY-AMC by hu i-20S (A) or c-20S (B) in the absence or presence of HT2106 at 0.78–100 μM. The curves were fit to equation I: P = (Vi/kobs) × [1 – exp(−kobst)], to determine the apparent first-order rate constant kobs values. Inset: kobs vs [I] for i-20S in panel A and for c-20S in panel B. (C) Plot of kobs on inhibitor concentrations yielded kinact and KI by fitting to equation II: kobs = kinact/(1 + Ki/[inhibitor]).
Table 1. Inhibition Constants of Oxathiazolones versus hu i-20S β5i and hu c-20S β5c.
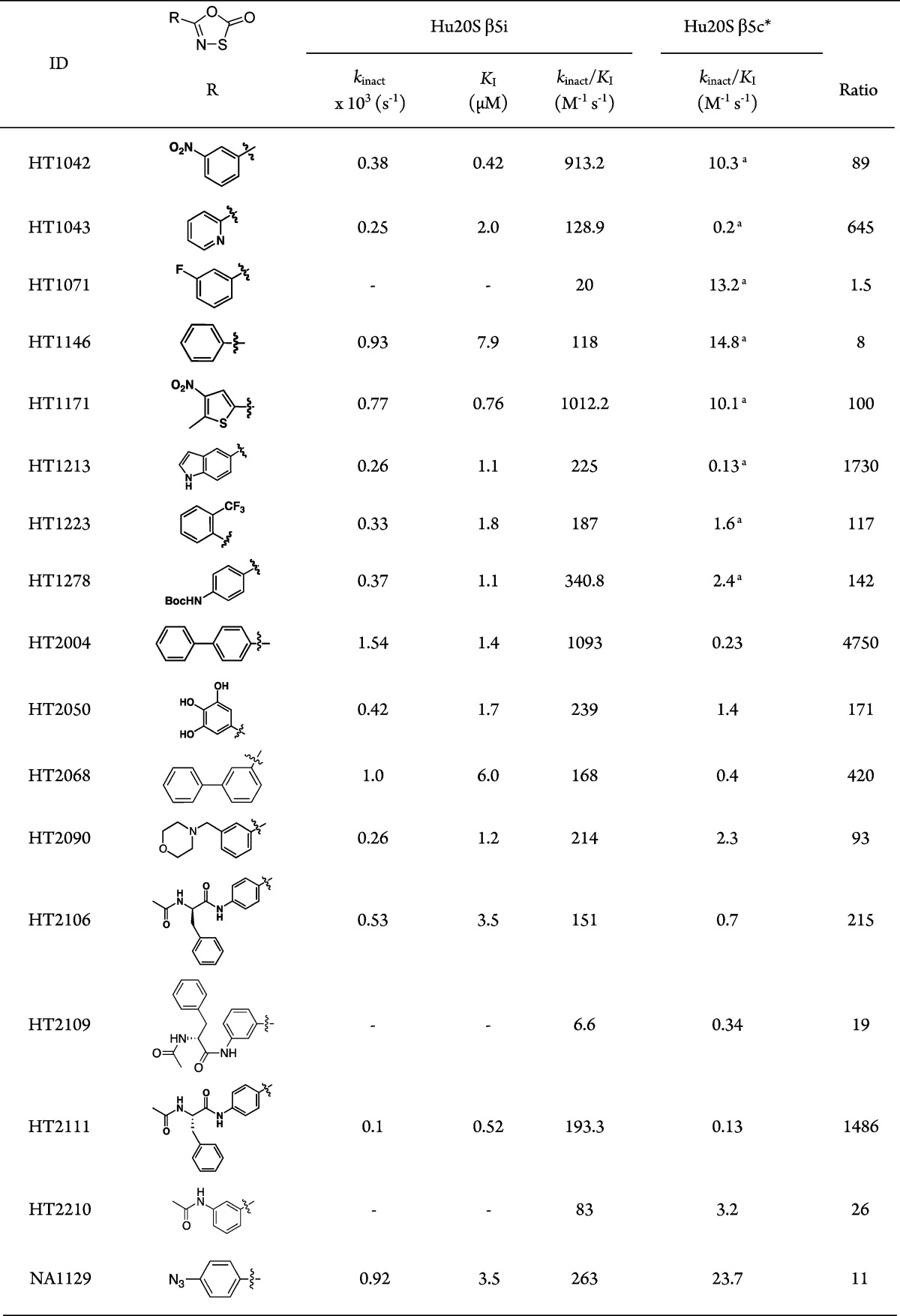
The plots of kobs vs [I] for hu c-20S were linear. Individual kinact and KI cannot be derived; instead, kinact/KI values were derived from the slopes of the plots.
Data from ref (23).
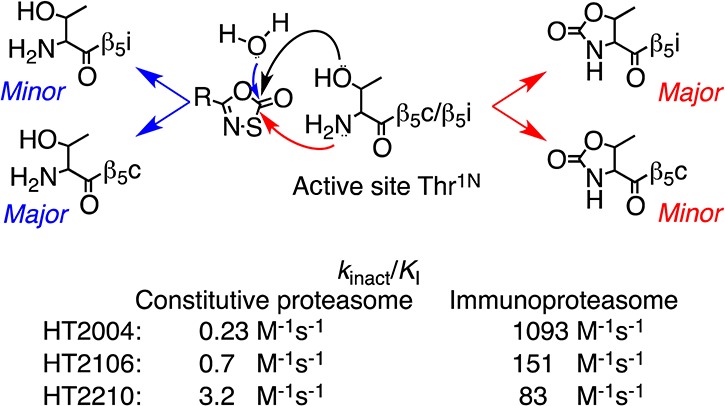
Given the similarity of the inhibition of hu i-20S to that of Mtb 20S by oxathiazolones, we propose the following mechanism of hu i-20S inhibition by oxathiazolones (Figure 4): the hydroxyl group of the active site Thr1N of the i-20S in the encounter complex of i-20S–oxathiazolone attacks the carbonyl group of the oxathiazolones, resulting in formation of a carbonate–enzyme or carbonthioate–enzyme intermediate. The activated NH2 group of the Thr1N then attacks the carbonyl group of the carbonate or carbonthioate, which leads to the cyclic carbonylation of the β-OH and α-NH2 of the Thr1N active site. The carbonate–enzyme or carbonthioate–enzyme intermediate can undergo reactivation via attack on the carbonyl group by water, which resembles the reactivation step from an acyl enzyme intermediate that is formed during the hydrolysis of an oligopeptide by the proteasomes. In the case of the reaction of the c-20S and oxathiazolones, the reactivation step must be much faster than cyclization.
Figure 4.

Proposed mechanism of inactivation of human i-20S by 1,3,4-oxathiazol-2ones.
As the half-life of oxathiazolones in aqueous solution ranges from 7 min to a few hours,23 their therapeutic potential may depend on parenteral administration and rapid access to target cells. We chose a human lymphoma cell line, Karpas 1106p, which constitutively expresses i-20S without stimulation by interferon-γ or TNF-α for biological testing.24 In a preliminary screen, HT2210 and HT2106 were the most active of the oxathiazolones we tested against Karpas cells (unpublished results). Their t1/2s in tissue culture medium were 81.8 and 52.7 min, respectively. To determine if HT2210 and HT2106 were able to inhibit i-20S in the presence of other cytosolic components, we incubated cell free extracts of Karpas 1106p cells with HT2210 or HT2106 at various concentrations. At 1 μM, HT2210 and HT2106 inhibited 60% and 40% of β5i activity, respectively. At 10 μM, HT2210 inhibited >90% of β5i activity, compared to 65% by HT2106. At 100 μM, both oxathiazolones completely inhibited β5i activity. In comparison, bortezomib led to 58% inhibition of β5i activity at 1 nM and 90–100% inhibition at 10 and 100 nM, respectively (Figure 5A). Using a cell-based Proteasome-Glo assay, we determined that the EC50s of HT2210 and HT2106 were 4.2 and 15.4 μM, respectively, for the 26S proteasome in the intact cells, indicating that oxathiazolones are permeable to mammalian cells (Figure 5B). The apparent discrepancy between the EC50s and IC50s likely reflects the mixed population of proteasomes in Karpas 1106p cells. The inhibition also led to accumulation of polyubiquitinylated proteins (Figure 4C), which may result from the inhibition of both proteasomes.
Figure 5.

HT2210 and HT2106 inhibit i-20S β5i activity in cell free extracts and intact cells, leading to accumulation of polyubiquitinated proteins. (A) Inhibition of i-20S β5i activity in Karpas 1106p cell lysates; (B) Inhibition of 26S proteasomal activity in Karpas 1106p cells: ●, HT2210; ○, HT2106. (C) Western blot analysis of polyubiquitinated proteins of Karpas 1106p cells treated with HT2210, HT2106, or bortezomib at indicated concentrations for 4 h at 37 °C. Data were the representative of three independent experiments.
In conclusion, select oxathiazolones are covalent and presumably irreversible inhibitors of human immunoproteasome β5i with a remarkable degree of selectivity for β5i over constitutive β5 active sites. Kinetic studies suggested that oxathiazolones inhibit i-20S β5i through a mechanism-based inactivation. Oxathiazolones can enter human cells and inhibit immunoproteasomes within them. However, at present, the instability of the oxathiazolones in aqueous solution hampers their development as therapeutics in their current form. A systematic effort to improve stability while maintaining potency and selectivity may produce compounds suitable for biological testing. Future structural studies will address the hypothesis that hu β5i, like Mtb 20S, undergoes a conformational change during reaction with oxathiazolones that accounts for its susceptibility to suicide-substrate inhibition and could provide structural rationale for design of β5i selective compounds that can take advantage of the difference at the interface between S4–H1 of β5i and the adjacent β6 subunits.
Acknowledgments
We thank Dr. Christopher Tsu at Millennium Pharma Inc. for the gift of specific β5i substrate Ac-ANW-AMC. We thank Ms. Rong Wang, Ms. Hui Fang, and Dr. George Sukenick at Memorial Sloan Kettering Cancer Center for mass spectrometry assistance.
Glossary
ABBREVIATIONS
- AMC
7-amino-4-methylcoumarin
- c-20S
human constitutive 20S proteasome
- hu
human
- i-20S
human immuno-20S proteasome
- Mtb
Mycobacterium tuberculosis
- UPS
ubiquitin–proteasome system
Supporting Information Available
Reagents, assays, synthesis, Figure S1, and Table S1. This material is available free of charge via the Internet at http://pubs.acs.org.
Supported by NIH 1R21AI101393 (GL), R56AI080618 (CN), Alliance for Lupus Research (255848), and the Milstein Program in Chemical Biology and Translational Medicine. The Department of Microbiology and Immunology is supported by the William Randolph Hearst Foundation.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Bedford L.; Lowe J.; Dick L. R.; Mayer R. J.; Brownell J. E. Ubiquitin-like protein conjugation and the ubiquitin–proteasome system as drug targets. Nat. Rev. Drug Discovery 2011, 10129–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nalepa G.; Rolfe M.; Harper J. W. Drug discovery in the ubiquitin–proteasome system. Nat. Rev. Drug Discovery 2006, 57596–613. [DOI] [PubMed] [Google Scholar]
- Baumeister W.; Walz J.; Zuhl F.; Seemuller E. The proteasome: paradigm of a self-compartmentalizing protease. Cell 1998, 923367–380. [DOI] [PubMed] [Google Scholar]
- Adams J. The proteasome: a suitable antineoplastic target. Nat. Rev. Cancer 2004, 45349–360. [DOI] [PubMed] [Google Scholar]
- Muchamuel T.; Basler M.; Aujay M. A.; Suzuki E.; Kalim K. W.; Lauer C.; Sylvain C.; Ring E. R.; Shields J.; Jiang J.; Shwonek P.; Parlati F.; Demo S. D.; Bennett M. K.; Kirk C. J.; Groettrup M. Erratum: A selective inhibitor of the immunoproteasome subunit LMP7 blocks cytokine production and attenuates progression of experimental arthritis. Nat. Med. 2009, 15111333. [DOI] [PubMed] [Google Scholar]
- Neubert K.; Meister S.; Moser K.; Weisel F.; Maseda D.; Amann K.; Wiethe C.; Winkler T. H.; Kalden J. R.; Manz R. A.; Voll R. E. The proteasome inhibitor bortezomib depletes plasma cells and protects mice with lupus-like disease from nephritis. Nat. Med. 2008, 147748–755. [DOI] [PubMed] [Google Scholar]
- Kincaid E. Z.; Che J. W.; York I.; Escobar H.; Reyes-Vargas E.; Delgado J. C.; Welsh R. M.; Karow M. L.; Murphy A. J.; Valenzuela D. M.; Yancopoulos G. D.; Rock K. L. Mice completely lacking immunoproteasomes show major changes in antigen presentation. Nat. Immunol. 2012, 132129–135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guillaume B.; Stroobant V.; Bousquet-Dubouch M. P.; Colau D.; Chapiro J.; Parmentier N.; Dalet A.; Van den Eynde B. J. Analysis of the processing of seven human tumor antigens by intermediate proteasomes. J. Immunol. 2012, 18973538–3547. [DOI] [PubMed] [Google Scholar]
- Frohlich K.; Holle J. U.; Aries P. M.; Gross W. L.; Moosig F. Successful use of bortezomib in a patient with systemic lupus erythematosus and multiple myeloma. Ann. Rheum. Dis. 2011, 7071344–1345. [DOI] [PubMed] [Google Scholar]
- Raghavan R.; Jeroudi A.; Achkar K.; Gaber A. O.; Patel S. J.; Abdellatif A. Bortezomib in kidney transplantation. J. Transplant. 2010, 2010, 698594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muchamuel T.; Basler M.; Aujay M. A.; Suzuki E.; Kalim K. W.; Lauer C.; Sylvain C.; Ring E. R.; Shields J.; Jiang J.; Shwonek P.; Parlati F.; Demo S. D.; Bennett M. K.; Kirk C. J.; Groettrup M. A selective inhibitor of the immunoproteasome subunit LMP7 blocks cytokine production and attenuates progression of experimental arthritis. Nat. Med. 2009, 157781–787. [DOI] [PubMed] [Google Scholar]
- Ichikawa H. T.; Conley T.; Muchamuel T.; Jiang J.; Lee S.; Owen T.; Barnard J.; Nevarez S.; Goldman B. I.; Kirk C. J.; Looney R. J.; Anolik J. H. Beneficial effect of novel proteasome inhibitors in murine lupus via dual inhibition of type I interferon and autoantibody-secreting cells. Arthritis Rheum. 2012, 642493–503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalim K. W.; Basler M.; Kirk C. J.; Groettrup M. Immunoproteasome subunit LMP7 deficiency and inhibition suppresses Th1 and Th17 but enhances regulatory T cell differentiation. J. Immunol. 2012, 189, 4182–4193. [DOI] [PubMed] [Google Scholar]
- Singh A. V.; Bandi M.; Aujay M. A.; Kirk C. J.; Hark D. E.; Raje N.; Chauhan D.; Anderson K. C. PR-924, a selective inhibitor of the immunoproteasome subunit LMP-7, blocks multiple myeloma cell growth both in vitro and in vivo. Br. J. Hamaetol. 2011, 1522155–163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kisselev A. F.; van der Linden W. A.; Overkleeft H. S. Proteasome inhibitors: an expanding army attacking a unique target. Chem. Biol. 2012, 19199–115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma L. K.; Lee N. R.; Jang E. R.; Lei B.; Zhan C. G.; Lee W.; Kim K. B. Activity-based near-infrared fluorescent probe for LMP7: a chemical proteomics tool for the immunoproteasome in living cells. ChemBioChem 2012, 13131899–1903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ho Y. K.; Bargagna-Mohan P.; Wehenkel M.; Mohan R.; Kim K. B. LMP2-specific inhibitors: Chemical genetic tools for proteasome biology. Chem. Biol. 2007, 144419–430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Basler M.; Lauer C.; Moebius J.; Weber R.; Przybylski M.; Kisselev A. F.; Tsu C.; Groettrup M. Why the structure but not the activity of the immunoproteasome subunit low molecular mass polypeptide 2 rescues antigen presentation. J. Immunol. 2012, 18941868–1877. [DOI] [PubMed] [Google Scholar]
- Kuhn D. J.; Hunsucker S. A.; Chen Q.; Voorhees P. M.; Orlowski M.; Orlowski R. Z. Targeted inhibition of the immunoproteasome is a potent strategy against models of multiple myeloma that overcomes resistance to conventional drugs and nonspecific proteasome inhibitors. Blood 2009, 113194667–4676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin G.; Tsu C.; Dick L.; Zhou X. K.; Nathan C. Distinct specificities of Mycobacterium tuberculosis and mammalian proteasomes for N-acetyl tripeptide substrates. J. Biol. Chem. 2008, 2834934423–34431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huber E. M.; Basler M.; Schwab R.; Heinemeyer W.; Kirk C. J.; Groettrup M.; Groll M. Immuno- and constitutive proteasome crystal structures reveal differences in substrate and inhibitor specificity. Cell 2012, 1484727–738. [DOI] [PubMed] [Google Scholar]
- Lin G.; Chidawanyika T.; Tsu C.; Warrier T.; Vaubourgeix J.; Blackburn C.; Gigstad K.; Sintchak M.; Dick L.; Nathan C. N,C-Capped dipeptides with selectivity for mycobacterial proteasome over human proteasomes: role of S3 and S1 binding pockets. J. Am. Chem. Soc. 2013, 135279968–9971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin G.; Li D.; de Carvalho L. P.; Deng H.; Tao H.; Vogt G.; Wu K.; Schneider J.; Chidawanyika T.; Warren J. D.; Li H.; Nathan C. Inhibitors selective for mycobacterial versus human proteasomes. Nature 2009, 4617264621–626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blackburn C.; Gigstad K. M.; Hales P.; Garcia K.; Jones M.; Bruzzese F. J.; Barrett C.; Liu J. X.; Soucy T. A.; Sappal D. S.; Bump N.; Olhava E. J.; Fleming P.; Dick L. R.; Tsu C.; Sintchak M. D.; Blank J. L. Characterization of a new series of non-covalent proteasome inhibitors with exquisite potency and selectivity for the 20S beta5-subunit. Biochem. J. 2010, 4303461–476. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.