

Abstract
The 2-(2-arylphenyl)benzoxazole moiety has been found to be a new and selective ligand for the enzyme cyclooxygenase-2 (COX-2). The 2-(2-arylphenyl)benzoxazoles 3a–m have been synthesized by Suzuki reaction of 2-(2-bromophenyl)benzoxazole. Further synthetic manipulation of 3f and 3i led to 3o and 3n, respectively. The compounds 3g, 3n, and 3o selectively inhibited COX-2 with selectivity index of 3n much better than that of the COX-2 selective NSAID celecoxib. The in vivo anti-inflammatory potency of 3g and 3n is comparable to that of celecoxib and the nonselective NSAID diclofenac at two different doses, and 3o showed better potency compared to these clinically used NSAIDs.
Keywords: 2-(2-Arylphenyl)benzoxazoles, novel anti-inflammatory scaffold, 3D QSAR, cyclooxygenase-2 selective, in vivo potency
Inflammation is the natural defense mechanism of the body to deal with infection and tissue damage. However, uncontrolled inflammatory cascades is responsible for various diseases such as chronic asthma, rheumatoid and osteo-arthritis, multiple sclerosis, inflammatory bowl diseases and psoriasis,1 diabetic nephropathy,2 tumor initiation, and malignant progression.3 Pain is the most prevalent inflammatory symptom needing medical attention with an estimated amount of 105 million of affected people in the US amounting to the financial burden of US$ 100 billion per annum to the health care expenditure.4 Rheumatoid arthritis (RA) and osteoarthritis (OA) are the sever forms of inflammatory pain with a decade span of the disease shortening the life expectancy.5,6 The major complication of RA is its association with acceleration of cardiovascular diseases (CVDs).7 The nonsteroidal anti-inflammatory drugs (NSAIDs) are used worldwide for therapeutic intervention of pain and inflammation. These exert their anti-inflammatory activity by inhibition of cyclooxygenase-1 (COX-1) and cyclooxygenase-2 (COX-2) enzymes.8 The side effects such as gastric ulcer and gastrointestinal bleeding associated with the traditional NSAIDs (nonselective inhibitors of COX-1 and COX-2) led to the development of selective COX-2 inhibitors as a new class of NSAIDs generally recognized as coxibs.9 The recent withdrawal of COX-2 selective inhibitors rofecoxib and valdecoxib due to adverse cardiovascular side effects10 fuelled a debate about the increased cardiovascular risk associated with existing COX-2 inhibitors11 pressing the need for novel anti-inflammatory scaffold. The additional findings on therapeutic benefit of COX-2 inhibition for new therapeutic areas such as diabetes,2 cancer,3,12−16 and kidney dysfunction17 rejuvenate the medicinal chemists to search for novel COX-2 selective anti-inflammatory agents to address the concern over the safe use of currently approved drugs that are associated with poor tolerability, unfavorable side effects, long-term safety, abuse, and inconvenience of use.18,19 The COX-2 isoenzyme plays a central role in the development of colorectal cancer through acceleration of angiogenesis, increased invasiveness, and antiapoptotic effects.20 The role of COX-2 in tumor growth and/or metastases has been demonstrated through exhaustive preclinical and clinical data.21 The ability of COX-2 inhibitors to prevent colorectal cancer has been demonstrated by several in vitro and in vivo clinical studies.22,23 In view of the mortality associated with many cancers and limited current treatment options using agents such as adriamycin that induces direct cardiovascular damage in several patients, COX-2 remains a very important drug target to treat debilitating diseases such as RA and OA and a preventive measure for colon cancer. However, it is worth mentioning that selective COX-2 inhibitors can also exhibit their anticarcinogenic effects through COX-independent pathway.24−26 Herein we disclose 2-(2-arylphenyl)benzoxazole as a novel scaffold for selective inhibition of COX-2 and discover three new compounds in this structural class as potential new drug candidates for RA/OA with two of them having anti-inflammatory potential comparable to the clinically used diclofenac and celecoxib and the other being better than these therapeutic agents.
Design
Scaffold-hopping27 has been a popular approach to discover structurally novel potent compounds with improved properties. The current therapeutic regime for RA is dominated by the 1,2-diaryl heterocycle28 class of COX-2 selective inhibitors such as celecoxib, rofecoxib, valdecoxib, and etoricoxib commonly termed as coxibs generated through scaffold-hopping by heterocycle replacement of the nonselective inhibitor oxaprozin.29 Thus, the structural modification of nonselective NSAIDs has been a proven strategy to evolve COX-2 selective inhibitors29 as exemplified by transformation of the (i) nonselective flurbiprofen to its COX-2 selective diethoxy derivative, (ii) indomethacin to the corresponding ester/amide and reverse ester/amide for selective inhibition of COX-2, and (iii) diclofenac to the most highly COX-2 selective agent lumiracoxib. Under the present study we derived a topological model (Figure 1) that contains a central aromatic ring with adjacently disposed heteroaryl and aryl rings. The heteroaryl moiety is represented by benzoxazole (in analogy with the central oxazole ring of the prototype oxaprozin), and the other aryl ring provides scope for diversity generation through incorporation of various substituents/functionalities. The new pharmacophore model was rationalized through computational studies (3D QSAR modeling) that indicated it to be a ligand selective for the COX-2 active site with the ligand–receptor binding interaction energy comparable to that of the COX-2 selective NSAID celecoxib and exhibited docking pose in the COX-2 active site similar to that of celecoxib, rofecoxib, and etoricoxib.29
Figure 1.

Topological model for selective COX-2 inhibition through scaffold-hopping.
Synthesis
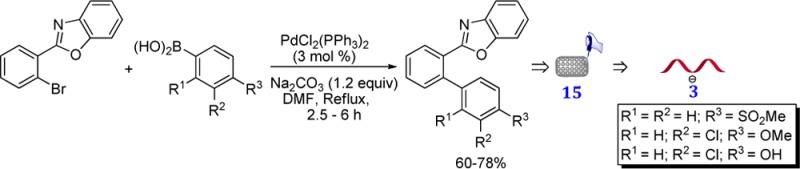
The compounds 3a–m belonging to the scaffold II were synthesized in 60–78% yields by Suzuki cross-coupling reaction of 2-(2-bromophenyl)-benzoxazole 1(30,31) with the arylboronic acids 2a–m bearing various functional groups, e.g., OCH3, SCH3, OC5H9, OiPr, OCH2Ph, CF3, CHO, CN, and Cl (Scheme 1).
Scheme 1. Synthesis of 3a–m.

Reagents and conditions: (a) Pd(PPh3)4 (3 mol %), Na2CO3 (1.2 equiv), DMF, reflux, 2.5–6 h.
Biological evaluation
The compounds 3a–m were subjected to COX-1 and COX-2 enzyme inhibitory assay (Table 1).
Table 1. In Vitro COX-1 and COX-2 Inhibitory Potency of 2-Benzoxazolyl Biarylsa.
| % inhibitionb,c |
|||||
|---|---|---|---|---|---|
| entry | compd | COX-1 | COX-2 | COX-2/COX-1 | SId |
| 1 | 3a | 23.98 ± 1.41 | 10.61 ± 1.41 | 0.44 ± 0.09 | |
| 2 | 3b | 19.08 ± 2.12 | 14.85 ± 2.83 | 0.78 ± 0.24 | |
| 3 | 3c | 58.23 ± 4.24 | 86.62 ± 1.41 | 1.49 ± 0.08 | |
| 4 | 3d | 74.87 ± 3.54 | 59.85 ± 2.12 | 0.80 ± 0.07 | |
| 5 | 3e | 17.13 ± 4.24 | 14.85 ± 3.54 | 0.87 ± 0.01 | |
| 6 | 3f | 4.07 ± 2.12 | 43.45 ± 2.12 | 10.68 ± 6.07 | |
| 7 | 3g | 6.45 ± 1.41 | 81.36 ± 2.83 | 12.61 ± 2.33 | |
| 46.78 ± 0.02e | 0.67 ± 0.04e | 69.82 ± 3.71 | |||
| 8 | 3h | 2.19 ± 0.71 | 33.94 ± 3.54 | 15.50 ± 6.65 | |
| 9 | 3i | 10.28 ± 2.12 | 16.97 ± 2.12 | 1.65 ± 0.55 | |
| 10 | 3j | 1.65 ± 0.55 | 8.46 ± 2.12 | 1.57 ± 0.42 | |
| 11 | 3k | 3.45 ± 1.41 | 11.67 ± 3.54 | 3.38 ± 2.41 | |
| 12 | 3l | 6.58 ± 2.12 | 57.95 ± 2.83 | 8.81 ± 2.43 | |
| 13 | 3m | 8.46 ± 2.12 | 40.85 ± 2.83 | 4.83 ± 1.54 | |
| 14 | 3n | 7.56 ± 4.24 | 94.90 ± 4.24 | 12.55 ± 7.57 | |
| 46.25 ± 0.04e | 0.41 ± 0.02e | 112.80 ± 5.62 | |||
| 15 | 3o | 10.80 ± 2.12 | 75.12 ± 2.12 | 6.96 ± 1.17 | |
| 45.06 ± 0.04e | 1.34 ± 0.05e | 33.62 ± 1.21 | |||
| 16 | 4 | 26.68 ± 2.12 | 100 | 3.75 ± 0.30 | |
| 27.25 ± 0.04e | 0.29 ± 0.02e | 92.87 ± 6.46 | |||
The enzyme inhibition studies were performed using COX inhibitor screening assay kit available commercially (catalog number 560131, Cayman chemical company) as per the manufacturer’s protocol (www.caymanchem.com).
Results are expressed as means of two determinations ± SD, and deviation from the mean is <10% of the mean value.
Calculated at 10 μM concentration of the compound.
The COX-2 selectivity index (SI) [IC50 (COX-1)/IC50 (COX-2)] are expressed as means of two determinations ± SD.
The IC50 values are expressed as means of two determinations ± SD.
While, in general, all of these inhibited the COX enzymes, 3g was the most potent and selective inhibitor of COX-2. The analogous O-benzylated compound 3i that exhibited poor enzyme inhibitory potential on conversion to the hydroxyl derivative 3n (Scheme 2) showed COX-2 inhibitory activity and selectivity exceedingly superior to those of 3i and better than that of 3g. The transformation of 3f to the corresponding methane sulfonate derivative 3o (Scheme 2) drastically increased the COX-2 inhibitory potency and selectivity.
Scheme 2. Synthetic Modification of 3i and 3f to Form 3n and 3o, Respectively.

Reagents and conditions: (a) Pd/C (10% ww/w), H2, 40 psi, EtOH, rt, 4 h, 80%; (b) aq. oxone (3 equiv), 1,4-dioxane, rt, 4 h, 80%.
The in vivo anti-inflammatory activity of 3g, 3n, and 3o was evaluated (Table 2) by carrageenan-induced rat paw edema using Wistar albino rats32 at two different doses (12.5 and 25 mg/kg).
Table 2. In Vivo Anti-Inflammatory Activity of 3g, 3n, 3o, Celecoxib (4), and Diclofenac (5).
| entry | group (mg/kg) | dose (mL)a | rat paw volume | % inhibition |
|---|---|---|---|---|
| 1 | carrageenan (control) | vehicle treated | 0.87 ± 0.20 | |
| 2 | 3g | 12.5 | 0.40 ± 0.04b | 53.85 |
| 3 | 3g | 25 | 0.31 ± 0.06b | 64.42 |
| 4 | 3n | 12.5 | 0.28 ± 0.04b | 68.27 |
| 5 | 3n | 25 | 0.15 ± 0.04b | 82.69 |
| 6 | 3o | 12.5 | 0.32 ± 0.04b | 62.50 |
| 7 | 3o | 25 | 0.22 ± 0.05b | 75.00 |
| 8 | 4 | 12.5 | 0.34 ± 0.04b | 60.57 |
| 9 | 4 | 25 | 0.22 ± 0.05b | 74.61 |
| 10 | 5 | 12.5 | 0.28 ± 0.04b | 67.31 |
| 11 | 5 | 25 | 0.13 ± 0.03b | 84.62 |
Carrageenan induced rat paw edema model, using six animals in each group of Wistar albino rats at 3 h. Results are expressed as mean ± SD and analyzed by two-way ANOVA followed by Tukey’s multiple comparison test.
p < 0.05 vs carrageenan control considered to be statistically significant.
The anti-inflammatory activity of 3g and 3o was found to be comparable to that of the COX-2 selective NSAID 4, and the traditional (nonselective inhibitor COX-1 and COX-2) NSAID diclofenac 5 and 3n proved to be more potent than 4 and 5 as anti-inflammatory agent (Figure 2).
Figure 2.

Comparison of the COX enzyme inhibitory activity/selectivity and anti-inflammatory potency of 3g, 3n, and 3o with those of the clinically used NSAIDs 4 and 5.
The COX-2 selectivity of 3g, 3n, and 3o was rationalized through computational studies (3D QSAR).29 All of these three novel COX-2 inhibitors were found to be very good ligands of the COX-2 (6COX) active site with docking score comparable to that of celecoxib. The compounds 3g, 3n, and 3o exhibited a V-shaped docking pose similar to that of celecoxib, rofecoxib, and etoricoxib in the COX-2 active site and did not exhibit any significant interaction with the COX-1 (3KK6) active site that justified their COX-2 selectivity.
The other derivatives (3b–o) were also docked inside the active site of COX-2 , and all were observed to have similar V-shaped docking poses.29 The in silico (docking) studies support the better activity/selectivity of compounds 3g, 3n, and 3o compared to the other benzoxazole derivatives (3b–o). The docking scores of 3g, 3n, and 3o are higher than those of the remaining benzoxazoles and showed some distinct interactions with the active site of the COX-2 enzyme.29 The compound 3g showed weak interaction of the hydrogen atom of the methoxy group with the carbonyl (C=O) oxygen atom of Gln192. The compound 3n showed hydrogen bonding interaction through the hydrogen atom of the hydroxyl group with the carbonyl (C=O) oxygen atom of Gln192 (O–H···O=C, Gln192, 3.0 Å). The compound 3o showed strong hydrogen bonding interaction through the oxygen atom of SO2Me group with the hydrogen atom of the C=NH of the guanidine moiety of Arg513 (S=O···H–N, Arg512, 2.8 Å). The marketed COX-2 selective drugs, having methyl sulfonyl moieties, e.g., rofecoxib and etoricoxib, were docked into the active site of COX-2, and similar types of interactions were observed. The devoid of the amino acid Arg513 in COX-1 provides the advantage for COX-2 selectivity. However, the corresponding interactions in the cases of 3b–o are weak.
In conclusion, a new pharmacophoric model for selective inhibition of COX-2 has been devised through scaffold-hopping to generate novel compounds for controlling inflammation as potential therapeutic agents for the treatment of RA and OA. The synthesized compounds exhibited COX inhibitory potential, and three of them emerged as selective in vitro COX-2 inhibitors with in vivo anti-inflammatory potency better than that of the standard anti-inflammatory drugs diclofenac and celecoxib. The COX-2 selectivity of the newly generated compounds representing a novel structural scaffold was rationalized through computational studies (3D QSAR) that revealed the various noncovalent interaction with the COX-2 (6COX) active site. The lack of any significant interaction with the COX-1 (3KK6) active site and V-shaped docking pose of these three COX-2 inhibitors as well as of celecoxib, rofecoxib, and etoricoxib established their COX-2 selectivity. Thus, the present work unfolds a new chemical class and next generation selective COX-2 inhibitors as anti-inflammatory agents. In view of the adverse side effect of some of the existing COX-2 selective anti-inflammatory drugs that led to the withdrawal of some of them from the market, the present findings of new anti-inflammatory scaffold should help finding more effective therapeutics for the treatment of RA and OA. With the newer clinical indication of NSAIDs with cancer risk reduction in chronic dialysis patients, this new generation of COX-2 selective inhibitors may also provide newer and alternate cancer therapeutics.
Supporting Information Available
Spectral data and scanned spectra (1H and 13C NMR, HPLC); 3D QSAR. This material is available free of charge via the Internet at http://pubs.acs.org.
Author Present Address
∥ Department of Pharmacology, School of Pharmaceutical Sciences, Shoolini University, Solan 173212, HP, India.
Author Contributions
A.K.C. conceived the project, designed the experiments, analyzed the data, and wrote the paper. K.S., S.K.G., R.K., and P.P. performed the synthesis and enzyme assays. V.S.M. and U.C.B. were associated with the enzyme inhibitory studies. R.G. performed the in vivo experiments.
K.S. and S.K.G. thank CSIR and UGC (New Delhi, India), respectively, for senior research fellowships. Financial support from DST (New Delhi) (SR/S1/OC-33/2008) is also gratefully acknowledged.
The authors declare no competing financial interest.
Supplementary Material
References
- Simmons D. L. What makes a good anti-inflammatory drug target?. Drug Discovery Today 2006, 11, 210–219. [DOI] [PubMed] [Google Scholar]
- Navarro-González J. F.; Mora-Fernández C.; de Fuentas M. M.; García-Pérez J. Inflammatory molecules and pathways in the pathogenesis of diabetic nephropathy. Nat. Rev. Nephrol. 2011, 7, 327–340. [DOI] [PubMed] [Google Scholar]
- Mantovani A.; Allavena P.; Sica A.; Balkwill F. Cancer-related inflammation. Nature 2008, 454, 436–444. [DOI] [PubMed] [Google Scholar]
- Melnikova I. Pain market. Nat. Rev. Drug Discovery 2010, 9, 589–590. [DOI] [PubMed] [Google Scholar]
- Naz S. M.; Symmons D. P. M. Mortality in established rheumatoid arthritis. Best. Pract. Res. Clin. Rheumatol. 2007, 21, 871–883. [DOI] [PubMed] [Google Scholar]
- Gabriel S. E. Why do people with rheumatoid arthritis still die prematurely?. Ann. Rheum. Dis. 2008, 67Suppl. 3iii30–iii34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Summers G. D.; Metsios G. S.; Stavropoulos-Kalinoglou A.; Kitas G. D. Rheumatoid cachexia and cardiovascular disease. Nat. Rev. Rheumatol. 2010, 6, 445–451. [DOI] [PubMed] [Google Scholar]
- Marnett L. J.; Rowlinson S. W.; Goodwin D. C.; Kalgutkar A. S.; Lanzo C. A. Arachidonic acid oxygenation by COX-1 and COX-2. J. Biol. Chem. 1999, 274, 22903–22906. [DOI] [PubMed] [Google Scholar]
- Masferrer J. L.; Zweifel B. S.; Manning P. T.; Hauser S. D.; Leahy K. M.; Smith W. G.; Isakson P. C.; Seibert K. Selevtive inhibition of inducible cyclooxygenase 2 in vivo is anti-inflammatory and nonulcerogenic. Proc. Natl. Acad. Sci. U.S.A. 1994, 91, 3228–3232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dogné J. M.; Supuran C. T.; Pratico D. Adverse cardiovascular effects of the coxibs. J. Med. Chem. 2005, 48, 2251–2257. [DOI] [PubMed] [Google Scholar]
- Cannon C. P.; Cannon P. J. COX-2 inhibitors and cardiovascular risk. Science 2012, 336, 1386–1387. [DOI] [PubMed] [Google Scholar]
- Shono T.; Tofilon P. J.; Bruner J. M.; Owolabi O.; Lang F. F. Cyclooxygenase-2 expression in human gliomas: prognostic significance and molecular correlations. Cancer. Res. 2001, 61, 4375–4381. [PubMed] [Google Scholar]
- Karim A.; Mccarthy K.; Jawahar A.; Smith D.; Willis B.; Nanda A. Differential cyclooxygenas-2 enzyme expression in radiosensitive versus redioresistant glioblastoma multiforme cell lines. Anticancer Res. 2005, 25, 675–679. [PubMed] [Google Scholar]
- Bijnsdorp I. V.; van den Berg J.; Kuipers G. K.; Wedekind L. E.; Slotman B. J.; van Rijn J.; Lafleur M. V. M.; Sminia P. Radiosensitizing potential of the selective cyclooxygenase-2 (COX-2) inhibitor meloxicam on human glioma cells. J. Neurooncol. 2007, 85, 25–31. [DOI] [PubMed] [Google Scholar]
- Kang K. B.; Wang T. T.; Woon C. T.; Cheah E. S.; Moore X. L.; Zhu C.; Wong M. C. Enhancement of glioblastoma radioresponse by a selective COX-2 inhibitor celecoxib: inhibition of tumor angeiogenesis with extensive tumor necrosis. Int. Radiat. Oncol. Biol. Phys. 2007, 67, 888–896. [DOI] [PubMed] [Google Scholar]
- Höcherl K.; Schmidt C.; Bucher M. COX-2 inhibition attenuates endotoxin-induced downregulation of organic anion transporters in the rat renal cortex. Kidney Int. 2009, 75, 373–380. [DOI] [PubMed] [Google Scholar]
- Schneider F.; Meziani F.; Chartier C.; Alt M.; Jaeger A. Fatal allergic vasculitis associated with celecoxib. Lancet 2002, 359, 852–853. [DOI] [PubMed] [Google Scholar]
- Woodcock J. A difficult balance: pain management, drug safety, and the FDA. N. Engl. J. Med. 2009, 361, 2105–2107. [DOI] [PubMed] [Google Scholar]
- Dannenberg A. J.; Altorki N. K.; Boyle J. O.; Dang C.; Howe L. R.; Weksler B. B.; Subbaramaiah K. Cyclooxogenase 2: a pharmacological target for prevention of cancer. Lancet Oncol. 2001, 2, 544–551. [DOI] [PubMed] [Google Scholar]
- Thun M. J.; Henley S. J.; Patrono C. Nonsteroidal anti-inflammatory drugs as anticancer agents: mechanistic, pharmacologic, and clinical issues. J. Natl. Cancer. Ist. 2002, 94, 252–266. [DOI] [PubMed] [Google Scholar]
- Arber N.; DuBois R. N. Nonsteroidal anti-inflammatory drugs and prevention of colorectal cancer. Curr. Gastroenterol. Rep. 1999, 1, 441–448. [DOI] [PubMed] [Google Scholar]
- Gupta R. A.; DuBois R. N. Colorectal cancer prevention and treatment by inhibition of cyclooxygenase-2. Nat. Rev. Cancer. 2001, 1, 11–21. [DOI] [PubMed] [Google Scholar]
- Chakraborti A. K.; Garg S. K.; Kumar R.; Motiwala H. F.; Jadhavar P. S. Progress in COX-2 inhibitors: a jounney so far. Curr. Med. Chem. 2010, 17, 1563–1593. [DOI] [PubMed] [Google Scholar]
- Rigas B.; Kashfi K. Cancer prevention: a new era beyond cyclooxygenase-2. J. Pharmacol. Exp. Ther. 2005, 314, 1–8. [DOI] [PubMed] [Google Scholar]
- Grösch S.; Maier T. J.; Schiffmann S.; Geisslinger G. Cyclooxygenase-2 (COX-2)-independent anticarcinogenic effects of selective COX-2 inhibitors. J. Nat. Cancer. Inst. 2006, 98, 736–747. [DOI] [PubMed] [Google Scholar]
- Kaur J.; Vaish V.; Sanyal S. N. COX-2 as a molecular target of colon cancer chemoprevention: promise and reality. Biomed. Aging Pathol. 2012, 2, 67–72. [Google Scholar]
- Sun H.; Tawa G.; Wallqvist A. Classification of scaffold-hopping approaches. Drug Discovery Today 2012, 17, 310–324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blobaum A. L.; Marnett L. J. Structural and functional basis of cyclooxygenase inhibition. J. Med. Chem. 2007, 50, 1425–1441. [DOI] [PubMed] [Google Scholar]
- Please see the Supporting Information.
- Kumar D.; Rudrawar S.; Chakraborti A. K. One-pot synthesis of 2-substituted benzoxazoles directly from carboxylic acids. Aust. J. Chem. 2008, 61, 881–887. [Google Scholar]
- Kumar R.; Selvam C.; Kaur G.; Chakraborti A. K. Microwave-assisted direct synthesis of 2-substituted benzoxazoles from carboxylic acids under catalyst and solvent free conditions. Synlett 2005, 1401–1404. [Google Scholar]
- Winter C. A.; Risley E. A.; Nuss G. W. Anti-inflammatory and antipyretic activities of indomethacin, 1-(p-chlorobenzoyl)-5-methoxy-2-methylindole-3-acetic acid. J. Pharmacol. Exp. Ther. 1963, 141, 369–376. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.