

Abstract
Objectives
Epidemiological and experimental evidence have indicated potential health benefits of vitamin E supplementation on coronary heart disease (CHD), but several clinical trials have reported no benefit from vitamin E supplementation on CHD. We hypothesized that supplemental intake of vitamin E from an early age may prevent or retard the development and progression of atherosclerosis and CHD mortality.
Methods
To test this hypothesis, 300 Ldlr-/- mice were divided into groups receiving Western style high fat/cholesterol (HFHC), moderate fat/cholesterol (MFMC), or low fat/cholesterol (LFLC) diets all containing 50 IU of vitamin E. These dietary groups were further subdivided into four sub-groups (N=25) receiving their respective diets with no vitamin E supplementation or additionally supplemented with vitamin E (500 IU/kg diet) starting at the early age of 5 wks, or 6 mo, or 12 mo. All mice remained on their assigned diets until age 18 mo. Body weight, health status and survival rate of mice were monitored and recorded. After 18 mo of dietary treatments, mice were sacrificed.
Results
Body weight was the highest in HFHC groups and the lowest in LFLC groups. Plasma concentration of cholesterol and triglycerides was high in all dietary groups, and plasma vitamin E was high in vitamin E supplemented groups. Fifty percent of mice fed Western style HFHC diet and 53% of mice fed MFMC diet survived during the 18 mo, whereas 75% of mice fed LFLC diet survived during the 18 mo dietary treatments. At the age of 18 mo, all the Ldlr-/- mice, regardless of dietary treatments, had several advanced atherosclerotic lesions in both aortic root and aortic tree. Within the LFLC groups, those that received vitamin E supplements from age 5 wks up to 18 mo had a significantly higher survival rate of 88% (p=0.04) and lower mortality (12%) compared to mice that did not receive vitamin E supplements (64%). This lower mortality rate and higher survival rate coincided with significantly (p=0.03) fewer aortic lesions in the vitamin E supplemented LFLC group (50%) compared to LFLC mice that did not receive vitamin E supplements in their diets (65%). Subjective immunohistochemical evaluation of aortic valves showed that LFLC mice that received vitamin E supplements for 18 mo had less intima media thickness compared to LFLC mice that did not receive vitamin E supplements in their diet. The LFLC mice that were supplemented with vitamin E for 18 mo had the lowest mRNA expression of inflammatory markers such as VCAM-1, MCP-1 and CD36 in samples obtained from lesion and non-lesion areas.
Conclusion
In conclusion, 500 mg vitamin E/kg diet in Ldlr-/- mice is not effective at reducing mortality and atherosclerosis when the diet contained high or medium levels of fat and cholesterol. However, a relatively low dose and long-term vitamin E supplementation started from an early age is effective in reducing mortality and atherosclerotic lesions in genetically prone Ldlr-/- mice fed LFLC diet.
Keywords: Vitamin E, atherosclerosis, Ldlr-/- mouse, survival, fatty lesion, long-term supplementation
Introduction
Atherosclerosis is an inflammatory disease of the major arteries and is the major cause of morbidity and mortality from CHD in Western countries [1-3]. Over the past two decades, the health benefits of vitamin E have been examined in numerous studies, many of which have demonstrated a clear reduction in the relative risk of CHD with vitamin E; however, several of the large, multicenter clinical trials have reported no benefit from vitamin E supplementation. The LDL oxidation and oxidative injury hypothesis proposed by Steinberg et al. [4] has been the main stimulus for the investigation of vitamin E and other antioxidants for the prevention of CHD. Evidence from several observational and mechanistic animal and in vitro studies [5-9] have supported the concept that a high dietary intake of vitamin E prevents and reduces the risk of CHD. However, the results of vitamin E intervention in clinical trials of primary and secondary prevention in patients with diagnosed CHD have been equivocal with several of trials not showing a benefit from vitamin E supplementation [10-16]. The discrepancy between the results of observational and mechanistic studies and clinical trials suggests that vitamin E may not be effective once disease is already established whereas it might be effective when a vitamin E deficiency is present [17], or it might be effective in preventing the development of disease when oxidative stress is present [17-19]. For example, a prospective double blind trial of vitamin E supplementation reduced cardiovascular events in individuals with diabetes mellitus and haptaglobulin 2-2 genotype [20, 21]. Since the formation of fatty streak lesions in the vascular bed starts from an early age [22-25] and then matures to full blown atheroma lesions at midor later life causing heart attack, it is plausible that the prevention of fatty streak formation from early life may prevent or retard the development of atherosclerosis and CHD in later life [22]. In fact, the most effective approach for the prevention of CHD in adulthood suggests that the development of disease should be prevented beginning at an early age or even earlier during the fetal stage [22, 25-30], however; most primary prevention studies with vitamin E were conducted in middle-aged subjects [10, 15, 31]. The benefit of lowering fat and cholesterol intake to reduce hypercholesterolemia from early life has been well recognized [32]. In addition, an animal study has suggested the benefit of vitamin E supplementation during pregnancy for the prevention of atherosclerosis in later life [28]. The majority of vitamin E supplementation trials are conducted on middle-aged or older patients who were not supplemented with vitamin E at a younger age or prior to the clinical manifestation of CHD [10, 15, 31]. No control studies have been conducted to date to demonstrate the preventive role of vitamin E supplementation on atherosclerosis at middle-age when supplementation is initiated at an early age. We hypothesized that “supplementation with vitamin E from an early age prevents or retards the development of atherosclerosis and the risk of CHD in individuals who have Western dietary habits (high fat, high cholesterol) or who reduce their intake of fat and cholesterol, as suggested by American Heart Association” [33]. Since long-life vitamin E supplementation studies in humans are rather costly and require a clinical trial of a very long duration, this hypothesis was tested in an animal model of atherosclerosis.
Materials And Methods
Animals and diets
We used male LDL receptor deficient (Ldlr-/-) mice to determine the effect of vitamin E supplementation initiated at early, middle, and older ages and its interaction with high, moderate, and low fat/cholesterol diet on the development of atherosclerotic lesions in the aorta. This mouse model is homozygous for the LdlrtmlHer mutation, and LDL and IDL are the major carriers of blood cholesterol [34]. A chow-fed Ldlr-/- mouse has a lipoprotein-cholesterol distribution similar to that of normolipidemic humans and responds to a high fat/cholesterol diet, which makes it a useful model for studying lipid metabolism and atherosclerosis development as well as for examining a variety of dietary and pharmacological interventions [35]. A total of 300, four-wk-old, male Ldlr-/- mice were purchased from Jackson Laboratory (Bar Harbor, ME). After one wk of acclimation, one group of mice (n=100) was fed with a pelleted semi-purified Western-type, high fat and cholesterol diet (HFHC), formulated to contain 0.1% cholesterol, 20% fat (Dyets, Bethlehem, PA). The composition of this diet is shown in Table 1(supplement), which is adapted from Lichtman et al. [36]. We used AIN-93 mineral and vitamin mixes [37]. The 20% fat in the HFHC diet is composed of 17.7% cocoa butter, a saturated fat without cholesterol, and 2.8% soybean oil. This level of fat provides ∼41% of total calories in this diet. Soybean oil is a polyunsaturated fatty acid (PUFA) and provides essential fatty acids (linoleic acid). The ratio of PUFA to saturated fatty acids (P/S) in this diet was 0.16. Since a lower level of soybean oil (28.5 g) was used in this and other formulations in this study compared to AIN-93 formula (40 g), we reduced the level of vitamin E in the basal diet from 75 IU (the level in AIN-93 formula) to 50 IU. The vitamin mix contained all-rac-α-tocopheryl acetate and provided 50 IU of vitamin E/ kg of diet, of which 1/8 was as RRR-α-tocopherol, the natural form of vitamin E with the highest biological activity [38]. This Western-style diet has been used in this mouse model before and induces hypercholesterolemia as in humans consuming a Western-type diet containing high fat and cholesterol [39, 40].
Another group of mice (n=100) was fed a moderately high fat and cholesterol semi-purified pelleted diet (MFMC). This diet contained 0.05% cholesterol and10% fat providing ∼24% of total calories and composed of 7.8% cocoa butter and 2.5 % soybean oil (Table 1, supplement). The reduced fat calories were substituted isocalorically with cornstarch, a complex carbohydrate. We hypothesized that cutting the cholesterol level in the diet of the MFMC groups by 50% to 0.05%, reducing fat calories from 41% to 24%, and increasing the P/S ratio to 0.32 in this diet would substantially reduce the increase of plasma cholesterol levels and thus attenuate the progression of atherosclerosis in these groups of mice.
Another group of mice (n=100) were fed with a diet containing low fat and low cholesterol semi-purified pelleted diet (LFLC). This diet contained 0.02% cholesterol and basal levels of 4.3% fat with a P/S ratio of 1.25 and provided ∼11% of total calories (Table 1, supplement), the level that is commonly present in regular mouse chow.
Each one of the dietary fat/cholesterol groups was divided into four subgroups (n=25/ subgroup), and their respective diets were supplemented with 500 IU vitamin E (all-rac-α-tocopheryl acetate) either a) started from age 5 wk and receiving a supplemented diet to the age of 18 mo (18E), b) started from age 6 mo and receiving a supplemented diet for 12 mo to the age of 18 mo (12E), c) started from age 12 mo and receiving a supplemented diet for 6 mo to the age of 18 mo (6E), or d) started from age 5 wk to the age of 18 mo and receiving a diet not supplemented with vitamin E (0E). The combination of 3 dietary fat/cholesterol (HFHC, MFMC, LFLC) groups and four categories of vitamin E supplementation (0E, 6E,12E,18E) resulted in a total of 12 different dietary interventions (3×4 factorial design). The inclusion of 25 mice in each dietary regimen was spread out over a period of 5 weeks.
Each mouse was housed singly in a shoebox type polycarbonate cage. Food and water were provided ad libitum. All dietary treatments were continued to the age of 18 mo. Body weight of mice was recorded weekly at early age, then monthly thereafter. Mice were observed daily for general health by the animal caretaker. Moribund animals were killed and samples were collected; animals found dead in their cages were documented.
This study was reviewed and approved by the Animal Care and Use Committee of the Jean Mayer USDA Human Nutrition Research Center on Aging at Tufts University according to the NIH Guidelines for the Care and Use of Laboratory Animals.
Organ isolation and preparation
At the end of a feeding period, mice were anesthetized by Metafane (Pitman-Moore, Washington Crossing, NJ) inhalation. Subsequently, the thoracic cavity was opened, a blood sample was collected, and the aorta was perfused by injecting buffered saline through the left ventricle of the heart.
In 10 mice, this procedure was followed by perfusion with buffered formaldehyde. Since there were advanced atherosclerotic lesions in all groups of deletion mice, the lesions were readily visible under the dissecting microscope even without opening the aortas (Figure 4A). The whole aortic tree was opened longitudinally and pinned down on black wax platforms (Figure 4B and C). The aortic segments were examined en face under a Zeiss dissecting microscope equipped with fiber optic illuminators. Digital images of the aortic surfaces were captured for quantification of lesion surface area using NIH Scion image analysis software. The ratio of the total lesion area was calculated as the percent of total luminal surface of the aortic tree.
Figure 4.
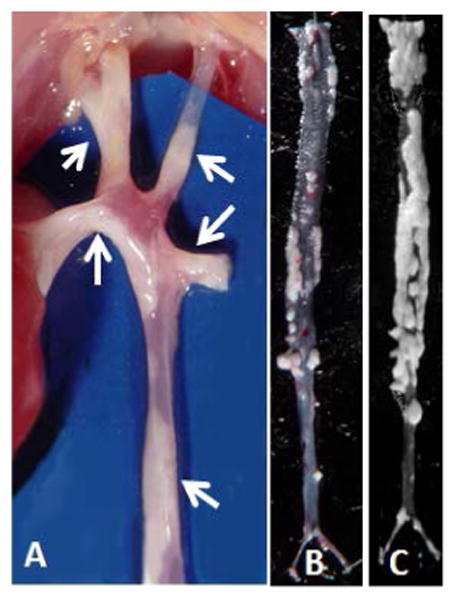
Aortic arc, brachial and carotid branches and descending aorta of an Ldlr-/- mouse fed MFMC diet. Atherosclerotic fatty deposit lesions are observable from serosal side without opening the arteries (A). Representative photomicrographs of en-face atherosclerotic lesions in descending aortic tree of Ldlr-/- mice fed LFLC diet supplemented with vitamin E for 18 mo (B), versus extent of lesions in aortic tree of a mouse fed LFLC diet without vitamin E supplementation (C).
In 6 animals, aortas were perfused with 4 % (wt/vol.) paraformaldehylde solution (Merck, Whitehouse Station, NJ) in PBS (GIBCO, Rockville, MD). This step preserves the endothelial cells. After a brief fixation, the upper portion of the heart including aortic valves, sinuses and aortic arc to the second intercostal region up to the carotids was dissected, removed en bloc, rinsed thoroughly with PBS, quickly embedded in optimal cutting temperature (OCT) compound (Miles Laboratories Inc., Elkhart, IN), and frozen in liquid nitrogen. From 30 to 40 serial sections were cut from each tissue sample using Leica Microsystem CM1850 Cryostat, (Leica, Nussloch Germany) at -28°C. Sections were captured using Fisher brand white Superfrost Plus microscope slides. Samples were air dried for 30 minutes and stored at -80°C until further analysis.
The aortas of 6 animals following perfusion with PBS were excised 1 mm above the aortic valve and at the bifurcation of the descending aorta. After clearing the aorta from the adventitia, the aorta was minced and flash frozen in liquid nitrogen and stored at -80°C for RNA isolation. Due to the small size of aortic arteries, limited amounts of RNA can be obtained from each mouse.
Immunohistochemistry
The 6-μm frozen tissue sections were air-dried for 30 minutes and fixed for 2 minutes in HPLC-UV grade acetone. Slides were washed in 1x phosphate buffered saline (PBS) 3 times for 5 minutes. Endogenous peroxidases were quenched using 3% H2O2. Slides were again washed in PBS 3 times for 5 minutes then incubated with 10% normal goat blocking serum. Slides were incubated overnight at 4°C with monoclonal antibody against CD31 and polyclonal antibodies against α-actin, F4/80, von Willebrand factor (vWF), CD36, and MCP-1, as well as normal rat and normal rabbit control serum. After rinsing off the primary antibodies, the slides were incubated for 30 minutes with the appropriate biotinylated secondary antibodies (goat anti-rat, goat anti-rabbit, 1:50 dilution in PBS). Slides were washed and incubated with avidin-biotin horseradish peroxidase complex (Santa Cruz Biotechnology, Inc.) for 30 minutes. After rinsing, the slides were developed with a diaminobenzidine (DAB) substrate kit (Vector Laboratories, Inc.) and counter-stained with Gill's hematoxylin.
Laser capture microdissection (LCM)
After obtaining sections of aortic sinuses, slides were air-dried for 30 minutes then dehydrated using a sequence of 50%, 95% and 100% ethanol solution, and 100% xylene, and then air-dried again. Lesion and non-lesion portions of the sections were independently microdissected using an Arcturus Pixcell II laser capture microdissection instrument by following the instructions supplied with the instrument. Separate parameters were used for each type of sample removed under arid conditions. For lesion-areas, the following parameters were used: a 15 um spot size, a power setting of 80mW, and 3.0 ms duration. For non-lesion-areas, the parameters set were: a 7.5 um spot size, a power setting of 45mW, and duration of 1.5 ms. Samples were captured on Arcturus Capsure Macro LCM caps and placed into Eppendorf tubes containing 50μL RNA extraction buffer (PicoPure RNA Isolation Kit, Arcturus).
LCM-qPCR
The total RNA from the laser-captured cells was extracted using PicoPure RNA Isolation Kit (Arcturus, Mountain View, CA), following the manufacturer's instructions. Briefly, the cells with 50 μL Extraction Buffer in a 0.5 mL microcentrifuge tube were incubated for 30 minutes at 42°C. Next, the cell extract was mixed with 50 μL of 70% ethanol, and the cell extract and ethanol mixture was then added into the preconditioned purification column. After centrifugation, the column was washed with Wash Buffer. The total RNA was eluted in 11 μL elution buffer. The quantity of total RNA was determined using the NanoDrop ND-1000 spectrophotometer (NanoDrop Technologies, Wilmington, DE). Approximately 200ng total RNA can be extracted from lesion and non-lesion areas of a single mouse aortic section.
Analysis of Gene Expression by Real-Time Quantitative RT-PCR
The mRNA levels of IL-1β, TNF-α, MCP-1/JE, CD36, and VCAM-1 in both the lesion and non-lesion areas of mouse aortic tissue were determined using real-time quantitative RT-PCR as previously reported [41, 42]. Briefly, first strand cDNA synthesis was performed with the Superscript III Reverse Transcriptase (Invitrogen Life Technologies, Carlsbad, CA) by using 100 ng total RNA in 20 μL total volume. Subsequently, real-time quantitative PCR was carried out using ABI Prism 7900HT Sequence Detection System (Applied Biosystems, Foster City, CA) with amplification reactions performed in a final volume of 10 μL containing 5.0 μL of 2x QuantiTect SYBR Green PCR Master Mix (Qiagen, Valencia, CA), 0.6 μM of the primers, and 1μL cDNA solution. Each sample was assayed in triplicate. The primer sequences listed below were designed by using Primer Express version 2.0 for Windows (Applied Biosystems, Foster City, CA). To reduce variability between replicates, PCR premixes, which contained all reagents except cDNA, were prepared and aliquoted into a 384-wall-plate. The glyceraldehyde-3-phosphate dehydrogenase (GAPDH) gene was used as a housekeeping gene to normalize levels of the transcripts.
| Primers for real-time RT-PCR | ||
|
| ||
| Gene | Forward primer | Reverse primer |
|
| ||
| IL-1β | 5′-GCCCATCCTCTGTGACTCAT-3 | 5′-AGGCCACAGGTATTTTGTCG-3′ |
| TNF-α | 5′-CGTCAGCCGATTTGCTATCT-3′ | 5′-CGGACTCCGCAAAGTCTAAG-3′ |
| MCP-1/JE | 5′-AGCAGCAGGTGTCCCAAAGAA-3′ | 5′-CATTTGGTTCCGATCCAGGTT-3′ |
| CD36 | 5′-TGGCCAAGCTATTGCGACAT-3′ | 5′-CAAAGGCATTGGCTGGAAGA-3′ |
| VCAM-1 | 5′-TCCCCTGAATACAAAACGATCG-3′ | 5′-GTCCTCACCTTCGCGTTTA-3′ |
| GAPDH | 5′-TGCAGTGGCAAAGTGGAGAT-3′ | 5′-GTTGAATTTGCCGTGAGTGG-3′ |
|
| ||
Cholesterol and triglyceride analysis
Total cholesterol and triglyceride in blood serum were measured using enzymatic reagents on an Abbott Diagnostics Spectrum Analyzer [43].
Vitamin E analysis
The concentration of vitamin E in plasma was measured by high performance chromatography (HPLC, Waters, Milford, MA) using Tocol as an internal standard and the C18 column according to our earlier procedure [44] Concentration of vitamin E is expressed as μmol/mmol of cholesterol.
Statistical analysis
The Kaplan-Meier survival curves of each group of mice over 74 weeks of dietary fat and vitamin E treatments were plotted to compare each survival curve within each dietary fat intervention group (LFLC, MFMC, HFHC), which is affected by the duration of vitamin E supplementation (Time). Statistical analysis was carried out by the Lifetest Procedure using SAS statistical program. Data were tested for homogeneity of survival curves, and Kaplan-Meier curves were plotted with 4 strata for duration of vitamin E supplementation (Time: 0, 6, 12, and 18 mo) (see Figure 3). The general linear model procedure was used to carry out multiple comparisons for the percent of atherosclerotic lesions present in the descending aorta within each dietary fat group as affected by duration of vitamin E supplementation. We performed a 2-way analysis of variances for the effect of dietary fat/cholesterol and the duration of vitamin E supplementation on serum triglyceride, cholesterol, and vitamin E levels. The arithmetic means of data were calculated and expressed as mean ± SEM and p<0.05 was considered to be significant.
Figure 3.
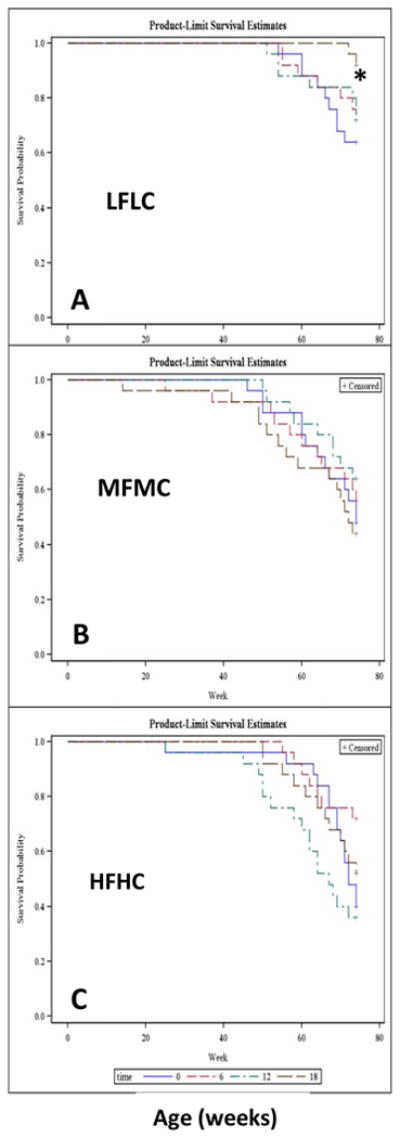
Kaplan-Meier survival curves. Effect of starting age and duration of vitamin E supplementation on survival probability of Ldlr-/- mice over 18 mo period. 25 Ldl-/- mice were assigned to LFLC (panel A), MFMC (panel B) and HFHC (Panel C) and their respective diets were either not supplemented with vitamin E (blue line) or supplemented with 500 IU/kg diet from the age of 12 mo to the age of 18 mo (6 mo vitamin E supplementation, red line), or from the age of 6 mo to the age of 18 mo (12 mo vitamin E supplementation, green line), or from the age of 5wk to the age of 18 mo (18 mo vitamin E supplementation, black line). Note that when LFLC mice received vitamin E supplement in the diet from the early age of 5-wk, they significantly survived better (* p=0.03) than other dietary MFMC and HFHC groups who also received vitamin E supplement for 18 mo (panel A compared to panels B and C).
Results
Body Weight
The body weight of Ldlr-/- mice at the age of 5 wks (start of study) was between 17-18 g. The body weight of mice fed the HFHC diet reached a maximum body weight of 50 g at 14 mo, and thereafter declined slightly until age 18 mo (Figure 1). The body weight of mice fed the MFMC diet reached a maximum of 45 g by age 15 mo and then declined slightly (Figure 1). The body weight of mice fed the LFLC diet reached a maximum of 42 g at 17 mo without subsequent decline. Thus, consumption of the HFHC diet containing 20% fat by weight resulted in a higher body weight gain than the mice fed the LFLC diet (p<0.05).
Figure 1.

Effect of high fat and cholesterol (HFHC), medium fat and cholesterol (MFMC), and low fat and cholesterol (LFLC) diets without and with vitamin E supplementation (500 IU/kg) of diets on body weights gain of Ldlrl-/- mice over 18 months.
Plasma triglyceride, cholesterol and tocopherol
The total cholesterol and triglyceride plasma levels at the end of the 18 mo dietary treatments were relatively high in all groups. Two-way ANOVA showed a significant effect of duration of vitamin E supplementation (Time effect, p=0.057) and its significant interaction with dietary fat/cholesterol (p=0.003) on plasma cholesterol levels. Post-hoc analysis showed significant differences in plasma cholesterol levels in HFHC groups supplemented with vitamin E compared to LFLC groups (p=0.05). Only plasma cholesterol levels of mice fed MFMC and supplemented with vitamin E for 18 mo had significantly higher total cholesterol levels compared to the LFLC groups (Table 2). There was also a significant effect of Time with vitamin E supplementation (p=0.018) and dietary fat/cholesterol (p=0.039) on plasma triglyceride levels, but there was no significant interaction with Time and dietary fat/cholesterol on plasma triglyceride levels. However, post-hoc analysis showed significant differences in plasma triglycerides of HFHC-fed mice compared to those of LFLC-fed mice. There was also a lower level of triglycerides in the plasma of MFMC-fed mice who did not receive vitamin E supplements compared to LFLC mice (Table 2). The mice receiving vitamin E supplements for 6, 12 or 18 mo had significantly (p<0.05) higher plasma levels of tocopherol (expressed per total cholesterol levels) compared to those mice that did not receive vitamin E supplements in their diet (Figure 2).
Table 2.
Plasma cholesterol and triglyceride concentration at the end of 18 months dietary interventions.
| LFLC | MFMC | HFHC | ||||
|---|---|---|---|---|---|---|
|
|
|
|
||||
| Chol mg/dl | TG mg/dl | Chol mg/dl | TG mg/dl | Chol mg/dl | TG mg/dl | |
|
|
|
|
|
|
|
|
| No E suppl. | 568 ± 59 | 208 ± 23 | 670 ± 104 | 176 ± 25† | 794 ± 77* | 245 ± 37 |
| 6mo E suppl. | 875 ± 82‡ | 257 ± 35 | 606 ± 60 | 156 ± 26 | 839 ± 106 | 246 ± 28† |
| 12mo E suppl. | 994 ± 61 | 304 ± 30 | 731 ± 78 | 259 ± 32 | 845 ± 84 | 236 ± 37† |
| 18mo E suppl | 536 ± 67# | 175 ± 28 | 877 ± 76* | 244 ± 19 | 886 ±130* | 326 ± 54† |
Data are mean ± SEM. N= 8-18. ANOV: Significant (p=0.057) effect of duration of vitamin E supplementation (Time), and Time X dietary Fat/Cholesterol interaction (p= 0.003) on plasma cholesterol levels. Significant effect of Time (p=0.018) and dietary Fat/Cholesterol (p=0.039) on plasma triglyceride levels, but no significant interaction. Post-hoc analysis:
Significantly different from LFLC.
Significantly different from LFLC.
Significantly different from No E supplement.
Significantly different from 12mo E supplment.
Figure 2.

Plasma concentration of vitamin E (μmol of α-tocopherol/mmol of cholesterol) in groups of Ldlr-/- mice consumed three different diets containing LFLC, MFMC, and HFHC either without vitamin E supplement or with 500 IU vitamin E/kg diet starting from the early age of 5wk or 6 mo and 12 mo to the age of 18 mo. Data are mean ± SE, n= 10-15, * p<0.05 compared to vitamin E supplemented groups.
Survival rate
Survival data over the 18 mo period of dietary intervention were plotted as a Kaplan-Meier survival curve (Figure 3). Regardless of vitamin E supplementation, mice treated with HFHC and MFMC diets overall had a lower survival rate (52% and 53%) compared to a 75% survival rate in mice fed the LFLC diet (Figure 3, panel A vs panels B and C). The group of mice fed the LFLC diet supplemented with vitamin E from age 5 wks to the age of 18 mo lost only 2 mice from the 25 mice assigned to this group (Figure 3, panel A). The 92% survival rate for this group was significantly (p=0.009) higher than the 64% survival rate of the LFLC-fed mice who did not receive vitamin E supplements in their diet (Figures 3, panel A, black curve vs. blue curve). Further, this high survival rate of 92% was also significantly (p=0.0006) higher than the 52% survival rate of the mice fed HFHC, and it was higher than 44% survival rate of mice fed MFMC and supplemented with vitamin E for 18 mo from the early age of 5 wks (Figure 3, black curves in panel A vs. black curves in panels B and C).
Where mice began receiving vitamin E supplementation from the age of 6 mo to18 mo (i.e., 12 mo supplementation), the LFLC-fed mice had a significantly (p=0.006) better survival rate (72%) than the mice fed HFHC diet (40%) or MFMC diet (56%) (Figure 3, green line in panel A vs.green lines in panels B and C). There were no significant survival differences between the groups who received vitamin E supplements from12 mo age, i.e., supplemented for only 6 mo, as denoted by red lines. Overall, vitamin E supplementation was not effective in reducing mortality in the groups of mice fed MFMC or HFHC diets. It was mainly effective in the survival of Ldlr-/- mice fed the LFLC diet.
Atherosclerotic lesions
Regardless of dietary treatments, Ldlr-/- mice, which are genetically prone to atherosclerosis, had several advanced atherosclerotic lesions in the lumen of the descending aorta at age 18 mo. Further, fatty lesions were observable without opening and exposing the luminal side of descending aorta (Figures 4A), and they were clearly observable without staining during en-face examination on black wax background (Figures 4B&C). The extent of atherosclerotic lesions was calculated as a percent of the lumen surface area of the descending aorta covered by the lesion(s). The percent of the lesion area was different between and within the HFHC- and MFMC-fed mice (Figure 5). However, within the groups of mice fed the LFLC diet, the extent of atherosclerotic lesions in the descending aorta was significantly lower in the groups of mice who received vitamin E supplements in their diet from age 5 wk to age 18 mo compared to those who did not receive vitamin E supplements in their diet (50% vs. 65% aortic lesions, p=0.03), or compared to those who received vitamin E supplements for only 6 mo. i.e., from middle age of 12 mo (50% vs 66%, p=0.02). Vitamin E supplementation for 12 mo (started from the age 6 mo to the age of 18 mo) in LFLC-fed mice was also effective at reducing lesion development compared to those who did not receive vitamin E or those received vitamin E for only 6 mo (50% vs,65%, p=0.03, and vs. 66%, p=0.05, respectively).
Figure 5.

Effect of type of diets (LFLC, MFMC, HFHC), starting age and duration of vitamin E (500 IU/kg) supplementation on percent of aortic atherosclerotic lesions development in Ldl-/-mice over 18 mo. Diet not supplemented with vitamin E (0), or supplemented for 6 mo from 12 to the age of 18 mo (6), or supplemented for 12 mo from 6 mo age to the age of 18 mo (12), or supplemented for 18 mo from the age of 5 wk to the age of 18 mo (18). N= 10-22. Data are mean ± SE, *p< 0.05 compared to 0 and 6 mo supplementation.
Since we found that vitamin E's effects on survival rates and aortic lesions was only observable in LFLC-fed mice, we therefore further examined the atherosclerotic lesions in cross sections of frozen samples of the aortic sinuses from the LFLC-fed mice who received vitamin E supplement for 18 mo and the mice who did not received any vitamin E. We used specific antibodies for endothelial cells, macrophages, smooth muscle cells (SMC), and macrophage scavenger receptors. In our subjective evaluation, the vitamin E-supplemented animals appeared overall to have relatively less thickening of intima/media than the un-supplemented mice (Figure 6, 4X, see supplement). Immunohistochemistry of aortic sinus sections from LFLC mice revealed the presence of large numbers of lipid-laden macrophages detected by CD31 (Figure 6a) antibody. There was also a strong expression of both F4/80, a trans-membrane protein present on the surface of macrophages (Figure 6b), along with strong expression of CD36, a scavenger receptor on macrophages (Figure 6c), as well as the presence of several vascular smooth muscle cells as indicated by α-actin (Figure 6d). The presence of the von Willebrand factor (Figure 6e) and diffused expression of MCP-1 (Figure 6f) in the aortic sinuses indicated a high state of inflammation in LFLC-fed mice regardless of their vitamin E status.
LCM, qRT-PCR
To determine the extent of vitamin E supplementation on the recruitment of macrophages and the expression of inflammatory mediators, we first collected whole descending aorta samples from 6 mice in each dietary group. Tissue samples taken from 2 mice from each group were pooled and homogenized. RNA was extracted and qRT-PCR was performed. Since aortic samples contained both non-lesion and lesion tissues, the analysis showed no noticeable differences between the vitamin E supplemented and non-supplemented groups on mRNA expression of IL-1β, TNF-α, MCP-1/JE, CD36, and VCAM-1 genes (data not shown). We assumed that non-lesion healthy tissue might have diluted the changes in gene expression and masked the vitamin E effects. Therefore, we used laser capture micro-dissecting (LCM) techniques to obtain samples from atherosclerotic lesion areas and non-lesion areas from 6 μm frozen sections of aortic sinuses for gene expression of TNF-a, MCP-1/JE, CD36, and VCAM-1 by qRT-PCR.
The mRNA expression of inflammatory markers VCAM-1, MCP-1, and CD36 in samples obtained from lesion and non-lesion areas of aortic valves are presented in Figure 7, panels A and B. As shown, there are distinct patterns of increased mRNA expression of inflammatory markers in non-lesion areas as affected by the time and duration of vitamin E supplementation. For example, the mRNA expression of all three inflammatory markers was highest in the samples obtained from mice fed a diet supplemented with vitamin E for only 6 mo starting at the late age of 12 mo (Figure 7 A). This increase was alleviated by increased duration of vitamin E supplementation so that after 18 mo vitamin E intake, CD36 expression was not detectable and mRNA expression of VCAM-1 and MCP-1 was lowest. In contrast, mRNA expression of these inflammatory markers in the aortic samples obtained from lesion areas showed different patterns (Figure 7B). With increased duration of vitamin E supplementation, a clear reduction of mRNA expression of VCAM-1 and MCP-1 was apparent in the lesion area. However, mRNA expression of CD36 followed the pattern observed in the non-lesion area. Since we did not notice any defined trends or patterns of mRNA expression of IL-1β or TNFα in lesion and non-lesion areas, these data are not presented.
Figure 7.

mRNA expression of VCAM-1, MCP-1/JE and CD36 in the non-lesion (panel A) and lesion areas (panel B) of aortic valves of LFLC-fed mice not supplemented or supplemented with 500 IU vitamin E /Kg diet for different periods of time. Data are mean ± SE, n=5.
Discussion
Most of the clinical intervention studies on vitamin E's effects on coronary heart disease (CHD) have been conducted in middle-aged or older patients who were not supplemented with vitamin E at a younger age or prior to the clinical manifestation of CHD [10-12, 14, 15, 31] No control studies have been conducted to date on vitamin E supplementation initiated at an early age in order to demonstrate the preventive role of vitamin E supplementation on atherosclerosis developed during old age. We hypothesized that “supplementation with vitamin E from an early age prevents or retards the development of atherosclerosis and the risk of CHD in individuals with either Western dietary habits (high fat, high cholesterol) or a reduced fat and cholesterol intake as suggested by American Heart Association” [33]. Since life-long vitamin E intervention studies in humans require a clinical trial of very long duration, we tested our hypothesis in the Ldlr-/- mouse model, which has been shown to exhibit accelerated atherosclerosis when fed a high fat and cholesterol diet [35]. Using this and other mouse models, the effectiveness of dietary antioxidants on prevention of atherosclerosis has been tested. Short term supplementation of a high fat diet in 5 wk old Ldlr-/- mice with a relatively high dose of a dietary antioxidant mixture including vitamin E (1,000 mg/kg), β-carotene (5000mg/kg) and vitamin C (500 mg/Kg) for 8 wks has been shown to reduce fatty streak lesion in aortic sinuses [45]. A high dose of 2000 IU vitamin E/ kg diet for 3 months was also effective at reducing the progression of atheroma lesions in high fat-fed Ldlr-/- mice [7]. Vitamin E at a very high dose of 5,000 mg/kg diet was also effective at increasing the survival of CD-1/UCadiz male mice [46]. However, long-term supplementation with a low dose of vitamin E from the early age of 4-5 wks to adulthood and older age has not been investigated.
Due to the lack of an LDL receptor in Ldlr-/- mice, they carry high levels of cholesterol and triglycerides in circulation, and thus they develop atherosclerosis. In our study, those mice receiving a low fat and low cholesterol diet (LFLC group) had 568±166 mg /dL cholesterol, which is three times more than the levels found in C57BL/J6 mice (157±7 mg/dL), the background mice strain to Ldlr-/- mice [47, 48]. Thus, our Ldlr-/- mice were already under lipid stress, which was then compounded by the additional stress imposed on them by a HFHC or MFMC diet. This lipid stress manifested itself as advanced atheroma lesions in the aortic tree with significant amounts of lipid deposits (Figure 4). We found that their survival rate up to the age of 18 mo was about 50% when they were fed Western style diet (HFHC); however, when the fat and cholesterol levels in the diet were reduced by 50% (MFMC), the survival rate was only 47%. These results indicated that cutting the fat content by half in the diet of Ldlr-/- mice is not enough to prevent the mortality that is potentially associated with atherosclerosis. By age 18 mo, both the HFHC and MFMC mice had developed well-advanced lesions in the aortic sinuses, aortic arc, descending aorta, and in all branching points of the major arteries. However, the survival rate was relatively high when Ldlr-/- mice were fed a diet low in fat and cholesterol (LFLC). Of great interest, the survival rate in these groups of mice was much higher when the LFLC diet of mice was supplemented with 500 mg vitamin E/kg diet from the early age of 5 wks to 18 mo age, but, when vitamin E supplementation started at later ages of 6 mo or 12 mo, survival rate was not increased over the 18 mo study period. Further, this effect of vitamin E supplementation from early age in LFLC mice was also reflected in low numbers of atherosclerotic lesions in the aortic sinuses, descending aorta as well as at the branching points to major arteries. Therefore, it appears that the overload of lipid stress in Ldlr-/- mice resulting from HFHC or MFMC diets was so high that vitamin E supplementation at 500 mg/kg diet was not effective at overcoming the overload of lipid stress associated with a high intake of fat/cholesterol and a deficiency of LDL receptors, even when the diet was supplemented with vitamin E from the young age of 5 wks. In contrast, even though vitamin E supplemented at 500 mg/kg diet was not able to totally eliminate atherosclerotic lesion development in LFLC mice, it was effective however at reducing the progression of atherosclerosis and probably the associated mortality when it was included in the diet from a young age.
It is also important to point out that while we did not mechanistically identify the association of atherosclerosis with the mortality prior to 18 mo age in this study, the number of our observed mortalities suggests that the development of advanced atheroma lesions induced by high fat and cholesterol diets might be responsible for the significant mortality in high fat-fed mice. In support of our notion, it is worth pointing out that when C57BL/J6 mice, the background mice strain of Ldlr-/-, are fed regular chow diet, which contains less than 5% fat, they live up to 30 mo of age with only a few mice dying prior to 18 mo or younger [47, 48], whereas in our study, substantial numbers of Ldlr-/- mice in HFHC and MFMC groups died before the age of 18 mo, probably due to advanced atheroma lesions.
In this study, we did not intend to elucidate how consumption of a high fat, Western style diet with or without vitamin E supplementation may lead to the development of atherosclerosis. However, as already well-established, the consumption of high fat and cholesterol increases blood levels of lipids, lipid stress, and the risk of atherogenesis. This effect of a high fat/cholesterol diet has been well-documented in Ldlr-/- mice and as expected, vitamin E supplementation had generally no significant effect on blood cholesterol and triglyceride levels. Nonetheless, a trend of increased serum cholesterol and triglyceride levels was noted with vitamin E supplementation for 18 mo in mice fed HFHC and MFMC diets.
Evidence from several cell culture and animal studies has also suggested and delineated that in addition to antioxidant activity, vitamin E through modulation of molecular signaling might play a major role in the prevention of atherogenesis in humans; however, evidence from animal models of human atherosclerosis is stronger. To cite some, vitamin E has been shown to slow the progression of hypercholesterolemia-induced oxidative stress in the heart, liver and kidney of rabbits [49, 50]. Vitamin E has been shown to suppress LDL oxidation, reduce the overall oxidative stress, suppress expression of adhesion molecules, reduce expression of pro-inflammatory cytokines, macrophage activation, inflammation, reduce proliferation of SMC, and suppress thromboxane production [7, 8, 51-59].
We observed that some of the moribund mice had hind limb paralysis just prior to death. The gross postmortem pathology revealed no tumors or any macroscopic lesions other than well-advanced, fatty atherosclerotic lesions in the aortic artery, at the branching points to the nephritic arteries, and at the point of bifurcation to iliac arteries. The hind limb paralysis might have resulted from ischemic neuropathy due to reduced blood circulation to the hind limbs or due to internal iliac artery thrombosis.
After 18 mo of dietary interventions, all the Ldl-/- mice, regardless of dietary treatment groups, had several advanced atherosclerotic lesions in the sinuses, in the lumen of the descending aorta, and in the branching points; however, the extent of atherosclerosis and mortality in LFLC mice as indicated above was relatively lower, therefore, we focused on these groups of mice. Since atherosclerosis is an inflammatory disease of the arteries, we sought to learn the status of some of the inflammatory cytokines in the aortic trees [60, 61]. Since the results of qRT-PCR analysis of RNA isolated from the whole aortic tree did not reveal the differential effects of vitamin E supplementation, we therefore used the LCM technique to dissect and separate out lesion from non-lesion areas in the frozen sections of aortic sinuses where we could see the extent of the pathology. Due to the high variation in the expression of inflammatory markers, no statistical differences were observed between the lesion and non-lesion areas; however, there were distinct patterns of differential expression of VCAM-1, CD36, and MCP-1 in non-lesion and lesion areas, which are worth discussing briefly. It appears that the expression of these genes in non-lesion areas was influenced by the inflammation of the lesion areas and was expressed more with short term vitamin E supplementation. However, 18 mo vitamin E supplementation suppressed the expression of VCAM-1, MCP-1, and CD36 genes in both lesion and non-lesion areas.
The low expression of CD36 in non-lesion areas relative to lesion areas may reflect an overall suppressed expression of this scavenger receptor in non-lesion areas, particularly in those mice that received a vitamin E-supplemented diet for a long period of time (18 mo). Perhaps vitamin E's antioxidant activity in non-lesion areas prevented LDL oxidation and attenuated the expression of CD36 by macrophages. Vitamin E and other antioxidants have been reported to attenuate CD36 expression by macrophages [56, 62-64]. In contrast, the expression of VCAM-1 and MCP-1 was high in the non-lesion areas of mice supplemented with vitamin E for 6 or 12 mo and was low in lesion areas. The low expression of these inflammatory markers in lesions might be due to a high accumulation of fat/cholesterol in the core of lesions, where a majority of the cells were dead, and thus they expressed fewer inflammatory markers compared to the non-lesion areas, where the cells were undergoing inflammation and expressing more of these inflammatory genes while developing advanced lesions [54, 65-67]. Overall, the expression CD36, VCAM-1, and MCP-1 was low in both lesion and non-lesion areas of aortic valves of those mice receiving vitamin E-supplemented diets for 18 mo, which is in agreement with a lower percentage of lesions in the aortic tree and a low mortality rate in this group.
In conclusion, vitamin E supplementation in Ldlr-/- mice, which are genetically predisposed to dyslipidemia and atherosclerosis, is not effective at reducing high lipid stress and atheroma lesion development and its associated mortality when the mouse diet contained high or even medium levels of fat and cholesterol. Importantly, reducing fat and cholesterol levels without vitamin E supplementation was not sufficient enough to suppress development of atherosclerosis. However, a relatively low dose and long-term vitamin E supplementation initiated from the early age of 5 wks was effective at reducing atherosclerotic lesions and mortality in genetically predisposed Ldlr-/- mice when they were fed a diet containing low levels of fat and cholesterol.
Supplementary Material
Figure 6 (supplement): Representative photomicrographs of sections of atheroma lesions from aortic sinus of a Ldlrl-/- mouse fed LFLC diet without vitamin E supplementation (A: upper panels) and from a mouse fed LFLC diet supplemented with 500 mg vitamin E/kg diet from early age of 5 wk for 18 mo (B: lower panels). Expression of CD-31 (6a) and immunohistochemistry of F4/80, a trans-membrane protein present on the surface of macrophages are stained brown and shown by red arrows in the 40X magnification of rectangle insets (6b). Expression of CD36, the scavenger receptor (6c) α-actin as an indicator of SMC (6d) and expression of Von Willebrand factor as an indicator of adverse changes in the endothelium (6e) are shown by red arrows. Expression of MCP-1 is stained immunologically and is shown in brown pigment (6f). The extent of atheroma lesions (macrophages with lipid deposits and SMC) in the aortic sinus of LFLC mice supplemented with vitamin E for 18 mo (6f, 4X lower panel) was relatively less than that of a mouse fed a diet not supplemented with vitamin E.
Highlights.
Medium, or high fat/cholesterol accelerates atherogenesis and death in Ldlr-/- mice.
Vitamin E (E) is not effective to reduce atherogenesis, even started from early age.
E supplementation also is not effective to reduce fat/cholesterol-induced mortality.
E is only effective when fat/cholesterol is reduced and E started from an early age.
Acknowledgments
This publication is based on work supported by grant R01 HL069897 from the National Heart, Lung and Blood Institute of National Institute of Health and by the Agricultural Research Services of United States Department of Agriculture under agreement # 58-1950-0-014. The authors would like to thank Stephanie Marco for her assistance in the preparation of the manuscript. Any opinions, findings, conclusions, or recommendations expressed in this publication are those of the author(s) and do not necessarily reflect the view of the U.S. Department of Agriculture.
Supported by NIH grant 1R01HL069897-01A1 and Agricultural Research Services of United States Department of Agriculture, under agreement # 58-1950-0-014.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Roger VL, et al. Heart disease and stroke statistics--2011 update: a report from the American Heart Association. Circulation. 2011;123:e18–e209. doi: 10.1161/CIR.0b013e3182009701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Thorn TJ, Epstein FH, Feldman JJ, Leaverton PE, Wolz M. National Institutes of Health. Betesda, MD: 1992. Total mortality and morbidity from heart disease, cancer, and stroke from 1950 to 1987 in 27 countries. NIH publication no.92-3088. [Google Scholar]
- 3.American Heart Association. Heart and stroke facts: 1994 statistical supplement. Dallas, TX: American Heart Association; 1994. pp. 1–22. [Google Scholar]
- 4.Steinberg D, Parthasarathy S, Carew TE, Khoo JC, Witztum JL. Beyond Cholesterol: modifications of low density lipoprotein that increase its atherogenicity. New Engl J Med. 1989;320:915–924. doi: 10.1056/NEJM198904063201407. [DOI] [PubMed] [Google Scholar]
- 5.Pryor WA. Vitamin E and heart disease: basic science to clinical intervention trials. Free Rad Biol Med. 2000;28:141–64. doi: 10.1016/s0891-5849(99)00224-5. [DOI] [PubMed] [Google Scholar]
- 6.Meydani M. Vitamin E and prevention of heart disease in high-risk patients. Nutr Rev. 2000;58:278–281. doi: 10.1111/j.1753-4887.2000.tb01881.x. [DOI] [PubMed] [Google Scholar]
- 7.Cyrus T, Yao Y, Rokach J, Tang LX, Pratico D. Vitamin E reduces progression of atherosclerosis in low-density lipoprotein receptor-deficient mice with established vascular lesions. Circulation. 2003;107:521–3. doi: 10.1161/01.cir.0000055186.40785.c4. [DOI] [PubMed] [Google Scholar]
- 8.Pratico D, Tangirala RK, Rader DJ, Rokach J, FitzGerald GA. Vitamin E suppresses isoprostane generation in vivo and reduces atherosclerosis in ApoE-deficient mice. Nature Med. 1998;4:1189–92. doi: 10.1038/2685. [DOI] [PubMed] [Google Scholar]
- 9.Azzi A, Gysin R, Kempna P, Munteanu A, Negis Y, Villacorta L, Visarius T, Zingg JM. Vitamin E mediates cell signaling and regulation of gene expression. Ann N Y Acad Sci. 2004;1031:86–95. doi: 10.1196/annals.1331.009. [DOI] [PubMed] [Google Scholar]
- 10.Sesso HD, Buring JE, Christen WG, Kurth T, Belanger C, MacFadyen J, Bubes V, Manson JE, Glynn RJ, Gaziano JM. Vitamins E and C in the prevention of cardiovascular disease in men: the Physicians' Health Study II randomized controlled trial. JAMA. 2008;300:2123–33. doi: 10.1001/jama.2008.600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hoogwerf BJ, Young JB. The HOPE study. Ramipril lowered cardiovascular risk, but vitamin E did not. Cleve Clin J Med. 2000;67:287–93. doi: 10.3949/ccjm.67.4.287. [DOI] [PubMed] [Google Scholar]
- 12.Lonn E, Bosch J, Yusuf S, Sheridan P, Pogue J, Arnold JM, Ross C, Arnold A, Sleight P, Probstfield J, Dagenais GR. Effects of long-term vitamin E supplementation on cardiovascular events and cancer: a randomized controlled trial. JAMA. 2005;293:1338–47. doi: 10.1001/jama.293.11.1338. [DOI] [PubMed] [Google Scholar]
- 13.Marchioli R, Levantesi G, Macchia A, Marfisi RM, Nicolosi GL, Tavazzi L, Tognoni G, Valagussa F. Vitamin E increases the risk of developing heart failure after myocardial infarction: Results from the GISSI-Prevenzione trial. J Cardiovasc Med. 2006;7:347–50. doi: 10.2459/01.JCM.0000223257.09062.17. [DOI] [PubMed] [Google Scholar]
- 14.GISSI-Prevenzione Investigators. Dietary supplementation with n-3 polyunsaturated fatty acids and vitamin E after myocardial infarction: results of the GISSI-Prevenzione trial. Lancet. 1999;354:447–455. [PubMed] [Google Scholar]
- 15.Heart protection collaborative group. MRC/BHF Heart Protection Study of antioxidant vitamin supplementation in 20536 high-risk individuals: a randomised placebo-controlled trial. Lancet. 2002;360:23–33. doi: 10.1016/S0140-6736(02)09328-5. [DOI] [PubMed] [Google Scholar]
- 16.Chae CU, Albert CM, Moorthy MV, Lee IM, Buring JE. Vitamin E supplementation and the risk of heart failure in women. Circulation Heart failure. 2012;5:176–82. doi: 10.1161/CIRCHEARTFAILURE.111.963793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Suarna C, Wu BJ, Choy K, Mori T, Croft K, Cynshi O, Stocker R. Protective effect of vitamin E supplements on experimental atherosclerosis is modest and depends on preexisting vitamin E deficiency. Free Radic Biol Med. 2006;41:722–30. doi: 10.1016/j.freeradbiomed.2006.05.013. [DOI] [PubMed] [Google Scholar]
- 18.Shirpoor A, Norouzi L, Khadem Ansari MH, Ilkhanizadeh B, Gharaaghaji R. Vasoprotective effect of vitamin E: Rescue of ethanol-induced atherosclerosis and inflammatory stress in rat vascular wall. International immunopharmacology. 2013;16:498–504. doi: 10.1016/j.intimp.2013.04.024. [DOI] [PubMed] [Google Scholar]
- 19.Goldenstein H, Levy NS, Lipener YT, Levy AP. Patient selection and vitamin E treatment in diabetes mellitus. Expert review of cardiovascular therapy. 2013;11:319–26. doi: 10.1586/erc.12.187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Farbstein D, et al. Vitamin E therapy results in a reduction in HDL function in individuals with diabetes and the haptoglobin 2-1 genotype. Atherosclerosis. 2011;219:240–4. doi: 10.1016/j.atherosclerosis.2011.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Milman U, et al. Vitamin E supplementation reduces cardiovascular events in a subgroup of middle-aged individuals with both type 2 diabetes mellitus and the haptoglobin 2-2 genotype: a prospective double-blinded clinical trial. Arterioscler Thromb Vasc Biol. 2008;28:341–7. doi: 10.1161/ATVBAHA.107.153965. [DOI] [PubMed] [Google Scholar]
- 22.Van Horn L. Fiber, lipids, and coronary heart disease. A statement for healthcare professionals from the Nutrition Committee, American Heart Association. Circulation. 1997;95:2701–4. doi: 10.1161/01.cir.95.12.2701. [DOI] [PubMed] [Google Scholar]
- 23.McGill HCJ, McMahan CA, Zieske AW, Sloop GD, Walcott JV, Troxclair DA, Malcom GT, Tracy RE, Oalmann MC, Strong JP. Associations of coronary heart disease risk factors with the intermediate lesion of atherosclerosis in youth. The Pathobiological Determinants of Atherosclerosis in Youth (PDAY) Research Group. Arterioscler Thrombo Vasc Biol. 2000;20:1998–2004. doi: 10.1161/01.atv.20.8.1998. [DOI] [PubMed] [Google Scholar]
- 24.Burke GL, Cresanta JL, Shear CL, Miner MH, Berenson GS. Cardiovascular risk factors and their modification in children. Cardiol Clin. 1986;4:33–46. [PubMed] [Google Scholar]
- 25.Giannini C, de Giorgis T, Scarinci A, Cataldo I, Marcovecchio ML, Chiarelli F, Mohn A. Increased carotid intima-media thickness in pre-pubertal children with constitutional leanness and severe obesity: the speculative role of insulin sensitivity, oxidant status, and chronic inflammation. Eur J Endocrinol. 2009;161:73–80. doi: 10.1530/EJE-09-0042. [DOI] [PubMed] [Google Scholar]
- 26.Bekkers MB, Brunekreef B, Smit HA, Kerkhof M, Koppelman GH, Oldenwening M, Wijga AH. Early-Life Determinants of Total and HDL Cholesterol Concentrations in 8-Year-Old Children; The PIAMA Birth Cohort Study. PLoS One. 2011;6:e25533. doi: 10.1371/journal.pone.0025533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bao W, Srinivasan S, Valdez R, Greenlund K, Wattigney W, Berenson G. Longitudinal changes in cardiovascular risk from childhood to young adulthood in offspring of parents with coronary artery disease: the Bogalusa Heart Study. JAMA. 1997;278:1749–1754. [PubMed] [Google Scholar]
- 28.Napoli C, Glass C, Witztum JL, Deutsch R, D'Armiento FP, Palinski W. Influence of maternal hypercholesterolaemia during pregnancy on progression of early atherosclerotic lesions in childhood: Fate of Early Lesions in Children (FELIC) study. Lancet. 1999;354:1234–41. doi: 10.1016/S0140-6736(99)02131-5. [DOI] [PubMed] [Google Scholar]
- 29.Napoli C, Witztum JL, Calara F, de Nigris F, Palinski W. Maternal hypercholesterolemia enhances atherogenesis in normocholesterolemic rabbits, which is inhibited by antioxidant or lipid-lowering intervention during pregnancy. An experimental model of atherogenic mechanisms in human fetuses. Circ Res. 2000;87:946–952. doi: 10.1161/01.res.87.10.946. [DOI] [PubMed] [Google Scholar]
- 30.Juonala M, et al. Influence of age on associations between childhood risk factors and carotid intima-media thickness in adulthood: the Cardiovascular Risk in Young Finns Study, the Childhood Determinants of Adult Health Study, the Bogalusa Heart Study, and the Muscatine Study for the International Childhood Cardiovascular Cohort (i3C) Consortium. Circulation. 2010;122:2514–20. doi: 10.1161/CIRCULATIONAHA.110.966465. [DOI] [PubMed] [Google Scholar]
- 31.Walsh PC. Effects of long-term vitamin E supplementation on cardiovascular events and cancer: a randomized controlled trial. J Urol. 2005;174:1823–4. doi: 10.1097/01.ju.0000183077.52404.ec. [DOI] [PubMed] [Google Scholar]
- 32.Gidding SS, Dennison BA, Birch LL, Daniels SR, Gillman MW, Lichtenstein AH, Rattay KT, Steinberger J, Stettler N, Van Horn L. Dietary recommendations for children and adolescents: a guide for practitioners: consensus statement from the American Heart Association. Circulation. 2005;112:2061–75. doi: 10.1161/CIRCULATIONAHA.105.169251. [DOI] [PubMed] [Google Scholar]
- 33.Lichtenstein AH, et al. Diet and lifestyle recommendations revision 2006: a scientific statement from the American Heart Association Nutrition Committee. Circulation. 2006;114:82–96. doi: 10.1161/CIRCULATIONAHA.106.176158. [DOI] [PubMed] [Google Scholar]
- 34.Ishibashi S, Brown MS, Goldstein JL, Gerard RD, Hammer RE, Herz J. Hypercholesterolemia in low density lipoprotein receptor knockout mice and its reversal by adenovirus mediated gene delivery. J Clin Invest. 1993;92:883–93. doi: 10.1172/JCI116663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Smith JD, Breslow JL. The emergence of mouse models of atherosclerosis and their relevance to clinical research. J Intern Med. 1997;242:99–109. doi: 10.1046/j.1365-2796.1997.00197.x. [DOI] [PubMed] [Google Scholar]
- 36.Lichtman AH, Clinton SK, liyama K, Connelly PW, Libby P, Cybulsky MI. Hyperlipidemia and atherosclerotic lesion development in LDL receptor-deficient mice fed defined semipurified diets with and without cholate. Artherioscler Thromb VasC Biol. 1999;19:1938–44. doi: 10.1161/01.atv.19.8.1938. [DOI] [PubMed] [Google Scholar]
- 37.Reeves PG, Nielsen FH, Fahey GC. AIN-93 purified diets for laboratory rodents: Final report of the American Institute of Nutrition ad hoc writing committee in the reformulation of the AIN-76A rodent diet. J Nutr. 1993;123:1939–1951. doi: 10.1093/jn/123.11.1939. [DOI] [PubMed] [Google Scholar]
- 38.Dietary reference intakes for vitamin C, vitamin E, selenium, and carotenoids. National Academy Press; Washington, D.C: 2000. A report of the Panel on Dietary Antioxidants and Related Compounds, Vitamin E; pp. 186–283. [Google Scholar]
- 39.Palinski W, Tangirala RK, Miller E, Young SG, Witztum JL. Increased autoantibody titers against epitopes of oxidised LDL in LDL receptor-deficient mice with increased atherosclerosis. Artherioscler Thromb VasC Biol. 1995;15:1569–76. doi: 10.1161/01.atv.15.10.1569. [DOI] [PubMed] [Google Scholar]
- 40.Tangirala RK, Pratico D, FitzGerald GA, Chun S, Tsukamoto K, Maugeais C, Usher DC, Pure E, Rader D. Reduction of isoprostanes and regression of advanced atherosclerosis by apolipoprotein E. J Biol Chem. 2001;276:261–266. doi: 10.1074/jbc.M003324200. [DOI] [PubMed] [Google Scholar]
- 41.Guo W, Wise ML, Collins FW, Meydani M. Avenanthramides, polyphenols from oats, inhibit IL-1 beta-induced NF-kappaB activation in endothelial cells. Free Radic Biol Med. 2008;44:415–29. doi: 10.1016/j.freeradbiomed.2007.10.036. [DOI] [PubMed] [Google Scholar]
- 42.Rodriguez SK, Guo W, Liu L, Band MA, Paulson EK, Meydani M. Green tea catechin, epigallocatechin-3-gallate, inhibits vascular endothelial growth factor angiogenic signaling by disrupting the formation of a receptor complex. Int J Cancer. 2006;118:1635–44. doi: 10.1002/ijc.21545. [DOI] [PubMed] [Google Scholar]
- 43.McNamara JR, Schaefer EJ. Automated enzymatic standardized lipid analyses for plasma and lipoprotein fractions. Clin Chem Acta. 1987;166:1–8. doi: 10.1016/0009-8981(87)90188-4. [DOI] [PubMed] [Google Scholar]
- 44.Meydani M, Evans WJ, Handelman G, Biddle L, Feilding RA, Meydani SN, Burrill J, Fiatarone MA, Blumberg JB, Cannon JG. Protective effect of vitamin E on exercise-induced oxidative damage in young and older adults. Am J Physiol. 1993;33:R992–R998. doi: 10.1152/ajpregu.1993.264.5.R992. [DOI] [PubMed] [Google Scholar]
- 45.Crawford RS, Kirk E, Rosenfeld ME, LeBoeuf RC, Chart A. Dietary antioxidants inhibit developmen of fatty streak lesions in the LDL receptor-deficient mouse. Artherioscl Thromb Vascul Biol. 1998;18:1506–13. doi: 10.1161/01.atv.18.9.1506. [DOI] [PubMed] [Google Scholar]
- 46.Navarro A, Gomez C, Sanchez-Pino MJ, Gonzalez H, Bandez MJ, Boveris AD, Boveris A. Vitamin E at high doses improves survival, neurological performance, and brain mitochondrial function in aging male mice. American journal of physiology Regulatory, integrative and comparative physiology. 2005;289:R1392–9. doi: 10.1152/ajpregu.00834.2004. [DOI] [PubMed] [Google Scholar]
- 47.Ejaz A, Wu D, Kwan P, Meydani M. Curcumin inhibits adipogenesis in 3T3-L1 adipocytes and angiogenesis and obesity in C57/BL mice. J Nutr. 2009;139:919–25. doi: 10.3945/jn.108.100966. [DOI] [PubMed] [Google Scholar]
- 48.Bruell JH, Daroczy AF, Hellerstein HK. Strain and sex differences in serum cholesterol levels of mice. Science. 1962;135:1071–2. doi: 10.1126/science.135.3508.1071. [DOI] [PubMed] [Google Scholar]
- 49.Prasad K, McNair ED, Qureshi AM, Casper-Bell G. Vitamin E slows the progression of hypercholesterolemia-induced oxidative stress in heart, liver and kidney. Molecul Cell Biochem. 2012;368:181–7. doi: 10.1007/s11010-012-1358-z. [DOI] [PubMed] [Google Scholar]
- 50.Abbas AM, Sakr HF. Simvastatin and vitamin E effects on cardiac and hepatic oxidative stress in rats fed on high fat diet. J Physiol Biochem. 2013 doi: 10.1007/s13105-013-0250-y. [DOI] [PubMed] [Google Scholar]
- 51.Huang ZG, Liang C, Han SF, Wu ZG. Vitamin E ameliorates ox-LDL-induced foam cells formation through modulating the activities of oxidative stress-induced NF-kappaB pathway. Molecul Cell Biochem. 2012;363:11–9. doi: 10.1007/s11010-011-1153-2. [DOI] [PubMed] [Google Scholar]
- 52.Devaraj S, Tang R, Adams-Huet B, Harris A, Seenivasan T, de Lemos JA, Jialal I. Effect of high-dose alpha-tocopherol supplementation on biomarkers of oxidative stress and inflammation and carotid atherosclerosis in patients with coronary artery disease. Am J Clin Nutr. 2007;86:1392–8. doi: 10.1093/ajcn/86.5.1392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Koga T, Claycombe K, Meydani M. Homocysteine increases monocyte and T-cell adhesion to human aortic endothelial cells. Atherosclerosis. 2002;161:365–74. doi: 10.1016/s0021-9150(01)00670-0. [DOI] [PubMed] [Google Scholar]
- 54.Koga T, Kwan P, Zubik L, Ameho C, Smith D, Meydani M. Vitamin E supplementation suppresses macrophage accumulation and endothelial cell expression of adhesion molecules in the aorta of hypercholesterolemic rabbits. Atherosclerosis. 2004;176:265–72. doi: 10.1016/j.atherosclerosis.2004.05.034. [DOI] [PubMed] [Google Scholar]
- 55.Hasty AH, Gruen ML, Terry ES, Surmi BK, Atkinson RD, Gao L, Morrow JD. Effects of vitamin E on oxidative stress and atherosclerosis in an obese hyperlipidemic mouse model. J Nutr Biochem. 2007;18:127–33. doi: 10.1016/j.jnutbio.2006.03.012. [DOI] [PubMed] [Google Scholar]
- 56.Singh U, Devaraj S, Jialal I. Vitamin E, oxidative stress, and inflammation. Annu Rev Nutr. 2005;25:151–74. doi: 10.1146/annurev.nutr.24.012003.132446. [DOI] [PubMed] [Google Scholar]
- 57.Guyton AC. Textbook of Medical Physiology. WB Saunders Company; 1971. [Google Scholar]
- 58.Meydani M, Meydani SN, Blumberg JB. Modulation by dietary vitamin E and selenium of clotting whole blood thromboxane A2 and aortic prostacyclin synthesis in rats. Nutr Biochem. 1993;4:322–326. [Google Scholar]
- 59.Baldi S, Innocenti M, Frascerra S, Nannipieri M, Lippi A, Rindi P, Ferrannini E. Effects of hemodialysis and vitamin E supplementation on low-density lipoprotein oxidizability in end-stage renal failure. J Nephrol. 2013;26:549–55. doi: 10.5301/jn.5000190. [DOI] [PubMed] [Google Scholar]
- 60.Libby P, Ridker PM, Maseri A. Inflammation and atherosclerosis. Circulation. 2002;105:1135–43. doi: 10.1161/hc0902.104353. [DOI] [PubMed] [Google Scholar]
- 61.Stoll G, Bendszus M. Inflammation and atherosclerosis: novel insights into plaque formation and destabilization. Stroke; a journal of cerebral circulation. 2006;37:1923–32. doi: 10.1161/01.STR.0000226901.34927.10. [DOI] [PubMed] [Google Scholar]
- 62.Kawai Y, Tanaka H, Murota K, Naito M, Terao J. (-) -Epicatechin gallate accumulates in foamy macrophages in human atherosclerotic aorta: implication in the anti-atherosclerotic actions of tea catechins. Biochem Biophys Res Commun. 2008;374:527–32. doi: 10.1016/j.bbrc.2008.07.086. [DOI] [PubMed] [Google Scholar]
- 63.Ricciarelli R, Azzi A ZJ. Vitamin E reduces the uptake of oxidized LDL by inhibiting CD36 scavenger receptor expression in cultured aortic smooth muscle cells. Circulation. 2000:82–7. doi: 10.1161/01.cir.102.1.82. [DOI] [PubMed] [Google Scholar]
- 64.Lecompte S, Szabo de Edelenyi F, Goumidi L, Maian G, Moschonis G, Widhalm K, Molnar D, Kafatos A, Spinneker A, Breidenassel C, Dallongeville J, Meirhaeghe A, Borel P. Polymorphisms in the CD36/FAT gene are associated with plasma vitamin E concentrations in humans. Am J Clin Nutr. 2011;93:644–51. doi: 10.3945/ajcn.110.004176. [DOI] [PubMed] [Google Scholar]
- 65.Murphy N, Grimsditch DC, Vidgeon-Hart M, Groot PH, Overend P, Benson GM, Graham A. Dietary antioxidants decrease serum soluble adhesion molecule (sVCAM-1, slCAM-1) but not chemokine (JE/MCP-1, KC) concentrations, and reduce atherosclerosis in C57BL but not apoE*3 Leiden mice fed an atherogenic diet. Dis Markers. 2005;21:181–90. doi: 10.1155/2005/394152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Peluzio MC, Miguel E, Jr, Drumond TC, Cesar GC, Santiago HC, Teixeira MM, Vieira EC, Arantes RM, Alvarez-Leite JI. Monocyte chemoattractant protein-1 involvement in the alpha-tocopherol-induced reduction of atherosclerotic lesions in apolipoprotein E knockout mice. Br J Nutr. 2003;90:3–11. doi: 10.1079/bjn2003870. [DOI] [PubMed] [Google Scholar]
- 67.Rayment SJ, Shaw J, Woollard KJ, Lunec J, Griffiths HR. Vitamin C supplementation in normal subjects reduces constitutive ICAM-1 expression. Biochem Biophys Res Commun. 2003;308:339–45. doi: 10.1016/s0006-291x(03)01383-4. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure 6 (supplement): Representative photomicrographs of sections of atheroma lesions from aortic sinus of a Ldlrl-/- mouse fed LFLC diet without vitamin E supplementation (A: upper panels) and from a mouse fed LFLC diet supplemented with 500 mg vitamin E/kg diet from early age of 5 wk for 18 mo (B: lower panels). Expression of CD-31 (6a) and immunohistochemistry of F4/80, a trans-membrane protein present on the surface of macrophages are stained brown and shown by red arrows in the 40X magnification of rectangle insets (6b). Expression of CD36, the scavenger receptor (6c) α-actin as an indicator of SMC (6d) and expression of Von Willebrand factor as an indicator of adverse changes in the endothelium (6e) are shown by red arrows. Expression of MCP-1 is stained immunologically and is shown in brown pigment (6f). The extent of atheroma lesions (macrophages with lipid deposits and SMC) in the aortic sinus of LFLC mice supplemented with vitamin E for 18 mo (6f, 4X lower panel) was relatively less than that of a mouse fed a diet not supplemented with vitamin E.