

Abstract
Yeast Atg1 initiates autophagy in response to nutrient limitation. The Ulk gene family encompasses the mammalian orthologs of yeast ATG1. We created mice deficient for both Ulk1 and Ulk2 and found that the mice die within 24 h of birth. When found alive, pups exhibited signs of respiratory distress. Histological sections of lungs of the Ulk1/2 DKO pups showed reduced airspaces with thickened septae. A similar defect was seen in Atg5-deficient pups as both Ulk1/2 DKO and Atg5 KO lungs show numerous glycogen-laden alveolar type II cells by electron microscopy, PAS staining, and increased levels of glycogen in lung homogenates. No abnormalities were noted in expression of genes encoding surfactant proteins but the ability to incorporate exogenous choline into phosphatidylcholine, the major phospholipid component of surfactant, was increased in comparison to controls. Despite this, there was a trend for total phospholipid levels in lung tissue to be lower in Ulk1/2 DKO and Atg5 KO compared with controls. Autophagy was abundant in lung epithelial cells from wild-type mice, but lacking in Atg5 KO and Ulk1/2 DKO mice at P1. Analysis of the autophagy signaling pathway showed the existence of a negative feedback loop between the ULK1 and 2 and MTORC1 and 2, in lung tissue. In the absence of autophagy, alveolar epithelial cells are unable to mobilize internal glycogen stores independently of surfactant maturation. Together, the data suggested that autophagy plays a vital role in lung structural maturation in support of perinatal adaptation to air breathing.
Keywords: Ulk1/2 DKO mice, Atg5 KO mice, perinatal mortality, glycogen, lung development
Introduction
Autophagy has emerged as an essential mechanism for cell survival in the face of metabolic stress. In addition to playing an important role in normal cell maintenance as a way for cells to rid themselves of damaged organelles, autophagy is involved in many disease states.1-4 As first described in yeast, multiple genes are involved in the autophagic cascade. Mouse models have provided considerable insight into the functions of autophagy genes in mammals. Targeted deletions of individual autophagy genes lead to either embryonic or perinatal lethality.5 Targeted deletion of Becn1, the mammalian ortholog of yeast VPS30/ATG6, leads to early embryonic lethality around embryonic day (ED)7.5.6,7 However, targeted deletions of many other Atg genes, including Atg3, Atg5, Atg7, Atg9, and Atg16l1, result in perinatal lethality.8-13 These knockout models share several similar features, which may contribute to the high perinatal mortality, including low birth weight. Consistent with this, low amino acid levels in plasma and tissues, particularly in the heart, were noted at ED19.5 following Caesarean section.8-10 These results indicate that degradation of protein to amino acids via autophagy can provide an energy source in the neonatal period. The demonstration of glycogen containing autophagosomes in the liver of newborn, normal rats also indicates that mobilization of glycogen stores from the liver in the neonatal period by autophagy serves as a mechanism to maintain glucose homeostasis.14 At birth hypoglycemia develops which triggers glycogen autophagy.15 Administration of glucose to the newborn rat prevents the mobilization of glycogen and the development of autophagic vacuoles. Together, these features suggest that autophagy plays an important role in providing nutrients during the transition from embryonic life to the neonatal period before efficient nursing has been established. Interestingly, while Caesarean delivery and feeding with artificial milk prolonged survival of atg5−/− pups from 12 to 24 h, all atg5−/− mice were dead by about 40 h, whereas a majority of the control mice were alive past 60 h at the end of the experiment, indicating that factors other than nutrient deprivation may contribute to the perinatal mortality in autophagy-deficient mice.9
ULK1 and ULK2 are the mammalian orthologs of yeast ATG1, a kinase that initiates autophagy in response to nutrient limitation. We have previously reported the phenotypes of ulk1−/− and ulk2−/− mice.16,17 ulk1−/− mice are viable with a normal life span and exhibit a mild autophagy defect manifested by defective mitochondria clearance during erythrocyte differentiation. ulk2−/− mice exhibit a normal life span with no overt phenotype. As ULK1 and ULK2 may have redundant functions, we generated mice deficient for both Ulk1 and Ulk2. ulk1−/− ulk2−/− double-knockout mice (Ulk1/2 DKO) display neonatal mortality, as previously described for Atg3, Atg5, Atg7, Atg9, and Atg16l1-deficient mice. Pathological examination of Ulk1/2 DKO pups revealed a defect in lung development manifested by the presence of glycogen-laden alveolar type II cells despite the expression of the genes that normally accompanies surfactant production and morphological conversion to type I alveolar cells. To determine if this defect was unique to Ulk1/2 DKO, the lungs of Atg5-deficient (Atg5 KO) mice were also examined perinatally and found to have the same defect in lung development. We demonstrated both by immunohistochemistry and western blotting that autophagy is active in normal, neonatal lung tissue but absent in Ulk1/2 DKO and Atg5 KO lungs. Thus, our studies of Ulk1/2 DKO and Atg5 KO mice suggested that autophagy plays a role in perinatal lung adaptation that is distinct from surfactant maturation, and may contribute to the perinatal lethality seen in many autophagy-deficient mice.
Results
High perinatal mortality of Ulk1/2 DKO mice
To study the fate of Ulk1/2 DKO mice, ulk1+/− ulk2−/− males and females were mated. Initially, genotyping of litters at weaning did not yield any Ulk1/2 DKO mice. Therefore, consecutive litters were sacrificed and genotyped as soon as the pups were found in the breeding cages. Often several dead pups were observed in each litter and tissue was also collected from these. As shown in Table 1, out of 23 Ulk1/2 DKO pups found on postnatal day 1 (P1), 21 were found dead. The 2 mice surviving past P1 were severely growth-retarded and died within weeks. The observed frequency of Ulk1/2 DKO pups was lower than the expected Mendelian frequency. However, this could be due to cannibalization of the dead Ulk1/2 DKO pups that were thus missed in the analysis. In support of early cannibalization, Table 1 demonstrates close to normal Mendelian frequency of litters at ED18.5. Thus, as shown for other autophagy-deficient strains, Ulk1/2 DKO mice show a high perinatal mortality. Similar results were noted if the mice used for breeding were ulk1−/− ulk2+/− (data not shown).
Table 1.Ulk1/2 DKO pups show high perinatal mortality.
| Offspring from Ulk1+/−ulk2−/− parents | ||||
|---|---|---|---|---|
| Genotype | Total | Observed (%) | Expected (%) | |
| ED18.5 | 1+/+2−/− | 38 | 27.7 | 25.0 |
| 1+/−2−/− | 71 | 51.8 | 50.0 | |
| 1−/−2−/− | 28 | 20.4 | 25.0 | |
| P1 | 1+/+2−/− | 56 (2 dead day 1) | 35.6 | 25.0 |
| 1+/−2−/− | 80 (3 dead day 1) | 50.0 | 50.0 | |
| 1−/−2−/− | 23 (21 dead day 1) | 14.4 | 25.0 | |
All pups in consecutive litters from 16 breeding pairs were included in the analysis for the mice at P1.
As shown in Table 2, the birth weight of the Ulk1/2 DKO was significantly lower than their littermate controls, as previously noted for other autophagy-deficient strains. Low body weights were also seen at ED 18.5 for Ulk1/2 DKO mice.
Table 2. Weights of Ulk1/2 DKO and litter mate controls at ED18.5 and P1.
| ED18.5 | P1 | |
|---|---|---|
| Genotype | Weight (g) | Weight (g) |
| 1−/−2−/− | 0.99 ± 0.13 (n = 8) | 1.26 ± 0.10 (n = 12) |
| 1+/−2−/− | 1.20 ± 0.12 (n = 10) | 1.50 ± 0.22 (n = 23) |
| 1+/+2−/− | 1.18 ± 0.19 (n = 8) | 1.37 ± 0.09 (n = 12) |
P value < 0.05 when weights were compared between 1−/−2−/− and 1+/−2−/− or 1+/+2−/− at ED18.5 and P1. Only litters containing Ulk1/2 DKO mice at P1 were included in the analysis.
The Ulk1/2 DKO newborn pups when found alive displayed signs of respiratory distress and in some cases cyanosis. To further investigate the abnormalities in these mice, live-born pups were sacrificed and subjected to whole body embedding. As shown in Figure 1, the lungs of Ulk1/2 DKO mice exhibited reduced airspace size and thickened septae, indicating that a defect in lung development could be a contributing factor to the high perinatal mortality seen for Ulk1/2 DKO mice. Intra-alveolar blood was frequently observed in the lungs of Ulk1/2 DKO mice.
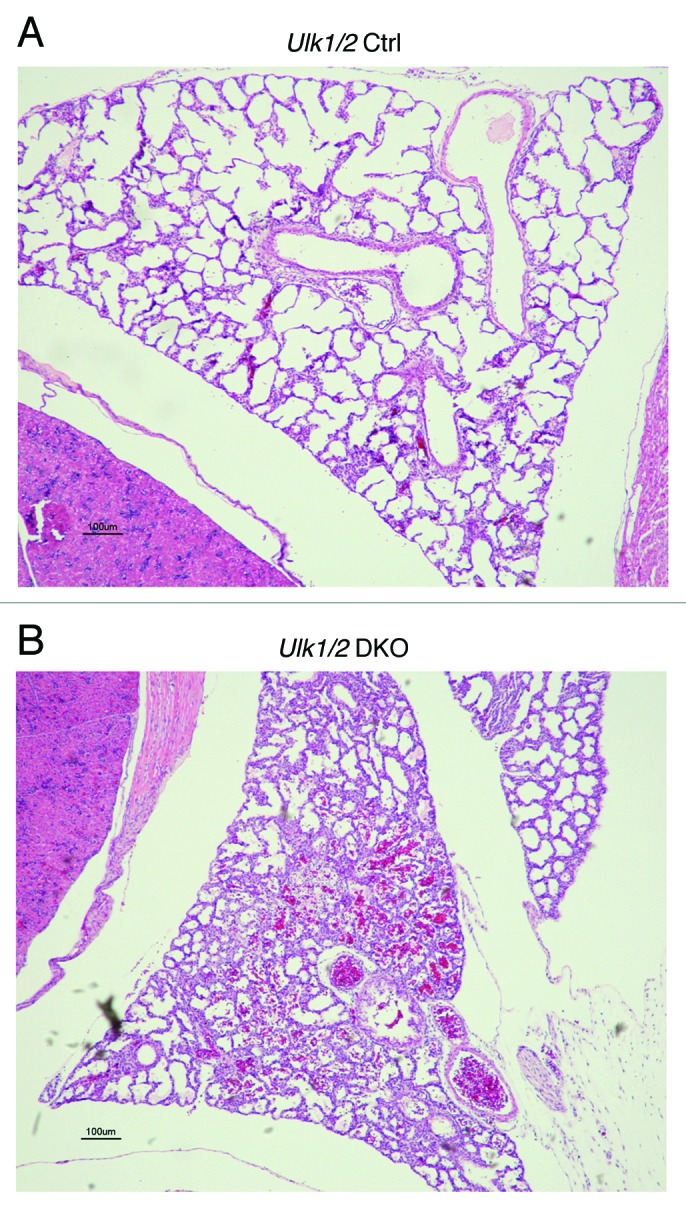
Figure 1. Lungs of Ulk1/2 DKO mice at P1 show reduced airspace. Ulk1/2 littermate control (Ulk1/2 Ctrl) and Ulk1/2 DKO mice were sacrificed at birth and subjected to whole-body fixation and embedding in paraffin followed by sagittal sectioning and HE staining. Chest cavity with lung lobe is shown. Scale bar: 100 μm.
Targeted deletion of the Rb1cc1 (RB1-inducible Coiled Coil 1) gene product, a ULK1-interacting protein, leads to embryonic lethality due to in part, a heart defect characterized by left ventricular dilation, thin trabecular myocardium and absence of the compact subepicardial myocardium. Histological examination of the hearts of Ulk1/2 DKO mice did not display any defects of the myocardium, as shown in Figure S1.
Glycogen-filled alveolar epithelial type II cells found in lungs of autophagy-deficient mice
To further investigate the lung defect noted in Ulk1/2 DKO pups, electron microscopic analysis was performed on lungs from ED18.5 embryos. Atg5 KO embryos were included in the analysis to determine whether the lung defect was evident in other autophagy-deficient strains of mice. As seen in Figure 2A, the lungs of both Ulk1/2 DKO and Atg5 KO embryos contain numerous enlarged alveolar epithelial type II cells with cytoplasmic glycogen, indicated by arrowheads, that were not evident in littermate controls (type I and type II alveolar epithelial cells in controls are indicated by arrows in the upper left and right panels). Despite the glycogen-filled cytoplasm, the type II cells displayed normal appearing intracytoplasmic and secreted lamellar bodies in both Ulk1/2 DKO and Atg5 KO lungs (Fig. 2B, indicated by arrows). To confirm the presence of glycogen in the type II cells and to determine if the glycogen-filled cells persisted past birth, Periodic Schiff Acid (PAS) staining was performed on histological sections of P1 lungs of Ulk1/2 DKO, Atg5 KO pups and their littermate controls (Fig. 3A). Numerous glycogen-filled cells could be seen in the lungs of mutant mice. Furthermore, glycogen levels in lung homogenates from Ulk1/2 DKO and Atg5 KO mice were elevated above those in littermate controls (Fig. 3B). Glycogen is a substrate for surfactant phospholipid synthesis, and the ubiquitous presence of glycogen-filled type II cells suggests that there is lung epithelial dysfunction in mobilizing glycogen in both Ulk1/2 DKO and Atg5 KO mice.

Figure 2. Structural analysis of lungs from Ulk1/2 DKO and Atg5 KO mice. Lungs from Ulk1/2 Ctrl; Ulk1/2 DKO; Atg5 Ctrl and Atg5 KO embryos at ED18.5 were prepared for transmission electron microscopy as described in Materials and Methods. (A) Ulk1/2 DKO and Atg5 KO mice display glycogen-filled alveolar type II cells, indicated by arrowheads. Alveolar type I and II cells are indicated by arrows in upper left and right panel (Ulk1/2 and Atg5 Ctrl). Scale bar: 2 μm. (B) The alveolar type II cells of Ulk1/2 DKO and Atg5 KO mice contain lamellar bodies (see arrows) that are similar in number and structure to those found in alveolar type II cells of littermate controls. Scale bar: 500 nm.

Figure 3. PAS staining of lungs from Ulk1/2 DKO and Atg5 KO mice at P1 show persistence of glycogen-filled cells. (A) Lungs from Ulk1/2 Ctrl; Ulk1/2 DKO; Atg5 Ctrl and Atg5 KO mice at P1 were fixed, embedded in paraffin, sectioned, and subjected to PAS staining. Scale bar: 10 μm. (B) Increased glycogen levels in lungs from Ulk1/2 DKO and Atg5 KO mice at P1. Lungs from Ulk1/2 Ctrl; Ulk1/2 DKO; Atg5 Ctrl and Atg5 KO mice at P1 were homogenized and glycogen content was measured as described in Materials and Methods. The box and whisker plot shows glycogen levels as ng glycogen/μg protein, with the whiskers set to the minimum and maximum values in each group and the box displaying the 25th and 75th percentiles around the median. Data were normalized within each genetic background (Ulk1/2 Ctrl with DKO, and Atg5 Ctrl with KO), with n = 6 for Ulk1/2 Ctrl, n = 6 for Ulk1/2 DKO, n = 7 for Atg5 Ctrl and n = 5 for Atg5 KO. * indicates P value < 0.05 determined by ANOVA with Bonferonni’s Multiple Comparison test.
Glycogen levels in heart and liver as well as blood glucose were also examined as shown in Figure S2. As the primary goal of this study was to investigate the lung phenotype of Ulk1/2 DKO and Atg5 KO pups, under physiological conditions after a natural birth, no attempt was made to control for food intake. Considering this caveat, glycogen levels were determined in hearts and livers of mutant mice and their littermate controls (Fig. S2A and S2B). Glycogen levels in the liver were similar in mutant and control pups but the glycogen levels in the hearts of Atg5 KO pups were significantly increased compared with littermate controls. No difference in heart glycogen was seen between the Ulk1/2 DKO and littermate controls. The glycogen concentration in liver and heart was variable as evidenced by the error bars. We also obtained blood from the pups at sacrifice and found that the blood glucose levels were generally lower in the mutant mice but reached statistical significance only for the Atg5 KO pups (Fig. S2C). Shown in Figure S3, are histological sections of livers from 2 wild-type, 2 Ulk1/2 DKO and 2 Atg5 KO pups stained with PAS demonstrating variable levels of glycogen in this organ, particularly for the mutant mice. As food intake was not controlled, these results may be due to the fact that the mutant mice because of their respiratory distress may not have been able to compete with their healthy littermates for access to milk from the mother.
Glycogen storage diseases are a well-described group of genetic disorders, which are due to deficiencies in enzymes of glycogen metabolism, and many of these enzymes are the targets of phosphorylation. Because ULK1/2 are kinases, there was a possibility that reduced phosphorylation of enzymes involved in glycogen metabolism in Ulk1/2 DKO lungs could result in increased glycogen in the alveolar type II cells of the lung. However, western blot analysis of lung lysates for several enzymes involved in glycogen metabolism including GYS1 (glycogen synthase), PHKG2 (phosphorylase kinase, gamma 2 [testis]), PYGB/M/L (phosphorylase, glycogen; brain/muscle/liver) and GSK3B (glycogen synthase kinase 3 β) failed to show any expression and/or phosphorylation differences between Ulk1/2 DKO and littermate controls (Fig. S4). In addition, the fact that ATG5 is not a kinase and that Atg5 KO mice show the same lung phenotype as Ulk1/2 DKO mice supports the conclusion that defects in phosphorylation of enzymes involved in glycogen metabolism, as seen in many glycogen storage disorders, are not the underlying cause of the lung defect seen in autophagy-deficient mice.
Analysis of the phospholipid component of surfactant in the lungs of Ulk1/2 DKO and Atg5 KO mice
Phospholipid (PL), largely phosphatidylcholine (PC), constitutes up to 70% of surfactant by weight and forms the surface active monolayer at the air-liquid interface. About 50% of surfactant PL is dipalmitoylphosphatidylcholine, a disaturated PL synthesized predominantly in the lung. As glycogen is an important substrate for PC synthesis, we next determined the incorporation of labeled choline into newly synthesized PC. Lungs of Ulk1/2 DKO and Atg5 KO mice show similar abilities to incorporate exogenous choline into PC compared with littermate controls at ED 18.5. In fact, after birth the ability to incorporate exogenous choline into PC was enhanced in lung tissue for both mutant strains in comparison to lung tissue from wild-type control mice (Fig. 4A and B). Thus, when provided with exogenous substrate, PC synthesis was not impaired. Despite an increased enzymatic ability to synthesize PC the extracts of lungs from Ulk1/2 DKO and Atg5 KO mice exhibited if anything reduced PL accumulation in comparison to littermate controls (Fig. 4C). At P1 the percent saturation of PC was similar for control and knockout lungs (50.8 ± 1.4% and 53.2 ± 1.6%, Ulk1/2 control and Ulk1/2 DKO, respectively; 47.2 ± 1.4% and 47.6 ± 2.2%, Atg5 control and Atg5 KO, respectively, n = 4–7). Thus, autophagy-deficient mice display a lung defect characterized by glycogen-laden alveolar epithelial type II cells without an evident defect in the enzymes required to synthesize PC from exogenous substrates.
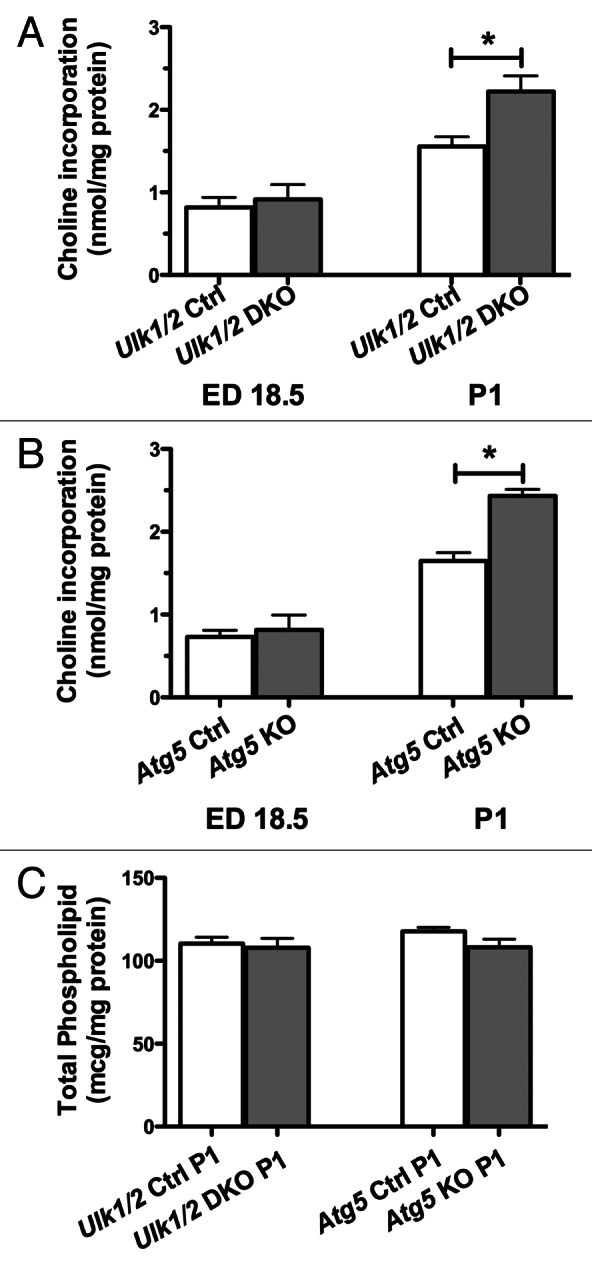
Figure 4. The PC synthetic rate is increased in the absence of increased phospholipid content in lungs from Ulk1/2 DKO and Atg5 KO mice. Lungs from (A) Ulk1/2 Ctrl; Ulk1/2 DKO at ED 18.5 (n = 7 for Ulk1/2 Ctrl and n = 3 for Ulk1/2 DKO) and P1 (n = 5 for Ulk1/2 Ctrl and Ulk1/2 DKO respectively) (B) Atg5 Ctrl and Atg5 KO at ED18.5 (n = 4 for Atg5 Ctrl and n = 3 for Atg5 KO) and P1 (n = 7 for Atg5 Ctrl and n = 4 for Atg5 KO) were harvested and incubated in vitro with medium containing 3H-choline followed by measurement of the incorporation of 3H choline into newly synthesized PC. Data are presented as the mean ± SEM * denotes P value < 0.05. (C) Total phospholipid content was measured at P1. Data are presented as mean ± SEM with n = 4 for each experimental group.
Analysis of lung alveolar epithelial cells in Ulk1/2 DKO and Atg5 KO mice
To assess the maturation of the alveolar epithelium, immunohistochemistry was performed on lung tissue from mutant mice and their littermate controls using antibodies specific for markers of alveolar epithelial type I and II cells. Staining for PDPN/T1-α (podoplanin), a marker for type I cells, revealed a pattern of distribution in mutant mice comparable to that seen in their littermate controls (Fig. 5A). Similarly, staining for SFTPB (surfactant protein B) and pro-SFTPC, the hydrophobic surfactant proteins that contribute to surfactant function, revealed a distribution and frequency of type II cells in the mutant mice that was similar to that seen in their littermate controls (Fig. 5B and C). Next, quantitative RT-PCR analysis of several genes involved in type II cell function was performed. The expression of all 4 genes encoding surfactant proteins was similar in the mutant mice compared with their littermate controls (Fig. 6A). Expression of genes encoding proteins associated with surfactant PL, specifically ABCA3 (ATP-binding cassette, sub-family A [ABC1], member 3) and FASN (fatty acid synthase), and of NKX2-1/TTF1 (NK2 homeobox 1/thyroid transcription factor 1), a transcription factor important for regulating surfactant gene expression, was also similar in both mutant mice and littermate controls (Fig. 6B). Because complete processing of SFTPB to its mature form is essential for normal surfactant function, we also performed western blotting of lung homogenates to assess the levels of SFTPB, SFTPB intermediates, and another key enzyme in SFTPB posttranslational processing, CTSH (cathepsin H). Western blot analysis for SFTPB presented as relative expression, normalized by litters, following densitometry of multiple western blots, with n = 11 for Ulk1/2 Ctrl, n = 8 for Ulk1/2 DKO, n = 6 for Atg5 Ctrl and n = 6 for Atg5 KO and a representative blot are shown in Figure 6C. While the relative levels of mature SFTPB were reduced in Ulk1/2 DKO lungs, no significant differences were seen between Atg5 KO and littermate controls. Moreover, lung tissue levels of CTSH and of SFTPB intermediates did not indicate a block in SFTPB proteolytic processing that would account for reduced mature SFTPB. Although the levels of SFTPB were significantly reduced in Ulk1/2 DKO lung tissue to ~40% of wild-type expression, this is unlikely to contribute to the perinatal lethality as it has been previously shown that mice exhibit normal lung function until SFTPB levels are reduced to 25% of normal.18 A consistent finding was that SFTPB expression levels in Atg5 Ctrl as well as Atg5 KO lungs were significantly lower than in Ulk1/2 Ctrl lungs. This could be attributed to strain differences as Atg5 heterozygous mice are on the congenic C57Bl.6 background whereas the Ulk1/2 heterozygous mice are on a mixed C57Bl.6/129Sv background. Together, these data indicate that alveolar epithelial differentiation is relatively unperturbed in Ulk1/2 DKO and Atg5 KO compared with that seen in their littermate controls.

Figure 5. Immunohistochemical analysis of Ulk1/2 DKO and Atg5 KO lungs at P1 for (A) PDPN (B) SFTPB (C) pro-SFTPC. Lungs from Ulk1/2 Ctrl; Ulk1/2 DKO; Atg5 Ctrl and Atg5 KO at P1 were fixed, embedded in paraffin and sectioned followed by immunohistochemical analysis with the indicated antibodies, as described in Materials and Methods. Scale bar: 10 µm.

Figure 6. RT-PCR and western blot analysis of Ulk1/2 DKO and Atg5 KO lungs. RT-PCR analysis of genes encoding (A) surfactant proteins SFTPA, SFTPB, SFTPC and SFTPD, (B) ABCA3, FASN and NKX2-1 were measured at ED18.5 in Ulk1/2 Ctrl; Ulk1/2 DKO; Atg5 Ctrl and Atg5 KO lung RNA. Data are presented as mean ± SEM where n = 4 for Ulk1/2 Ctrl, n = 5 for Ulk1/2 DKO, n = 3 for Atg5 Ctrl and Atg5 KO respectively for the analysis of SFTPA, SFTPC, SFTPD, and NKX2-1. For analysis of SFTPB, ABCA3, and FASN the n = 6 for Ulk1/2 Ctrl and Ulk1/2 DKO respectively and n = 3 for Atg5 Ctrl and Atg5 KO respectively. (C) Western blot analysis for SFTPB presented as relative expression, normalized by litters, following densitometry of multiple western blots, with n = 11 for Ulk1/2 Ctrl, n = 8 for Ulk1/2 DKO, n = 6 for Atg5 Ctrl and n = 6 for Atg5 KO. * indicates P value < 0.05 determined by ANOVA with Bonferonni’s Multiple Comparison test. A representative blot is shown for SFTPB, SFTPB intermediates detected by the NFlank antibody (SFTPB/proSP-B top, 25-kDa intermediate bottom arrowhead), CTSH, and GAPDH.
Autophagy is impaired in the lungs of Ulk1/2 DKO and Atg5 KO mice
In examining autophagy in organs of neonatal, wild-type GFP-MAP1LC3 (LC3) transgenic mice, Kuma et al.9 observed a transient increase in LC3 puncta in the lung, peaking at 3 to 12 h after birth. Using an antibody specific for LC3, Figure 7A shows multiple autophagic puncta in histological sections in the alveolar epithelial cells of wild-type mice at P1, indicated by arrowheads. Clara cells in the distal bronchioles in wild-type newborn mice also displayed numerous puncta (data not shown). Clara cells, the nonciliated columnar epithelial cells of bronchioles, also contain glycogen during fetal life, and Clara cells lose cytoplasmic glycogen in the perinatal period.19 In contrast, LC3 staining of lung tissue from Ulk1/2 DKO mice at P1 shows a diffuse cytoplasmic staining pattern in epithelial cells typical of the LC3-I form (the arrow shows an example of such a cell), indicating absence of autophagy (Fig. 7B). Figure 7C examines the autophagy signaling pathways by western blotting in litters of wild-type pups at ED 18.5 and P1, and in Atg5 KO and Ulk1/2 DKO pups and their littermate controls at P1. In wild-type ED18.5 embryos there is no evidence of autophagy as judged by the ratio of LC3-I to LC3-II but at P1 there is conversion of LC3-I to LC3-II indicating active autophagy. However, examination of Atg5 KO and Ulk1/2 DKO pups at P1 show predominantly the LC3-I form indicative of the absence of autophagy in the lung tissue as opposed to the littermate controls where conversion to the LC3-II form is seen. In examining the autophagy signaling pathway, we demonstrate that in the Ulk1/2 DKO pups MTORC1 and 2 (mechanistic target of rapamycin complex 1 and 2) are active as evidenced by the elevated phosphorylation of downstream targets RPS6KB (p70 S6 kinase) at Thr389 (p-RPS6KB), and AKT at Ser473 (p-AKT). Thus, in the absence of ULK1 and ULK2 the MTOR complexes stay active and this indicates that the lung epithelial cells do not respond to changes in post-natal nutrient availability. This pattern is unchanged from ED18.5, where nutrients are known to be abundant. These results indicate that there is a feedback loop between both of the MTOR complexes and ULK1 and ULK2. The levels of p-PRKAA/AMPKα in ED18.5 and P1 wild-type lungs are similar as well as those in the Atg5 KO and littermate control. Although the levels of p-PRKAA are lower in the Ulk1/2 litter compared with the wild-type sample in lane 2, the levels of p-PRKAA in the Ulk1/2 DKO and Ulk1/2 CTRL are similar. In addition, for all the litters there is no correlation between the levels of p-PRKAA and autophagy. Our results indicated that induction of autophagy in neonatal lung is independent of increased PRKAA phosphorylation. In summary, we showed that autophagy is abundant in the normal, neonatal lung but is lacking in both Atg5 KO and Ulk1/2 DKO neonatal mice.

Figure 7. Analysis of autophagy in Ulk1/2 DKO and Atg5 KO neonatal lung tissue. Immunohistochemical staining of lung sections from (A) wild-type (WT) and (B) Ulk1/2 DKO mice at P1 with a LC3 antibody is shown after fixation, embedding in paraffin, and sectioning as described in Materials and Methods. Arrowheads in (A) show examples of alveolar epithelial cells with puncta. The arrow in (B) shows an example of an alveolar epithelial cell with diffuse, cytoplasmic LC3 staining. Scale bars: 20 μm. (C) Western blot analysis of protein lysates of lungs from wild-type mice at ED18.5 and P1, and from Atg5 KO mice and Ulk1/2 DKO mice and their respective littermate controls at P1 are shown. Blots were probed with antibodies specific for LC3, phospho-RPS6KB (Thr389), phospho-AKT (Ser473) and phospho-PRKAA/AMPKα (Thr172). Antibodies specific for total RPS6KB, AKT and PRKAA/AMPKα as well as ACTB were included as controls. Wild-type MEFs, which were serum starved and replated in medium with 10% FCS in the presence of Torin1 at 0.25 μM, were included as a positive control for autophagy.
Discussion
It is well established that during lung development large amounts of glycogen accumulate in the undifferentiated epithelial cells and that these glycogen stores are depleted as the epithelium matures, utilized intracellularly to provide substrates for the enhanced synthesis of surfactant phospholipids.20,21 Specifically, it was found in normal mice that PC synthesis started to increase between ED17 to 18 and glycogen depletion started at ED18 as the synthetic rate of PC continued to rise through the newborn period. Due to this inverse relationship between glycogen levels and PC synthetic rate, it has been suggested that glycogen is the substrate for the surfactant lipids, of which PC is the predominant species.22-24 However, it remains controversial whether failure to utilize glycogen is sufficient to disrupt PC synthesis and reduce surfactant PC levels sufficiently to impair perinatal transition to air breathing.
Our data confirmed that from ED18.5 to the newborn period, there is a sharp increase in maximal lung PC synthetic rate in normal mice. The similar content of total lung PL in Ulk1/2 DKO, Atg5 KO and control mice lungs at P1, suggests no deficiency of surfactant PC in autophagy-deficient mice that might be contributing to perinatal lethality in Ulk1/2 DKO or Atg5 KO mice. Likewise, the increase in incorporation of exogenously supplied choline between ED18.5 and P1 shows a normal developmental increase for all groups regardless of genotype, albeit the increase is greater for the autophagy-deficient mice. Furthermore, the similarities in the percentage of newly synthesized saturated PC between the groups support the capacity for normal surfactant PC synthesis in both autophagy-deficient mice and their littermate controls. The elevated rate of incorporation of exogenous choline into PC seen in autophagy-deficient mice compared with their littermate controls in the perinatal period may reflect a compensatory mechanism resulting from an inability to generate synthetic intermediates from intracellular metabolism of glycogen stores.
The striking increase in cellular thickness in alveolar epithelial cells during the perinatal period in autophagy-deficient mice suggests that a significant barrier to gas exchange and/or pulmonary ventilation results from the failure of alveolar epithelial cells to clear their embryonically accumulated glycogen stores. Despite their ability to undergo differentiation and to induce the enzymes for surfactant phospholipid production, in the absence of autophagy, alveolar epithelial cells are unable to clear cytoplasmic glycogen. The Ulk1/2 and Atg5 KO mouse models suggest that autophagy is required to prevent persistent glycogen deposition within the alveolar epithelium, thereby facilitating the thinning of the alveolocapillary membrane that is necessary for effective gas exchange during the transition to air breathing. The respiratory distress of autophagy-deficient mice is further compounded by the lack of nutrients in the neonatal period which is normally provided via more global autophagy of glycogen in other tissues before efficient nursing is established.
The well-described gsd/gsd rat has a glycogen storage disease with increased glycogen levels in liver as well as in the lungs due to a deficiency of the glycogenolytic enzyme phosphorylase b kinase resulting in significantly reduced kinase activity.25-28 Reduced viability during the perinatal period has been reported for the gsd/gsd rat but interestingly a significant portion of the pups survive to adulthood.26 The gsd/gsd rat exhibits a one-day delay in peak PC synthesis, at P1 as opposed to before birth in littermate controls. As in the Ulk1/2 DKO and Atg5 KO mice, increased PC synthesis in gsd/gsd rats was not accompanied by an increase in total phospholipid. In addition, the levels of the surfactant protein SFTPA are similar in control and gsd/gsd rats.26 These studies suggest that glycogen may not be the only intracellular constituent that must be cleared from alveolar epithelial cells during the perinatal period, and support the need for examination of lung phospholipids in mouse models demonstrating persistent glycogen in alveolar epithelium. This is especially important when the genes involved may play a more expanded role in lung development and/or type II cell differentiation. In addition, it should also be noted that glycogen storage disorders in humans are not associated with pulmonary symptoms in the neonatal period,29 indicating that there could be species-specific differences in the response to glycogen accumulation in alveolar type II cells.
Autophagy provides an important source of nutrients needed during the initial postnatal period.9 Amino acids generated from autophagic breakdown of proteins as well as glucose generated from glycogenolysis are sources of nutrients for the newborn animal. Normally, glycogen mobilization from the lung epithelium begins prenatally and continues through the early postnatal period. Our data strongly suggest that this mobilization of glycogen also involves autophagy although it may be initiated by signals different from the autophagy induced by systemic decline in extracellular nutrients that accompany maternal/placental separation. In support of this hypothesis we showed that in intact lung tissue autophagy takes place in neonatal lung epithelial cells of wild-type mice at P1, but that this does not occur in Atg5 KO and Ulk1/2 DKO lung epithelial cells. Interestingly, in the absence of ULK1 and ULK2, MTORC1 and MTORC2 stay active as evidenced by elevated phosphorylation of downstream targets. This suggests that a feedback loop exists between both MTOR complexes and ULK1 and ULK2. This is supported by experiments in Drosophila where overexpression of Atg1 leads to negative feedback of TOR activity.30 In vitro experiments, using ulk1−/− MEF and knockdown of Ulk1 in other cell types, show a similar negative feedback loop between ULK1 and MTORC1.31,32 Our experiments are the first to report that, in intact tissue, a feedback loop exists between both MTORC1 and MTORC2 and ULK1 and ULK2. Our data also indicate that the previously described ULK1/2-independent autophagy pathway is not active during neonatal lung development.17,33
In insects, autophagy has been shown to be required to remodel cells and tissues during developmental morphogenesis.34-37 This essential role for autophagy is separate from its role in supporting cell survival in response to nutrient starvation and/or cellular damage. The studies presented here suggested that mammalian species may also use autophagy to complete the physical morphogenesis of lung epithelium as the lung transits from a differentiating tissue into a gas-exchanging organ.
Materials and Methods
Mice
The generation of ulk1−/− and ulk2−/− mice has been described previously.16,17 Atg5+/− mice were generously provided by Dr Noboru Mizushima (Tokyo Medical and Dental Univeristy, Bunkyo-Ku, Japan). ulk1−/− ulk2−/− (Ulk1/2 DKO) mice were generated by matings of either ulk1+/− ulk2−/− males and females or ulk1−/− ulk2+/− males and females. atg5−/− mice were generated by the mating of atg5+/− males and females. For some experiments pregnant females were sacrificed at ED 18.5 and embryos were removed for further analysis. For other experiments newborn pups were sacrificed after birth. Mice were cared for in accordance with the Institutional Animal Care and Use Committees at the University of Pennsylvania and Memorial Sloan-Kettering Cancer Center.
Histological analysis
Organs were fixed in 10% buffered formalin or in some cases 4% paraformaldehyde overnight followed by dehydration, embedding, and sectioning following standard protocols. Sections were subjected to hematoxylin-eosin (HE) staining for routine analysis. Sections were also stained with Periodic Acid Schiff stain (PAS, Sigma-Aldrich, 395) followed by counterstaining with methyl green, to visualize intracellular glycogen according to the manufacturer’s instructions. Immunohistochemistry for PDPN/T1-α (Developmental Studies Hybridoma Bank at the University of Iowa, 8.1.1), pro-SFTPC (Chemicon, AB3786) and SFTPB (Chemicon, AB3780) were performed using standard protocols. Immunohistochemistry with an antibody specific for MAP1LC3B (LC3B) that shows strong reactivity with the type II form of LC3 (Cell Signaling, 2775) was performed as previously described.38 For electron microscopy, organs were finely minced and fixed in 2% glutaraldehyde in 0.1 M sodium cacodylate buffer overnight followed by washing in cacodylate buffer and incubation with 2% osmium tetroxide. Gradual dehydration of the samples was done with acetone followed by embedding in Epon. Blocks were stained with 2% uranyl acetate. Images were examined with a JEOL-1010 electron microscope (JEOL).
Glycogen assay
Glycogen assay kits were purchased from MBL International (JM-K646-100) and the assays were done according to the manufacturer’s instructions on freshly homogenized lungs from Ulk1/2 DKO and Atg5 KO mice and their littermate controls. Determination of protein concentrations of the homogenates were done with the BCA Protein Assay Kit (Thermo Fisher Scientific Inc., 23325) according to manufacturer’s instructions.
RT-PCR
Lungs from mutant mice and littermate controls were frozen and pulverized with a Bessman Tissue Pulverizer (Spectrum Chemical & Laboratory Products) and RNA was prepared using Qiashredder (Qiagen, 79654) and RNeasy Mini Kit (Qiagen, 74104) with on-column RNase-free DNase treatment (Qiagen, 79254).
A singleplex PCR strategy with an ABI Prism 7900 system (ABI) was used as described previously.39 The primer/probe sets used were: SFTPA/SP-A Mm00499170_m1, SFTPB/SP-B Mm00455681_m1, SFTPC/SP-C Mm00488144_m1, SFTPD/SP-D Mm00486060_m1, ABCA3 Mm00550501_m1, FASN/FAS Mm0125329_m1, TTF1 Mm00657018_m1, and 18s Hs99999901_s1. Results are depicted as relative quantities (RQ) of RNA after correcting for 18S to normalize for variability in loading, followed by normalizing to an appropriate littermate control sample.
Western blot analysis
Frozen lung tissue was pulverized and then solubilized in lysis buffer (1% Triton X-100, 150 mM NaCl, 50 mM TRIS-HCl, 5 mM ethylenediamine tetraacetic acid, 5% glycerol, pH 8.0) with 1X protease inhibitor (Roche Applied Bioscience, 05892988001). Samples containing equal amounts of total protein were immunoblotted using NuPAGE Bis-Tris gels (Invitrogen). Primary antibodies used were: SFTPB/SP-B40 (generously provided by Michael Beers, University of Pennsylvania), NFlank SFTPB/SP-B41 (prepared by S Guttentag). Secondary antibodies conjugated to Alexa Fluor 680 (Molecular Probes, A10043) or IRdye 800 (Rockland, 611-732-127) were used. Membranes were analyzed using the Odyssey infrared imaging system (Li-Cor). To study expression of enzymes involved in glycogen metabolism as well as autophagy signaling pathways by western blot, frozen lung tissue was pulverized, lysed, and homogenized in RIPA buffer (50 mM Tris-Cl pH 7.4, 150 mM NaCl, 1% NP-40, 0.5% deoxycholate, 0.1% SDS, 1 mM EDTA) supplemented with protease inhibitor cocktail (Roche Applied Bioscience, 05892988001) and phosphatase inhibitors (Sigma-Aldrich, P5726 and P0044). Lysates were resolved by SDS-PAGE and blots were probed using the following primary antibodies: anti-phospho-GYS1 (glycogen synthase [Epitomics, 1919-1]), anti-GYS1 (glycogen synthase [Cell Signaling, 3886]), anti-PHKG2 (phosphorylase kinase, gamma 2 [testis]) (GenWay Biotech, 18-003-42332), anti-PYGB/L/M (phosphorylase, glycogen; brain/liver/muscle) (Santa Cruz Biotechnology, sc-66913), anti-phospho-GSK3B (glycogen synthase kinase 3 β [Cell Signaling, 9323]), anti-GSK3B (Cell Signaling, 9315) and anti-ACTB (β-actin) (Sigma Aldrich, A5441). To study autophagy signaling pathways the following antibodies were used; anti-LC3 (generated by Quality Control Biochemicals), anti-phospho p70 S6 Kinase (RPS6KB) Thr389 (p-RPS6KB) (Cell Signaling, 9234), anti-RPS6KB (p70S6 kinase) (Cell Signaling, 2708), anti-phospho PRKAA/AMPKα Thr172 (p-PRKAA) (Cell Signaling, 2535), anti-PRKAA/AMPK-α (PRKAA) (Cell Signaling, 2532), anti-phospho AKT Ser473 (p-AKT) (Cell Signaling, 4060), and anti-AKT (AKT) (Cell Signaling, 9272). Bands were detected using horseradish peroxidase-labeled secondary antibodies and enhanced chemiluminescence detection kit (GE Healthcare, RPN2135).
Phospholipid analysis
Each mouse lung was removed and stored at −70 °C. Following genotyping each lung was sonicated in 1 ml 0.15 M NaCl, of which 100 μl was used for protein assay and the remainder extracted with chloroform/methanol for total lipid by the Bligh-Dyer method.42 The lipid extract was dried, resuspended in chloroform, and aliquots assayed by ultramicrophosphorus assay43 to quantitate total PL.
Phosphatidylcholine (PC) synthesis in mouse lung tissue
Each mouse lung was removed, stored overnight at 4 °C in 500 μl MEM media (Invitrogen, 11095). For PC synthesis study, each lung was dissected free of airway, and lobes cut apart, then lobes from each lung were placed in 300 μl Ham’s F12 (Invitrogen, 11765054) supplemented with 15 mM Hepes, 0.8 mM CaCl2, 0.25% BSA, plus the HITES combination (hydrocortisone, 10 nM; insulin, 5 ug/ml; transferrin,10 μg/ml; 17-B-estradiol, 10 nM; selenium, 30 nM), for ~1 h at 37 °C to equilibrate temperature and restore metabolism. After 1 h, fresh media was added containing 3H-choline precursor (choline chloride [methyl-3H]), (New England Nuclear, Net109111MC, 80.6 Ci/mmol) 6 μCi/ml media, 0.10 mM total choline] and incubation continued at 37 °C for 6 h, on a rocker platform in the incubator with 5% CO2 . After 6 h incorporation, labeled media was removed, tissue rinsed with PBS, and stored at −80 °C to allow time for genotyping. Lungs of selected genotypes were sonicated in 1 ml 0.9% NaCl, from which 100 μl was reserved for protein assay, and 900 μl was extracted with chloroform/methanol to extract total lipid (100 μg DPPC + 100 μg egg PC were added as carrier). An aliquot (1/6 in duplicate) was taken from which PC was isolated by thin-layer chromatography (TLC), as previously described44 and spots were visualized with 0.1% ANS (8-anilino-1-naphthalene-sulfonic acid, Sigma A-1028) aqueous spray and scraped into scintillation vials for total PC counts. Incorporation into total PC was expressed as nmol choline incorporated per 6 h per mg protein. To determine percent saturation of newly synthesized PC, aliquots of total lipid extract (1/6, in duplicate) were subjected to TLC to isolate total PC, which was eluted from the plate, treated with OsO4, and then further separated by TLC on borate-dipped plates to separate saturated PC from unsaturated PC which were scraped individually for counting as above.44
Blood glucose measurements
Blood glucose measurements were performed on serum samples using an YSI 2300 nutrient analyzer (YSI Life Sciences).
Statistical analysis
The Student 2-tailed t test and One-way Anova with Bonferroni’s multiple comparison test were used.
Supplementary Material
Acknowledgments
The expert technical assistance of Hongwei Yu, Raymond Meade, Min Min Lu, Peggy Zhang and Ping Wang and the Molecular Cytology Core Facility at MSKCC is gratefully acknowledged. The authors thank members of the Thompson Laboratory, Drs Edward Morrisey and Anthony Mancuso for technical assistance and helpful discussions. This study was funded in part by grants from the NCI (CBT) and by HL059959 (SHG).
Glossary
Abbreviations:
- CTSH
cathepsin H
- LC3
(MAP1LC3), microtubule-associated protein 1 light chain 3
- MEF
mouse embryonic fibroblast
- MTORC1 and 2
mechanistic target of rapamycin complex 1 and 2
- PC
phosphatidylcholine
- PL
phospholipid
Disclosure of Potential Conflicts of interest
CBT is a founder and consultant of Agios Pharmaceuticals and has a financial interest in Agios. CBT is also on the Board of Directors of Merck.
Supplemental Materials
Supplemental materials may be found here: www.landesbioscience.com/journals/autophagy/article/26505
Footnotes
Previously published online: www.landesbioscience.com/journals/autophagy/article/26505
References
- 1.Shintani T, Klionsky DJ. Autophagy in health and disease: a double-edged sword. Science. 2004;306:990–5. doi: 10.1126/science.1099993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Yang Z, Klionsky DJ. Eaten alive: a history of macroautophagy. Nat Cell Biol. 2010;12:814–22. doi: 10.1038/ncb0910-814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Mizushima N, Levine B, Cuervo AM, Klionsky DJ. Autophagy fights disease through cellular self-digestion. Nature. 2008;451:1069–75. doi: 10.1038/nature06639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Rabinowitz JD, White E. Autophagy and metabolism. Science. 2010;330:1344–8. doi: 10.1126/science.1193497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Mizushima N, Levine B. Autophagy in mammalian development and differentiation. Nat Cell Biol. 2010;12:823–30. doi: 10.1038/ncb0910-823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Yue Z, Jin S, Yang C, Levine AJ, Heintz N. Beclin 1, an autophagy gene essential for early embryonic development, is a haploinsufficient tumor suppressor. Proc Natl Acad Sci U S A. 2003;100:15077–82. doi: 10.1073/pnas.2436255100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Qu X, Yu J, Bhagat G, Furuya N, Hibshoosh H, Troxel A, Rosen J, Eskelinen EL, Mizushima N, Ohsumi Y, et al. Promotion of tumorigenesis by heterozygous disruption of the beclin 1 autophagy gene. J Clin Invest. 2003;112:1809–20. doi: 10.1172/JCI20039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sou YS, Waguri S, Iwata J, Ueno T, Fujimura T, Hara T, Sawada N, Yamada A, Mizushima N, Uchiyama Y, et al. The Atg8 conjugation system is indispensable for proper development of autophagic isolation membranes in mice. Mol Biol Cell. 2008;19:4762–75. doi: 10.1091/mbc.E08-03-0309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kuma A, Hatano M, Matsui M, Yamamoto A, Nakaya H, Yoshimori T, Ohsumi Y, Tokuhisa T, Mizushima N. The role of autophagy during the early neonatal starvation period. Nature. 2004;432:1032–6. doi: 10.1038/nature03029. [DOI] [PubMed] [Google Scholar]
- 10.Komatsu M, Waguri S, Ueno T, Iwata J, Murata S, Tanida I, Ezaki J, Mizushima N, Ohsumi Y, Uchiyama Y, et al. Impairment of starvation-induced and constitutive autophagy in Atg7-deficient mice. J Cell Biol. 2005;169:425–34. doi: 10.1083/jcb.200412022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Saitoh T, Fujita N, Hayashi T, Takahara K, Satoh T, Lee H, Matsunaga K, Kageyama S, Omori H, Noda T, et al. Atg9a controls dsDNA-driven dynamic translocation of STING and the innate immune response. Proc Natl Acad Sci U S A. 2009;106:20842–6. doi: 10.1073/pnas.0911267106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Saitoh T, Fujita N, Jang MH, Uematsu S, Yang BG, Satoh T, Omori H, Noda T, Yamamoto N, Komatsu M, et al. Loss of the autophagy protein Atg16L1 enhances endotoxin-induced IL-1beta production. Nature. 2008;456:264–8. doi: 10.1038/nature07383. [DOI] [PubMed] [Google Scholar]
- 13.Cadwell K, Liu JY, Brown SL, Miyoshi H, Loh J, Lennerz JK, Kishi C, Kc W, Carrero JA, Hunt S, et al. A key role for autophagy and the autophagy gene Atg16l1 in mouse and human intestinal Paneth cells. Nature. 2008;456:259–63. doi: 10.1038/nature07416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kotoulas OB, Kalamidas SA, Kondomerkos DJ. Glycogen autophagy in glucose homeostasis. Pathol Res Pract. 2006;202:631–8. doi: 10.1016/j.prp.2006.04.001. [DOI] [PubMed] [Google Scholar]
- 15.Kalamidas SA, Kondomerkos DJ. The administration of nonmetabolizable glucose analogues fails to suppress the development of glycogen autophagy in newborn rat hepatocytes. Microsc Res Tech. 2010;73:1009–14. doi: 10.1002/jemt.20825. [DOI] [PubMed] [Google Scholar]
- 16.Kundu M, Lindsten T, Yang CY, Wu J, Zhao F, Zhang J, Selak MA, Ney PA, Thompson CB. Ulk1 plays a critical role in the autophagic clearance of mitochondria and ribosomes during reticulocyte maturation. Blood. 2008;112:1493–502. doi: 10.1182/blood-2008-02-137398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cheong H, Lindsten T, Wu J, Lu C, Thompson CB. Ammonia-induced autophagy is independent of ULK1/ULK2 kinases. Proc Natl Acad Sci U S A. 2011;108:11121–6. doi: 10.1073/pnas.1107969108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Melton KR, Nesslein LL, Ikegami M, Tichelaar JW, Clark JC, Whitsett JA, Weaver TE. SP-B deficiency causes respiratory failure in adult mice. Am J Physiol Lung Cell Mol Physiol. 2003;285:L543–9. doi: 10.1152/ajplung.00011.2003. [DOI] [PubMed] [Google Scholar]
- 19.Ten Have-Opbroek AA, De Vries EC. Clara cell differentiation in the mouse: ultrastructural morphology and cytochemistry for surfactant protein A and Clara cell 10 kD protein. Microsc Res Tech. 1993;26:400–11. doi: 10.1002/jemt.1070260508. [DOI] [PubMed] [Google Scholar]
- 20.Brehier A, Rooney SA. Phosphatidylcholine synthesis and glycogen depletion in fetal mouse lung: developmental changes and the effects of dexamethasone. Exp Lung Res. 1981;2:273–87. doi: 10.3109/01902148109052323. [DOI] [PubMed] [Google Scholar]
- 21.Rinaudo MT, Curto M, Bruno R, Ponzetto C. Some aspects of carbohydrate metabolism in rat lung during the period of growth. Int J Biochem. 1981;13:571–5. doi: 10.1016/0020-711X(81)90182-8. [DOI] [PubMed] [Google Scholar]
- 22.Bourbon JR, Rieutort M, Engle MJ, Farrell PM. Utilization of glycogen for phospholipid synthesis in fetal rat lung. Biochim Biophys Acta. 1982;712:382–9. doi: 10.1016/0005-2760(82)90356-3. [DOI] [PubMed] [Google Scholar]
- 23.Carlson KS, Davies P, Smith BT, Post M. Temporal linkage of glycogen and saturated phosphatidylcholine in fetal lung type II cells. Pediatr Res. 1987;22:79–82. doi: 10.1203/00006450-198707000-00018. [DOI] [PubMed] [Google Scholar]
- 24.Ridsdale R, Post M. Surfactant lipid synthesis and lamellar body formation in glycogen-laden type II cells. Am J Physiol Lung Cell Mol Physiol. 2004;287:L743–51. doi: 10.1152/ajplung.00146.2004. [DOI] [PubMed] [Google Scholar]
- 25.Clark DG, Topping DL, Illman RJ, Trimble RP, Malthus RS. A glycogen storage disease (gsd/gsd) rat: studies on lipid metabolism, lipogenesis, plasma metabolites, and bile acid secretion. Metabolism. 1980;29:415–20. doi: 10.1016/0026-0495(80)90165-1. [DOI] [PubMed] [Google Scholar]
- 26.Rannels SR, Rannels SL, Sneyd JG, Loten EG. Fetal lung development in rats with a glycogen storage disorder. Am J Physiol. 1991;260:L419–27. doi: 10.1152/ajplung.1991.260.6.L419. [DOI] [PubMed] [Google Scholar]
- 27.Rannels SR. Impaired surfactant synthesis in fetal type II lung cells from gsd/gsd rats. Exp Lung Res. 1996;22:213–29. doi: 10.3109/01902149609050848. [DOI] [PubMed] [Google Scholar]
- 28.Liu L, Rannels SR, Falconieri M, Phillips KS, Wolpert EB, Weaver TE. The testis isoform of the phosphorylase kinase catalytic subunit (PhK-gammaT) plays a critical role in regulation of glycogen mobilization in developing lung. J Biol Chem. 1996;271:11761–6. doi: 10.1074/jbc.271.20.11761. [DOI] [PubMed] [Google Scholar]
- 29.Dimauro S, Garone C. Metabolic disorders of fetal life: glycogenoses and mitochondrial defects of the mitochondrial respiratory chain. Semin Fetal Neonatal Med. 2011;16:181–9. doi: 10.1016/j.siny.2011.04.010. [DOI] [PubMed] [Google Scholar]
- 30.Scott RC, Juhász G, Neufeld TP. Direct induction of autophagy by Atg1 inhibits cell growth and induces apoptotic cell death. Curr Biol. 2007;17:1–11. doi: 10.1016/j.cub.2006.10.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Jung CH, Seo M, Otto NM, Kim DH. ULK1 inhibits the kinase activity of mTORC1 and cell proliferation. Autophagy. 2011;7:1212–21. doi: 10.4161/auto.7.10.16660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Dunlop EA, Hunt DK, Acosta-Jaquez HA, Fingar DC, Tee AR. ULK1 inhibits mTORC1 signaling, promotes multisite Raptor phosphorylation and hinders substrate binding. Autophagy. 2011;7:737–47. doi: 10.4161/auto.7.7.15491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gammoh N, Florey O, Overholtzer M, Jiang X. Interaction between FIP200 and ATG16L1 distinguishes ULK1 complex-dependent and -independent autophagy. Nat Struct Mol Biol. 2013;20:144–9. doi: 10.1038/nsmb.2475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Beaulaton J, Lockshin RA. Ultrastructural study of the normal degeneration of the intersegmental muscles of Anthereae polyphemus and Manduca sexta (Insecta, Lepidoptera) with particular reference of cellular autophagy. J Morphol. 1977;154:39–57. doi: 10.1002/jmor.1051540104. [DOI] [PubMed] [Google Scholar]
- 35.Scharrer B. Ultrastructural study of the regressing prothoracic glands of blattarian insects. Z Zellforsch Mikrosk Anat. 1966;69:1–21. doi: 10.1007/BF00406264. [DOI] [PubMed] [Google Scholar]
- 36.Schin KS, Clever U. Lysosomal and free acid phosphatase in salivary glands of chironomus tentans. Science. 1965;150:1053–5. doi: 10.1126/science.150.3699.1053. [DOI] [PubMed] [Google Scholar]
- 37.Schin K, Laufer H. Studies of programmed salivary gland regression during larval-pupal transformation in Chironomus thummi. I. Acid hydrolase activity. Exp Cell Res. 1973;82:335–40. doi: 10.1016/0014-4827(73)90350-9. [DOI] [PubMed] [Google Scholar]
- 38.Holt SV, Wyspianska B, Randall KJ, James D, Foster JR, Wilkinson RW. The development of an immunohistochemical method to detect the autophagy-associated protein LC3-II in human tumor xenografts. Toxicol Pathol. 2011;39:516–23. doi: 10.1177/0192623310396903. [DOI] [PubMed] [Google Scholar]
- 39.Foster C, Aktar A, Kopf D, Zhang P, Guttentag S. Pepsinogen C: a type 2 cell-specific protease. Am J Physiol Lung Cell Mol Physiol. 2004;286:L382–7. doi: 10.1152/ajplung.00310.2003. [DOI] [PubMed] [Google Scholar]
- 40.Beers MF, Bates SR, Fisher AB. Differential extraction for the rapid purification of bovine surfactant protein B. Am J Physiol. 1992;262:L773–8. doi: 10.1152/ajplung.1992.262.6.L773. [DOI] [PubMed] [Google Scholar]
- 41.Korimilli A, Gonzales LW, Guttentag SH. Intracellular localization of processing events in human surfactant protein B biosynthesis. J Biol Chem. 2000;275:8672–9. doi: 10.1074/jbc.275.12.8672. [DOI] [PubMed] [Google Scholar]
- 42.Bligh EG, Dyer WJ. A rapid method of total lipid extraction and purification. Can J Biochem Physiol. 1959;37:911–7. doi: 10.1139/o59-099. [DOI] [PubMed] [Google Scholar]
- 43.Bartlett GR. Phosphorus assay in column chromatography. J Biol Chem. 1959;234:466–8. [PubMed] [Google Scholar]
- 44.Gonzales LW, Ballard PL, Ertsey R, Williams MC. Glucocorticoids and thyroid hormones stimulate biochemical and morphological differentiation of human fetal lung in organ culture. J Clin Endocrinol Metab. 1986;62:678–91. doi: 10.1210/jcem-62-4-678. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.