

Abstract
Mitophagy, the autophagic removal of mitochondria, occurs through a highly selective mechanism. In the yeast Saccharomyces cerevisiae, the mitochondrial outer membrane protein Atg32 confers selectivity for mitochondria sequestration as a cargo by the autophagic machinery through its interaction with Atg11, a scaffold protein for selective types of autophagy. The activity of mitophagy in vivo must be tightly regulated considering that mitochondria are essential organelles that produce most of the cellular energy, but also generate reactive oxygen species that can be harmful to cell physiology. We found that Atg32 was proteolytically processed at its C terminus upon mitophagy induction. Adding an epitope tag to the C terminus of Atg32 interfered with its processing and caused a mitophagy defect, suggesting the processing is required for efficient mitophagy. Furthermore, we determined that the mitochondrial i-AAA protease Yme1 mediated Atg32 processing and was required for mitophagy. Finally, we found that the interaction between Atg32 and Atg11 was significantly weakened in yme1∆ cells. We propose that the processing of Atg32 by Yme1 acts as an important regulatory mechanism of cellular mitophagy activity.
Keywords: mitochondrial protease, mitophagy, starvation, vacuole, yeast
Introduction
Mitophagy is the cellular process of autophagic degradation of mitochondria.1 The molecular mechanism and physiological significance of mitophagy have been extensively studied in recent years in both yeast and mammalian cells. And the importance of mitophagy as a primary mitochondrial quality control system has been gradually revealed.
Mitochondria are essential organelles, but they are costly to maintain. Oxidative phosphorylation inside mitochondria provides the cells with energy to support a variety of cellular activities, while reactive oxygen species, the inevitable byproduct of oxidative respiration, has the potential to cause severe damage to cellular components, with mitochondria themselves as the most vulnerable targets.2 Mitochondrial damage and dysfunction have severe cellular consequences and are linked with neurodegenerative diseases and aging.3 Therefore, the amount and activity of mitochondria need to be tightly controlled, and excessive or damaged mitochondria need to be removed in a timely manner.
In fact, mitochondria have their own quality control systems including a protein degradation system,4 a DNA repair system,5 and phospholipid hydroperoxide glutathione peroxidase that is protective against membrane-damaging lipid peroxidation.6 In particular, the highly conserved, intraorganellar proteolytic system conducts the surveillance of protein quality control within mitochondria. Molecular chaperones and energy-dependent proteases monitor the folding status of mitochondrial proteins and remove excess and damaged proteins.4 A variety of ATP-dependent proteases are present in different subcompartments of the mitochondria, recognizing non-native polypeptides and triggering their proteolysis to peptides that are further degraded by oligopeptidases.7
Despite these mitochondrial quality control systems, autophagy has been recently demonstrated to play important roles in mitochondrial maintenance by delivering excess and damaged mitochondria to the vacuole/lysosome for degradation.8 Autophagy is a conserved cellular pathway in which double-membrane phagophores that form in the cytosol have the capacity to sequester essentially any cellular component.9 In fact, mitochondria were one of the first documented autophagic cargos.10 Recent studies in both yeast and mammals have greatly expanded our knowledge of the selective autophagic degradation of mitochondria, or mitophagy.11-16
In mammalian cells, damaged mitochondria with disrupted membrane potential are selectively recognized and degraded by autophagy.15 During this process, the E3 ubiquitin ligase PARK2/Parkin is selectively recruited to damaged mitochondria to facilitate recognition by the autophagic machinery. In addition, during erythroid cell maturation, mitochondria are eliminated by autophagy when BNIP3L/Nix-dependent loss of membrane potential is induced.17 Mitochondria elimination is also observed during spermatogenesis to prevent paternal mitochondrial DNA transmission.18 These studies demonstrated that mitophagy plays important roles in both cellular homeostasis and development.
In yeast, mitophagy has been characterized as a type of selective autophagy, similar to the cytoplasm-to-vacuole targeting (Cvt) pathway and pexophagy.13 Mitophagy in yeast requires the scaffold protein Atg1113,19 and the receptor protein Atg32.11,12 Atg11 is the common scaffold protein for selective types of autophagy that links the cargo to the core autophagic machinery.20 In contrast, Atg32 is specific for mitophagy; it is a mitochondrial outer membrane protein that confers selectivity for mitophagy through its interaction with Atg11 under mitophagy-inducing conditions.11,12 Atg32 contains a single transmembrane domain with its N terminus facing the cytosol and the C terminus in the intermembrane space (IMS).
The N-terminal cytosolic domain of Atg32 mediates the interaction with Atg11 and is critical for mitophagy.21 In contrast, two recent studies suggested that the C-terminal 117 amino acids in the IMS are dispensable for mitophagy.21,22 Kondo-Okamoto et al. reported that an Atg32 mutant engineered such that the N terminus is targeted to mitochondria is sufficient to promote mitophagy. Similarly, Aoki et al.21 reported that deletion of up to 100 amino acids of the C terminus of Atg32 caused no defect in mitophagy activity. These results demonstrated that the C-terminal 20 amino acids next to the transmembrane domain are important for the mitochondrial targeting of Atg32, while the majority of the C-terminal domain in the IMS is not needed for mitophagy.
Atg32 is phosphorylated at its N-terminal domain by an unknown kinase.21 This phosphorylation is required for mitophagy and is affected by Hog1 and Pbs2, which are involved in the osmoregulatory signal transduction cascade, suggesting that cytosolic factors contribute to the regulation of mitophagy by modifying Atg32. Considering that the IMS domain of Atg32 could be a potential module that can communicate with other proteins or sense physiological changes inside the organelle, we decided to explore the function of this domain.
We found that the C-terminal IMS domain of Atg32 needs to be removed for proper mitophagy activity. We observed that Atg32 undergoes a molecular mass shift upon mitophagy induction, caused by the processing of its C terminus. Blocking this processing caused a significant defect in mitophagy. We determined that the mitochondrial i-AAA protease Yme1 is responsible for the proteolytic processing of Atg32, and that Yme1 activity is required for mitophagy. Our results suggest that mitophagy is regulated in part by factors inside mitochondria. In addition, since Yme1 is responsible for degradation of unfolded or misfolded mitochondrial gene products,23 these findings established a link between mitophagy and other mitochondrial quality control systems.
Results
Atg32 undergoes a starvation-dependent molecular mass shift
Mitophagy is induced when cells are cultured in medium using nonfermentable substrates such as lactate as the sole carbon source and then shifted to nitrogen starvation medium supplemented with glucose.13 The mitochondrial outer membrane protein Atg32 is essential for this process, acting as the receptor protein that confers selectivity for mitophagy.11,24 The protein level of endogenous Atg32 is low and therefore it is difficult to detect. Accordingly, we generated strains expressing N-terminal TAP tagged Atg32 under the control of the GAL1 promoter as described previously.25 The protein level of Atg32 in this construct is greatly increased and the TAP tag allows sensitive detection of the protein by western blotting. Overexpression of N-terminal tagged Atg32 has previously been shown to be functional for mitophagy.24 Nonetheless, we confirmed our results by also examining the endogenous level of Atg32; expression of the Cre recombinase makes it possible to remove the GAL1 promoter, leaving the N-terminal TAP tagged Atg32 under the control of its native promoter.
With the overexpression strain, we found that TAP-Atg32 generated a second band with faster migration upon mitophagy induction (Fig. 1A, arrowhead). The intensity of the upper band, corresponding to full-length Atg32, gradually decreased during starvation, while the lower band gradually increased, suggesting a precursor-product relationship. Considering that the TAP tag was added to the N terminus, we hypothesized that formation of the lower band might be caused by processing of the C-terminal region of Atg32. To test this hypothesis, we decided to make C-terminal truncations of Atg32 and monitor the gel migration of the mutated constructs. A previous study demonstrated that the C-terminal region of Atg32, consisting of 117 amino acids, is in the mitochondrial IMS.11 Accordingly, we generated Atg32 C-terminal truncations of 90, 100, or 117 amino acids and examined their migration patterns. No lower bands corresponding to a processed form of the truncated mutants was observed, suggesting that cleavage indeed occurs at the C terminus of Atg32 upon mitophagy induction (Fig. S1).
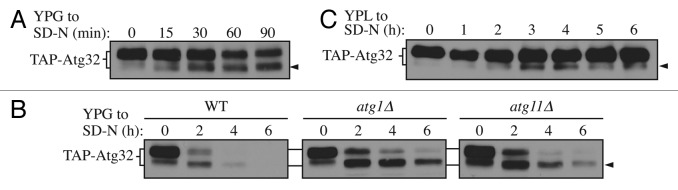
Figure 1. Atg32 undergoes starvation-dependent processing. (A) Cells expressing TAP tagged Atg32 under the control of the GAL1 promoter (KWY100) were cultured in YPG to mid-log phase and shifted to SD-N for the indicated times. TAP-Atg32 was monitored by immunoblotting with an antibody that recognizes PA. The arrowhead here and in the following panels indicates the processed band. (B) Cells expressing GAL1 promoter-driven TAP-Atg32 in wild-type (WT; KWY100), atg1∆ (KWY101), and atg11∆ (KWY104) backgrounds were cultured in YPG to mid-log phase and shifted to SD-N for the indicated times. (C) Cells expressing endogenous promoter-driven TAP-Atg32 in a WT background (KWY139) were cultured in YPL to mid-log phase and shifted to SD-N for the indicated times.
Atg32 is degraded along with the part of the degrading mitochondria during mitophagy.11,12 Therefore, we decided to examine the two forms of Atg32 during mitophagy. In wild-type cells, both forms of Atg32 were undetectable by 4 h after mitophagy induction (Fig. 1B). However, in atg1∆ and atg11∆ cells, which are defective for mitophagy, the lower band accumulated relative to the full-length protein, suggesting the faster-migrating species of Atg32 is the form that participates in mitophagic degradation, whereas the slower-migrating form of Atg32 serves as a precursor that is processed under mitophagy-inducing conditions to generate the faster-migrating form. Both forms of Atg32 were eventually degraded, even in autophagy mutants. This loss of signal could be explained by the fact that both autophagic and nonautophagic pathways are involved in Atg32 degradation, as previously reported.11
Next, we decided to validate our data by examining processing of Atg32 expressed at the endogenous level. To do this, we generated a strain expressing endogenous promoter-driven TAP-tagged Atg32 by removing the GAL1 promoter using the cre-loxp system as described previously.25 With this strain, a similar processed band was detected (Fig. 1C, arrowhead). The extent of processing was much weaker when Atg32 was expressed at the endogenous level, possibly reflecting a lower level of mitophagy induction (overexpression of Atg32 induces mitophagy12), and the fact that the level of mitochondrial turnover during mitophagy in a wild-type strain is relatively low, corresponding to a few percent of the total mitochondrial population. The processed band gradually accumulated upon shifting to glucose-containing medium lacking nitrogen, and then gradually degraded as starvation persisted, similar to what was observed with the overexpression strain.
Therefore, we concluded that the C terminus of Atg32 is proteolytically processed upon mitophagy induction, generating a form of the protein that is directly involved in mitophagy. Next, we decided to further explore the physiological meaning of this processing event.
C-terminal tagging of Atg32 blocks its processing and causes a mitophagy defect
A recent study reported that the C-terminal 100 amino acids of Atg32 are dispensable for mitophagy.21 This agrees with our data, which indicate that the C terminus is processed under mitophagy-inducing conditions. Accordingly, we hypothesized that the C terminus of Atg32 may play a negative, regulatory role in mitophagy, and that this part of the protein needs to be removed for proper mitophagy activity.
Previously, we had noted that the addition of a GFP tag to the C terminus of Atg32 resulted in a mitophagy defect (our unpublished data). Therefore, we decided to examine the effect of C-terminal tagging on processing of Atg32. When GFP was fused at the C terminus, there was a complete block in processing (Fig. 2A). When TAP-Atg32 was chromosomally tagged with a GFP tag at its C terminus, it remained as a single band upon mitophagy induction, in contrast to the C-terminal untagged TAP-Atg32 that generated the processed band (Fig. 2A, arrowhead).

Figure 2. C-terminal tagging of Atg32 interferes with its processing and mitophagy. (A) Strains expressing GAL1 promoter-driven TAP-Atg32 (KWY100) and TAP-Atg32-GFP (KWY110) were cultured as in Figure 1A. Proteins were monitored by immunoblotting with an antibody that detects PA. (B) Wild-type (WT; KWY90), atg32∆ (KWY22), and Atg32-GFP (KWY121) strains expressing mitoPho8∆60 were grown in YPL and shifted to SD-N for the indicated times. Samples were collected and protein extracts were assayed for mitoPho8∆60 activity. The results represent the mean and standard deviation (SD) of three independent experiments. (C) Strains expressing GAL1 promoter-driven N-terminal GFP-tagged Atg32 (KWY111) and C-terminal GFP-tagged Atg32 (KWY121) were cultured in YPGal to mid-log phase and stained with the mitochondrial marker MitoTracker Red. The localization of GFP and MitoTracker Red were visualized by fluorescence microscopy. DIC, differential interference contrast. Scale bar: 5 µm.
Therefore, we next tested whether mitophagy was affected by C-terminal tagging of Atg32. We used the mitoPho8∆60 assay to detect mitophagy activity. PHO8 encodes a vacuolar alkaline phosphatase, and its delivery into the vacuole through the secretory pathway is dependent on its N-terminal transmembrane domain.26 Pho8Δ60 is a truncated form that lacks the 60 N-terminal amino acid residues including the transmembrane domain; this altered protein is unable to enter the endoplasmic reticulum, remains in the cytosol and is delivered into the vacuole only through bulk autophagy.27 The mitoPho8∆60 construct is a modified version of Pho8∆60 that allows the quantitative measurement of mitophagy activity. If PHO8∆60 is fused with a mitochondrial targeting sequence, the encoded protein is specifically localized to mitochondria, and the ensuing alkaline phosphatase activity becomes an indicator of mitophagy.24
During a shift from rich medium containing lactate (YPL) to glucose-containing medium lacking nitrogen (SD-N) for 2, 4, and 6 h, wild-type cells showed a time-dependent increase of mitoPho8∆60-dependent alkaline phosphatase activity, indicating a robust induction of mitophagy (Fig. 2B, white bars). The mitophagy level in the atg32∆ strain showed a minor increase over time, and represented the background level (Fig. 2B, gray bars). Notably, there was not a complete block of mitophagy in the atg32∆ strain because nonspecific autophagy can also target a small portion of mitochondria for degradation.24 In cells harboring Atg32 that was C-terminally tagged with GFP, the mitoPho8∆60 activity was significantly reduced, being only slightly above the background level of the atg32∆ strain (Fig. 2B, black bars), suggesting a strong defect in mitophagy. Therefore, C-terminal tagging of Atg32 interferes with its function in mitophagy.
To exclude the possibility that C-terminally tagged Atg32 was not properly targeted to mitochondria, we checked the localization of Atg32-GFP by fluorescence microscopy. Since the protein level of endogenous Atg32 is too low to be observed by microscopy, we performed this experiment with overexpressed Atg32 under the control of the GAL1 promoter. With 8 h culturing in rich medium containing galactose (YPG), both N-terminally tagged and C-terminally tagged Atg32 were localized to mitochondria as determined by colocalization with the mitochondrial marker MitoTracker Red (Fig. 2C). This finding agrees with a previous report studying the topology of Atg32, which used both N-terminal 3HA- and C-terminal 2HA-tagged Atg32. In that case, both forms of Atg32 showed the expected localization and topology on mitochondria,11 suggesting that a C-terminal tag does not interfere with localization.
Taken together, these results support the conclusion that C-terminal tagging blocks Atg32 processing and affects its function in mitophagy, thus suggesting that the C-terminal region of Atg32 needs to be processed for proper mitophagy activity.
The mitochondrial i-AAA protease Yme1 mediates the processing of Atg32
To extend our analysis, we decided to investigate the mechanism of Atg32 processing. Considering the location of the C terminus in the IMS, we hypothesized that the processing could be mediated by a mitochondrial protease, and clearly one that had access to this subcompartment (i.e., not one present within the matrix). Therefore, we initially tested the effect of four mitochondrial proteases of the inner membrane, Oma1, Yta12, Pcp1, and Yme17 on Atg32 processing by deleting each corresponding gene. We found that the deletion of YME1, but not the genes encoding the other proteases, caused a complete block in Atg32 processing (Fig. S2), suggesting that Yme1 is the protease that mediates the proteolytic cleavage of Atg32.
To confirm the role of Yme1 in Atg32 processing, we further compared the processing of TAP-Atg32 in a yme1∆ strain with that in an isogenic wild-type strain over time (Fig. 3A). Although we could observe the processed form of Atg32 increase over the time course of mitophagy induction in wild-type cells, it was completely missing in the yme1∆ strain. In addition, the unprocessed Atg32 accumulated at a much higher level in the yme1∆ strain compared with the wild type, further suggesting a defect in processing, similar to the results with the atg1∆ and atg11∆ mutants (Fig. 1B).
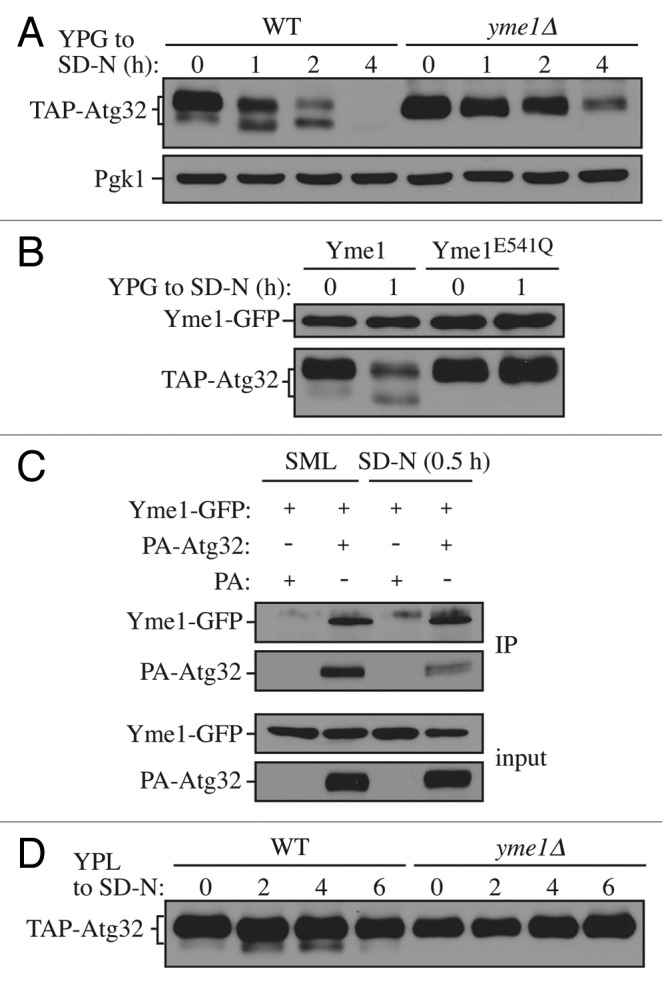
Figure 3. Yme1 mediates Atg32 processing. (A) Cells expressing GAL1 promoter-driven TAP-Atg32 in wild-type (WT; KWY100) and yme1∆ (KWY118) strains were cultured as in Figure 1A. TAP-Atg32 was monitored by immunoblotting with an antibody that binds to PA. Pgk1 was monitored by immunoblotting with anti-Pgk1 antibody as a loading control. (B) Cells expressing GAL1 promoter-driven TAP-Atg32 and Yme1-GFP (KWY134) or Yme1E541Q-GFP (KWY141) were cultured as in Figure 1A. TAP-Atg32 was monitored by immunoblotting with anti-Atg32 antiserum. Yme1-GFP was monitored by immunobloting with anti-YFP antibody. (C) A strain expressing Yme1-GFP (KWY133) was transformed with a plasmid expressing protein A only, or PA-Atg32 under the control of the CUP1 promoter, and cultured in SML medium to mid-log phase or starved in SD-N for 0.5 h. IgG-Sepharose was used to precipitate PA-Atg32 from cell lysates. The bottom two panels show the immunoblot of total cell lysates (input) and the upper two panels show the IgG precipitates (IP), which were probed with anti-YFP antibody and an antibody that recognizes PA. (D) Cells expressing endogenous promoter-driven TAP-Atg32 in WT (KWY139) and yme1∆ (KWY140) strains were cultured as in Figure 1C. TAP-Atg32 was monitored by immunoblotting with an antibody that recognizes protein A.
Yme1 is a mitochondrial i-AAA protease that functions in mitochondrial quality control by degrading unfolded or misfolded mitochondrial proteins.28 Yme1 has been reported to be the catalytic subunit of a protease complex with two other components, Mgr1 and Mgr3.29 Next, we tested whether these two other components are also required for Atg32 processing (Fig. S3). Compared with the yme1∆ strain, no apparent defect of Atg32 processing was observed in mgr1∆, mgr3∆, or mgr1∆ mgr3∆ strains. Mgr1 and Mgr3 function as adaptors that facilitate substrate binding to Yme1, but are not critical for its function.29
To further test whether the processing is dependent on the protease activity of Yme1, we generated a strain expressing a protease inactive mutant version harboring a point mutation, E541Q, in the proteolytic center.30 This version of Yme1 was also tagged with GFP at its C terminus so that the protein level could be detected by western blotting. In cells expressing chromosomally GFP tagged wild-type Yme1, Atg32 was properly processed in response to a shift to mitophagy-inducing conditions (Fig. 3B). This result also demonstrated that in the case of Yme1, C-terminal tagging did not interfere with its function. However, in cells expressing chromosomally GFP-tagged Yme1E541Q, the processing of Atg32 was completely blocked, comparable to yme1∆ cells, while the protein level of Yme1E541Q was comparable to the wild-type Yme1.
This result suggests that the processing of Atg32 by Yme1 depends on Yme1 protease activity, and implies that Atg32 could be a direct substrate of Yme1. Accordingly, we examined whether Atg32 directly interacts with Yme1. To test this possibility, we expressed PA-Atg32 in strains with Yme1 chromosomally tagged with GFP and performed protein A (PA) affinity isolation with immunoglobin G (IgG)-Sepharose (Fig. 3C). PA-Atg32 coprecipitated Yme1-GFP in both growing conditions (minimal medium with lactate, SML) and in SD-N, suggesting that Atg32 interacts with Yme1 in vivo.
Next, to test whether Yme1 is required for Atg32 processing at the endogenous level, we compared the processing of endogenous promoter-driven TAP-tagged Atg32 in wild-type and yme1∆ strains (Fig. 3D). Similar to the results with overexpressed Atg32, the yme1∆ strain displayed a complete block of Atg32 processing, suggesting that Yme1 is required for Atg32 processing at its physiological level. Taken together, these results demonstrated that the mitochondrial i-AAA protease Yme1 directly mediates Atg32 processing in response to mitophagy-inducing conditions.
Yme1 is specifically required for mitophagy
As C-terminal processing is required for mitophagy, and Yme1 mediates the processing, we next tested whether Yme1 is involved in mitophagy. We used the previously described mitoPho8∆60 assay to monitor mitophagy activity. As expected, the wild-type strain showed a gradual increase in mitoPho8∆60 activity in response to a shift to SD-N (Fig. 4A). In contrast, the yme1∆ strain displayed a significant block in mitophagy activity (~50% decrease at 6 h compared with wild type; however, if we account for the background level of activity represented by the atg32∆ strain, the absence of Yme1 caused an ~70% reduction).

Figure 4. Yme1 is specifically required for mitophagy. (A) Wild-type (WT; KWY20), yme1∆ (KWY136), and atg32∆ (KWY22) strains expressing mitoPho8∆60 were grown in YPL and shifted to SD-N for the indicated times. Samples were collected and protein extracts were assayed for mitoPho8∆60 activity. The results represent the mean and standard deviation (SD) of three independent experiments. (B) Wild-type (KWY20), yme1∆ (KWY136), atg32∆ (KWY22), and Yme1E541Q-GFP (KWY138) strains expressing mitoPho8∆60 were grown in YPL and shifted to SD-N for 6 h. Samples were collected and protein extracts were assayed for mitoPho8∆60 activity. The results represent the mean and standard deviation (SD) of three independent experiments. (C) Wild-type (WLY176), yme1∆ (KWY143), and atg1∆ (WLY192) strains expressing Pho8∆60 were cultured in YPD to mid-log phase and shifted to SD-N for 4 h. Samples were collected and protein extracts were assayed for Pho8∆60 activity. The results represent the mean and standard deviation (SD) of three independent experiments. (D) Wild-type (WLY176), yme1∆ (KWY143), and atg1∆ (WLY192) strains were cultured in YPD medium and analyzed for prApe1 maturation by immunoblotting to monitor the Cvt pathway during vegetative growth. The positions of precursor and mature Ape1 are indicated.
Since the protease activity of Yme1 is critical in its function of Atg32 processing, we further tested whether the mitophagy defect seen in the yme1∆ strain is dependent on its protease activity (Fig. 4B). The Yme1 protease inactive mutant (E541Q) displayed a mitophagy defect comparable to that of yme1∆ as determined by the mitoPho8∆60 assay. Therefore, these results demonstrated that proteolytically active Yme1 is required for mitophagy.
Next, we decided to test whether Yme1 is involved in other autophagic pathways, or is confined to a role in mitophagy. The Pho8∆60 assay is used to monitor the activity of nonspecific autophagy. This assay is similar to that used for monitoring mitophagy. In this case, Pho8∆60 remains cytosolic so that the Pho8∆60-dependent alkaline phosphatase activity represents the level of nonspecific autophagy.31 The wild-type strain showed increased Pho8∆60-dependent alkaline phosphatase activity following a shift from rich medium to SD-N, whereas the atg1∆ strain maintained a background level of activity (Fig. 4C). The yme1∆ strain displayed a level of autophagy activity comparable to wild type, demonstrating that Yme1 is not required for nonspecific autophagy.
The Cvt pathway is a type of selective autophagy that has been extensively characterized. This pathway is used for the delivery of the precursor form of the resident hydrolase Ape1 (prApe1) to the vacuole.32 We monitored the maturation of prApe1 in wild-type, atg1∆, and yme1∆ strains. The wild-type strain showed a substantial degree of maturation of prApe1 under vegetative growing conditions, while the atg1∆ strain was completely defective for prApe1 maturation (Fig. 4D). The yme1∆ strain displayed normal prApe1 maturation comparable to that of wild type, suggesting that Yme1 is not required for the Cvt pathway. Therefore, these results indicated that Yme1 is specifically required for mitophagy but not nonspecific autophagy or the Cvt pathway.
Yme1 is required for Atg32 processing and mitophagy in a post-log phase culture
In addition to nitrogen starvation in a glucose-containing medium, mitophagy can also be induced in a post-log phase culture.11,13 To test whether Atg32 processing is also occurring during post-log phase growth, we examined processing in the TAP-Atg32 overexpression strain cultured in YPG to post-log phase (Fig. S4A). In the wild-type strain a processed form of Atg32 was observed when cells were cultured for more than 12 h. Similar to the results in SD-N, Yme1 was required for this process since the yme1∆ strain showed a complete block in processing. Next, to test whether Yme1 and its protease activity are required for mitophagy induced at post-log phase, we used the mitoPho8∆60 assay to monitor mitophagy activity (Fig. S4B). The wild-type control strain displayed an increase in mitoPho8∆60-dependent alkaline phosphatase activity when cultured to post-log phase. In contrast, the yme1∆ strain and the Yme1E541Q-expressing strain showed a significant defect in mitophagy that was comparable to that observed during starvation-induced mitophagy. Therefore, Yme1 is also required for Atg32 processing and mitophagy during post-log phase growth-induced mitophagy.
Yme1 regulates the Atg32-Atg11 interaction
Atg32 interacts with Atg11 upon mitophagy induction11,12 and this interaction is critical for mitophagy activity.21,22 To provide a more mechanistic insight into how Yme1 regulates mitophagy, we decided to examine whether Atg32 and Atg11 interaction was affected in the yme1∆ strain. To do this, we expressed PA-Atg32 and HA-Atg11 in the wild-type and yme1∆ strains and performed PA affinity isolation with immunoglobin G (IgG)-Sepharose (Fig. 5). In wild-type cells, PA-Atg32 coprecipitated a significant amount of HA-Atg11 following a 1 h shift to mitophagy-inducing conditions (Fig. 5, lane 2). However, in the yme1∆ strain, the amount of HA-Atg11 coprecipitated by PA-Atg32 was significantly decreased (Fig. 5, lane 4), suggesting a strong block in the Atg32-Atg11 interaction. Therefore, Yme1 is required for the efficient interaction between Atg32 and Atg11.
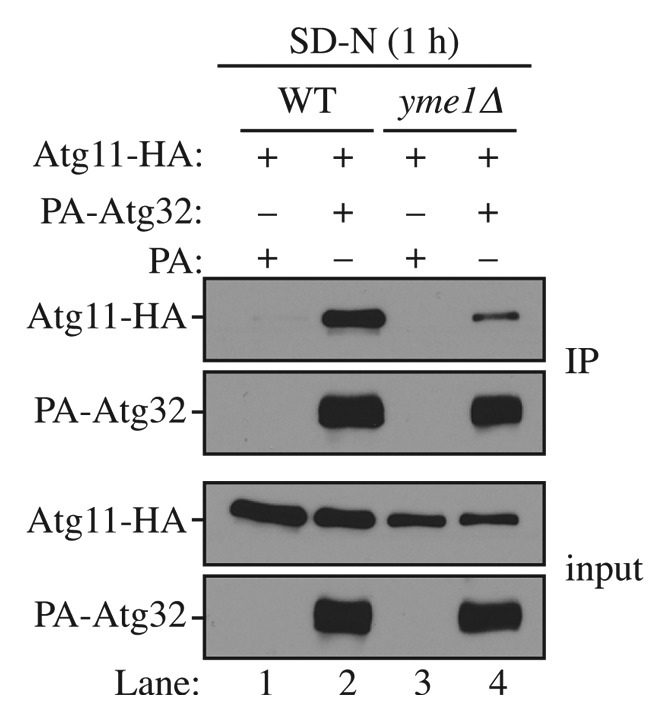
Figure 5. Yme1 regulates the interaction between Atg32 and Atg11. Wild-type (WT; SEY6210) and yme1∆ (KWY142) strains transformed with a plasmid expressing HA-tagged Atg11 together with a plasmid expressing protein A only or PA-Atg32 were cultured in SMD medium to mid-log phase or starved in SD-N for 1 h. IgG-Sepharose was used to precipitate PA-Atg32 from cell lysates. The bottom two panels show the immunoblot of total cell lysates (input) and the upper two panels show the IgG precipitates (IP), which were probed with anti-YFP antibody and an antibody that binds PA. All plasmids are under the control of the CUP1 promoter
Discussion
In the past few years there has been a steady increase in our knowledge of the process of mitophagy, in particular at the molecular level. The identification of the receptor protein Atg32 provided an important breakthrough regarding the mechanism of mitophagy in yeast;11,12 however, the identification of this protein also raised important questions as to how mitophagy is regulated. Considering that mitochondria are essential organelles, the proper regulation of mitophagy is critically important for mitochondrial and cellular homeostasis.
In this study, we characterized one aspect of mitophagy regulation. We demonstrated that Atg32 undergoes proteolytic processing of its C terminus upon mitophagy induction (Fig. 1), which is mediated by the mitochondrial i-AAA protease Yme1 (Fig. 3). This processing is important for mitophagy, as blocking the processing by C-terminal tagging of Atg32 caused a significant mitophagy defect (Fig. 2). In addition, Yme1 protease activity is required for efficient mitophagy (Fig. 4). Our results also suggested that the unprocessed Atg32 has lower binding affinity to Atg11, since the interaction between Atg32 and Atg11 was significantly reduced in a yme1∆ strain (Fig. 5). Therefore, our results suggested a model of mitophagy regulation where, upon induction, Yme1 processes Atg32 to allow its tight association with Atg11 and subsequent interaction with the core macroautophagic machinery.
This model of regulation is reminiscent of PINK1-PARK2-mediated mitophagy in mammalian cells. In steady-state cells, PINK1 is imported into the mitochondrial inner membrane in a membrane potential-dependent manner. The mitochondrial inner membrane presenilin associated, rhomboid-like/PARL protein mediates the cleavage and destabilization of PINK1.33 Upon mitochondria depolarization, PINK1 import into the inner membrane is impaired, leading to a rapid PINK1 accumulation on the outer membrane of damaged mitochondria. Accumulated PINK1 recruits PARK2 to the mitochondria, resulting in the ubiquitination of outer membrane proteins, which is required for the recognition of damaged mitochondria by the autophagic machinery. In this case, however, proteolytic processing of PINK1 leads to its destabilization and an inhibition of mitophagy. In addition, Pcp1, which is the yeast homolog of PARL, is not required for Atg32 processing (Fig. S2); however, Yme1 is well conserved in mammalian cells, and the function of YME1L, the homolog of Yme1, is largely unknown.7 It will be interesting to see whether YME1L plays a role in mitophagy in mammalian cells.
Through this study, we established a link between mitophagy and the mitochondrial protein quality control system. Yme1 is responsible for the degradation of unfolded or misfolded mitochondrial proteins,34 and is thus an important component of the mitochondrial protein quality control system. A recent study provided evidence that disruption of mitochondrial protein quality control promotes mitophagy,35 suggesting a crosstalk between these two types of mitochondrial quality control systems. Our study provided a molecular link for this crosstalk by showing that Yme1 recognizes the mitophagy receptor protein Atg32 as a substrate. If Atg32 was the only target of Yme1 that is relevant to mitophagy, expression of a truncated form of Atg32 lacking its C terminus should suppress the mitophagy defect of the yme1∆ strain; however, we found that a C-terminally truncated Atg32 did not complement the mitophagy defect seen in yme1∆ cells (our unpublished data). Therefore, we speculate that Yme1 has an additional target(s) and another role(s) during mitophagy besides the processing of Atg32. Furthermore, in our study, we monitored the role of Yme1 in removing superfluous mitochondria following a shift from a nonfermentable carbon source to glucose. It is reasonable to speculate that under other mitochondrial stress conditions, Yme1 actively degrades unfolded or misfolded mitochondrial proteins and at the same time activates Atg32 to trigger mitophagy to prevent the accumulation of damaged mitochondria.
Yme1 was first discovered to be involved in mitochondrial DNA transfer to the nucleus.36 Inactivation of Yme1 causes increased escape of DNA from mitochondria, which is dependent on vacuole function.37 However, based on our analyis, mitochondrial DNA transfer to the nucleus seemed to be an autophagy-independent process (our unpublished data). Thus, the involvement of Yme1 in mitophagy regulation may be distinct from its potential role in mitochondrial DNA escape.
What regulates Yme1 activity remains a mystery. Our results showed that Atg32 could be a direct substrate of Yme1, and the interaction between Atg32 and Yme1 is independent of culture conditions. Therefore, we speculate Yme1 is associated with Atg32 constitutively, but the processing only occurs upon mitophagy induction. What activates Yme1 to process Atg32 remains an interesting question for future study.
Why unprocessed Atg32 has a decreased affinity for Atg11 also needs to be further characterized. We speculate that the processing of Atg32 at its C terminus may cause a conformational change at its N terminus that favors its interaction with Atg11. If demonstrated, this will provide an example of how the status of the mitochondria, such as the potential across the inner membrane, can be transmitted to factors in the cytosol to initiate mitophagy. Such a mechanism has been characterized for various cell surface receptors.38 Upon binding to extracellular ligands, a conformational change is induced and the catalytic domain in the cytosol is activated. Structural studies of Atg32 are needed to explore this intriguing possibility that proteolytic processing can also play a role in signal transduction across the membrane.
Atg32 is phosphorylated by a still unknown kinase.21 This phosphorylation is required for mitophagy and the interaction between Atg32 and Atg11. We tried to explore the relationship between Atg32 processing and phosphorylation, but we were not able to detect a clear band shift corresponding to Atg32 phosphorylation. Nonetheless, it is clear that cellular mitophagy is a tightly controlled process and is regulated by multiple factors. So far, the regulation of mitophagy in yeast has largely focused on the receptor protein Atg32 and its interaction with Atg11. Apparently more experiments of a broader scope are needed to further elucidate the details of mitophagy regulation.
Materials and Methods
Strains and growth conditions
The yeast strains used in this study are listed in Table S1. Yeast cells were grown in rich medium (YPD; 1% yeast extract, 2% peptone, 2% glucose), lactate medium (YPL; 1% yeast extract, 2% peptone, 2% lactate), galactose medium (YPG; 1% yeast extract, 2% peptone, 2% galactose), synthetic minimal medium with glucose (SMD; 0.67% yeast nitrogen base, 2% glucose, amino acids, and vitamins), synthetic minimal medium with lactate (SML; 0.67% yeast nitrogen base, 2% lactate, amino acids, and vitamins), or synthetic minimal medium with galactose (SMG; 0.67% yeast nitrogen base, 2% galactose, amino acids, and vitamins). Starvation experiments were performed in synthetic minimal medium lacking nitrogen (SD-N; 0.17% yeast nitrogen base without amino acids, 2% glucose).
Plasmids and antibodies
The plasmids used to express PA-tagged Atg32 [pCuPA-Atg32(416)] and HA-tagged Atg11 [pCuHA-Atg11(414)] have been described previously.12 Monoclonal anti-YFP antibody clone JL-8 (Clontech, 632381), monoclonal anti-HA antibody clone-HA7 (Sigma-Aldrich, H3663), an antibody that binds to protein A with high affinity (no longer commercially available) and anti-Ape1 antiserum,39 were used for immunoblotting. Anti-Pgk1 antiserum was a kind gift of Dr Jeremy Thorner (University of California, Berkeley).
Assays for nonspecific autophagy and mitophagy
For monitoring nonspecific autophagy, the alkaline phosphatase activity of Pho8∆60 was performed as described previously.40 To monitor mitophagy, the alkaline phosphatase activity of mitoPho8∆60 was performed as described previously.24
Fluorescence microscopy
Yeast cells expressing fluorescent protein-fused chimeras were grown to mid-log phase or starved in the indicated media. To label mitochondria, cells were incubated in medium containing 1 µM MitoTracker Red CMXRos (Molecular Probes/Invitrogen, M7512) at 30 °C for 30 min. After being washed with medium, the cells were incubated in medium at 30 °C for 30 to 60 min. Fluorescence microscopy observations were performed as described previously.41
Supplementary Material
Acknowledgments
This work was supported by NIH grant GM053396 to DJK.
Glossary
Abbreviations:
- Ape1
aminopeptidase I
- Atg
autophagy-related
- Cvt
cytoplasm-to-vacuole targeting
- IgG
immunoglobulin G
- IMS
intermembrane space
- PA
protein A
- prApe1
precursor Ape1
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Supplemental Materials
Supplemental materials may be found here: www.landesbioscience.com/journals/autophagy/article/26281
Footnotes
Previously published online: www.landesbioscience.com/journals/autophagy/article/26281
References
- 1.Wang K, Klionsky DJ. Mitochondria removal by autophagy. Autophagy. 2011;7:297–300. doi: 10.4161/auto.7.3.14502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Wallace DC. A mitochondrial paradigm of metabolic and degenerative diseases, aging, and cancer: a dawn for evolutionary medicine. Annu Rev Genet. 2005;39:359–407. doi: 10.1146/annurev.genet.39.110304.095751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Yen W-L, Klionsky DJ. How to live long and prosper: autophagy, mitochondria, and aging. Physiology (Bethesda) 2008;23:248–62. doi: 10.1152/physiol.00013.2008. [DOI] [PubMed] [Google Scholar]
- 4.Tatsuta T, Langer T. Quality control of mitochondria: protection against neurodegeneration and ageing. EMBO J. 2008;27:306–14. doi: 10.1038/sj.emboj.7601972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Larsson NG, Clayton DA. Molecular genetic aspects of human mitochondrial disorders. Annu Rev Genet. 1995;29:151–78. doi: 10.1146/annurev.ge.29.120195.001055. [DOI] [PubMed] [Google Scholar]
- 6.Arai M, Imai H, Koumura T, Yoshida M, Emoto K, Umeda M, Chiba N, Nakagawa Y. Mitochondrial phospholipid hydroperoxide glutathione peroxidase plays a major role in preventing oxidative injury to cells. J Biol Chem. 1999;274:4924–33. doi: 10.1074/jbc.274.8.4924. [DOI] [PubMed] [Google Scholar]
- 7.Koppen M, Langer T. Protein degradation within mitochondria: versatile activities of AAA proteases and other peptidases. Crit Rev Biochem Mol Biol. 2007;42:221–42. doi: 10.1080/10409230701380452. [DOI] [PubMed] [Google Scholar]
- 8.Ashrafi G, Schwarz TL. The pathways of mitophagy for quality control and clearance of mitochondria. Cell Death Differ. 2013;20:31–42. doi: 10.1038/cdd.2012.81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Yorimitsu T, Klionsky DJ. Autophagy: molecular machinery for self-eating. Cell Death Differ. 2005;12(Suppl 2):1542–52. doi: 10.1038/sj.cdd.4401765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Clark SL., Jr. Cellular differentiation in the kidneys of newborn mice studies with the electron microscope. J Biophys Biochem Cytol. 1957;3:349–62. doi: 10.1083/jcb.3.3.349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Okamoto K, Kondo-Okamoto N, Ohsumi Y. Mitochondria-anchored receptor Atg32 mediates degradation of mitochondria via selective autophagy. Dev Cell. 2009;17:87–97. doi: 10.1016/j.devcel.2009.06.013. [DOI] [PubMed] [Google Scholar]
- 12.Kanki T, Wang K, Cao Y, Baba M, Klionsky DJ. Atg32 is a mitochondrial protein that confers selectivity during mitophagy. Dev Cell. 2009;17:98–109. doi: 10.1016/j.devcel.2009.06.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kanki T, Klionsky DJ. Mitophagy in yeast occurs through a selective mechanism. J Biol Chem. 2008;283:32386–93. doi: 10.1074/jbc.M802403200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Narendra DP, Jin SM, Tanaka A, Suen DF, Gautier CA, Shen J, Cookson MR, Youle RJ. PINK1 is selectively stabilized on impaired mitochondria to activate Parkin. PLoS Biol. 2010;8:e1000298. doi: 10.1371/journal.pbio.1000298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Narendra D, Tanaka A, Suen DF, Youle RJ. Parkin is recruited selectively to impaired mitochondria and promotes their autophagy. J Cell Biol. 2008;183:795–803. doi: 10.1083/jcb.200809125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kurihara Y, Kanki T, Aoki Y, Hirota Y, Saigusa T, Uchiumi T, Kang D. Mitophagy plays an essential role in reducing mitochondrial production of reactive oxygen species and mutation of mitochondrial DNA by maintaining mitochondrial quantity and quality in yeast. J Biol Chem. 2012;287:3265–72. doi: 10.1074/jbc.M111.280156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sandoval H, Thiagarajan P, Dasgupta SK, Schumacher A, Prchal JT, Chen M, Wang J. Essential role for Nix in autophagic maturation of erythroid cells. Nature. 2008;454:232–5. doi: 10.1038/nature07006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Al Rawi S, Louvet-Vallée S, Djeddi A, Sachse M, Culetto E, Hajjar C, Boyd L, Legouis R, Galy V. Postfertilization autophagy of sperm organelles prevents paternal mitochondrial DNA transmission. Science. 2011;334:1144–7. doi: 10.1126/science.1211878. [DOI] [PubMed] [Google Scholar]
- 19.Mijaljica D, Nazarko TY, Brumell JH, Huang W-P, Komatsu M, Prescott M, Simonsen A, Yamamoto A, Zhang H, Klionsky DJ, et al. Receptor protein complexes are in control of autophagy. Autophagy. 2012;8:1701–5. doi: 10.4161/auto.21332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Yorimitsu T, Klionsky DJ. Atg11 links cargo to the vesicle-forming machinery in the cytoplasm to vacuole targeting pathway. Mol Biol Cell. 2005;16:1593–605. doi: 10.1091/mbc.E04-11-1035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Aoki Y, Kanki T, Hirota Y, Kurihara Y, Saigusa T, Uchiumi T, Kang D. Phosphorylation of Serine 114 on Atg32 mediates mitophagy. Mol Biol Cell. 2011;22:3206–17. doi: 10.1091/mbc.E11-02-0145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kondo-Okamoto N, Noda NN, Suzuki SW, Nakatogawa H, Takahashi I, Matsunami M, Hashimoto A, Inagaki F, Ohsumi Y, Okamoto K. Autophagy-related protein 32 acts as autophagic degron and directly initiates mitophagy. J Biol Chem. 2012;287:10631–8. doi: 10.1074/jbc.M111.299917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Leonhard K, Stiegler A, Neupert W, Langer T. Chaperone-like activity of the AAA domain of the yeast Yme1 AAA protease. Nature. 1999;398:348–51. doi: 10.1038/18704. [DOI] [PubMed] [Google Scholar]
- 24.Kanki T, Klionsky DJ. Atg32 is a tag for mitochondria degradation in yeast. Autophagy. 2009;5:1201–2. doi: 10.4161/auto.5.8.9747. [DOI] [PubMed] [Google Scholar]
- 25.Puig O, Caspary F, Rigaut G, Rutz B, Bouveret E, Bragado-Nilsson E, Wilm M, Séraphin B. The tandem affinity purification (TAP) method: a general procedure of protein complex purification. Methods. 2001;24:218–29. doi: 10.1006/meth.2001.1183. [DOI] [PubMed] [Google Scholar]
- 26.Klionsky DJ, Emr SD. Membrane protein sorting: biosynthesis, transport and processing of yeast vacuolar alkaline phosphatase. EMBO J. 1989;8:2241–50. doi: 10.1002/j.1460-2075.1989.tb08348.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Noda T, Matsuura A, Wada Y, Ohsumi Y. Novel system for monitoring autophagy in the yeast Saccharomyces cerevisiae. Biochem Biophys Res Commun. 1995;210:126–32. doi: 10.1006/bbrc.1995.1636. [DOI] [PubMed] [Google Scholar]
- 28.Gerdes F, Tatsuta T, Langer T. Mitochondrial AAA proteases--towards a molecular understanding of membrane-bound proteolytic machines. Biochim Biophys Acta. 2012;1823:49–55. doi: 10.1016/j.bbamcr.2011.09.015. [DOI] [PubMed] [Google Scholar]
- 29.Dunn CD, Tamura Y, Sesaki H, Jensen RE. Mgr3p and Mgr1p are adaptors for the mitochondrial i-AAA protease complex. Mol Biol Cell. 2008;19:5387–97. doi: 10.1091/mbc.E08-01-0103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Graef M, Seewald G, Langer T. Substrate recognition by AAA+ ATPases: distinct substrate binding modes in ATP-dependent protease Yme1 of the mitochondrial intermembrane space. Mol Cell Biol. 2007;27:2476–85. doi: 10.1128/MCB.01721-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Klionsky DJ. Monitoring autophagy in yeast: the Pho8Delta60 assay. Methods Mol Biol. 2007;390:363–71. doi: 10.1007/978-1-59745-466-7_24. [DOI] [PubMed] [Google Scholar]
- 32.Lynch-Day MA, Klionsky DJ. The Cvt pathway as a model for selective autophagy. FEBS Lett. 2010;584:1359–66. doi: 10.1016/j.febslet.2010.02.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Jin SM, Lazarou M, Wang C, Kane LA, Narendra DP, Youle RJ. Mitochondrial membrane potential regulates PINK1 import and proteolytic destabilization by PARL. J Cell Biol. 2010;191:933–42. doi: 10.1083/jcb.201008084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Weber ER, Hanekamp T, Thorsness PE. Biochemical and functional analysis of the YME1 gene product, an ATP and zinc-dependent mitochondrial protease from S. cerevisiae. Mol Biol Cell. 1996;7:307–17. doi: 10.1091/mbc.7.2.307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Heo JM, Livnat-Levanon N, Taylor EB, Jones KT, Dephoure N, Ring J, Xie J, Brodsky JL, Madeo F, Gygi SP, et al. A stress-responsive system for mitochondrial protein degradation. Mol Cell. 2010;40:465–80. doi: 10.1016/j.molcel.2010.10.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Thorsness PE, White KH, Fox TD. Inactivation of YME1, a member of the ftsH-SEC18-PAS1-CDC48 family of putative ATPase-encoding genes, causes increased escape of DNA from mitochondria in Saccharomyces cerevisiae. Mol Cell Biol. 1993;13:5418–26. doi: 10.1128/mcb.13.9.5418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Campbell CL, Thorsness PE. Escape of mitochondrial DNA to the nucleus in yme1 yeast is mediated by vacuolar-dependent turnover of abnormal mitochondrial compartments. J Cell Sci. 1998;111:2455–64. doi: 10.1242/jcs.111.16.2455. [DOI] [PubMed] [Google Scholar]
- 38.Lemmon MA, Schlessinger J. Cell signaling by receptor tyrosine kinases. Cell. 2010;141:1117–34. doi: 10.1016/j.cell.2010.06.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Klionsky DJ, Cueva R, Yaver DS. Aminopeptidase I of Saccharomyces cerevisiae is localized to the vacuole independent of the secretory pathway. J Cell Biol. 1992;119:287–99. doi: 10.1083/jcb.119.2.287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Noda T, Klionsky DJ. The quantitative Pho8Delta60 assay of nonspecific autophagy. Methods Enzymol. 2008;451:33–42. doi: 10.1016/S0076-6879(08)03203-5. [DOI] [PubMed] [Google Scholar]
- 41.Monastyrska I, He C, Geng J, Hoppe AD, Li Z, Klionsky DJ. Arp2 links autophagic machinery with the actin cytoskeleton. Mol Biol Cell. 2008;19:1962–75. doi: 10.1091/mbc.E07-09-0892. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.