

Abstract
Resurgent sodium currents contribute to the regeneration of action potentials and enhanced neuronal excitability. Tetrodotoxin-sensitive (TTX-S) resurgent currents have been described in many different neuron populations, including cerebellar and dorsal root ganglia (DRG) neurons. In most cases, sodium channel Nav1.6 is the major contributor to these TTX-S resurgent currents. Here we report a novel TTX-resistant (TTX-R) resurgent current recorded from rat DRG neurons. The TTX-R resurgent currents are similar to classic TTX-S resurgent currents in many respects, but not all. As with TTX-S resurgent currents, they are activated by membrane repolarization, inhibited by lidocaine, and enhanced by a peptide-mimetic of the β4 sodium channel subunit intracellular domain. However, the TTX-R resurgent currents exhibit much slower kinetics, occur at more depolarized voltages, and are sensitive to the Nav1.8 blocker A803467. Moreover, coimmunoprecipitation experiments from rat DRG lysates indicate the endogenous sodium channel β4 subunits associate with Nav1.8 in DRG neurons. These results suggest that slow TTX-R resurgent currents in DRG neurons are mediated by Nav1.8 and are generated by the same mechanism underlying TTX-S resurgent currents. We also show that both TTX-S and TTX-R resurgent currents in DRG neurons are enhanced by inflammatory mediators. Furthermore, the β4 peptide increased excitability of small DRG neurons in the presence of TTX. We propose that these slow TTX-R resurgent currents contribute to the membrane excitability of nociceptive DRG neurons under normal conditions and that enhancement of both types of resurgent currents by inflammatory mediators could contribute to sensory neuronal hyperexcitability associated with inflammatory pain.
Keywords: action potential, hyperexcitability, nociceptor, resurgent sodium current, sodium current, voltage clamp
Introduction
Resurgent sodium currents activated during the repolarization phase of the action potential provide depolarizing drive for the generation of additional action potentials and, therefore, can contribute to repetitive firing of neurons (Raman et al., 1997; Theile and Cummins, 2011b). Direct evidence comes from the observation that cerebellar neuron excitability decreases when resurgent currents are reduced by knocking down the sodium channel β4 subunit (Bant and Raman, 2010), which has been characterized as the putative open-channel blocker underlying resurgent current generation (Grieco et al., 2005). According to this hypothesis, the cytoplasmic tail of the β4 subunit competes with the intrinsic sodium channel inactivation mechanism to interact with the inner pore region of open sodium channels during membrane depolarization. Upon repolarization, the β4 subunit tail dissociates from the open channels, resulting in a transient resurgence of inward sodium flux during the otherwise refractory repolarization phase of the action potential.
Since the initial report in cerebellar Purkinje neurons, resurgent currents have been characterized in neurons from the cerebellum (Raman and Bean, 1997), subthalamic nucleus (Do and Bean, 2003), perirhinal cortex (Castelli et al., 2007), calyx of Held (Kim et al., 2010), medial nucleus of trapezoid body (Leão et al., 2006), medial vestibular nucleus (Gittis and du Lac, 2008), trigeminal, and DRG (Cummins et al., 2005; Enomoto et al., 2006). Although they are observed in diverse neuronal populations, these currents exhibited similar kinetic and voltage-dependent characteristics and were blocked by nanomolar concentrations of tetrodotoxin (TTX). Under normal conditions, Nav1.6 seems to be the major contributor to these TTX-sensitive (TTX-S) resurgent currents observed in many central and peripheral neurons (Raman et al., 1997; Do and Bean, 2004; Cummins et al., 2005; Enomoto et al., 2007). However, in some neuronal populations, Nav1.6 is not the major contributor (Aman and Raman, 2007). In addition, toxins that slow the rate of sodium current inactivation in Nav1.6-null mouse neurons can induce resurgent currents (Grieco and Raman, 2004) and sodium channel mutations that slow the rate of sodium channel inactivation and are associated with paroxysmal extreme pain disorder, paramyotonia congenita, and long QT-3 substantially increased resurgent currents and neuronal excitability when expressed in DRG neurons (Jarecki et al., 2010). These findings suggested that the rate of channel inactivation is an important factor that influences resurgent current generation and, importantly, that isoforms other than Nav1.6 can generate resurgent currents under specific conditions.
DRG neurons are unusual in that they produce both fast-inactivating TTX-S and slow-inactivating TTX-resistant (TTX-R) sodium currents. In this report, we explored whether the TTX-R channel Nav1.8, which exhibits 3- to 5-fold slower channel inactivation than Nav1.6, might also generate resurgent currents. We identified a TTX-R resurgent current in rat DRG neurons and characterized the functional properties of this unique current. Furthermore, we show that resurgent currents generated by both TTX-S and TTX-R channels are enhanced by inflammatory mediators. Our results suggest that the slow TTX-R resurgent current reported in this study is mediated by Nav1.8, and both TTX-S and TTX-R resurgent currents could be important in modulating the membrane excitability of nociceptive DRG neurons and inflammatory pain sensations.
Materials and Methods
Cell culture.
Adult rat DRG neurons were dissociated and cultured as previously described (Caffrey et al., 1992; Cummins et al., 2000). Animal procedures were approved by the Indiana University School of Medicine Institutional Animal Care and Use Committee. Briefly, young adult Sprague Dawley rats were rendered unconscious by exposure to CO2 and decapitated. Lumbar DRG (L4-L6) were excised and then incubated in DMEM containing collagenase (1 mg/ml) and protease (1 mg/ml). The ganglia were sequentially triturated in DMEM supplemented with 10% FBS and plated on glass coverslips coated with poly-d-lysine and laminin. Cultures supplemented with 30 ng/ml NGF were maintained at 37°C in a humidified 95% air and 5% CO2 incubator.
Electrophysiology.
DRG recordings were obtained from cells 14–28 h after dissociation. Whole-cell patch-clamp recordings were conducted in either voltage-clamp or current-clamp mode at room temperature (∼22°C) using a HEKA EPC-10 amplifier. Data were acquired on a Windows-based Pentium IV computer using the Pulse program (version 8.80; HEKA Elektronik). Fire-polished electrodes (0.7–1.0 mΩ) were fabricated from 1.7 mm capillary glass using a Sutter P-97 puller (Novato), and the tips were coated with sticky wax (KerrLab) to minimize capacitive artifacts and enable increased series resistance compensation. For voltage-clamp recording, the standard electrode solution consisted of 140 mm CsF, 10 mm NaCl, 1.1 mm EGTA, and 10 mm HEPES, pH 7.3. The standard extracellular bathing solution contained 130 mm NaCl, 30 mm TEA chloride, 1 mm MgCl2, 3 mm KCl, 1 mm CaCl2, 0.05 mm CdCl2, 10 mm HEPES, and 10 mm d-glucose, pH 7.3. For current-clamp recording, the pipette solution contained 140 mm KCl, 0.5 mm EGTA, 5 mm HEPES, and 3 mm Mg-ATP, pH 7.3. The bathing solution contained 140 mm NaCl, 3 mm KCl, 2 mm MgCl2, 2 mm CaCl2, 10 mm HEPES, and 500 nm TTX, pH 7.3. Recording solutions were adjusted using d-glucose and NaOH to maintain physiological pH and osmolality values.
Cells on glass coverslips were transferred to a recording chamber containing 250 μl bathing solution; 500 nm TTX (Alomone Labs) was used to isolate TTX-R sodium currents. Series resistance errors were compensated by ≥80%. Leak currents were linearly canceled by digital P/−5 subtraction. Cells were held at a membrane potential of −80 mV. Membrane currents were sampled at 20 kHz, filtered online at 5 kHz, and further filtered at 1 kHz digitally in Pulsefit. Whole-cell currents were not recorded before 3 min after whole-cell configuration had been established to allow adequate time for the electrode solution and cytoplasmic milieu to equilibrate. Resurgent currents were assayed with a two-step protocol that initially depolarized the membrane to 30 mV for 20 ms before testing for inward resurgent sodium currents by hyperpolarizing the membrane potential in −5 mV increments from 15 mV to −75 mV, for 400 ms, before returning to the holding potential. For current-clamp recording, 500 nm TTX was used to block TTX-S sodium channels. Small DRG neurons were left at their resting membrane potentials (ranging from −48 to −74 mV) before current injection. A series of positive 2 s current steps ranging from 0 pA to 3× rheobase were injected into neurons. Neurons that fired two or more action potentials at any level of current injection were classified as “multiple.” Neurons that fired only one action potential over current range from 1× to 3× rheobase were classified as “single.” Experimental data were analyzed using Pulsefit (version 8.80; HEKA Elektronik), Origin (version 8.0; OriginLab), and Microsoft Excel software programs.
Averaged data were presented as Mean ± SEM. Student's t test or χ2 test was used to examine the statistical significance.
Immunoprecipitation.
Rat DRG and whole-brain tissue were harvested from male Sprague Dawley rats: whole brain from 1 rat and lumbar to cervical DRG from 7 rats were used. Rat DRG neurons were prepared in the same way as for electrophysiological experiments. DRG were left in culture for 21 h in media supplemented with 30 ng/ml NGF. Samples were solubilized in a buffer containing the following: 0.32 m sucrose, 10 mm HEPES, 2 mm EDTA, pH 8.0, 1.25% Triton X-100, and supplemented with 50 U/ml benzonase, and Pierce proteinase and phosphatase inhibitor mini-tablets. Samples were dounce homogenized, sonicated, incubated on ice for 1 h, centrifuged at 5500 × g in a refrigerated microcentrifuge for 20 min, and the supernatants were recovered for immunoprecipitations. A total of 15 μg of antibody covalently coupled to Dynabeads M-270 Epoxy was added to 5 mg of rat brain and DRG lysate. Antibody-lysate matrix was incubated at 4°C rotating for 22 h. Bead matrix was washed 4× with modified RIPA buffer (0.6% NP40, 0.6% DOC, 20 mm Tris-HCl, and 20 mm NaCl, supplemented with Pierce proteinase and phosphatase inhibitor mini-tablets). The immunoprecipitated lysate was eluted from bead matrix with pH 2.0 sodium citrate and immediately diluted in 1× lithium dodecyl sulfate sample buffer. Samples were run in 4–20% Criterion™ TGX™ polyacrylamide gels (Bio-Rad Laboratories) polyacrylamide gels and transferred to PVDF membrane. Blots were blocked for 2 h at room temperature in 5% dry milk in TBS Tween 20, incubated with primary antibody (either anti-pan NaV or anti-β4) overnight at 4°C, incubated with secondary antibody (either anti-mIgG HRP or anti-RbIgG HRP) for 1 h at room temperature, and developed with equal volumes of SuperSignal-West Femto ECL reagents (Thermo Scientific).
Monoclonal pan-sodium channel antibody (#S8809-K58/35) was purchased from Sigma, polyclonal anti-NaV1.8 antibody (ASC-016) was purchased from Alomone Labs, and polyclonal anti-β4 antibody (#Ab80539) was purchased from Abcam. The specificity of the NaV1.8 antibody was previously characterized by Hudmon et al. (2008). Rabbit anti-IgG antibody (EMD Millipore) was used as a control for immunoprecipitation. Secondary antibodies for Western blot analysis, anti-rabbit IgG, and anti-mouse IgG coupled to HRP were purchased from Cell Signaling Technology. Dynabeads M-270 Epoxy (Invitrogen) were purchased and precoupled to anti-NaV1.8 according to the manufacturer's instructions enclosed in the Dynabeads coimmunoprecipitation kit (Invitrogen).
Computational simulations.
Simulations were performed to explore variables that might impact properties of resurgent currents generated by voltage-gated sodium channels. Established models of TTX-S and TTX-R sodium channels were implemented in the ion channel simulator IChSim (IFUASLP) software program. TTX-R currents were simulated using a model developed for Nav1.8 currents (Sheets et al., 2007). TTX-S sodium currents were simulated using a model developed to investigate resurgent current generation (Raman and Bean, 2001). The decay time constants and time to onset for resurgent currents were measured using the Origin (version 8.0; OriginLab) software program.
Results
Identification of slow TTX-R resurgent currents in DRG neurons
Resurgent currents are typically elicited in voltage-clamp recordings by repolarizing steps to intermediate potentials (i.e., between −10 and −60 mV) that follow a depolarizing step to 30 mV. These resurgent currents are distinct from tail currents (which are best elicited after short depolarizations that produce incomplete inactivation) as the depolarization is long enough (i.e., 20 ms) to allow apparently complete inactivation of the peak transient sodium currents. TTX-S currents with classic resurgent current properties can be recorded from ∼50% of medium-diameter DRG neurons dissociated from adult rats and mice (Fig. 1B) (Cummins et al., 2005). However, in some cells, we observed a slower resurgent current that persisted in the presence of TTX (Fig. 1A). This slow TTX-R resurgent current was observed only in medium-diameter neurons that also exhibited peak transient TTX-R currents with Nav1.8-like kinetics. The presence of these slower, TTX-R resurgent currents was not associated with the presence of Nav1.9-like TTX-R currents, which are rarely observed in medium-diameter neurons and run down quickly in the whole-cell voltage-clamp mode (Cummins et al., 1999). The TTX-R resurgent current was best observed when the duration of the repolarization step was >200 ms (note the different time scales in Fig. 1A,B). Although both TTX-R and TTX-S resurgent currents exhibited a similar U-shaped current–voltage relationship with the peak currents seen at moderately repolarizing voltages (Fig. 1C), the voltage dependence of TTX-R resurgent currents was more positive compared with TTX-S resurgent currents. TTX-R resurgent currents were evident beginning at repolarization steps to 10 mV and peaked at ∼−20 mV, whereas classic TTX-S resurgent currents are not evident until −10 mV and peak at ∼−40 mV.
Figure 1.

DRG neurons exhibit TTX-R and TTX-S resurgent currents. A, B, Representative TTX-R and TTX-S sodium currents were recorded from medium-sized DRG neurons (30–45 μm) using a voltage-protocol (as shown at bottom) designed for recording resurgent sodium currents. TTX-R resurgent currents were recorded in the presence of 500 nm TTX. The repolarization voltages for colored current traces are listed according to the color and relative top-down position of colored traces. The peak resurgent current traces are black. The voltages for peak resurgent currents are labeled nearby. C, Current–voltage relationship of TTX-R and TTX-S resurgent currents. The peak voltage of TTX-R resurgent currents is more depolarized than that of TTX-S resurgent currents. D, Effects of lidocaine (n = 5), CsCl (n = 5), and A803467 (1 μm, n = 7; 5 μm, n = 6) on TTX-R resurgent currents. The peak current amplitude in the presence of each compound was normalized to its own control. Data are mean ± SEM. **p < 0.01 (Student's t test).
Both TTX-R and TTX-S resurgent currents showed a slow onset and a slow decay (Fig. 1A,B). This feature of resurgent currents differs from tail currents that are instantaneously activated and typically decay faster. However, as noted above, the TTX-R resurgent currents had much slower kinetics compared with TTX-S resurgent currents. For example, the time to peak and decay time constant of TTX-R resurgent currents at −20 mV are ∼37 and 15 times larger than those of TTX-S resurgent currents (130 ± 12 vs 3.5 ± 0.5 ms for time to peak, 796 ± 66 vs 55 ± 7 ms for decay time constant). By contrast, the time to peak and decay time constant for peak transient currents recorded at the same voltage were only 4–5 times slower for TTX-R compared with TTX-S peak transient currents (2.68 ± 1.11 vs 0.67 ± 0.03 ms for time to peak, 3.81 ± 0.66 vs 0.81 ± 0.05 ms for decay time constant).
TTX-S resurgent currents are not typically observed in small-diameter DRG neurons (Cummins et al., 2005), but we recorded TTX-R resurgent currents from small-diameter DRG neurons (data not shown). These currents were similar to those in medium-sized DRG neurons, with slow kinetics compared with classic TTX-S resurgent currents. The voltage for the peak TTX-R resurgent current, however, was more negative for currents recorded from small DRG neurons compared with medium DRG neurons (−29.2 ± 2.6 mV, n = 12 vs − 22.9 ± 1.3 mV, n = 17, respectively; p < 0.05). On the other hand, the relative magnitude of the resurgent current, determined by normalizing the peak resurgent current amplitude to maximal peak transient current amplitude, was similar for small and medium DRG neurons (0.028 ± 0.006, n = 12 vs 0.029 ± 0.005, n = 17, respectively).
The TTX-R resurgent currents were abolished by the broad-spectrum sodium channel blocker lidocaine (1 mm) and significantly inhibited by the Nav1.8 blocker A803467 at 1 and 5 μm levels but not inhibited by the HCN channel inhibitor CsCl (5 mm) (Fig. 1D). Together, these results indicate that the TTX-R resurgent currents in DRG neurons most likely are generated by Nav1.8 channels. Although the external solution contained 30 mm TEA, we also investigated whether the 3 mm K+ included in the standard external solution or Cs+ in the internal solution might affect the TTX-R resurgent currents. We therefore compared the TTX-R resurgent currents recorded in the standard condition (3 mm K+ in external solution, no TEA in internal solution) with those recorded with 0 mm K+ in the external solution and no TEA in the internal solution (condition A) or 0 mm K+ in the external solution and 4.5 mm TEA in the internal solution (condition B). We found that there were no significant differences among the three conditions in the ratio of peak resurgent current (0.022 ± 0.002, n = 11 for control; 0.026 ± 0.005, n = 10 for condition A; and 0.027 ± 0.004, n = 7 for condition B) or voltage of peak resurgent current (−21.8 ± 1.5 mV, n = 11 for control; −24.0 ± 1.5 mV, n = 10 for condition A; and −22.1 ± 1.5 mV, n = 7 for condition B). These results indicate that the TTX-R resurgent currents were not significantly affected by 3 mm external K+ or internal Cs+ in the current study.
Activation mechanism of slow TTX-R resurgent currents
During membrane depolarization, sodium channels transition to a nonconducting, inactivated configuration as the isoleucine, phenylalanine, and methionine (IFM) inactivation gate interacts with the pore to obstruct ion flux. This sequence of conformational transitions differs from the hypothesized mechanism for sodium resurgent current, where the channel is inactivated by an intracellular segment of the sodium channel β4 subunit at depolarized voltages and open-channel block is relieved upon repolarization to intermediate voltages (Grieco et al., 2005; Theile and Cummins, 2011b). The hypothesis is partially based on the observations that a β4 intracellular segment-mimetic peptide, often called the β4 peptide, is able to induce resurgent currents in neurons and heterologous expression systems that do not otherwise produce resurgent currents (Grieco et al., 2005; Theile and Cummins, 2011a).
To investigate whether the TTX-R resurgent currents in DRG neurons might be mediated by an interaction between Nav1.8 α and β4 subunits, we first conducted coimmunoprecipitation experiments on Nav1.8 α subunit in DRG and whole brain (Fig. 2). We observed selective precipitation of a Nav1.8 band in the DRG lysates but not in whole-brain lysates, indicating that the Nav1.8 antibody did not pick up sodium channel isoforms expressed in brain, including Nav1.6 (Fig. 2A,B). Furthermore, we found that Nav1.8 in DRG was associated with β4 subunits, which indicated that Nav1.8 and β4 subunits might interact with each other in DRG neurons, and this interaction could contribute to the TTX-R resurgent currents we recorded (Fig. 2B). If an interaction between Nav1.8 and the cytoplasmic tail of β4 subunits indeed produces the TTX-R resurgent currents in DRG neurons, increasing this interaction by adding intracellular β4 peptide should increase the TTX-R resurgent currents. We found that in DRG neurons intracellular application of β4 peptide (200 μm) significantly enhanced TTX-R slow resurgent currents (Fig. 3A,B,E). The β4 peptide also significantly enhanced classic TTX-S resurgent currents recorded in medium-sized DRG neurons that exhibited TTX-S but not TTX-R peak transient currents (Fig. 3C–E). Importantly, the β4 peptide did not induce significant changes in the voltage where peak resurgent currents were observed or the current kinetics for both TTX-R and TTX-S groups (Fig. 3F). These results suggest that the slow TTX-R resurgent currents likely share the same mechanism that has been proposed to underlie TTX-S resurgent currents in cerebellar neurons (Grieco et al., 2005) and thus should be considered as resurgent currents.
Figure 2.

Association of NaV1.8 with β4 subunits in DRG neurons. A, Immunoblot results from the coimmunoprecipitation experiment from rat brain lysate. Top, Sodium channel α subunit bands (200–260 kDa) are observed in the lysate input indicating that CNS NaV protein is present in the IP input lysate. Bottom, Sodium channel β4 subunit protein band is observed in the lysate input indicating that β4 subunit protein was present in the IP input lysate. Right, Neither NaV1.8 channel nor β4 subunit protein was coimmunoprecipitated from the rat brain (rBrain) lysate matrix using either the control IgG(Rb) or NaV1.8 antibody. B, Immunoblot results from the coimmunoprecipitation experiment from rat DRG lysate. Top, Sodium channel α subunit bands (200–260 kDa) are observed in the lysate input indicating that peripheral NaV protein is present in the IP input lysate. Bottom, β4 subunit protein band is observed in the lysate input indicating that β4 subunit protein was present in the IP input lysate. Right, NaV1.8 channel and β4 subunit protein are coimmunoprecipitated from rat DRG (rDRG) lysate only by the anti-NaV1.8 Asc-016 antibody.
Figure 3.

Increase of TTX-R and TTX-S resurgent currents by a β4 peptide in DRG neurons. Representative TTX-R (A, B) and TTX-S (C, D) resurgent currents were recorded from medium-sized DRG neurons in the absence and presence of 200 μm β4 peptide in the patch pipettes. The currents were recorded using the same voltage protocol in Fig. 1. Currents are scaled to reflect the ratio resurgent currents. The ratio resurgent currents were calculated by normalizing peak resurgent currents to peak transient currents recorded in the same cells. E, F, Summary of ratio resurgent current and voltage at which peak resurgent currents were recorded (TTX-R control, n = 16; TTX-R plus peptide, n = 8; TTX-S control, n = 12; TTX-S plus peptide, n = 6). White bars represent control; black bars represent plus peptide. Data are mean ± SEM. *p < 0.05 (Student's t test). **p < 0.01 (Student's t test).
There are striking differences between TTX-S and TTX-R resurgent currents in terms of voltage dependence (Fig. 1C), rate of onset, and rate of decay (Fig. 4A). To explore whether these differences are dictated by differences in inactivation kinetics or voltage-dependent gating, we used computational models to simulate TTX-S and TTX-R sodium currents. TTX-S sodium currents were modeled using a kinetic scheme developed to investigate resurgent current generation (Raman and Bean, 2001). TTX-R currents were modeled using a kinetic scheme developed for Nav1.8 currents (Sheets et al., 2007). The decay rate of simulated peak transient currents elicited by a step depolarization to −20 mV were approximately eightfold slower for the TTX-R model currents than for the TTX-S model currents (Fig. 4B), and the midpoint for the voltage dependence of activation is ∼15 mV more depolarized for the model TTX-R currents (−16 mV) than for the model TTX-S currents (−31 mV). Resurgent currents were elicited in the simulated TTX-S and TTX-R channels using the kinetic scheme for the resurgent current particle developed by Raman and Bean (2001). Example traces elicited at −20 mV are shown in Figure 4C. The rate of onset was similar for the simulated TTX-S and TX-R resurgent currents, and the rate of decay differed only twofold between simulated TTX-R and TTX-S resurgent currents (70 vs 37 ms for decay time constants, respectively). Although the kinetics did not resemble those of the DRG resurgent currents, the voltage dependence of the resurgent currents (Fig. 4D) do mimic those of the endogenous currents (compare withFig. 1C). To determine the impact of only altering the rate of inactivation on resurgent current kinetics, the decay time constant for peak transient currents was increased by fivefold and 10-fold for the TTX-S model. This slowing of inactivation increased the decay time constant of the simulated TTX-S resurgent current by 2.5-fold and 3.3-fold, respectively (Fig. 4E). However, no substantial change was observed in the rate of onset for the simulated TTX-S resurgent currents, and no change was observed in the voltage dependence of the resurgent currents (data not shown). Because a higher affinity for the resurgent current particle is likely to impact the onset of resurgent currents (Lewis and Raman, 2011), we next asked whether increasing the affinity for the resurgent current particle would slow the onset of the resurgent current. Reducing the rate constant for exit from the open-channel blocked state induced by the resurgent current particle by fivefold, which effectively simulates an increased affinity, increased the time to peak for the simulated TTX-R resurgent by 2.5-fold (from 9.5 to 23 ms) and increased the time constant for decay by fourfold (Fig. 4F). These results suggest that resurgent currents in Nav1.8 channels may differ from TTX-S resurgent currents because of differences in the voltage dependence of channel gating, slower inactivation kinetics, and a higher affinity interaction with the cytoplasmic tail of the β4 subunit.
Figure 4.
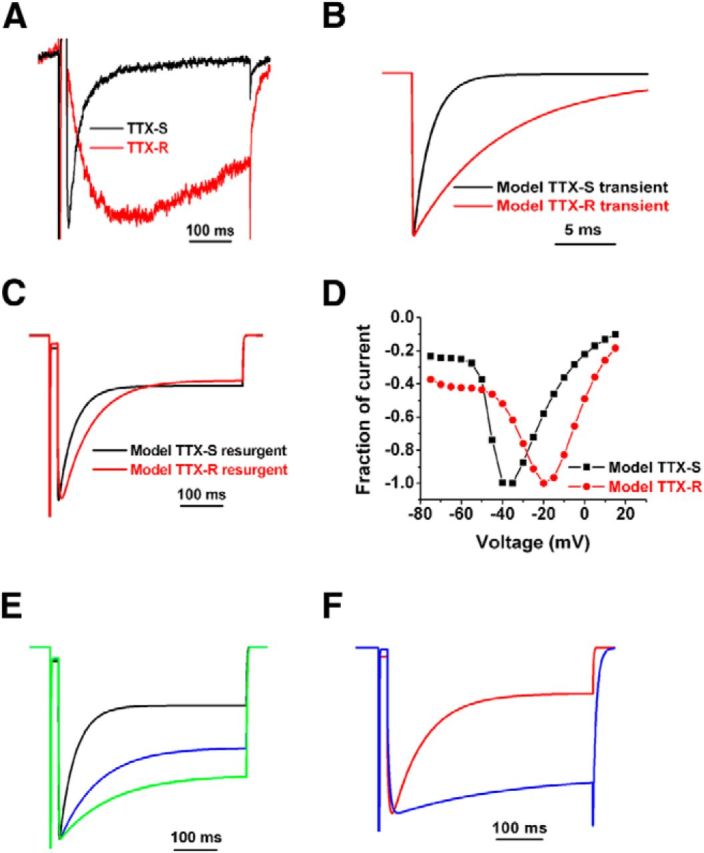
Simulations of TTX-R and TTX-S resurgent currents. A, Comparison of TTX-R (red trace) and TTX-S (black trace) resurgent currents recorded from DRG neurons. Traces were elicited with a −20 mV repolarization step after a 20 ms depolarization to 30 mV. B, Comparison of simulated TTX-S (black trace) and TTX-R (red trace) transient currents elicited with a step depolarization to −20 mV from a holding potential of −100 mV. C, Comparison of simulated TTX-S (black trace) and TTX-R (red trace) resurgent currents elicited with a −20 mV repolarization step after a 20 ms depolarization to 30 mV. D, Current–voltage relationship of simulated TTX-R and TTX-S resurgent currents. The peak voltage of TTX-R resurgent currents is more depolarized than that of TTX-S resurgent currents. E, Increasing the time constant of TTX-S inactivation slows the decay of simulated resurgent currents. Resurgent currents generated with control TTX-S model (black trace) and TTX-S channels with fivefold (blue trace) and 10-fold (green trace) longer time constants for inactivation are shown. F, Reducing the rate constant for exit from open-channel block by fivefold (blue trace) decreased both the onset of TTX-R resurgent current and the decay time constant for the resurgent current.
Modulation of TTX-R resurgent currents by inflammatory mediators
Inflammatory mediators released during tissue injury cause membrane hyperexcitability of DRG neurons and therefore contribute to enhanced pain sensations during inflammation (Richardson and Vasko, 2002; Hucho and Levine, 2007). One mechanism by which inflammatory mediators can sensitize DRG neurons is through increasing expression and enhancing activation of the TTX-R, slow-inactivating sodium channel Nav1.8 (Khasar et al., 1998; Lai et al., 2004; Cummins et al., 2007). We tested whether inflammatory mediators would enhance TTX-R slow resurgent currents in DRG neurons. We found a soup of inflammatory mediators, consisting of 1 μm bradykinin, 10 μm 5-HT, 10 μm histamine, 10 μm PGE2, and 5 μm ATP, significantly increased TTX-R and TTX-S resurgent currents in DRG neurons (Fig. 5A–E). In addition, the same inflammatory soup also shifted the voltage for peak TTX-R but not for peak TTX-S resurgent currents (Fig. 5F).
Figure 5.

Increase of TTX-R resurgent currents by inflammatory mediators (IMs) in DRG neurons. Representative TTX-R (A, B) and TTX-S (C, D) resurgent currents were recorded from medium-sized DRG neurons in the absence and presence of IMs. IMs (1 μm bradykinin, 10 μm 5-HT, 10 μm histamine, 10 μm PGE2, and 5 μm ATP) were pretreated for 5 min in the recording chamber before recording begins. The currents were recorded using the same voltage protocol in Fig. 1. Currents are scaled to reflect the ratio resurgent currents. The ratio resurgent currents were calculated by normalizing peak resurgent currents to peak transient currents recorded in the same cells. E, F, Summary of ratio resurgent current and voltage at which peak resurgent currents were recorded (TTX-R control n = 14; TTX-R plus IMs n = 10; TTX-S control n = 12; TTX-S plus IMs n = 19). White bars represent control; black bars represent plus IMs. Data are mean ± SEM. *p < 0.05 (Student's t test).
TTX-S resurgent sodium currents are generally accepted to facilitate repetitive firing of action potentials in CNS and DRG neurons (Khaliq et al., 2003; Jarecki et al., 2010). To test whether increased TTX-R resurgent currents would lead to increased membrane excitability in DRG neurons, we recorded action potentials in small DRG neurons in the presence of 500 nm TTX to block TTX-S sodium channels. We found intracellular β4 peptide (200 μm) significantly increased the number of DRG neurons that fired multiple action potentials in response to suprathreshold current injections (6 of 17 in control vs 11 of 16 in the β4 peptide group; p < 0.01 χ2 test) (Fig. 6A–C). On the other hand, the resting membrane potentials, input resistances, and action potential rheobases were not significantly changed by β4 peptide (−64.4 ± 1.8 mV, 317 ± 51 mΩ, and 717 ± 95 pA in control; −67.9 ± 1.0 mV, 371 ± 50 mΩ, and 588 ± 52 pA in the β4 peptide group). These results suggest that the enhancing effects of inflammatory mediators on the TTX-R and TTX-S resurgent currents would likely contribute to the hyperexcitability of DRG neurons that is associated with inflammation.
Figure 6.

Increase of TTX-R excitability in small DRG neurons by a β4 peptide. Small DRG neurons were examined under current-clamp conditions; 500 nm TTX was included in the bath solution to block TTX-S sodium channels. A series of depolarizing current steps (0 pA up to 3× rheobase with a 100 pA increment) were injected into small DRG neurons from their resting membrane potentials. Representative membrane responses to current injection from 0 pA to rheobase, 2× rheobase, and 3× rheobase were shown in control (A) and 200 μm β4 peptide (B). Compared with control, β4 peptide significantly (p < 0.01, χ2 test) increased the percentage of small DRG neurons that fire multiple action potentials after suprathreshold current injection up to 3× rheobase (C).
Discussion
Here we identified and characterized a TTX-R resurgent current in adult rat DRG neurons. This current was activated by membrane repolarization, inhibited by the broad-spectrum sodium channel modulator lidocaine as well as the Nav1.8 channel blocker A803467, augmented by a peptide mimetic of the β4 cytoplasmic tail, and enhanced by inflammatory mediators. In addition, we found that Nav1.8 α subunits were able to associate with β4 subunits in rat DRG neurons. Together, these data provide compelling evidence that the slow TTX-R resurgent current is mediated by TTX-R Nav1.8 sodium channels and indicate that it is most likely activated by a similar mechanism as proposed for classic TTX-S resurgent currents.
Nav1.8 is one of the major sodium channel isoforms expressed in nociceptive DRG neurons (Djouhri et al., 2003; Cummins et al., 2007; Liu and Wood, 2011). Nav1.8 sodium channels produce TTX-R, slow-inactivating sodium current with depolarized voltage gating properties. Here we determined that the slow resurgent current was most likely carried by Nav1.8 because (1) the slow resurgent currents were only observed in cells exhibiting Nav1.8 slow-inactivating currents, (2) the slow resurgent current was insensitive to TTX and the HCN blocker CsCl, and (3) the slow resurgent current was sensitive to lidocaine and the reported Nav1.8 selective inhibitor A80346. The voltage dependence of the TTX-R resurgent current was more depolarized than that of TTX-S resurgent currents, and the kinetics were substantially slower. These differences could either be explained by differences between TTX-S channels and Nav1.8 or, alternatively, indicate a distinct mechanism of action from TTX-S resurgent currents. However, inclusion of the β4 peptide in the intracellular recording solution substantially enhanced TTX-S resurgent current activity in cells expressing predominantly TTX-S currents and also substantially enhanced the slow resurgent current activity in cells exhibiting large Nav1.8-type currents. These data indicate that both the TTX-S and TTX-R resurgent currents, despite the large difference in kinetics, are generated by a similar relief-of-open-channel-block mechanism. Indeed, our coimmunoprecipitation experiments demonstrate that Nav1.8 in DRG sensory neurons can interact with endogenous β4 subunits. However, our data do not rule out the possibility that there are entities other than β4 involved in either TTX-S or TTX-R resurgent sodium currents in DRG neurons.
TTX-S and TTX-R resurgent currents differed in voltage dependence, rate of onset, and decay rate. Computer simulations indicated that the large differences in kinetics (37-fold slower time to peak and 15-fold slower decay for TTX-R resurgent currents) cannot be explained only by the different kinetics of transient current decay (4–5 times slower for TTX-R currents). In-depth studies of resurgent currents and TTX-S channels indicate that the strength of the interaction between the β4 subunit and the sodium channel α-subunit is an important determinant of the resurgent current onset (Aman and Raman, 2007; Lewis and Raman, 2011, 2013), and our computer simulations support the notion that prolonged unbinding of the β4 C-terminal from Nav1.8 contributes to the slower kinetics of the TTX-R resurgent currents. Furthermore, recovery from blockade by lidocaine, which also binds to the inner pore region as the β4 peptide does, is prolonged in Nav1.8 (Leffler et al., 2007), providing additional evidence that Nav1.8 channels interact differently with pore blockers. However, the difference in onset between TTX-S and TTX-R resurgent currents in DRG neurons is much larger than changes seen in previous studies. As we were unable to fully replicate the very slow onset kinetics of the TTX-R resurgent currents in our computer simulations, it seems that additional refinements in our mechanistic understanding are needed to adequately model the Nav1.8 current and its interaction with the resurgent current particle.
Functionally, Nav1.8 contributes to the majority of the upstroke of action potentials in nociceptive DRG neurons (Blair and Bean, 2002) and can generate action potentials in the presence of TTX and/or under sustained depolarization (Renganathan et al., 2001). TTX-R resurgent current produced by Nav1.8 would also likely contribute to action potential generation. Indeed, many small-diameter DRG neurons generate action potentials with a substantial shoulder on the repolarization phase of the action potential. Blair and Bean (2002) demonstrated that TTX-R sodium currents reactivate during this phase, providing the inward current underlying the action potential shoulder. Although it was proposed that incomplete inactivation of TTX-R sodium channels might underlie this shoulder current, the voltage dependence of the TTX-R current described by Blair and Bean (2002) is strikingly consistent with the voltage dependence of the TTX-R resurgent currents that we observe. Thus, TTX-R resurgent sodium currents might contribute to the broad action potentials observed in small-diameter DRG neurons and, as with TTX-S resurgent currents, likely would contribute to the generation of repetitive action potential firing.
Nav1.8 plays important roles in hyperexcitability of nociceptive DRG neurons under inflammatory pain conditions. Expression of Nav1.8 (both mRNA and protein levels) and slow-inactivating TTX-R sodium currents were significantly upregulated in carrageenan and complete Freund's adjuvant-treated animals (Tanaka et al., 1998; Yu et al., 2011). Inhibition of Nav1.8 in these models by antisense oligodeoxynucleotides or the selective channel blocker A803467 effectively reduced the thermal and mechanical hypersensitivity (Joshi et al., 2006; Jarvis et al., 2007; Yu et al., 2011). On the other hand, inflammatory mediators released during tissue injury enhance Nav1.8 function and neuronal excitability (Lai et al., 2004). Inflammatory soups as well as individual mediators can increase the amplitude and enhance activation of Nav1.8-mediated TTX-R slow-inactivating currents in sensory neurons (England et al., 1996; Gold et al., 1996; Cardenas et al., 2001). Interestingly, in the current study, we found that inflammatory mediators significantly increased TTX-R resurgent currents in rat DRG neurons. Because the β4 peptide increased TTX-R resurgent currents and TTX-R sodium current-dependent membrane excitability in DRG neurons, this increase would likely enhance the repetitive firing of nociceptive DRG neurons, contributing to neuronal hyperexcitability and inflammatory pain. In addition to TTX-R resurgent currents, the present study also found that inflammatory mediators increased TTX-S resurgent currents in DRG neurons. TTX-S resurgent sodium currents have been implicated in the neuronal hyperexcitability associated with seizures (Hargus et al., 2011) and cold-induced pain associated with the chemotherapeutic agent oxaliplatin (Sittl et al., 2012). The TTX-S resurgent currents in DRG are mostly expressed in medium- to large-sized DRG neurons and are mostly carried by Nav1.6 (Cummins et al., 2005). On the other hand, TTX-R resurgent currents carried by Nav1.8 are mostly expressed in small- to medium-sized DRG neurons. Therefore, inflammatory mediator-induced increases in TTX-R and TTX-S resurgent currents might contribute to enhanced neuronal excitability of multiple DRG neuron populations, and attenuating TTX-S and TTX-R resurgent sodium currents could reduce neuronal excitability and pain associated with inflammation.
Footnotes
This work was supported by a Lilly Research Award. T.R.C. was also supported in part by a National Institutes of Health Grant NS053422. We thank Dr. Jonathan Theile for help with preliminary experiments and James Jackson for help with cell cultures.
A.D.P., B.T.P., K.L.K., J.L.K., J.S.M., and E.S.N. are employees of Eli Lilly and Co. The remaining authors declare no competing financial interests.
References
- Aman TK, Raman IM. Subunit dependence of Na channel slow inactivation and open channel block in cerebellar neurons. Biophys J. 2007;92:1938–1951. doi: 10.1529/biophysj.106.093500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bant JS, Raman IM. Control of transient, resurgent, and persistent current by open-channel block by Na channel beta4 in cultured cerebellar granule neurons. Proc Natl Acad Sci U S A. 2010;107:12357–12362. doi: 10.1073/pnas.1005633107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blair NT, Bean BP. Roles of tetrodotoxin (TTX)-sensitive Na+ current, TTX-resistant Na+ current, and Ca2+ current in the action potentials of nociceptive sensory neurons. J Neurosci. 2002;22:10277–10290. doi: 10.1523/JNEUROSCI.22-23-10277.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caffrey JM, Eng DL, Black JA, Waxman SG, Kocsis JD. Three types of sodium channels in adult rat dorsal root ganglion neurons. Brain Res. 1992;592:283–297. doi: 10.1016/0006-8993(92)91687-A. [DOI] [PubMed] [Google Scholar]
- Cardenas LM, Cardenas CG, Scroggs RS. 5HT increases excitability of nociceptor-like rat dorsal root ganglion neurons via cAMP-coupled TTX-resistant Na(+) channels. J Neurophysiol. 2001;86:241–248. doi: 10.1152/jn.2001.86.1.241. [DOI] [PubMed] [Google Scholar]
- Castelli L, Biella G, Toselli M, Magistretti J. Resurgent Na+ current in pyramidal neurones of rat perirhinal cortex: axonal location of channels and contribution to depolarizing drive during repetitive firing. J Physiol. 2007;582:1179–1193. doi: 10.1113/jphysiol.2007.135350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cummins TR, Dib-Hajj SD, Black JA, Akopian AN, Wood JN, Waxman SG. A novel persistent tetrodotoxin-resistant sodium current in SNS-null and wild-type small primary sensory neurons. J Neurosci. 1999;19:RC43. doi: 10.1523/JNEUROSCI.19-24-j0001.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cummins TR, Black JA, Dib-Hajj SD, Waxman SG. Glial-derived neurotrophic factor upregulates expression of functional SNS and NaN sodium channels and their currents in axotomized dorsal root ganglion neurons. J Neurosci. 2000;20:8754–8761. doi: 10.1523/JNEUROSCI.20-23-08754.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cummins TR, Dib-Hajj SD, Herzog RI, Waxman SG. Nav1.6 channels generate resurgent sodium currents in spinal sensory neurons. FEBS Lett. 2005;579:2166–2170. doi: 10.1016/j.febslet.2005.03.009. [DOI] [PubMed] [Google Scholar]
- Cummins TR, Sheets PL, Waxman SG. The roles of sodium channels in nociception: implications for mechanisms of pain. Pain. 2007;131:243–257. doi: 10.1016/j.pain.2007.07.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Djouhri L, Fang X, Okuse K, Wood JN, Berry CM, Lawson SN. The TTX-resistant sodium channel Nav1.8 (SNS/PN3): expression and correlation with membrane properties in rat nociceptive primary afferent neurons. J Physiol. 2003;550:739–752. doi: 10.1113/jphysiol.2003.042127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Do MT, Bean BP. Subthreshold sodium currents and pacemaking of subthalamic neurons: modulation by slow inactivation. Neuron. 2003;39:109–120. doi: 10.1016/S0896-6273(03)00360-X. [DOI] [PubMed] [Google Scholar]
- Do MT, Bean BP. Sodium currents in subthalamic nucleus neurons from Nav1.6-null mice. J Neurophysiol. 2004;92:726–733. doi: 10.1152/jn.00186.2004. [DOI] [PubMed] [Google Scholar]
- England S, Bevan S, Docherty RJ. PGE2 modulates the tetrodotoxin-resistant sodium current in neonatal rat dorsal root ganglion neurones via the cyclic AMP-protein kinase A cascade. J Physiol. 1996;495:429–440. doi: 10.1113/jphysiol.1996.sp021604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Enomoto A, Han JM, Hsiao CF, Wu N, Chandler SH. Participation of sodium currents in burst generation and control of membrane excitability in mesencephalic trigeminal neurons. J Neurosci. 2006;26:3412–3422. doi: 10.1523/JNEUROSCI.5274-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Enomoto A, Han JM, Hsiao CF, Chandler SH. Sodium currents in mesencephalic trigeminal neurons from Nav1.6 null mice. J Neurophysiol. 2007;98:710–719. doi: 10.1152/jn.00292.2007. [DOI] [PubMed] [Google Scholar]
- Gittis AH, du Lac S. Similar properties of transient, persistent, and resurgent Na currents in GABAergic and non-GABAergic vestibular nucleus neurons. J Neurophysiol. 2008;99:2060–2065. doi: 10.1152/jn.01389.2007. [DOI] [PubMed] [Google Scholar]
- Gold MS, Reichling DB, Shuster MJ, Levine JD. Hyperalgesic agents increase a tetrodotoxin-resistant Na+ current in nociceptors. Proc Natl Acad Sci U S A. 1996;93:1108–1112. doi: 10.1073/pnas.93.3.1108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grieco TM, Raman IM. Production of resurgent current in NaV1.6-null Purkinje neurons by slowing sodium channel inactivation with beta-pompilidotoxin. J Neurosci. 2004;24:35–42. doi: 10.1523/JNEUROSCI.3807-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grieco TM, Malhotra JD, Chen C, Isom LL, Raman IM. Open-channel block by the cytoplasmic tail of sodium channel beta4 as a mechanism for resurgent sodium current. Neuron. 2005;45:233–244. doi: 10.1016/j.neuron.2004.12.035. [DOI] [PubMed] [Google Scholar]
- Hargus NJ, Merrick EC, Nigam A, Kalmar CL, Baheti AR, Bertram EH, 3rd, Patel MK. Temporal lobe epilepsy induces intrinsic alterations in Na channel gating in layer II medial entorhinal cortex neurons. Neurobiol Dis. 2011;41:361–376. doi: 10.1016/j.nbd.2010.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hucho T, Levine JD. Signaling pathways in sensitization: toward a nociceptor cell biology. Neuron. 2007;55:365–376. doi: 10.1016/j.neuron.2007.07.008. [DOI] [PubMed] [Google Scholar]
- Hudmon A, Choi JS, Tyrrell L, Black JA, Rush AM, Waxman SG, Dib-Hajj SD. Phosphorylation of sodium channel Na(v)1.8 by p38 mitogen-activated protein kinase increases current density in dorsal root ganglion neurons. J Neurosci. 2008;28:3190–3201. doi: 10.1523/JNEUROSCI.4403-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jarecki BW, Piekarz AD, Jackson JO, 2nd, Cummins TR. Human voltage-gated sodium channel mutations that cause inherited neuronal and muscle channelopathies increase resurgent sodium currents. J Clin Invest. 2010;120:369–378. doi: 10.1172/JCI40801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jarvis MF, Honore P, Shieh CC, Chapman M, Joshi S, Zhang XF, Kort M, Carroll W, Marron B, Atkinson R, Thomas J, Liu D, Krambis M, Liu Y, McGaraughty S, Chu K, Roeloffs R, Zhong C, Mikusa JP, Hernandez G, et al. A-803467, a potent and selective Nav1.8 sodium channel blocker, attenuates neuropathic and inflammatory pain in the rat. Proc Natl Acad Sci U S A. 2007;104:8520–8525. doi: 10.1073/pnas.0611364104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joshi SK, Hernandez G, Mikusa JP, Zhu CZ, Zhong C, Salyers A, Wismer CT, Chandran P, Decker MW, Honore P. Comparison of antinociceptive actions of standard analgesics in attenuating capsaicin and nerve-injury-induced mechanical hypersensitivity. Neuroscience. 2006;143:587–596. doi: 10.1016/j.neuroscience.2006.08.005. [DOI] [PubMed] [Google Scholar]
- Khaliq ZM, Gouwens NW, Raman IM. The contribution of resurgent sodium current to high-frequency firing in Purkinje neurons: an experimental and modeling study. J Neurosci. 2003;23:4899–4912. doi: 10.1523/JNEUROSCI.23-12-04899.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khasar SG, Gold MS, Levine JD. A tetrodotoxin-resistant sodium current mediates inflammatory pain in the rat. Neurosci Lett. 1998;256:17–20. doi: 10.1016/S0304-3940(98)00738-1. [DOI] [PubMed] [Google Scholar]
- Kim JH, Kushmerick C, von Gersdorff H. Presynaptic resurgent Na+ currents sculpt the action potential waveform and increase firing reliability at a CNS nerve terminal. J Neurosci. 2010;30:15479–15490. doi: 10.1523/JNEUROSCI.3982-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lai J, Porreca F, Hunter JC, Gold MS. Voltage-gated sodium channels and hyperalgesia. Annu Rev Pharmacol Toxicol. 2004;44:371–397. doi: 10.1146/annurev.pharmtox.44.101802.121627. [DOI] [PubMed] [Google Scholar]
- Leão RN, Naves MM, Leão KE, Walmsley B. Altered sodium currents in auditory neurons of congenitally deaf mice. Eur J Neurosci. 2006;24:1137–1146. doi: 10.1111/j.1460-9568.2006.04982.x. [DOI] [PubMed] [Google Scholar]
- Leffler A, Reiprich A, Mohapatra DP, Nau C. Use-dependent block by lidocaine but not amitriptyline is more pronounced in tetrodotoxin (TTX)-resistant Nav1.8 than in TTX-sensitive Na+ channels. J Pharmacol Exp Ther. 2007;320:354–364. doi: 10.1124/jpet.106.109025. [DOI] [PubMed] [Google Scholar]
- Lewis AH, Raman IM. Cross-species conservation of open-channel block by Na channel beta4 peptides reveals structural features required for resurgent Na current. J Neurosci. 2011;31:11527–11536. doi: 10.1523/JNEUROSCI.1428-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis AH, Raman IM. Interactions among DIV voltage-sensor movement, fast inactivation, and resurgent Na current induced by the NaVbeta4 open-channel blocking peptide. J Gen Physiol. 2013;142:191–206. doi: 10.1085/jgp.201310984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu M, Wood JN. The roles of sodium channels in nociception: implications for mechanisms of neuropathic pain. Pain Med. 2011;12(Suppl 3):S93–S99. doi: 10.1111/j.1526-4637.2011.01158.x. [DOI] [PubMed] [Google Scholar]
- Raman IM, Bean BP. Resurgent sodium current and action potential formation in dissociated cerebellar Purkinje neurons. J Neurosci. 1997;17:4517–4526. doi: 10.1523/JNEUROSCI.17-12-04517.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raman IM, Bean BP. Inactivation and recovery of sodium currents in cerebellar Purkinje neurons: evidence for two mechanisms. Biophys J. 2001;80:729–737. doi: 10.1016/S0006-3495(01)76052-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raman IM, Sprunger LK, Meisler MH, Bean BP. Altered subthreshold sodium currents and disrupted firing patterns in Purkinje neurons of Scn8a mutant mice. Neuron. 1997;19:881–891. doi: 10.1016/S0896-6273(00)80969-1. [DOI] [PubMed] [Google Scholar]
- Renganathan M, Cummins TR, Waxman SG. Contribution of Na(v)1.8 sodium channels to action potential electrogenesis in DRG neurons. J Neurophysiol. 2001;86:629–640. doi: 10.1152/jn.2001.86.2.629. [DOI] [PubMed] [Google Scholar]
- Richardson JD, Vasko MR. Cellular mechanisms of neurogenic inflammation. J Pharmacol Exp Ther. 2002;302:839–845. doi: 10.1124/jpet.102.032797. [DOI] [PubMed] [Google Scholar]
- Sheets PL, Jackson JO, 2nd, Waxman SG, Dib-Hajj SD, Cummins TR. A Nav1.7 channel mutation associated with hereditary erythromelalgia contributes to neuronal hyperexcitability and displays reduced lidocaine sensitivity. J Physiol. 2007;581:1019–1031. doi: 10.1113/jphysiol.2006.127027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sittl R, Lampert A, Huth T, Schuy ET, Link AS, Fleckenstein J, Alzheimer C, Grafe P, Carr RW. Anticancer drug oxaliplatin induces acute cooling-aggravated neuropathy via sodium channel subtype Na(V)1.6-resurgent and persistent current. Proc Natl Acad Sci U S A. 2012;109:6704–6709. doi: 10.1073/pnas.1118058109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanaka M, Cummins TR, Ishikawa K, Dib-Hajj SD, Black JA, Waxman SG. SNS Na+ channel expression increases in dorsal root ganglion neurons in the carrageenan inflammatory pain model. Neuroreport. 1998;9:967–972. doi: 10.1097/00001756-199804200-00003. [DOI] [PubMed] [Google Scholar]
- Theile JW, Cummins TR. Inhibition of Navbeta4 peptide-mediated resurgent sodium currents in Nav1.7 channels by carbamazepine, riluzole, and anandamide. Mol Pharmacol. 2011a;80:724–734. doi: 10.1124/mol.111.072751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Theile JW, Cummins TR. Recent developments regarding voltage-gated sodium channel blockers for the treatment of inherited and acquired neuropathic pain syndromes. Front Pharmacol. 2011b;2:54. doi: 10.3389/fphar.2011.00054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu YQ, Zhao F, Guan SM, Chen J. Antisense-mediated knockdown of Na(V)1.8, but not Na(V)1.9, generates inhibitory effects on complete Freund's adjuvant-induced inflammatory pain in rat. PloS One. 2011;6:e19865. doi: 10.1371/journal.pone.0019865. [DOI] [PMC free article] [PubMed] [Google Scholar]