

Abstract
Intramembrane proteases hydrolyze peptide bonds within the membrane as a signaling paradigm universal to all life forms and with implications in disease. Deciphering the architectural strategies supporting intramembrane proteolysis is an essential but unattained goal. We integrated a new, quantitative and high-throughput thermal light-scattering technology, reversible equilibrium un/refolding, and quantitative protease assays to interrogate rhomboid architecture with 151 purified variants. Rhomboid proteases maintain low intrinsic thermodynamic stability (ΔG=2.1-4.5kcal/mol) resulting from a multitude of generally-weak transmembrane packing interactions, making them highly-responsive to their environment. Stability is consolidated by two buried glycines and several packing leucines, with a few multifaceted hydrogen bonds strategically-deployed to two peripheral regions. Opposite these regions lie transmembrane segment 5 and connected loops that are notably exempt of structural responsibility, suggesting intramembrane proteolysis involves considerable but localized protein dynamics. Our analyses provide a comprehensive ‘heat map’ of the physio-chemical anatomy underlying membrane-immersed enzyme function at unprecedented resolution.
Integral membrane proteins rule the border between the cell and the outside world; they mediate diverse forms of communication and transport across the membrane, develop intimate contacts with the cell’s surroundings, and become the front lines during pathogen infection. The ability of membrane proteins to achieve their remarkable functions depends on their underlying protein architecture1,2. By virtue of being membrane-immersed, integral membrane proteins experience a fundamentally different chemical environment relative to soluble proteins. This makes them intriguingly unlike soluble proteins, while presenting challenges to mechanistic study.
Over the past decade, proteases that catalyze the hydrolysis of peptide bonds within the membrane have been discovered to regulate a remarkable array of signaling functions. These intramembrane proteases are universal to all forms of life, and play central roles in devastating human diseases, including γ-secretase in Alzheimer’s disease and cancer, rhomboid in malaria and Parkinson’s disease, and site-2 protease in tuberculosis and cystic fibrosis infection3-7. In total these enzymes have already been implicated in over a dozen pathological events. Intriguingly, rhomboid architecture is useful even beyond intramembrane proteolysis: Derlins and inactive rhomboid proteins lack catalytic residues but maintain an intact rhomboid architecture that facilitates ER-associated degradation8,9.
Biochemical reconstitution and structural analyzes have finally started to reveal the mechanisms underlying enzymatic function4. Particularly instructive were the structural analyses of the membrane core of GlpG, the rhomboid protease from Escherichia coli10-12. GlpG is comprised of helices with different lengths and relative angles, resulting in a compact but highly-asymmetric bundle. The long L1 loop forms a lateral hairpin comprised of three short helices, and lies protruding into the outer leaflet of the membrane. Transmembrane segment (TM) 4 rests in the enzyme center, entering the protease as an extended peptide chain, abruptly forming a short helix starting at the catalytic serine and continuing slanted relative to the other TMs. At this catalytic core lies the hydrogen-bonded S201-H254 catalytic pair contributed by TMs 4 and 6. Water enters the open catalytic cavity from the extracellular side10,11, while substrates enter laterally through opening of a gate formed by TM5 and the L5 loop11,13-15.
While these seminal studies have revealed the detailed three-dimensional geometry of the molecule, the chemical principles that underlie the function and stability of this architecture remain unclear. The associated difficulty in deciphering these principles should not be underestimated: only a handful of quantitative assays have been developed for assessing membrane protein stability, and all are laborious, not amenable to high throughput, and not uniformly applicable to different membrane proteins16,17. In fact, quantitative measurements of stability have only been achieved for half a dozen multi-spanning helical membrane proteins of the thousands that exist2,16. While it is tempting to extrapolate principles from model studies, the modern challenge is to understand how stabilizing forces are deployed within the architecture of specific membrane proteins to allow them to accomplish their particular functions.
We have integrated a new, robustly-quantitative and high-throughput thermostability technology with equilibrium unfolding and protease activity assays. Through the systematic analysis of 151 variants we derived the first comprehensive structural stability and activity ‘heat maps’ of any intramembrane protease. The picture that emerges details how rhomboid is constructed and uncovers its functional properties, including globally low stability that’s responsive to its environment, as well as strategic regions for maintaining stability, and localized elements deployed for dynamic functions during proteolysis.
RESULTS
Rhomboid Proteases Display Low Thermodynamic Stability
GlpG is a robust enzyme that has been crystallized over a dozen times in different space groups, implying rhomboid proteases harbor a particularly stable architecture. Moreover, the recent structure of GlpG in a membrane bicelle revealed surprisingly low root-mean-square-deviation relative to the detergent state (0.66Å along all Cα atoms)15, suggesting that much of GlpG stability is intrinsic to the protein architecture itself. We examined the thermodynamic stability of four pure rhomboid proteases in detergent micelles using an SDS-mediated unfolding assay (the only equilibrium method established for helical membrane proteins16,18). We followed protease activity, endogenous tryptophan fluorescence, and helicity by circular dichroism, as a function of SDS concentration during unfolding (Fig. 1a, Supplementary Results, Supplementary Fig. 1). Monitoring tryptophan fluorescence and protease activity resulted in comparable unfolding curves, while secondary structure was maintained at all SDS concentrations, as often observed with helical membrane proteins. We could restore fluorescence (Supplementary Fig. 1d) and protease activity (Fig. 1b) upon dilution from fully-denatured conditions for all four rhomboid proteases, revealing that unfolding was reversible.
Figure 1. Thermodynamic assessment of rhomboid protease stability.
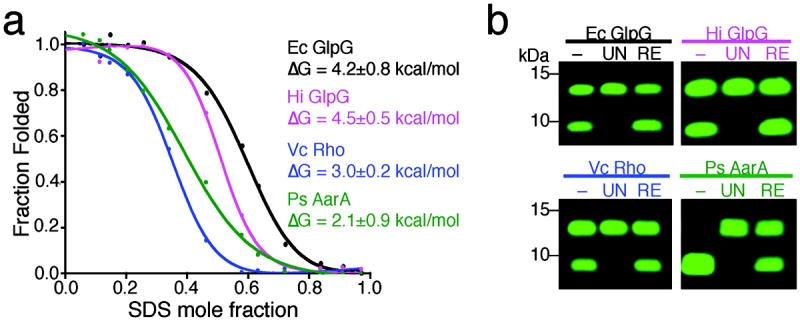
(a) Chemical unfolding of wildtype rhomboid proteases in 2mM DDM at 25°C monitored by protease activity. The fraction folded was plotted against SDS mole fraction and the curves were fit with a two-state model according to the Santoro-Bolen equation. Ec is Escherichia coli, Vc is Vibrio cholerae, Hi is Haemophilus influenzae, and Ps is Providencia stuartii. (b) Activity analysis of refolded rhomboid proteases. ‘–’ denotes enzyme prior to treatment, ‘UN’ the fully-unfolded state (0.9 mole fraction SDS), and ‘RE’ the refolded state (by dilution from 0.9 to 0.1 mole fraction SDS). Shown are infrared fluorescence western analyses in which the top band is the substrate while the lower band is the cleaved product. Refolding restored 100% activity for all rhomboid proteases tested except AarA (~50%).
We achieved an excellent fit of the SDS unfolding data to a two-state unfolding reaction with linear dependence of ΔG on SDS concentration (Supplementary Fig. 1e). However, using tryptophan fluorescence to monitor unfolding was unreliable for most rhomboid enzymes due to sloping and/or unstable baselines, while the protease activity data was much more robust. Therefore, to derive the ΔG of stability for each rhomboid protease in this equilibrium system, we fit the unfolding curves monitored by protease activity with the Santoro-Bolen equation to take both baselines into account 19 (Fig. 1a). The ΔG of stability for the four rhomboid proteases ranged from only 2.1 to 4.5 kcal/mol, revealing that rhomboid proteases as a family maintain low intrinsic thermodynamic stability.
Probing GlpG Architecture with Thermal Light Scattering
We next sought to understand how structure is sustained at the molecular level, and whether different regions have specialized functions, by interrogating on a comprehensive scale all contacts predicted in the crystal structures. To accomplish this goal, we needed a quantitative unfolding assay that is robust for helical membrane proteins and capable of moderate throughput. Thermostability assays using fluorescent probes to monitor unfolding of soluble proteins are now routine, but similar approaches are tricky with membrane proteins because of their hydrophobic nature20. Instead, we turned to a recently-developed differential static light scattering technology in 384-well format that has been used predominantly to detect changes in protein stability upon ligand binding21.
Pure GlpG was heated from 25.0°C to 85.0°C, and scattering of ~620nm light was quantified every 0.5°C (Fig. 2a). The result was a two-state Boltzmann curve with a sharp transition and a transition midpoint (Tm) of 71.0±0.48°C (n=24). We repeated this thermal unfolding, monitoring protease activity (Supplementary Fig. 2a), and secondary structure by circular dichroism, both of which revealed nearly identical curves (Fig. 2a). Notably this system is irreversible: conducting protease assays directly at elevated temperatures resulted in the same amount of activity loss as preincubating the enzyme at the elevated temperature followed by cooling and protease activity analysis at its natural 37°C (Fig. 2b).
Figure 2. Differential static light scattering as a probe of rhomboid stability.

(a) Static light scattering quantified every 0.5°C was plotted against temperature (°C) for E. coli GlpG. The transition midpoint (Tm) for wildtype GlpG was 71.0±0.48°C (n=24). Note that the red line denotes loss of protease activity (not light scattering) and the purple line monitors helicity (ellipticity at 222 nm) as a function of temperature. The lower panel shows a western analysis of GlpG protease activity after pretreatment at varying temperatures. (b) Thermal denaturation of GlpG is irreversible: black bars show protease activity (mean ± standard deviation) conducted at the designated temperatures for 20 minutes, while the blue bars denote GlpG preincubated at the designated temperatures for 20 minutes, cooled, and tested for activity at 37°C. (c) Effect of protein concentration and rate of heating on the Tm (± standard deviation) of wildtype GlpG. (d). Quantified change in Tm (ΔTm) of all engineered GlpG variants plotted against primary structure (residue numbers). Shown are substitutions to alanine and glycine-to-valine mutants in each residue. Colors and segment numbering correlate with tertiary structure (lateral view from membrane, cytoplasm down). The five most perturbed variants (R137A, H145A, G162V, E166A, G261V) are highlighted with red bars (ΔTm>-20°C).
The similarity of the three thermal curves raised the possibility that light scattering was synonymous with monitoring GlpG unfolding. This could be true if GlpG unfolding is rate-limiting, with aggregation/light-scattering proceeding immediately after unfolding. To evaluate this further, we examined concentration- and time-dependence of the light scattering signal. If unfolding is unimolecular and rate-limiting, then changes in concentration and heating rate should not have major effects on Tm. Indeed, a 6-fold range of GlpG concentrations and 5-fold change in heating rate resulted in statistically-indistinguishable Tm values (Fig. 2c), revealing light scattering serves as a valid proxy for monitoring GlpG unfolding. In fact, this thermostability assay was remarkably reproducible, with <1°C standard deviation across the plate and between independent runs, and robust to pH (5.5 to 8.5), ionic strength (50-250mM), divalent ions (0-10mM), and detergent concentration (0.025%-1%). Only salt ≤25mM and detergent below its critical micelle concentration (≤0.01%) perturbed GlpG stability (Supplementary Fig. 2b). GlpG purified using different methods, or harboring N-terminal hemagglutinin or C-terminal His tags, produced indistinguishable transition curves (Fig. 2a). Of the methods that we examined, this assay was by far the most robust, reproducible, and high-throughput.
The transmembrane core of GlpG is a compact helical bundle of 181 structured residues. To interrogate this architecture comprehensively, we ultimately engineered and purified nearly 150 variants from 1L cultures to >90% purity (see Supplementary Figures 3 and 4 for gel analyses of >150 purified proteins), and subjected each to quantitative stability and protease activity analyses. The thermal light scattering approach achieved a remarkably high resolving power with this mutant collection (Fig. 2d). Tms of different mutants spanned a ~31°C range (+3°C to -28°C relative to wildtype GlpG) with an average standard deviation of 0.42°C (also see Supplementary Dataset 1). Finally, the correlation coefficient for deriving Tms by following unfolding using proteolysis versus light scattering was 0.99 with seven mutants that covered the entire spread of ΔTm values (Supplementary Fig. 2c), thereby validating light scattering as a powerful assay with which to interrogate GlpG mutants on a global scale.
Hydrogen bonds are deployed to stabilize only two regions
Hydrogen bonds have traditionally been thought to contribute greatly to membrane protein stability because of the low dielectric of the membrane: even a single polar residue can drive TM helix association 22,23. Elegant studies with bacteriorhodopsin challenge this view24, although it is unclear whether they are representative because bacteriorhodopsin stability comes largely from the covalently-bound retinal cofactor25. GlpG structural models suggest 26 side-chain hydrogen bonds, including several charge-assisted (‘salt bridges’), throughout the TM core. However, we found most contribute little to the structural stability of GlpG. In particular, intramembrane inter-helical hydrogen bonds of W158 with TM1, Y160 with TM5, and Y138 on the L1 loop that should be enhanced in the hydrophobic environment, had little, if any, stabilizing effect on GlpG structure (Fig. 3a). Moreover, two salt bridges, which are commonly stabilizing in soluble proteins, also did not strongly enhance GlpG stability. Instead, structurally-critical hydrogen bonds are deployed to stabilize two relatively peripheral regions.
Figure 3. Only two regions of hydrogen bonding are critical for GlpG stability.

(a) The hydrogen bonds (in pink throughout) made by the TM2 residue E166 (inset) including to T97 and S171 make critical contributions to GlpG thermostability, while intramembrane hydrogen bonds Y138, W158, Y160, and T178 make only minor contributions to stability and protease activity. Mutant effects on GlpG thermostability are color-matched between structure diagrams and ΔTm (lower) graphs throughout according to the heat-map legend below (threshold for statistical significance is a 3°C change, which corresponds to p<0.015). Effects on catalysis are only color-coded in the upper graphs (not structures). All values are mean ± standard deviation. (b) Hydrogen bond network on the cytosolic surface of GlpG make small contributions to GlpG stability or activity, with the exception of the D268, K173 and Y210 triad interaction. ‘Hex’ is the R168A+K173A+R214A+D218A+S221A+D268A mutant. (c) Lower L1 loop residues hydrogen bond to the upper L1 polypeptide backbone to provide an important stabilizing influence that’s also important for protease activity. (d) Helix capping residues provide little stabilizing influence, while hydrogen bonding between the L1 loop and protease core (through H141, H145, S147) are critical for stability and proteolysis.
The first region mediates interaction of TM helices near the cytosolic face of GlpG. Mutation of E166 that is located near the bottom of TM2 had the greatest destabilizing effect of any hydrogen-bonding residue, decreasing GlpG Tm by a remarkable 21.3±0.44°C (Fig. 3a). E166 makes four hydrogen bonds, two to the backbone nitrogens of V96 and T97 on TM1, and two to the sidechains of T97 on TM1 and S171 on TM3. These interactions collate TMs 1, 2, and 3 into an apex at the bottom of the GlpG molecule. Although E166 makes three hydrogen bonds to TM1, removing just the bond to T97 (leaving two backbone bonds) has a stronger effect (-11.9±0.77°C) than removing the only bond to TM3 via S171 (-5.8±0.57°C), perhaps because TM2 and TM3 are covalently linked by the short L2 loop.
The cytosolic underside of GlpG further harbors a network of 9 hydrogen bonds that bridge TMs 2, 3, 4, 5 and 6 (Fig. 3b). Remarkably, however, the vast majority of these hydrogen bonds contributed little to GlpG stability. In fact, removing all bonds in a hextuple mutant had the same effect as just mutating D268 alone to alanine (16.9±1.6°C decrease). The D268A mutant on TM6 abrogates a salt bridge to K173 on TM2 and hydrogen bonding with Y210 on TM4. Therefore, structurally-key interactions bring together TMs 1, 2, and 3 through E166, and TMs 6, 3 and 4 via D268. The two hydrogen bonds that link to TM5 play no significant role in GlpG stability.
The lateral L1 loop forms the second region in which hydrogen bonds play key stabilizing functions. A series of residues on the lower helix of the L1 loop extend upwards to make hydrogen bonds with the loop’s upper helix, thereby stabilizing its lateral hairpin conformation (Fig. 3c). R137 makes 5 hydrogen bonds within the L1 loop, and its mutation to alanine decreased Tm by 20.3±0.95°C. Allowing a single hydrogen bond by replacing R137 with lysine mildly rescued stability (Tm decrease of 16.1±0.52°C). Two charge-assisted hydrogen bonds from R137 are to the universally conserved E134, however, mutating E134 to alanine had an unexpectedly mild Tm decrease (7.0±0.36°C). Mutating neighboring W136 or T140 decreased Tm by ~10°C, indicating that additional hydrogen bonding to the upper L1 polypeptide backbone oxygens enhances GlpG stability, albeit much less so than by R137.
Surprisingly, L1 loop residues that had perhaps the strongest destabilizing effect on GlpG were H141 and H145 (Fig. 3d). The importance of these residues has escaped attention until now. H141 and H145 mediate hydrogen bonds between the L1 loop and the internal face of TM3 and TM2, respectively. H145 plays a particularly important role, because its mutation to alanine decreased both stability by 22.2±1.65°C and protease activity >30-fold. However, the basis of this effect is not fully clear, because mutation of N154 to which H145 hydrogen bonds in some structures was only mildly destabilizing. Substituting H145 with glutamine or phenylalanine did not rescue the stability defect to any significant degree.
Lastly, while residues that cap TM helices are often thought to play significant roles in structural stability, we found that they have little if any stabilizing influence in GlpG (Fig. 3d). The only exception is the interaction between S147 on the L1 loop and TM2, although this probably aids L1 loop stability.
Key glycine packing: a helix bend and a GxxxGxxxA motif
TM segments are evolutionarily constrained because of extensive van der Waals packing interactions, but their exact contributions to stability are uncertain26,27. In some instances, strong packing interactions have been discovered28, while other studies have found packing in membrane proteins are generally weak and not unlike those of soluble proteins29. Interestingly, the most destabilized GlpG variants that we discovered involved two transmembrane glycines. The first variant was G162V (-24.6±0.27°C), which provides the only TM helix bend in GlpG and lies just above the critical E166 residue (Fig. 4a). Abrogating additional local packing by L174 in TM3, which buries its sidechain inwards, also decreased stability by a notable 16.3±0.97°C. Mutating G170 (to alanine), around which the L2 loop bends, also strongly decreased GlpG stability by 11.1±0.09°C.
Figure 4. A small number of van der Waals interactions are critical for GlpG architecture and catalysis.
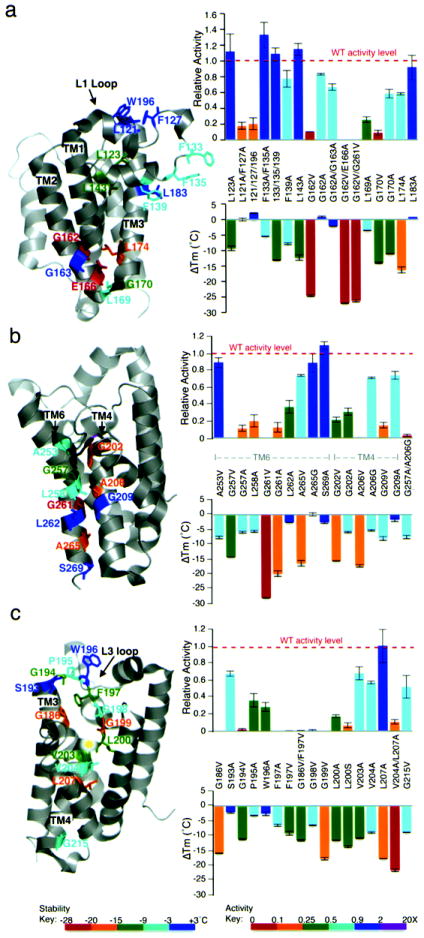
(a) TM2 helix bending around G162, and packing by L174, were essential for GlpG architecture. Packing interactions made by L1 loop residues L123 and L143 onto neighboring TMs in particular enhanced stability. Mutant effects on GlpG thermostability are color-matched between structure diagrams and ΔTm (lower) graphs according to the heat-map legend below. Effects on catalysis are only color-coded in the upper graphs (not structures). All values are mean ± standard deviation. (b) Close interface between TM6 and TM4, which contribute the catalytic H254 and S201, respectively, was essential for GlpG architecture. (c) Central cavity packing by L3/TM4 residues, especially G186, G199, L200 and L207, contributed to architectural stability. Many of these interactions were particularly important to catalysis (upper graph).
The second key glycine, G261 in TM6, decreased Tm by 28.1±0.08°C when mutated to valine (Fig. 4b). This critical residue is part of a GxxxG dimerization motif that allows close apposition of peptide backbones30. The GlpG GxxxG mediates a less common heterotypic interaction (with TM4). Mutating G257, the first glycine of this motif, also resulted in a 14.4±0.95°C decrease in Tm. In both cases, even alanine substitutions resulted in strong destabilization (only ~8°C less than valine), and reduced proteolytic activity by 90% (valine variants were inactive). One helical turn following GxxxG lies A265, and although A265 has not been considered to be part of this GxxxG association motif in GlpG, its mutation to valine decreased stability by 15.1±0.37°C, revealing that it plays a more stabilizing role than G257. However, this mutation decreased proteolytic activity by only 25%, suggesting that structural perturbations near the catalytic histidine, rather than their severity on structural stability globally, have a more dramatic effect on activity. Mutating A265 to glycine to produce two tandem motifs (GxxxGxxxG) did not increase thermostability or activity. Finally, A253, which lies one helical turn above G257 and does not make any obvious contacts, decreased thermostability (by 7.7±0.43°C) when mutated to valine.
Beyond the glycine residues of the GxxxG motif, β-branched residues following each glycine and a serine/threonine one helical turn away have been defined to play central roles in stabilizing some GxxxG interactions28,31. However, mutating the correspondingly-positioned (albeit not β-branched) and conserved L258, L262, and S269 (one helical turn below A265) in GlpG resulted in almost no detectable change in stability. The corresponding interface on TM4 was also sensitive to mutation, albeit less so (Fig. 4b). Mutation of A206, which lies closest to G261, to valine decreased stability by 17.4±0.39°C, while mutation of G202 or G209, which lie one helical turn above or below A206, respectively, to valine decreased Tm by 15.7±0.09°C and 8.1±0.66°C, respectively. Therefore, the close, heterotypic apposition of TMs 4 and 6 plays a critical role in the structural stability of GlpG (with side-chain hydrogen bonds adding no further stability).
Contributions from sidechain packing interactions
Interaction of most GlpG helices are mediated by a large sidechain of one helix interdigitating between two adjacent sidechains of its neighbor. We therefore generated over 30 mutants in which we mutated large residues to alanine, and discovered that single mutations were generally weakly-destabilizing, but with notable exceptions in two localized regions.
L3/TM4 runs diagonally through the center of GlpG, making a series of stabilizing interactions including with two leucines (Fig. 4c). Of particular importance was L207, which lies on TM4 and contacts TM2. Mutating L207 to alanine alone decreased stability by a striking 17.7±0.02°C, while a double mutant with V204 decreased stability by 21.6±0.41°C. L200, which immediately precedes the catalytic serine, buries its sidechain into the floor of the protease, and its mutation to alanine decreased stability by 11.6±0.1°C. Other L3/TM4 residues also made stabilizing interactions, including packing of F197 with G186 on TM3, and V203 and V204, which follow the catalytic serine on TM4, each mutant decreasing stability by ~10°C. Also intriguing were L3 glycines, especially G194 and G199 (but not G198) that decreased Tm by 11.2±0.19°C and 17.7±0.42°C, respectively, when mutated to valine. Each of these three mutants reduced protease activity ≥50-fold (Fig. 4c). Conversely, L3 residues P195, around which the L3 loop bends, and W196, had little, if any, effect on stability.
Where the L1 loop nestles onto the underlying TMs 1 and 3 proved a second region sensitive to altered packing interactions. Mutation of L143 in particular, but also L123 or F139, which lie on the L1 loop and buttress it by making contact with TM1 or TM3, decreased stability by 12.2±0.89°C, 9.2±0.7°C, and 7.7±0.28°C, respectively (Fig. 4a). This is not a common effect, since two large residues, F133 and F135, which are on the same face of the L1 helix but distant from TM1/3, decreased stability by only 5.3±0.06°C when mutated together. Packing in other parts of the L1 loop, such as with its top helix, had no measurable effect on stability even in the triple L121A+F127A+W196A mutant (although activity was reduced).
Besides the small regions of stronger packing, interactions between TM helices encircling GlpG showed small but consistent effects on stability (Fig. 5a), suggesting that these many weak interactions make GlpG quite flexible. Nevertheless, double alanine mutants on the same helix increased severity in an approximately additive manner, while quadruple interfacial mutations targeting the interface between TM1/TM2, and TM3/TM6 (two on each interacting helix) decreased GlpG stability by nearly 20°C (Fig. 5a). These measurements indicate that most outer transmembrane packing interactions contribute little to GlpG stability individually, but in sum exert a considerable stabilizing influence. This was true for all regions of the protease except one, namely TM5.
Figure 5. Quantitative assessment of peripheral TM interactions and catalytic residues: implications for dynamics.

(a) Peripheral packing interactions involving interdigitating large residues between neighboring TM segments had weak but additive effects on stability and catalysis. Displayed in color on the structures are thermostability effects of double mutants, but note that quarduple mutants had even greater effects. All values are mean ± standard deviation. (b) TM5, L4 and L5 mutants had no effect on GlpG stability, but dramatically enhanced protease activity. Mutant effects on GlpG thermostability are color-matched between structure diagrams and ΔTm (lower) graphs according to the heat-map legend below. Effects on catalysis are only color-coded in the upper graphs (not structures). (c) Polar residues lining the active site that could be thought to provide a ‘hydrophilic effect’ in the membrane make little or no contribution to GlpG architecture, but are essential for catalysis. (d) ‘Heat-map’ illustrating the effect of all GlpG mutants on intramembrane protease activity (color-matched to legend). Activating mutants (>2-fold, in purple) were only isolated on TM5 and its neighbor TM2. Conversely, note that many (but not all) variants that reduced activity >10-fold (in red) line the active site cavity. Of residues clearly involved in structural stability, decrease in protease activity often correlated with proximity of the mutant to catalytic residues, rather than its destabilizing nature globally.
TM5, its loops, and catalytic residues are not stabilizing
The only TM helix whose packing did not contribute at all to GlpG structural stability was TM5, which forms part of the proposed substrate gate (Fig. 5b). Single, double, triple or quadruple mutants on any side of TM5 (and its interacting neighbor TM2) had no or little effect on GlpG stability. Nevertheless, the effect of these mutations on protease activity was extraordinary, increasing proteolysis even up to ~15-fold, suggesting a rate-limiting function. In fact, of the ~150 variants, this was the only region of the protease in which we encountered activating (>2-fold) mutants. Mutations in the cytosolic L4 loop and the overlying L5 loop that connect TM5 to other TM segments also did not significantly impact structural stability, in contrast to all other loops. These observations indicate that packing interactions can evolve to have little effect on structural stability of a membrane protein, allowing specialization of function.
The most conspicuous hydrogen bond in GlpG pairs catalytic S201 and H254. Interestingly, mutation of the serine decreased GlpG Tm by 6.2±0.84°C (Fig. 5c). However, this was not due to removing this hydrogen bond, because mutagenesis of the catalytic histidine did not alter stability. The double serine and histidine mutant rescued GlpG stability, suggesting that, in the absence of the serine, the histidine may make destabilizing interactions. H254 also normally stacks onto Y205, which emanates from TM4, but replacing Y205 with alanine increased thermostability slightly. Currently the best sidechain candidates for oxyanion stabilization are H150 and N154, and their mutation to alanine mildly decreased GlpG stability by ~6°C each. Therefore, residues lining the active-site cavity are important for activity but do not play a significant role in GlpG stability (Fig. 5d and Supplementary Fig. 5).
Thermodynamic assessment of key interactions
The thermal light-scattering assay is a new technology for studying the principles of membrane protein stability. We therefore evaluated our findings by subjecting 25 of our most informative GlpG mutants to the independent, SDS-mediated unfolding assay to measure their thermodynamic stability (Fig. 6a). Since most of these mutants lost protease activity, we followed their unfolding by tryptophan fluorescence. Despite the variability with this detection method, in the case of GlpG we found it agrees well (R=0.94) with using protease activity to monitor unfolding of five destabilized GlpG mutants that retained some protease activity (Fig. 6b).
Figure 6. Thermodynamic assessment of GlpG architectural mutants.

(a) Free energy change (ΔΔG) for 25 GlpG variants measured by tryptophan fluorescence was plotted against their transition temperature change (ΔTm) and analyzed by linear regression. Note that the line does not go through the origin because the SDS-mediated unfolding assay was unable to detect differences in mutants that destabilized GlpG structure by less than ~10°C. (b) Comparison of ΔG values derived for GlpG mutants using protease activity versus tryptophan fluorescence to monitor SDS-mediated unfolding. Note that only a few destabilized mutants retained sufficient protease activity to permit analysis, but the data were in good correlation (R=0.94) with tryptophan fluorescence data. (c) Change in relative protease activity and change in structural stability (ΔTm) display a weak correlation. Shown by a disconnect on the right are activating mutants (>2-fold), which were included into the correlation analysis. (d) The strongest two hydrogen bonds (E166-T97 and D268-K173) were analyzed by double-mutant cycle analysis. The ΔG of the individual hydrogen bond was calculated by subtracting the ΔΔG value of the double mutant from the sum of ΔΔG values (± standard deviation) of the two corresponding single mutants.
The five most important residues identified by thermostability analysis also provide the strongest free energy interactions to stabilize GlpG in the SDS unfolding assay (Fig. 6a); the critical glycines, G162 and G261, contribute free energies of -2.61±0.37 and -2.88±0.31 kcal/mol, while the three key hydrogen-bonding residues R137, H145, and E166, contribute -2.71±0.20, -2.46±0.01, -1.83±0.04 kcal/mol, respectively, to GlpG stability. Moreover, as evident with the thermostability analysis, weak packing interactions also combine to provide strong support; both the quadruple TM1-TM2 peripheral packing mutant, and the double TM4 mutant V204A+L207A, resulted in a -2.29±0.60 kcal/mol and -2.49±0.19 kcal/mol change in free energy, respectively. Overall, we found the change in Tm using the thermal light scattering assay and the change in ΔG in the SDS assay have a correlation coefficient of 0.92 with our 25 mutants (Fig. 6a). This is particularly remarkable, since the assays differ in denaturant (heat versus chemical), readout (light scattering versus tryptophan fluorescence), and reversibility. As such, our global thermostability analysis is revealing the basis of GlpG architecture without a major bias from assay method. We were, however, surprised to find that the correlation between mutant effect on structural stability and protease activity was alarmingly weak, with R=0.24 (Fig. 6c). This poor correlation reveals that measuring decrease in protease activity relative to wildtype is not a valid way to gauge the effect of mutation on structural stability; this has implications since most attempts at interrogating intramembrane protease architecture currently rely on measuring loss of protease activity as the only available assay to report on structure. However, this should not be confused with using residual protease activity of mutants (relative to themselves) to follow unfolding in the presence of denaturants (e.g. Fig. 1a), which is the most robust way to monitor unfolding.
Multifaceted, not single, hydrogen bonds confer stability
Since several residues that we identified as crucial for GlpG stability make multiple hydrogen bonds, we used double mutant cycle analysis with SDS-mediated unfolding to evaluate the contributions of individual hydrogen bonds24. This analysis compares the ΔΔG resulting from single versus double mutants, allowing other factors to cancel, leaving the ΔG of the bond itself. While we could not use this approach to analyze all individual hydrogen bonds (because several are to backbone atoms), we successfully analyzed 7 distinct bonds through 15 mutants. This analysis revealed that all individual hydrogen bonds are indeed strikingly weak, with the E166-T97 and D268-K173 bonds yielding the largest values, which were nevertheless only -0.58 and -0.31 kcal/mol, respectively (Fig. 6d). Therefore, it is the multifaceted interactions made by individual sidechains that strengthen their stabilizing effects.
Natural diversity in the stability of rhomboid proteases
Finally, we examined the thermostability of three other bacterial rhomboid proteases. Although all are robustly active enzymes, all had Tm values considerably lower than E. coli GlpG, with the lowest being 23.0±0.81°C below that of E. coli GlpG (Fig. 7a). The correlation between Tm and δG of stability for these four rhomboid enzymes was 0.92 (Supplementary Fig. 6a). Since these rhomboid enzymes have all of the conserved residues required for activity, this result indicates that variation in non-conserved residues provides a great deal of natural diversity in rhomboid stability. Of particular interest was GlpG from Haemophilus influenzae, which has been crystallized and harbors five natural variants in residues that we discovered contribute to E. coli GlpG stability (Fig. 7b). Mutating one of these, V122, which corresponds to L207 on TM4 of E. coli GlpG, to leucine alone increased thermostability to E. coli GlpG levels (Supplementary Fig. 6b). The strong effect of this single mutant is particularly remarkable because E. coli and H. influenzae GlpG proteins are only ~37% identical.
Figure 7. Natural diversity in the thermostability of rhomboid proteases.
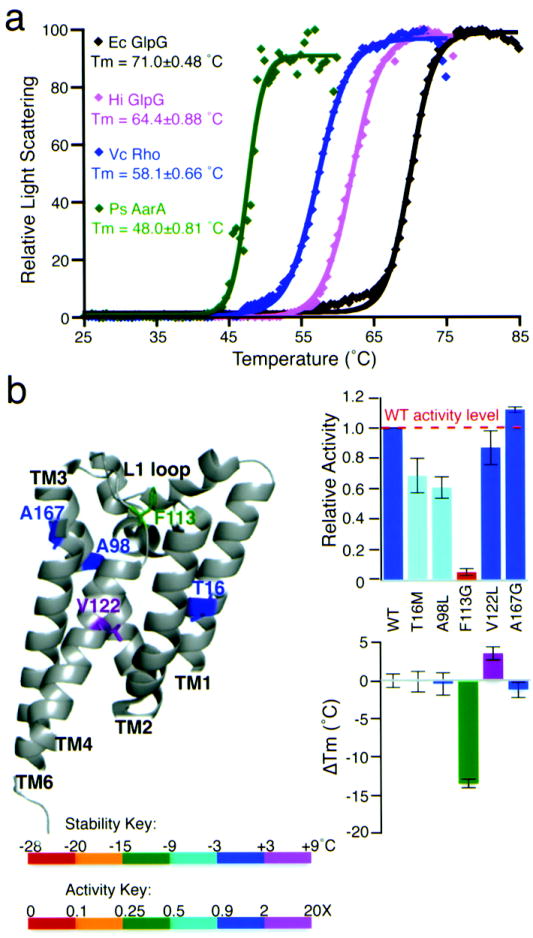
(a) Thermostability analysis of four diverse wildtype rhomboid enzymes: relative light scattering data are shown by discrete points, while the Boltzmann curve fits are depicted by lines (R2 values of the shown fits range from 0.984-0.999). Ec is E. coli, Vc is Vibrio cholerae, Hi is Haemophilus influenzae, and Ps is Providencia stuartii. (b) Quantitative protease and thermostability analysis of five natural variants of H. influenzae GlpG that were mutated to their corresponding E. coli GlpG residues. All values are mean ± standard deviation. The V122L mutant of H. influenzae GlpG increased thermostability to a level that was statistically indistinguishable from that of E. coli GlpG (also see Supplementary Fig. 6b).
DISCUSSION
Architectural properties underlie protein function, but in contrast to their soluble cousins, membrane proteins remain poorly understood1,2. This discrepancy results largely from a paucity of tools with the necessary resolution for studying membrane proteins. We integrated three quantitative methods to achieve a comprehensive view of how intramembrane protease chemistry underlies structural stability and enzyme function for rhomboid proteases.
By engineering and quantitatively interrogating ~150 variants that targeted interactions in an unbiased way, we reveal for the first time the functional anatomy in its entirety of an intramembrane protease (Fig. 8). We discovered GlpG stability is consolidated by a minimal number of localized interactions: only five residues decrease GlpG stability by >20°C (>1.8 kcal/mol). Despite widespread interest, four of these critical residues, H145, G162, E166, and G261, have not been studied experimentally. The critical residues fall into four discrete regions on the primary structure that we term ‘keystones’ (Fig. 8). Strong packing interactions are deployed to solidify the TM4:TM6 interaction via an asymmetric GxxxGxxxA motif on TM6 and a corresponding GxxxAxxG interface on TM4. This interaction is heterotypic, and does not depend on surrounding residues. The opposite face of TM4, together with the extended L3, forms a second critical area of packing, although here mediated predominantly by large residues. While together these interactions secure the core of the molecule, they are not sufficient for enzyme stability: GlpG further deploys a series of hydrogen bonds to two more-peripheral areas (Fig. 8). The first is the L1 loop near the extracellular face of the enzyme, while the second is localized to the cytosolic surface and collates TMs 1, 2, 3 and TMs 3, 4, 6. These hydrogen bonds are strong because they are multifaceted, and are further buttressed by neighboring packing interactions.
Figure 8. Architectural principles underlying rhomboid protease function.
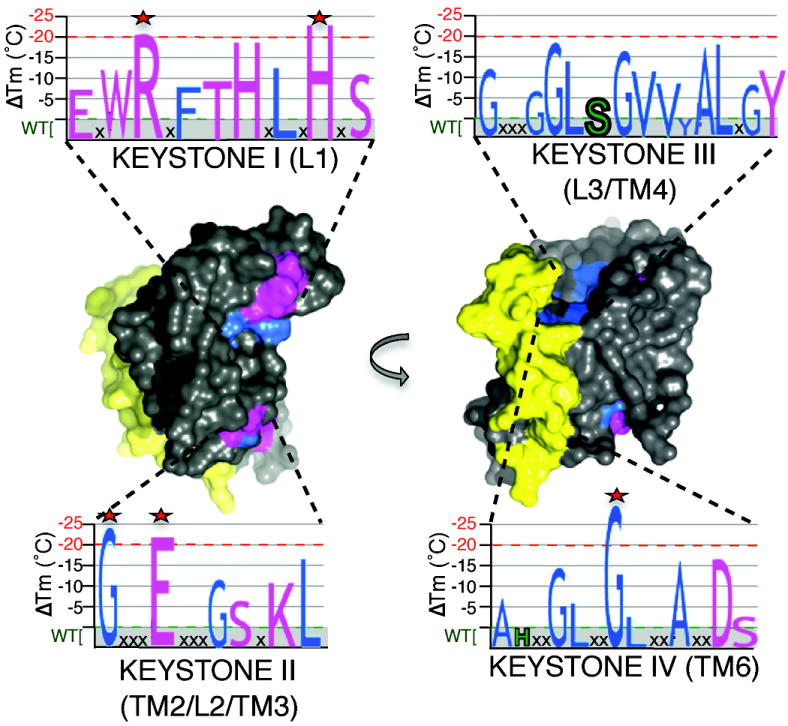
Space-fill model highlighting architectural properties underlying GlpG function. Regions in grey represent a multitude of weak van der Waals interactions that contribute to the low thermodynamic stability and high environmental responsiveness of rhomboid proteases. Two regions in which hydrogen-bonding residue interactions predominate (pink) are deployed to the upper (L1) and lower (TM2/L2/TM3, with peripheral residues from TM4 and 6) areas of GlpG. Strong residue-packing interactions are illustrated in blue. Yellow highlights the localized region relinquished from structural roles and poised for dynamic functions during proteolysis. Graphs represent the four primary sequence ‘keystone’ regions in which both packing (blue letters) and hydrogen bonding (pink letters) interactions are deployed to stabilize the structure. The destabilizing effects of each mutant in each region is denoted quantitatively by the size of each letter (catalytic serine and histidine are in green, and the 5 most important stabilizing residues are highlighted with stars). Note that keystones III and IV are in the internal core of the molecule and are only partly visible.
The strategic placement of these keystones allows one lateral face of GlpG (Fig. 8) and its inner cavity to be reserved for non-structural roles. This region centers on the TM5/L5 substrate gate11,13,32 in which mutating packing interactions on TM5 actually increases protease activity up to >15-fold while leaving structural stability completely unperturbed. This overall strategy allows TM5/L5 to be poised for bending away from the protease core without compromising structural stability. However, even the underlying L4 loop and lower part of TM5, neither believed to function in gating, are devoid of structural responsibility. Since loops can play important roles in membrane protein structure33, this discrepancy implies that rhomboid proteolysis may involve greater protein dynamics than anticipated. Based on modeling studies, Derlins also appear to maintain an intact rhomboid architecture and deploy residues analogous to those that we found consolidate GlpG stability.
The localized nature of these stability keystones nevertheless allows GlpG to harbor low intrinsic thermodynamic stability globally, being constructed largely from a myriad of weak van der Waals interactions (Fig. 8). This low intrinsic thermodynamic stability is notably unlike that of well-studied membrane protein paradigms: bacteriorhodopsin (20 kcal/mol)17, diacyglycerol kinase (16 kcal/mol)18, KcsA (30 kcal/mol)34, and the aquaporin GlpF (16.4 kcal/mol)35. However, such apparent meta-stability may have advantages: this property results in a high degree of thermostability yet makes GlpG globally malleable and thus responsive to its chemical environment. In fact, rhomboid protease activity is dramatically influenced by membrane composition36,37. More recent investigations have uncovered two other proteins, E. coli GalP38 and DsbB39, that also may maintain low intrinsic stability (<5 kcal/mol), raising the possibility that some membrane enzymes are deliberately less stable to accommodate their function.
Low global stability resulting from many weak interactions also provides a coherent explanation for why mutants dispersed throughout GlpG’s structure strongly affect protease activity13. In fact, our large-scale analysis of >140 E. coli GlpG variants found ~75% reduced protease activity. This is intriguingly similar to >100 Familial Alzheimer’s Disease mutations that are unexpectedly distributed throughout the presenilin molecule, the catalytic center of γ-secretase, and lower protease activity3,40,41. One possible explanation is that presenilin may also maintain low global stability by relying on a myriad of weak interactions. Consistent with this hypothesis, γ-secretase activity is also dramatically affected by its membrane environment42. While GlpG has been used as a model for distilling the general principles of intramembrane proteolysis and such comparisons are instructive4, they must be approached with caution because of the obvious evolutionary and chemical differences between GlpG and presenilin.
Our approach further offers general applications that might prove important. First, while protein engineering is an indispensible strategy for interrogating enzyme mechanisms, the inability to distinguish effects on function from unintended effects on structure seriously limits the accuracy of current studies of membrane proteases. The thermostability methodology provides a much-needed means to untangle these effects. This observation also has potential biological caveats; although all published reports use catalytic serine mutants as inactive rhomboid controls, our stability analysis revealed that this simple mutant is partly destabilized structurally. As such, over-expression of this mutant could result in unintended effects including increasing protein-aggregation load in cells. This might at least partly explain why a dominant-negative approach to uncovering rhomboid function in Toxoplasma parasites yielded an unexpected defect in parasite replication43. Our current observations also offer a simple solution: a serine and histidine double alanine mutant is both structurally unperturbed and devoid of enzymatic activity.
Our results also raise the exciting possibility that this thermal light-scattering assay could prove more widely applicable to studying helical membrane protein architecture on a much-needed quantitative and high-throughput scale. In fact, this method was much less prone to variation and much more sensitive at detecting small structural perturbations than the SDS-mediated unfolding assay. It could therefore allow a comprehensive analysis directly of how different interactions are used to stabilize each particular class of membrane proteins, and the unbiased distilling of those rare and informative mutants that are likely to yield key insights for further in-depth studies.
METHODS
DNA constructs
We used rhomboid proteases cloned into the pGEX-6P-1 vector and, in the case of E. coli GlpG and P. stuartii AarA, harboring a single hemagglutinin (HA) tag at their N terminus for detection by western analysis. Specific amino acid substitutions in GlpG were generated by site-directed mutagenesis using Pfu Ultra (Agilent Technologies, La Jolla, CA), and confirmed by sequencing the entire rhomboid ORF.
Protein expression and moderate-throughput purification strategy
Wild-type GlpG and engineered variants were expressed as N-terminal GST-fusion proteins in 1L cultures of E. coli C43(DE3), purified from DDM-solubilized membranes by glutathione-sepharose affinity chromatography, and eluted by on-column cleavage to remove the GST tag, as described previously13,36. Protein purity (>90%) and yield were quantified by infrared scanning (Li-Cor Odyssey) of Coomassie-stained polyacrylamide gels (see Supplementary Fig. 4 for gels of the purified proteins). To maintain stability, each mutant was expressed in cultures grown at 16°C, purified at 4°C, and the final state of the purified mutants was analyzed by monitoring light scattering at 25°C.
SDS-mediated unfolding assay
0.4 μM GlpG in 50 mM Tris pH 7.4, 150 mM NaCl, and 2mM (0.1%) DDM was treated for 30 minutes with 0.065-69.4 mM SDS to achieve a bulk mole fraction of 0.031 to 0.972. Intrinsic tryptophan fluorescence (excitation at 280nm and emission at 320nm) was measured at 25°C using a Synergy H4 microplate reader (BioTek). We achieved excellent fits to the SDS unfolding data using the simplest model, which assumes a two-state unfolding reaction and linear dependence of ΔG on the concentration of SDS. Equilibrium unfolding data were individually fit in Prism to the Santoro-Bolen equation19 to account for the baselines, with YN, YU, mN, mU, ΔGN-U, and m as fitted parameters using:
The parameters resulting from the curve fits were YN = 1.01, mN = -0.01, YU = -0.28, mU = 0.28, ΔGN-U = 4.17, m = 6.85 for Ec GlpG, YN = 0.91, mN = 0.46, YU = -0.27, mU = 0.31, ΔGN-U = 4.46, m = 8.75 for Hi GlpG, YN = 0.99, mN = 0.00, YU = -0.07, mU = 0.10, ΔGN-U = 2.99, m = 8.38 for Vc Rho, and YN = 1.07, mN = 0.00, YU = -0.15, mU = 0.14, ΔGN-U = 2.10, m = 5.08 for Ps AarA.
CD Spectroscopy
CD analysis of GlpG was conducted using a J-810 spectropolarimeter (Jasco) at 25°C by averaging ten 260-185 nm scans at 1nm/sec. For thermal unfolding, GlpG was heated at 1°C/minute and ellipticity at 222nm was monitored at each degree by averaging three scans. To ensure that the CD signal corresponded to the GlpG structure, we used the same construct that was used for crystallization10-12.
Thermostability analysis
Pure GlpG at a concentration of ~2.5 μM in 50 mM Tris pH 7.4, 100 mM NaCl, 1 mM EDTA, 1 mM DTT, 0.1% DDM was subjected to thermal denaturation analysis in a Stargazer instrument (Harbinger Biotech, Toronto, Canada). Protein samples overlayed with mineral oil in clear-bottom 384-well black plates were heated from 25°C to 85°C at a rate of 1°C per minute (or 0.5°C per minute for the 5 most perturbed mutants) and light scattering from a 620 nm LED source was detected by a CCD camera every 0.5°C. The intensity of scattered light in each image was quantified and plotted against temperature. Transition temperatures were derived from fitting the data to a two-state Boltzmann curve using Stargazer BioActive software. While we achieved an assay window of ΔTms >80 standard deviations wide, to err on the side of caution, we set our threshold for statistical significance to a 3°C change, which corresponds to p<0.015 (by applying the student t-test at a standard deviation of 0.5°C with each mutant being analyzed twice, although most were analyzed ≥4 times). The changes were then binned into five categories and the resulting stability ‘heat map’ of different mutants was color-coded manually in MacPyMOL2.
Protease activity analysis
GlpG proteolytic activity was measured using the recombinant C-terminal Flag-tagged C100Spitz substrate as described previously 13,36. Briefly, 0.5 μM GlpG was incubated with ~3 μM substrate in 0.1% DDM at 37°C for 1 and 2 hours. Reactions were resolved by SDS-PAGE, and HA-tagged GlpG and Flag-tagged substrate/products were quantified by two-color western analysis on a Li-Cor Odyssey infrared fluorescence scanner. Protease activity was monitored under denaturing conditions with a FITC-tagged Providencia stuartii TatA construct as above, except that the reaction products were resolved on 16% Tricine SDS-PAGE gels and quantified on a Typhoon phosphorimager (GE Healthcare). Protease activity was corrected for denaturant stimulation when encountered.
Supplementary Material
Acknowledgments
We are grateful to all members of the Urban lab and Daniel Otzen for stimulating scientific discussions, and to the Malaria Research Institute Biophysics Core for use of their CD spectropolarimeter. This work was supported by the Howard Hughes Medical Institute and the David and Lucile Packard Foundation.
Footnotes
AUTHOR CONTRIBUTIONS
RB designed research, performed all biochemical experiments (except CD spectroscopy), analyzed data, and prepared all Figures; SU designed research, made all DNA constructs, performed CD spectroscopy, analyzed the data, and wrote the paper.
COMPETING FINANCIAL INTERESTS STATEMENT
The authors declare no conflict of interest exists.
References
- 1.Bowie JU. Solving the membrane protein folding problem. Nature. 2005;438:581–9. doi: 10.1038/nature04395. [DOI] [PubMed] [Google Scholar]
- 2.Booth PJ, Curnow P. Folding scene investigation: membrane proteins. Curr Opin Struct Biol. 2009;19:8–13. doi: 10.1016/j.sbi.2008.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.De Strooper B, Annaert W. Novel research horizons for presenilins and gamma-secretases in cell biology and disease. Annu Rev Cell Dev Biol. 2010;26:235–60. doi: 10.1146/annurev-cellbio-100109-104117. [DOI] [PubMed] [Google Scholar]
- 4.Wolfe MS. Intramembrane proteolysis. Chem Rev. 2009;109:1599–612. doi: 10.1021/cr8004197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Brown MS, Ye J, Rawson RB, Goldstein JL. Regulated intramembrane proteolysis: a control mechanism conserved from bacteria to humans. Cell. 2000;100:391–8. doi: 10.1016/s0092-8674(00)80675-3. [DOI] [PubMed] [Google Scholar]
- 6.Urban S. Making the cut: central roles of intramembrane proteolysis in pathogenic microorganisms. Nature Reviews Microbiology. 2009;7:411–23. doi: 10.1038/nrmicro2130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Fluhrer R, Steiner H, Haass C. Intramembrane proteolysis by signal peptide peptidases: a comparative discussion of GXGD-type aspartyl proteases. J Biol Chem. 2009;284:13975–9. doi: 10.1074/jbc.R800040200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Greenblatt EJ, Olzmann JA, Kopito RR. Derlin-1 is a rhomboid pseudoprotease required for the dislocation of mutant alpha-1 antitrypsin from the endoplasmic reticulum. Nat Struct Mol Biol. 2011;18:1147–1152. doi: 10.1038/nsmb.2111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zettl M, Adrain C, Strisovsky K, Lastun V, Freeman M. Rhomboid family pseudoproteases use the ER quality control machinery to regulate intercellular signaling. Cell. 2011;145:79–91. doi: 10.1016/j.cell.2011.02.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wang Y, Zhang Y, Ha Y. Crystal structure of a rhomboid family intramembrane protease. Nature. 2006;444:179–80. doi: 10.1038/nature05255. [DOI] [PubMed] [Google Scholar]
- 11.Wu Z, et al. Structural analysis of a rhomboid family intramembrane protease reveals a gating mechanism for substrate entry. Nat Struct Mol Biol. 2006;13:1084–91. doi: 10.1038/nsmb1179. [DOI] [PubMed] [Google Scholar]
- 12.Ben-Shem A, Fass D, Bibi E. Structural basis for intramembrane proteolysis by rhomboid serine proteases. Proc Natl Acad Sci U S A. 2007;104:462–6. doi: 10.1073/pnas.0609773104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Baker RP, Young K, Feng L, Shi Y, Urban S. Enzymatic analysis of a rhomboid intramembrane protease implicates transmembrane helix 5 as the lateral substrate gate. Proc Natl Acad Sci U S A. 2007;104:8257–62. doi: 10.1073/pnas.0700814104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Urban S. Taking the plunge: integrating structural, enzymatic and computational insights into a unified model for membrane-immersed rhomboid proteolysis. Biochem J. 2010;425:501–12. doi: 10.1042/BJ20090861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Vinothkumar KR. Structure of rhomboid protease in a lipid environment. J Mol Biol. 2011;407:232–47. doi: 10.1016/j.jmb.2011.01.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hong H, Joh NH, Bowie JU, Tamm LK. Methods for measuring the thermodynamic stability of membrane proteins. Methods Enzymol. 2009;455:213–36. doi: 10.1016/S0076-6879(08)04208-0. [DOI] [PubMed] [Google Scholar]
- 17.Curnow P, Booth PJ. Combined kinetic and thermodynamic analysis of alpha-helical membrane protein unfolding. Proc Natl Acad Sci U S A. 2007;104:18970–5. doi: 10.1073/pnas.0705067104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lau FW, Bowie JU. A method for assessing the stability of a membrane protein. Biochemistry. 1997;36:5884–92. doi: 10.1021/bi963095j. [DOI] [PubMed] [Google Scholar]
- 19.Santoro MM, Bolen DW. Unfolding free energy changes determined by the linear extrapolation method. 1. Unfolding of phenylmethanesulfonyl alpha-chymotrypsin using different denaturants. Biochemistry. 1988;27:8063–8. doi: 10.1021/bi00421a014. [DOI] [PubMed] [Google Scholar]
- 20.Yeh AP, McMillan A, Stowell MH. Rapid and simple protein-stability screens: application to membrane proteins. Acta Crystallogr D Biol Crystallogr. 2006;62:451–7. doi: 10.1107/S0907444906005233. [DOI] [PubMed] [Google Scholar]
- 21.Senisterra GA, et al. Assessing the stability of membrane proteins to detect ligand binding using differential static light scattering. J Biomol Screen. 2010;15:314–20. doi: 10.1177/1087057109357117. [DOI] [PubMed] [Google Scholar]
- 22.Choma C, Gratkowski H, Lear JD, DeGrado WF. Asparagine-mediated self-association of a model transmembrane helix. Nat Struct Biol. 2000;7:161–6. doi: 10.1038/72440. [DOI] [PubMed] [Google Scholar]
- 23.Zhou FX, Merianos HJ, Brunger AT, Engelman DM. Polar residues drive association of polyleucine transmembrane helices. Proc Natl Acad Sci U S A. 2001;98:2250–5. doi: 10.1073/pnas.041593698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Joh NH, et al. Modest stabilization by most hydrogen-bonded side-chain interactions in membrane proteins. Nature. 2008;453:1266–70. doi: 10.1038/nature06977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Curnow P, Booth PJ. The contribution of a covalently bound cofactor to the folding and thermodynamic stability of an integral membrane protein. J Mol Biol. 2010;403:630–42. doi: 10.1016/j.jmb.2010.09.003. [DOI] [PubMed] [Google Scholar]
- 26.Eilers M, Shekar SC, Shieh T, Smith SO, Fleming PJ. Internal packing of helical membrane proteins. Proc Natl Acad Sci U S A. 2000;97:5796–801. doi: 10.1073/pnas.97.11.5796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Oberai A, Joh NH, Pettit FK, Bowie JU. Structural imperatives impose diverse evolutionary constraints on helical membrane proteins. Proc Natl Acad Sci U S A. 2009;106:17747–50. doi: 10.1073/pnas.0906390106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Doura AK, Kobus FJ, Dubrovsky L, Hibbard E, Fleming KG. Sequence context modulates the stability of a GxxxG-mediated transmembrane helix-helix dimer. J Mol Biol. 2004;341:991–8. doi: 10.1016/j.jmb.2004.06.042. [DOI] [PubMed] [Google Scholar]
- 29.Joh NH, Oberai A, Yang D, Whitelegge JP, Bowie JU. Similar energetic contributions of packing in the core of membrane and water-soluble proteins. J Am Chem Soc. 2009;131:10846–7. doi: 10.1021/ja904711k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Russ WP, Engelman DM. The GxxxG motif: a framework for transmembrane helix-helix association. J Mol Biol. 2000;296:911–9. doi: 10.1006/jmbi.1999.3489. [DOI] [PubMed] [Google Scholar]
- 31.Schneider D, Engelman DM. Motifs of two small residues can assist but are not sufficient to mediate transmembrane helix interactions. J Mol Biol. 2004;343:799–804. doi: 10.1016/j.jmb.2004.08.083. [DOI] [PubMed] [Google Scholar]
- 32.Urban S, Baker RP. In vivo analysis reveals substrate-gating mutants of a rhomboid intramembrane protease display increased activity in living cells. Biological Chemistry. 2008;389:1107–15. doi: 10.1515/BC.2008.122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Tastan O, Klein-Seetharaman J, Meirovitch H. The effect of loops on the structural organization of alpha-helical membrane proteins. Biophys J. 2009;96:2299–312. doi: 10.1016/j.bpj.2008.12.3894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Barrera FN, et al. Protein self-assembly and lipid binding in the folding of the potassium channel KcsA. Biochemistry. 2008;47:2123–33. doi: 10.1021/bi700778c. [DOI] [PubMed] [Google Scholar]
- 35.Veerappan A, Cymer F, Klein N, Schneider D. The tetrameric alpha-helical membrane protein GlpF unfolds via a dimeric folding intermediate. Biochemistry. 2011;50:10223–30. doi: 10.1021/bi201266m. [DOI] [PubMed] [Google Scholar]
- 36.Urban S, Wolfe MS. Reconstitution of intramembrane proteolysis in vitro reveals that pure rhomboid is sufficient for catalysis and specificity. Proc Natl Acad Sci U S A. 2005;102:1883–8. doi: 10.1073/pnas.0408306102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Bondar AN, del Val C, White SH. Rhomboid protease dynamics and lipid interactions. Structure. 2009;17:395–405. doi: 10.1016/j.str.2008.12.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Findlay HE, Rutherford NG, Henderson PJ, Booth PJ. Unfolding free energy of a two-domain transmembrane sugar transport protein. Proc Natl Acad Sci U S A. 2010;107:18451–6. doi: 10.1073/pnas.1005729107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Otzen DE. Folding of DsbB in mixed micelles: a kinetic analysis of the stability of a bacterial membrane protein. J Mol Biol. 2003;330:641–9. doi: 10.1016/s0022-2836(03)00624-7. [DOI] [PubMed] [Google Scholar]
- 40.Fluhrer R, et al. Intramembrane proteolysis of GXGD-type aspartyl proteases is slowed by a familial Alzheimer disease-like mutation. J Biol Chem. 2008;283:30121–8. doi: 10.1074/jbc.M806092200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Page RM, et al. Generation of Abeta38 and Abeta42 is independently and differentially affected by familial Alzheimer disease-associated presenilin mutations and gamma-secretase modulation. J Biol Chem. 2008;283:677–83. doi: 10.1074/jbc.M708754200. [DOI] [PubMed] [Google Scholar]
- 42.Osenkowski P, Ye W, Wang R, Wolfe MS, Selkoe DJ. Direct and potent regulation of gamma-secretase by its lipid microenvironment. J Biol Chem. 2008;283:22529–40. doi: 10.1074/jbc.M801925200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Santos JM, Ferguson DJ, Blackman MJ, Soldati-Favre D. Intramembrane cleavage of AMA1 triggers Toxoplasma to switch from an invasive to a replicative mode. Science. 2011;331:473–7. doi: 10.1126/science.1199284. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.