

Abstract
A series of α-ketooxazoles containing heteroatoms embedded within conformational constraints in the C2 acyl side chain of 2 (OL-135) were synthesized and evaluated as inhibitors of fatty acid amide hydrolase (FAAH). The studies reveal that the installation of a heteroatom (O) in the conformational constraint is achievable, although the potency of these novel derivatives is reduced slightly relative to 2 and the analogous 1,2,3,4-tetrahydronaphthalene series. Interestingly, both enantiomers (R and S) of the candidate inhibitors bearing a chiral center adjacent to the electrophilic carbonyl were found to effectively inhibit FAAH.
INTRODUCTION
Fatty acid amide hydrolase (FAAH)1,2 hydrolyzes and terminates the signaling of several endogenous lipid amides3–6 including the endocannabinoid anandamide (1a)7–9 and the sleep-inducing lipid oleamide (1b)10–12 (Figure 1). The distribution of FAAH in the central nervous system is consistent with its role in regulating these neuromodulating fatty acid amides6 at their sites of action.3 FAAH is a member of amidase signature family of serine hydrolases. While there are a number of prokaryotic enzymes in this family, FAAH is the only well-characterized mammalian enzyme bearing the family’s diagnostic Ser–Ser–Lys catalytic triad.13–16
Figure 1.

Substrates of fatty acid amide hydrolase.
There has been wide-spread interest17 in the development of selective inhibitors of FAAH given their therapeutic potential18–20 for the treatment of pain,21–23 inflammation,24 or sleep disorders.13,25 FAAH inhibition potentiates an activated pathway, increasing the endogenous levels of a released lipid signaling molecule only at its site of action, thus providing a temporal and spatial pharmacological control not available with classical cell surface receptor agonists. Following the initial characterization of the enzyme, the endogenous sleep-inducing molecule 2-octyl α-bromoacetoacetate26 was found to be a potent, reversible FAAH inhibitor (Ki = 0.8 μM). Subsequent work led to the disclosure of nonselective, substrate-inspired, reversible inhibitors with an electrophilic ketone (e.g., trifluoromethyl ketone-based inhibitors)27–30 and irreversible inhibitors (e.g., fluorophosphonates and sulfonyl fluorides).31–36 Since that time and with notable exceptions,50 two generalized classes of inhibitors with promising therapeutic potential have been identified. One class is the reactive aryl carbamates and ureas39–49 that irreversibly acylate a FAAH active site serine. The second class consists of the α-ketoheterocycle-based inhibitors that bind to an active site serine in FAAH by reversible hemiketal formation.51–53 Many members of this class have been shown to be potent FAAH inhibitors and selective for FAAH over other mammalian serine hydrolases and several have been shown to be efficacious in vivo.54
Within this class, 2 (OL-135)55 emerged as an important lead inhibitor for further development.55 This compound is a potent (Ki = 4.7 nM)55 and selective (>60–300 fold)30 FAAH inhibitor that exhibits antinociceptive and anti-inflammatory activity and increases levels of anandamide in vivo.54 No antinociception was observed in FAAH knockout mice, indicating FAAH is the target responsible for the in vivo effects.54 Significantly, 2 does not bind to cannabinoid (CB1 and CB2) or vanilloid receptors and does not inhibit the human ether-a-go-go related gene (hERG) or common P450 metabolism enzymes (3A4, 2C9, 2D6). Further, the antinociceptive effects are not associated with the side effects observed with opioid administration (respiratory depression and dosing desensitization) or cannabinoid agonists (increased feeding and decreased motor control).54
Because of its therapeutic potential, we have conducted a series of systematic structure–activity relationship (SAR) studies exploring the 4- and 5-position of the central oxazole,55,57,58,62 the C2 acyl side chain,55,59,60 and the central heterocycle56,61 (Figure 2). Each of these sites were found to independently impact inhibitor potency or selectivity.62
Figure 2.

Progression of the inhibitor series.
In the most recent of these studies, we disclosed63,64 a series of inhibitors including 3 with added conformational constraints in the flexible C2 acyl side chain of 2 (Figure 2). This latest series showed improved or comparable enzyme inhibition potency relative to 2, indicating that removing many of the rotatable bonds in the C2 acyl side chain can simultaneously enhance target binding affinity and improve the drug-like characteristics of the inhibitor. Although many may be under the impression such inhibitors bearing electrophilic carbonyls may be metabolically labile, we have found that they are subject to competitive reduction/reoxidation metabolism that sets up a steady-state equilibrium between the two states (ketone/alcohol) with the true in vivo fate of the candidate inhibitors being determined by other features of the molecule.63 Moreover, the added conformational constraints in the C2 acyl chain and the increased steric hindrance surrounding the electrophilic ketone both slow the rate of equilibration and improve the ketone/alcohol ratio in vivo.63 As a result, the lead 1,2,3,4-tetrahydronaphthalene analogue 3 exhibited robust, long acting analgesic activity when administered orally in mouse models of thermal hyperalgesia and neuropathic pain in preliminary in vivo studies.63 Herein, we report our continued exploration of such inhibitors with conformationally restricted C2 acyl side chains. To probe the tolerance for additional modifications, two series of analogues were designed and prepared: one with an oxygen atom α to the electrophilic carbonyl (X = O, the 6-phenoxychroman-2-yl-2-ketooxazoles) and a second with an oxygen atom β to the carbonyl (Y = O, the 7-arylchroman-3-yl-2-ketooxazoles). The former change was expected to substantially increase the electrophilic character of the activated ketone by virtue of the inductive electron-withdrawing α-alkoxy substituent (X = O). Potentially, this could improve FAAH inhibitor potency provided the change does not competitively promote gem-diol formation, an attribute not observed with 2 or 3 and related compounds that would compete with active site adduct formation. An analogous, but dampened effect might be expected of the introduction of an ether oxygen β to the activated ketone (Y = O). However, the impact of the added heteroatom on the intrinsic FAAH active site binding through introduction of either destabilizing electrostatic or stabilizing H-bonding interactions with the proximal active site residues was difficult to a priori predict.
CHEMISTRY
The synthesis of the 6-phenoxychroman-2-yl-2-ketooxazole series of analogues is described in Scheme 1. The carboxylic acid 4 was prepared in five steps from commercially available 4-phenoxyphenol according to literature procedure.65 The carboxylic acid was converted to the corresponding aldehyde 6 through reduction of the Weinreb amide 5 (96%), accessed by coupling 4 with N,O-dimethylhydroxyamine (67%).
Scheme 1.

Vedejs oxazole metalation66 and condensation with aldehyde 6 yielded the secondary alcohol 7 (70%). The secondary alcohol was protected as its TBS ether to give 8 (95%). Selective C5-lithiation67 and quench with Bu3SnCl generated the tributylstannane intermediate 9 (72%). Stille coupling68 with either 2-bromopyridine (76%) or methyl 6-bromopicolinate (80%) produced the C5-aryl oxazoles 10 and 11, respectively. Subsequent TBS ether deprotection (Bu4NF) provided 12 and 13, and oxidation with Dess–Martin periodinane69 provided the desired α-ketooxazoles 14 and 15. Because ketones 14 and 15 were too polar for facile resolution by chiral HPLC, the chromatographic enantiomer separation was conducted at an earlier stage of the synthesis. Specifically, the TBS ethers 10 and 11 were separated by flash chromatography (SiO2) into their two diastereomers and each diastereomer was resolved by chiral HPLC to provide the two enantiomers (ChiralPak AD for 10 and ChiralCel OD for 11). This enantiopure material was then carried through the deprotection and oxidation steps described above to generate each single enantiomer of the ketone. The unsubstituted parent α-ketooxazole 16 was generated by the Dess–Martin periodinane oxidation of the alcohol 7 (71%), followed by resolution on a ChiralPak AD HPLC column.
The synthesis of the 7-arylchroman-3-yl-2-ketooxazole series of analogues began with commercially available 4-benzyloxy-2-hydroxybenzaldehyde, which was transformed into the tert-butyl ester 17 in two steps according to a literature procedure.70 One of three side chains (R1 = Ph, OPh, or OBn) was then added to the chromane core by reaction of the free phenol of 17 (Scheme 2). For the 7-phenyl series (R1 = Ph), the phenol 17 was converted to the corresponding triflate intermediate 18 (91%) and coupled with phenylboronic acid under Suzuki conditions to provide 19 (88%). Alkylation of the phenol with BnBr (cat. Bu4NI, 2 equiv K2CO3) provided 20 (93%) for accessing the 7-benzyloxy series (R1 = OBn). Alternatively, 17 was subjected to a modified Ullmann reaction68 with phenylboronic acid to produce 21 (63%) needed to prepare the 7-phenoxy series (R1 = OPh). For 19–21, the tert-butyl esters were reduced to the corresponding alcohols 22–24 with LiAlH4 and subsequently oxidized with Dess–Martin periodinane69 to provide the corresponding aldehydes 25–27.
Scheme 2.
With the aldehydes in hand, the synthesis of the α-ketooxazole inhibitors proceeded as described for the chroman-2-yl-2-ketooxazole series. Vedejs C2 metalation66 of oxazole was followed by condensation with the aldehydes to yield the secondary alcohols. The alcohols were TBS protected before selective C5-lithiation67 of the oxazole and quench with Bu3SnCl to generate the tributylstannane intermediates. Stille coupling71 with either 2-bromopyridine or methyl 6-bromopicolinate generated the C5-aryl oxazoles. Subsequent TBS ether deprotection (Bu4NF) and oxidation using Dess–Martin periodinane69 provided the desired α-ketooxazoles. For the 7-benzyloxy and 7-phenyl derivatives, each separated diastereomer of the corresponding TBS ethers were chromatographically resolved into the two enantiomers and independently carried forward to the enantiomerically pure α-ketooxazoles. Only the 7-phenoxy derivatives could be resolved into its enantiomers as the final ketone on a ChiralPak AD column. The parent α-ketooxazoles were generated by the Dess–Martin periodinane oxidation69 of the secondary alcohols.
ENZYME ASSAY
Enzyme assays were performed at 20–23 °C with purified recombinant rat FAAH (rFAAH) expressed in Escherichia coli as previously described.72 The initial rates of hydrolysis were monitored using enzyme concentrations (typically 1 nM) at least three times below the measured Ki by following the breakdown of 14C-oleamide, and the Ki values were established as described (Dixon plot).28
RESULTS AND DISCUSSION
Previous studies revealed that the C2 acyl side chain of 2 and related inhibitors bind in a hydrophobic channel of the FAAH active site reserved for the unsaturated lipid chain of the fatty acid amide substrates. Both the hydrophobic character and π-saturation of these inhibitors mimic the nature, location, and unsaturation (e.g., the oleyl Δ9,10 double bond) of the fatty acid chain of the endogenous substrates. Removing many of the rotatable bonds and introducing additional π-saturation led to a series of inhibitors that exhibited enhanced binding affinity and drug-like properties.63 In early work59 exploring conformational constraints in the C2 acyl side chain, three terminal aryl substituents displayed improved potency relative to 2 following the trend of phenyl > benzyloxy > phenoxy (Figure 2). With the later 1,2,3,4-tetrahydronaphthalene series exemplified by 3, a subtle reordering of aryl substituent preference was observed, following the trend phenoxy > benzyloxy > phenyl.63 Previous work also established that the aryl C5 oxazole substituents exhibit well-defined potency trends (2-Pyr > 3-Pyr > 4-Pyr > Ph) that correlate with the location and H-bond acceptor capabilities of the weakly basic heterocycle substituent.58 The addition of electron-withdrawing and water-solubilizing substituents onto the pyridine ring (6-CO2Me or 6-CO2H) weakly modulated the potency of these inhibitors, improved water solubility, and did not appear to impact CNS penetration.58 These optimized C5 oxazole substituents and the phenoxy aryl substituent were combined with the candidate C2 or C3 chromane cores. With the C3 chromane series, all three improved side chains (R1 = Ph, OBn, OPh) were synthesized and tested.
6-Phenoxychroman-2-yl-2-ketooxazole Series
For each derivative, the racemic mixture and the pure enantiomers were prepared and tested in the in vitro enzyme inhibition assay. The results (Ki) for this series are reported in Figure 3.
Figure 3.

FAAH inhibition, Ki (nM).
In each case, it was the faster eluting enantiomer obtained from the chiral phase chromatographic resolution that was the more potent. Unlike derivatives of 2 itself but like the observations made with 3, there was no improvement in potency upon introduction of the C5-pyridine substituent (14 vs 16), rather there was an analogous 2-fold reduction in activity. Analogous to the behavior of 2, which experienced a 13-fold loss in activity with the introduction of an α-oxygen atom,59 the introduction of the α-oxygen to 3 led to an 8-fold loss in activity (3 vs 14). Thus, the intrinsic electron-withdrawing effect of an α-oxygen atom that would be expected to enhance the electrophilic reactivity of the activated ketone does not productively improve the FAAH inhibition. The distinguishing feature of the series was that the activity of the slower eluting enantiomer was only 2–4 fold less potent than the faster eluting more potent enantiomer. This is in contrast to the 1,2,3,4-tetrahydronaphthalene series, where the more potent (S) enantiomer was an average of 70-fold more potent than the less active (R) enantiomer.
One might suspect that the proton on the stereogenic center adjacent to the ketone could be sufficiently activated such that epimerization of this center might take place during synthetic procedures or under the conditions of the enzyme inhibition assay. The enantiopurity of each sample was measured before the sample was tested in vitro. In each case, the enantiomeric excess (ee) of the sample was ≥95%, indicating little erosion in enantiopurity during the deprotection, oxidation reaction, and purification. To establish whether the compounds were epimerizing under the conditions of the assay (pH = 9.1), the sample was dissolved in DMSO and placed in the assay buffer without enzyme. The enantiopurity of the sample was monitored at 4-minute intervals for 30 minutes, the time course of the assay. For the parent inhibitor 16 and the C5-pyridine substituted inhibitor 14, the total change in ee was only −6.8% and −4.4%, respectively. This suggests that the 2 to 3 fold differences in the inhibitor enantiomer potencies is reflective of their activity and not significantly impacted by competitive epimerization in the assay although that cannot be conclusively ruled out. To further characterize the potential for racemization at this center, we monitored the enantiopurity of the sample after dissolution in a variety of solvents. In this case, only the presence of base (5% Et3N in EtOAc) led to significant racemization within the first hour of study, and continued exposure to these conditions led to complete epimerization within 48 h (Figure 4).
Figure 4.
Change in enantiomeric excess for 14 in solution.
It was previously observed that 2 and related α-ketooxazoles preferentially exist in solution in their ketone form and do not adopt a hydrated (gem diol) state. Similarly, 3 showed no detectable hydrate or hemiketal formation in CD3OD and 7% D2O–DMSO-d6. In contrast and consistent with an expected enhanced electrophilic character, significant hemiketal formation was observed for the parent α-ketooxazole 16 (38%), 14 (34%), and 15 (45%) in CD3OD. These three compounds also displayed slow, time-dependent hydrate formation (7% D2O–DMSO-d6) with no gem diol detected initially, but with a significant amount observed at 24 h: 16 (13%), 14 (25%), 15 (27%). It is conceivable that this competitive hydrate formation contributes to the less effective FAAH inhibition by 14–16 relative to 3 and 2.
7-Arylchroman-3-yl-2-ketooxazole Series
For each derivative, the racemic mixture and the pure enantiomers were prepared and tested in the in vitro enzyme inhibition assay. The results (Ki) for this series are reported in Figure 5.
Figure 5.
FAAH inhibition, Ki (nM).
The most interesting C5-pyridyloxazole series (45, 46, and 47) were found to be 2- to 4-fold less potent than 3 against FAAH, indicating that the introduction of the β-oxygen atom only slightly reduced the inhibitor activity. This is in sharp contrast to the analogous impact within the structure of 2, where the introduction of β-oxygen atom lead to a 20-fold loss in activity.59 With the exception of 48, the difference in potency between the two enantiomers (average of 1.5-fold) was less than that observed for the 6-phenoxychroman-2-yl-2-ketooxazole series and distinct from the 1,2,3,4-tetrahydronaphthalene series including 3. Unlike in the C2 chromane series (Figure 3), the installation of the pyridine substituent at C5 provided a roughly equipotent (e.g. 46) or slightly more potent ((S)-45) inhibitor than the unsubstituted parent oxazoles.
In order to unambiguously establish the absolute stereochemistry of each enantiomer, the inhibitor 54 in the series with an iodo substituent at the C5 position of the oxazole was prepared and resolved (ChiralPak AD, 20% iPrOH/hexane, α = 1.18, Scheme 3). A single-crystal X-ray crystal structure determination73 conducted on a heavy atom derivative established that the slower eluting enantiomer from the chiral phase HPLC separation and the less potent enantiomer possesses the (S)-configuration. The absolute configuration of the additional inhibitors 45, 46, 47, and 48 were tentatively assigned based on their analogous chromatographic mobility on the ChiralPak AD column with the slower eluting enantiomer assigned the (S)-configuration. Confirming this assignment for 47 and consistent with the additional tentative assignments, Stille coupling of the faster eluting (R)-enantiomer of 54 with 2-tributylstannylpyridine provided (R)-47 (faster eluting enantiomer). Interestingly, the two enantiomers of 54 were only 1.6-fold different in potency in inhibiting FAAH.
Scheme 3.

To determine if the results in Figure 5 could be attributed to epimerization of the stereocenter during the enzyme inhibition assay, the enantiopurity of the samples were monitored by chiral HPLC analysis over the course of a 30 min exposure to the assay buffer (pH 9.1). For the 5-pyridyloxazole inhibitors, the loss of enantiopurity was modest and followed the trend (greatest to least) of 7-phenyl 45 (−7.7%), 7-benzyloxy 46 (−4.8%), and 7-phenoxy 47 (−1.7%). Similarly, the unsubstituted parent oxazoles were only modestly susceptible to epimerization under the assay conditions: 7-phenyl 49 (−9.5%) and 7-benzyloxy 50 (−6.6%). Thus, it seems unlikely that the results in Figure 5 reflect sufficient racemization under the assay conditions to account for the near equivalent activities of the inhibitor enantiomers although that cannot be conclusively ruled out. To further explore the potential for racemization, the enantiopurity of 45 was monitored after exposure to a variety of solvents. Only exposure to 5% Et3N in EtOAc led to any significant racemization within the first hour of study (Figure 6). For all the inhibitors in this series, no detectable hydrate (gem diol) or hemiketal formation was observed in CD3OD and 7% D2O–DMSO-d6.
Figure 6.
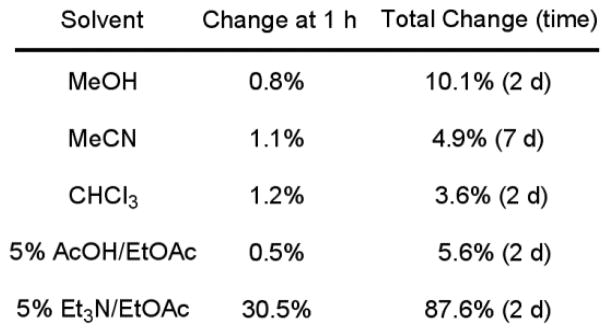
Change in enantiopurity of 45 in solution.
Interestingly, there appears little preference for the (R) or (S) configuration of the inhibitors although there are subtle trends that track with the C7 aryl substituent. The C7-phenoxy (R-enantiomer) and C7-phenyl (S-enantiomer) series appear to switch the modest enantioselectivity (45 vs 47), whereas the 7-benzyloxy series exhibit no selectivity (46). Although this may appear unusual at first glance, it may simply reflect a 180° flip of the chromane core in the active site to preserve the key anchoring interactions of the C2 acyl chain terminal phenyl group. In our earlier 1,2,3,4-tetrahydronapthalene series, the X-ray structure of 3 bound to the enzyme revealed that the enantiomeric selectivity is similarly imposed by the spatial relationship of the anchoring C6-phenoxy substituent relative to the chiral center adjacent to the electrophilic carbonyl.63,74,75
Most interesting of these unusual observations is the potential that the racemic inhibitors like 46 may serve as potent FAAH inhibitors just as effective as either enantiomer precluding the need for resolution, that they approach the activity of 2 and 3 (Ki = 4.7 and 4.4 nM vs 17 nM for 46), and that they may be expected to maintain the attributes of the conformationally-constrained inhibitor 3 (orally active, long acting FAAH inhibitor).
Inhibitor Selectivity
One inhibitor from each series was examined for FAAH selectivity in an activity-based protein profile (ABPP) assay for serine hydrolases76 conducted using the mouse brain proteome. Previous studies30 have shown that the C5-pyridyl α-ketooxazoles are exquisitely selective for FAAH over other proteome-wide serine hydrolases, although other derivatives display offsite activity against the membrane associated hydrolase KIAA1363, monoacylglycerol lipase (MAGL), and/or α,β-hydrolase containing domain 6 (ABHD6). Both enantiomers of the inhibitors 14 and 47 were tested for their effects on the fluorophosphonate (FP)-rhodamine probe labeling of serine hydrolases in the mouse brain proteome at concentrations ranging from 10 nM to 100 μM (Figure 7). Each inhibitor showed superb selectivity for FAAH over all other serine hydrolases in the mouse brain proteome including KIAA1363, MAGL, and ABHD6 (>200-fold).
Figure 7.

ABPP screen of (a) 14 and (b) 47 in mouse brain proteome (1 mg/mL) with FP-rhodamine (100 nM). Inhibitors were preincubated with proteome for 10 min prior to 10 min treatment with probe.
CONCLUSIONS
Herein, we report the synthesis and evaluation of a series of α-ketooxazoles as candidate FAAH inhibitors based on 3, incorporating an oxygen heteroatom into a conformationally constrained C2 acyl side. This modification led to slightly diminished potency against FAAH relative to 2 and 3 despite the inductive electron-withdrawing effect of the heteroatom insertion, and maintained their exquisite selectivity for FAAH. Unlike the observations made with 3, both enantiomers of the candidate FAAH inhibitors proved to be effective. Representative of these observations, 46 was found be only 4-fold less active than either 2 or 3, and the two enantiomers proved to be equally active (Ki = 18 and 17 nM).
EXPERIMENTAL SECTION
Inhibitors
Full experimental for the synthesis, characterization, and purity of the candidate inhibitors is provided in Supporting Information. The purity of each tested compound (>95%) was determined on an Agilent 1100 LC/MS instrument using a ZORBAX SB-C18 column (3.5 mm, 4.6 mm × 50 mm, with a flow rate of 0.75 mL/min and detection at 220 and 253 nm) with a 10–98% acetonitrile/water/0.1% formic acid gradient and 50–98% acetonitrile/water/0.1% formic acid gradient.
FAAH Inhibition
14C-labeled oleamide was prepared from 14C-labeled oleic acid as described.13 The truncated rat FAAH (rFAAH) was expressed in E. coli and purified as described.72 The inhibition assays were performed as described.13 In brief, the enzyme reaction was initiated by mixing 1 nM rFAAH with 20 μM of 14C-labeled oleamide in 500 μL reaction buffer (125 mM TrisCl, 1 mM EDTA, 0.2% glycerol, 0.02% Triton X-100, 0.4 mM Hepes, pH 9.0) at room temperature in the presence of three different concentrations of the inhibitor. The enzyme reaction was terminated by transferring 20 μL of the reaction mixture to 500 μL of 0.1 N HCl at three different time points. The 14C-labeled oleamide (substrate) and oleic acid (product) were extracted with EtOAc and analyzed by TLC as detailed.13 The Ki of the inhibitor was calculated using a Dixon plot as described.13
The purity of each inhibitor (>95%) was determined on an Agilent 1100 LC/MS instrument on a ZORBAX SB-C18, 3.5 mm × 50 mm, with a flow rate of 0.75 mL/min and detection at 220 and 254 nm, with a 10–98% acetonitrile/water/0.1% formic acid gradient and a 50–98% acetonitrile/water/0.1% formic acid gradient (see Supporting Information).
Preparations of Mouse Tissue Proteomes
Tissues were Dounce-homogenized in PBS, pH 7.5, followed by a low-speed spin (1,400 g, 5 min) to remove debris. The supernatant was then subjected to centrifugation (64,000 g, 45 min) to provide the cytosolic fraction in the supernatant and membrane fraction as a pellet. The pellet was washed and resuspended in PBS buffer by sonication. Total protein concentration in each fraction was determined using a protein assay kit (Bio-Rad).
ABPP Studies
Tissue proteomes, diluted to 1 mg/mL in PBS, were preincubated with inhibitors (10–10,000 nM, DMSO stocks) for 10 min and then treated with rhodamine-tagged fluorophosphonate (FP-rhodamine 100 nM, DMSO stock) at 25 °C for 10 min. Reactions were quenched with SDS-PAGE loading buffer, subjected to SDS-PAGE, and visualized in-gel using a flatbed fluorescence scanner (MiraBio). Labeled proteins were quantified by measuring integrated band intensities (normalized for volume); control samples (DMSO alone) were considered 100% activity and inhibitor-treated samples were expressed as a percentage of remaining activity. IC50 values were determined from dose-response curves from three trials at each inhibitor concentration using Prism software (GraphPad).
Supplementary Material
Acknowledgments
We gratefully acknowledge the financial support of the National Institutes of Health (Grant DA015648, D.L.B.). We thank Raj. K. Chadha for the X-ray crystal structure of (S)-54 and B. F. Cravatt for the supply of FAAH used in the enzymatic assays.
Footnotes
Supporting Information. Full experimental procedures, characterization and purities of the candidate inhibitors, and enzyme inhibition measurement standard deviations for Figures 3, 5 and 7 and Scheme 3. This material is available free of charge via the Internet at xxxxxx.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Cravatt BF, Giang DK, Mayfield SP, Boger DL, Lerner RA, Gilula NB. Nature. 1996;384:83–87. doi: 10.1038/384083a0. [DOI] [PubMed] [Google Scholar]
- 2.Giang DK, Cravatt BF. Proc Natl Acad Sci USA. 1997;94:2238–2242. doi: 10.1073/pnas.94.6.2238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Patricelli MP, Cravatt BF. Vitam Horm. 2001;62:95–131. doi: 10.1016/s0083-6729(01)62002-8. [DOI] [PubMed] [Google Scholar]
- 4.Egertova M, Cravatt BF, Elphick MR. Neuroscience. 2003;119:481–496. doi: 10.1016/s0306-4522(03)00145-3. [DOI] [PubMed] [Google Scholar]
- 5.Boger DL, Fecik RA, Patterson JE, Miyauchi H, Patricelli MP, Cravatt BF. Bioorg Med Chem Lett. 2000;10:2613–2616. doi: 10.1016/s0960-894x(00)00528-x. [DOI] [PubMed] [Google Scholar]
- 6.Ezzili C, Otrubova K, Boger DL. Bioorg Med Chem Lett. 2010;20:5959–5968. doi: 10.1016/j.bmcl.2010.08.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Devane WA, Hanus L, Breuer A, Pertwee RG, Stevenson LA, Griffin G, Gibson D, Mandelbaum A, Etinger A, Mechoulam R. Science. 1992;258:1946–1949. doi: 10.1126/science.1470919. [DOI] [PubMed] [Google Scholar]
- 8.Martin BR, Mechoulam R, Razdan RK. Life Sci. 1999;65:573–595. doi: 10.1016/s0024-3205(99)00281-7. [DOI] [PubMed] [Google Scholar]
- 9.Schmid HHO, Schmid PC, Natarajan V. Prog Lipid Res. 1990;29:1–43. doi: 10.1016/0163-7827(90)90004-5. [DOI] [PubMed] [Google Scholar]
- 10.Boger DL, Henriksen SJ, Cravatt BF. Curr Pharm Des. 1998;4:303–314. [PubMed] [Google Scholar]
- 11.Cravatt BF, Lerner RA, Boger DL. J Am Chem Soc. 1996;118:580–590. [Google Scholar]
- 12.(a) Cravatt BF, Prospero–Garcia O, Suizdak G, Gilula NB, Henriksen SJ, Boger DL, Lerner RA. Science. 1995;268:1506–1509. doi: 10.1126/science.7770779. [DOI] [PubMed] [Google Scholar]; (b) Lerner RA, Siuzdak G, Prospero–Garcia O, Henriksen SJ, Boger DL, Cravatt BF. Proc Natl Acad Sci USA. 1994;91:9505–9508. doi: 10.1073/pnas.91.20.9505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.(a) Patricelli MP, Cravatt BF. Biochemistry. 1999;38:14125–14130. doi: 10.1021/bi991876p. [DOI] [PubMed] [Google Scholar]; (b) Patricelli MP, Cravatt BF. J Biol Chem. 2000;275:19177–19184. doi: 10.1074/jbc.M001607200. [DOI] [PubMed] [Google Scholar]; (c) Patricelli MP, Lovato MA, Cravatt BF. Biochemistry. 1999;38:9804–9812. doi: 10.1021/bi990637z. [DOI] [PubMed] [Google Scholar]
- 14.McKinney MK, Cravatt BF. J Biol Chem. 2003;278:37393–37399. doi: 10.1074/jbc.M303922200. [DOI] [PubMed] [Google Scholar]
- 15.McKinney MK, Cravatt BF. Ann Rev Biochem. 2005;74:411–432. doi: 10.1146/annurev.biochem.74.082803.133450. [DOI] [PubMed] [Google Scholar]
- 16.Bracey MH, Hanson MA, Masuda KR, Stevens RC, Cravatt BF. Science. 2002;298:1793–1796. doi: 10.1126/science.1076535. [DOI] [PubMed] [Google Scholar]
- 17.Otrubova K, Ezzili C, Boger DL. Bioorg Med Chem Lett. 2011;21:4674–4685. doi: 10.1016/j.bmcl.2011.06.096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Clement AB, Hawkins EG, Lichtman AH, Cravatt BF. J Neurosci. 2003;23:3916–3923. doi: 10.1523/JNEUROSCI.23-09-03916.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.(a) Cravatt BF, Lichtman AH. Curr Opin Chem Biol. 2003;7:469–475. doi: 10.1016/s1367-5931(03)00079-6. [DOI] [PubMed] [Google Scholar]; (b) Ahn K, McKinney MK, Cravatt BF. Chem Rev. 2008;108:1687–1707. doi: 10.1021/cr0782067. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Ahn K, Johnson DS, Cravatt BF. Exp Opin Drug Discov. 2009;4:763–784. doi: 10.1517/17460440903018857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lambert DM, Fowler CJ. J Med Chem. 2005;48:5059–5087. doi: 10.1021/jm058183t. [DOI] [PubMed] [Google Scholar]
- 21.Cravatt BF, Demarest K, Patricelli MP, Bracey MH, Giang DK, Martin BR, Lichtman AH. Proc Natl Acad Sci USA. 2001;98:9371–9376. doi: 10.1073/pnas.161191698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lichtman AH, Shelton CC, Advani T, Cravatt BF. Pain. 2004;109:319–327. doi: 10.1016/j.pain.2004.01.022. [DOI] [PubMed] [Google Scholar]
- 23.Cravatt BF, Saghatelian A, Hawkins EG, Clement AB, Bracey MH, Lichtman AH. Proc Natl Acad Sci USA. 2004;101:10821–10826. doi: 10.1073/pnas.0401292101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Karsak M, Gaffal E, Date R, Wang–Eckhardt L, Rehnelt J, Petrosino S, Starowicz K, Steuder R, Schlicker E, Cravatt BF, Mechoulam R, Buettner R, Werner S, Di Marzo V, Tueting T, Zimmer A. Science. 2007;316:1494–1497. doi: 10.1126/science.1142265. [DOI] [PubMed] [Google Scholar]
- 25.(a) Huitrón–Reséndiz S, Gombart L, Cravatt BF, Henriksen SJ. Exp Neurol. 2001;172:235–243. doi: 10.1006/exnr.2001.7792. [DOI] [PubMed] [Google Scholar]; (b) Huitrón–Reséndiz S, Sanchez–Alavez M, Wills DN, Cravatt BF, Henriksen SJ. Sleep. 2004;27:857–865. doi: 10.1093/sleep/27.5.857. [DOI] [PubMed] [Google Scholar]
- 26.Patricelli MP, Patterson JP, Boger DL, Cravatt BF. Bioorg Med Chem Lett. 1998;8:613–618. doi: 10.1016/s0960-894x(98)00073-0. [DOI] [PubMed] [Google Scholar]
- 27.Koutek B, Prestwich GD, Howlett AC, Chin SA, Salehani D, Akhavan N, Deutsch DG. J Biol Chem. 1994;269:22937–22940. [PubMed] [Google Scholar]
- 28.Patterson JE, Ollmann IR, Cravatt BF, Boger DL, Wong CH, Lerner RA. J Am Chem Soc. 1996;118:5938–5945. [Google Scholar]
- 29.Boger DL, Sato H, Lerner AE, Austin BJ, Patterson JE, Patricelli MP, Cravatt BF. Bioorg Med Chem Lett. 1999;9:265–270. doi: 10.1016/s0960-894x(98)00734-3. [DOI] [PubMed] [Google Scholar]
- 30.Leung D, Hardouin C, Boger DL, Cravatt BF. Nat Biotech. 2003;21:687–691. doi: 10.1038/nbt826. [DOI] [PubMed] [Google Scholar]
- 31.De Petrocellis L, Melck D, Ueda N, Maurelli S, Kurahashi Y, Yamamoto S, Marino G, Di Marzo V. Biochem Biophys Res Commun. 1997;231:82–88. doi: 10.1006/bbrc.1997.6000. [DOI] [PubMed] [Google Scholar]
- 32.Deutsch DG, Omeir R, Arreaza G, Salehani D, Prestwich GD, Huang Z, Howlett A. Biochem Pharmacol. 1997;53:255–260. doi: 10.1016/s0006-2952(96)00830-1. [DOI] [PubMed] [Google Scholar]
- 33.Deutsch DG, Lin S, Hill WAG, Morse KL, Salehani D, Arreaza G, Omeir RL, Makriyannis A. Biochem Biophys Res Commun. 1997;231:217–221. doi: 10.1006/bbrc.1997.6072. [DOI] [PubMed] [Google Scholar]
- 34.Edgemond WS, Greenberg MJ, McGinley PJ, Muthians S, Campbell WB, Hillard CJ. J Pharmacol Exp Ther. 1998;286:184–190. [PubMed] [Google Scholar]
- 35.Fernando SR, Pertwee RG. Br J Pharmacol. 1997;121:1716–1720. doi: 10.1038/sj.bjp.0701303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Du W, Hardouin C, Cheng H, Hwang I, Boger DL. Bioorg Med Chem Lett. 2005;15:103–106. doi: 10.1016/j.bmcl.2004.10.025. [DOI] [PubMed] [Google Scholar]
- 37.Kathuria S, Gaetani S, Fegley D, Valino F, Duranti A, Tontini A, Mor M, Tarzia G, La Rana G, Calignano A, Giustino A, Tattoli M, Palmery M, Cuomo V, Piomelli D. Nat Med. 2003;9:76–81. doi: 10.1038/nm803. [DOI] [PubMed] [Google Scholar]
- 38.Jayamanne A, Greenwood R, Mitchell VA, Aslan S, Piomelli D, Vaughan CW. Br J Pharmacol. 2006;147:281–288. doi: 10.1038/sj.bjp.0706510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Biancalani C, Giovanni MP, Pieretti S, Cesari N, Graziano A, Vergeli C, Cilibrizzi A, Di Gianuario A, Colucci M, Mangano G, Garrone B, Polezani L, Dal Piaz V. J Med Chem. 2009;52:7397–7409. doi: 10.1021/jm900458r. [DOI] [PubMed] [Google Scholar]
- 40.Clapper JR, Vacondio F, King AR, Duranti A, Tontini A, Silva C, Sanchini S, Tarzia G, Mor M, Piomelli D. ChemMedChem. 2009;4:1505–1513. doi: 10.1002/cmdc.200900210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.(a) Ahn K, Johnson DS, Fitzgerald LR, Liimatta M, Arendse A, Stevenson T, Lund ET, Nugent RA, Normanbhoy T, Alexander JP, Cravatt BF. Biochemistry. 2007;46:13019–13030. doi: 10.1021/bi701378g. [DOI] [PubMed] [Google Scholar]; (b) Ahn K, Johnson DS, Mileni M, Beidler D, Long JZ, McKinney MK, Weerapana E, Sadagopan N, Liimatta M, Smith SE, Lazerwith S, Stiff C, Kamtekar S, Bhattacharya K, Zhang Y, Swaney S, Van Becelaere K, Stevens RC, Cravatt BF. Chem Biol. 2009;16:411–420. doi: 10.1016/j.chembiol.2009.02.013. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Johnson DS, Stiff C, Lazerwith SE, Kesten SR, Fay LK, Morris M, Beidler D, Liimatta MB, Smith SE, Dudley DT, Sadogopan N, Bhattachar SN, Kesten SJ, Nomanbhoy TK, Cravatt BF, Ahn K. ACS Med Chem Lett. 2011;2:91–96. doi: 10.1021/ml100190t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Meyers JM, Long AS, Pelc JM, Wang LJ, Bowen JS, Schweitzer AB, Wilcox CM, McDonald J, Smith ES, Foltin S, Rumsey J, Yang SY, Walker CM, Kamtekar S, Beidler F, Torarensen A. Bioorg Med Chem Lett. 2011;21:6545–6553. doi: 10.1016/j.bmcl.2011.08.048. [DOI] [PubMed] [Google Scholar]
- 43.(a) Sit SY, Conway C, Bertekap R, Xie K, Bourin C, Burris K, Deng H. Bioorg Med Chem Lett. 2007;17:3287–3291. doi: 10.1016/j.bmcl.2007.04.009. [DOI] [PubMed] [Google Scholar]; (b) Sit SY, Conway CM, Xie K, Bertekap R, Bourin C, Burris KD. Bioorg Med Chem Lett. 2010;20:1272–1277. doi: 10.1016/j.bmcl.2009.11.080. [DOI] [PubMed] [Google Scholar]
- 44.(a) Keith JM, Apocada R, Xiao W, Seierstad M, Pattabiraman K, Wu J, Webb M, Karbarz MJ, Brown S, Wilson S, Scott B, Tham C-S, Luo L, Palmer J, Wennerholm M, Chaplan S, Breitenbucher JG. Bioorg Med Chem Lett. 2008;18:4838–4843. doi: 10.1016/j.bmcl.2008.07.081. [DOI] [PubMed] [Google Scholar]; (b) Keith JM, Apodaca R, Tichenor M, Xiao W, Jones W, Pierce J, Seierstad M, Palmer J, Webb M, Karbarz M, Scott B, Wilson S, Luo L, Wennerholm M, Chang L, Brown S, Rizzolio M, Rynberg R, Chaplan S, Breitenbucher JG. ACS Med Chem Lett. 2012;3:823–827. doi: 10.1021/ml300186g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kono M, Matsumoto T, Kawamura T, Nishimura A, Kiyota Y, Oki H, Miyazaki J, Igaki S, Bahnke CA, Shimojo M, Kori M. Bioorg Med Chem. 2013;21:28–41. doi: 10.1016/j.bmc.2012.11.006. [DOI] [PubMed] [Google Scholar]
- 46.Moore SA, Nomikos GG, Dickason–Chesterfield AK, Sohober DA, Schaus JM, Ying BP, Xu YC, Phebus L, Simmons RM, Li D, Iyengar S, Felder CC. Proc Natl Acad Sci USA. 2005;102:17852–17857. doi: 10.1073/pnas.0507470102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Alexander JP, Cravatt BF. J Am Chem Soc. 2006;128:9699–9704. doi: 10.1021/ja062999h. [DOI] [PubMed] [Google Scholar]
- 48.Alexander JP, Cravatt BF. Chem Biol. 2005;12:1179–1187. doi: 10.1016/j.chembiol.2005.08.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Minkkila A, Myllymaki MJ, Saario SM, Castillo-Melendez JA, Koskinen AMP, Fowler CJ, Leppanen J, Nevalainene T. Eur J Med Chem. 2009;44:2294–3008. doi: 10.1016/j.ejmech.2009.01.007. [DOI] [PubMed] [Google Scholar]
- 50.For additional studies, see: Wang X, Sarris K, Kage K, Zhang D, Brown SP, Kolassa T, Surowy C, El Kouhen OF, Muchmore SW, Brioni JD, Stewart AO. J Med Chem. 2009;52:170–180. doi: 10.1021/jm801042a.Min X, Thibault ST, Porter AC, Gustin DJ, Carlson TJ, Xu H, Lindstrom M, Xu G, Uyeda C, Ma Z, Li Y, Kayser F, Walker NPC, Wang Z. Proc Natl Acad Sci. 2011;108:7379–7384. doi: 10.1073/pnas.1016167108.Tian G, Pachetto KA, Gharahdaghi F, Gordon E, Wilkens DE, Luo X, Scott CW. Biochemistry. 2011;50:6867–6878. doi: 10.1021/bi200552p.
- 51.Otrubova K, Boger DL. ACS Chem Neurosci. 2012;3:340–348. doi: 10.1021/cn2001206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Boger DL, Sato H, Lerner AE, Hedrick MP, Fecik RA, Miyauchi H, Wilkie GD, Austin BJ, Patricelli MP, Cravatt BF. Proc Natl Acad Sci USA. 2000;97:5044–5049. doi: 10.1073/pnas.97.10.5044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Boger DL, Miyauchi H, Hedrick MP. Bioorg Med Chem Lett. 2001;11:1517–1520. doi: 10.1016/s0960-894x(01)00211-6. [DOI] [PubMed] [Google Scholar]
- 54.(a) Lichtman AH, Leung D, Shelton CC, Saghatelian A, Hardouin C, Boger DL, Cravatt BF. J Pharmacol Exp Ther. 2004;311:441–448. doi: 10.1124/jpet.104.069401. [DOI] [PubMed] [Google Scholar]; (b) Schlosburg JE, Boger DL, Cravatt BF, Lichtman AH. J Pharmacol Exp Ther. 2009;329:314–323. doi: 10.1124/jpet.108.150136. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Kinsey SG, Long JZ, O’Neal ST, Abdulla RA, Poklis JL, Boger DL, Cravatt BF, Lichtman AH. J Pharmacol Exp Ther. 2009;330:902–910. doi: 10.1124/jpet.109.155465. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Booker L, Kinsey SG, Abdullah RA, Blankman JL, Long JZ, Boger DL, Cravatt BF, Lichtman AH. Br J Pharmacol. 2012;165:2485–2496. doi: 10.1111/j.1476-5381.2011.01445.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Boger DL, Miyauchi H, Du W, Hardouin C, Fecik RA, Cheng H, Hwang I, Hedrick MP, Leung D, Acevedo O, Guimaráes CRW, Jorgensen WL, Cravatt BF. J Med Chem. 2005;48:1849–1856. doi: 10.1021/jm049614v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Leung D, Du W, Hardouin C, Cheng H, Hwang I, Cravatt BF, Boger DL. Bioorg Med Chem Lett. 2005;15:1423–1428. doi: 10.1016/j.bmcl.2004.12.085. [DOI] [PubMed] [Google Scholar]
- 57.Romero FA, Hwang I, Boger DL. J Am Chem Soc. 2006;68:14004–14005. doi: 10.1021/ja064522b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Romero FA, Du W, Hwang I, Rayl TJ, Kimball FS, Leung D, Hoover HS, Apodaca RL, Breitenbucher BJ, Cravatt BF, Boger DL. J Med Chem. 2007;50:1058–1068. doi: 10.1021/jm0611509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Hardouin C, Kelso MJ, Romero FA, Rayl TJ, Leung D, Hwang I, Cravatt BF, Boger DL. J Med Chem. 2007;50:3359–3368. doi: 10.1021/jm061414r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Kimball FS, Romero FA, Ezzili C, Garfunkle J, Rayl TJ, Hochstatter DG, Hwang I, Boger DL. J Med Chem. 2008;51:937–947. doi: 10.1021/jm701210y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Garfunkle J, Ezzili C, Rayl TJ, Hochstatter DG, Hwang I, Boger DL. J Med Chem. 2008;51:4393–4403. doi: 10.1021/jm800136b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.DeMartino JK, Garfunkle J, Hochstatter DG, Cravatt BF, Boger DL. Bioorg Med Chem Lett. 2008;18:5842–5846. doi: 10.1016/j.bmcl.2008.06.084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Ezzili C, Mileni M, McGlinchey N, Long JZ, Kinsey SG, Hochstatter DG, Stevens RC, Lichtman AH, Cravatt BF, Bilsky EJ, Boger DL. J Med Chem. 2011;54:2805–2822. doi: 10.1021/jm101597x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Otrubova K, Brown M, McCormick MS, Han GW, O’Neil ST, Cravatt BF, Stevens RC, Lichtman AH, Boger DL. J Am Chem Soc. 2013;135:6289–6299. doi: 10.1021/ja4014997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Witiak DT, Heilman WP, Sankarappa SK, Cavestri RC, Newman HAI. J Med Chem. 1975;19:934–942. doi: 10.1021/jm00243a015. [DOI] [PubMed] [Google Scholar]
- 66.Vedejs E, Monahan SD. J Org Chem. 1996;61:5192–5193. [Google Scholar]
- 67.Hari Y, Obika S, Sakaki M, Morio K, Yamagata Y, Imanishi T. Tetrahedron. 2002;58:3051–3063. [Google Scholar]
- 68.Ley SV, Thomas AW. Angew Chem Int Ed. 2003;42:5400–5449. doi: 10.1002/anie.200300594. [DOI] [PubMed] [Google Scholar]
- 69.Dess DB, Martin JC. J Am Chem Soc. 1991;113:7277–7287. [Google Scholar]
- 70.Choi Y-M, Shin H, Ilankumaran P, Kim H-W. 2005/021540. Patent WO. 2005
- 71.Farina V, Krishnamurthy V, Scott WJ. Org React. 1997;50:1–652. [Google Scholar]
- 72.Patricelli MP, Lashuel HA, Giang DK, Kelly JW, Cravatt BF. Biochemistry. 1998;37:15177–15187. doi: 10.1021/bi981733n. [DOI] [PubMed] [Google Scholar]
- 73.The structure and absolute configuration of (S)-54 was established with single-crystal X-ray structure determination conducted on a colorless needle grown from EtOAc/CH2Cl2 (CCDC 975738).
- 74.(a) Mileni M, Garfunkle J, DeMartino JK, Cravatt BF, Boger DL, Stevens RC. J Am Chem Soc. 2009;131:10497–10506. doi: 10.1021/ja902694n. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Mileni M, Garfunkle J, Kimball FS, Cravatt BF, Stevens RC, Boger DL. J Med Chem. 2010;53:230–240. doi: 10.1021/jm9012196. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Mileni M, Garfunkle J, Ezzili C, Cravatt BF, Stevens RC, Boger DL. J Am Chem Soc. 2011;133:4092–4100. doi: 10.1021/ja110877y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Guimaráes CRW, Boger DL, Jorgensen WL. J Am Chem Soc. 2005;127:17377–17384. doi: 10.1021/ja055438j. [DOI] [PubMed] [Google Scholar]
- 76.(a) Evans MJ, Cravatt BF. Chem Rev. 2006;106:3279–3301. doi: 10.1021/cr050288g. [DOI] [PubMed] [Google Scholar]; (b) Liu YS, Patricelli MP, Cravatt BF. Proc Natl Acad Sci USA. 1999;96:14694–14699. doi: 10.1073/pnas.96.26.14694. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.