

Abstract
Circulating immunoreactive trypsinogen (IRT), a biomarker of exocrine pancreatic disease in cystic fibrosis (CF), is elevated in most CF newborns. In those with severe CF transmembrane conductance regulator (CFTR) genotypes, IRT declines rapidly in the first years of life, reflecting progressive pancreatic damage. Consistent with this progression, a less elevated newborn IRT measure would reflect more severe pancreatic disease, including compromised islet compartments, and potentially increased risk of CF-related diabetes (CFRD). We show in two independent CF populations that a lower newborn IRT estimate is associated with higher CFRD risk among individuals with severe CFTR genotypes, and we provide evidence to support a causal relationship. Increased loge(IRT) at birth was associated with decreased CFRD risk in Canadian and Colorado samples (hazard ratio 0.30 [95% CI 0.15–0.61] and 0.39 [0.18–0.81], respectively). Using Mendelian randomization with the SLC26A9 rs7512462 genotype as an instrumental variable since it is known to be associated with IRT birth levels in the CF population, we provide evidence to support a causal contribution of exocrine pancreatic status on CFRD risk. Our findings suggest CFRD risk could be predicted in early life and that maintained ductal fluid flow in the exocrine pancreas could delay the onset of CFRD.
Introduction
Cystic fibrosis (CF), a recessive disease caused by mutations in the CF transmembrane conductance regulator (CFTR) gene, affects organs including the lungs and pancreas. CF severity is partly determined by CFTR; for example, exocrine pancreatic insufficiency (PI) occurs primarily in individuals with two severe CFTR mutations, although the degree of pancreatic damage (PD) at birth and the age at which clinically defined PI develops are variable among patients with the same CFTR genotypes (1,2). CF-related diabetes (CFRD) develops later in life in a subset of these individuals who are PI but not in those who are pancreatic-sufficient (3). The relationship between PI and CFRD is not well-understood.
Prenatally, the healthy exocrine pancreas shows considerable growth and maturation during the third trimester. At birth, secretion of most proteases (e.g., trypsinogen) are almost at adult levels (4). In CF, pancreatic development is markedly compromised beginning in utero. Obstruction of small pancreatic ducts by precipitated proteinaceous acinar secretions and mucus lead to dilation of proximal ductal and acinar lumina that compromise the acini (5). This process continues postnatally, where it has been observed that the exocrine pancreas undergoes destruction and is replaced with fat and/or fibrotic tissue in patients with severe CFTR genotypes (6,7).
In many countries, increased circulating immunoreactive trypsinogen (IRT) levels at birth are used as a newborn screening (NBS) test for CF in which IRT values above the normal population level are a biomarker of CF. Given that the elevated serum IRT exhibits rapid decline in the first year of life in CF patients with severe CFTR genotypes (8), we presumed that a relatively less elevated IRT value (lower IRT) at birth reflected more severe early disease with extensive PD, reduction of acinar tissue, and less capacity to produce digestive enzymes.
CFRD prevalence increases with age, and by the fourth decade of life >40% of PI patients have CFRD (9). CFRD is associated with lower BMI, female sex, pulmonary exacerbations, and type 2 diabetes family history (10,11). Since CFRD is limited to PI individuals and primarily characterized by impaired secretion of insulin, it is hypothesized that progressive damage to the exocrine pancreas by inflammation and fibrosis may also compromise islet cells. In this regard, single nucleotide polymorphism rs7512462 in the solute carrier family gene SLC26A9 has been shown to associate with both IRT birth measurements and the risk of developing CFRD, but not with CF lung disease (11–13). SLC26A9, an apical epithelial cell transporter that interacts with CFTR (14,15) and is expressed in the lung and at lower levels in the pancreas and prostate (16), is hypothesized to influence ion transport (chloride and bicarbonate) in pancreatic ducts prior to destruction of the epithelial exocrine tissue. Electrophysiological studies further support epithelial expression of SLC26A9 (17); hence, we suggest that loss of acinar epithelial tissue is concordant with loss of SLC26A9. In contrast to the exocrine pancreas, epithelial solute carriers such as SLC26A9 are unlikely to have a direct role in endocrine compartments, as the endocrine pancreas delivers its protein load to the bloodstream and surrounding cells without a specialized ductal system.
In two independent samples, we show that IRT levels at birth and their rate of decline, which reflect loss of pancreatic acinar tissue, are associated with CFRD among CF patients with severe CFTR genotypes. However, given the observational design, this association may be due to other unmeasured factors (i.e., confounding). A clinical trial that randomizes patients to a more or less severe degree of early exocrine PD could eliminate unmeasured confounding and assess the causal relationship with CFRD but is not possible; instead, we use an application of instrumental variable analysis known as Mendelian randomization (MR) (18). Assuming rs7512462 is unrelated to confounding factors that may influence the relationship between IRT and CFRD, MR compares the relationship between rs7512462 and IRT at birth with the relationship between rs7512462 and CFRD to estimate the causal effect of exocrine PD on the risk of CFRD (18).
Research Design and Methods
In the Canadian Cystic Fibrosis Gene Modifier Study (CGS), DNA from 1,661 PI CF individuals (with known CFRD status) was genotyped on the Illumina 610-Quad array (Illumina). All CGS participants were diagnosed by characteristic clinical manifestations of CF. IRT measurements, including a baseline measure within 24 months of age, were available on 126 of these 1,661 individuals. The additional 1,535 CGS participants were genotyped at rs7512462 but were without IRT measurements.
A replication sample, without genotypes, included 260 CF PI patients from Colorado diagnosed through a statewide NBS program and voluntarily enrolled in a longitudinal study (8). Institutional review committees at all participating Canadian CF clinics and at the Children’s Hospital Colorado approved this study.
Data collection, IRT protocols, genotyping, and quality control procedures are reported elsewhere (8,19,20).
CFRD status was determined on chart review. Individuals required a physician’s diagnosis, no type 1 or type 2 diabetes, and one of the following: 1) daily treatment with insulin or oral diabetes medication; 2) abnormal 2-h glucose level (>11.1 mmol/L or >200 mg/dL) during modified oral glucose tolerance test; or 3) HbA1c ≥7%.
Statistical Methodology
In the CGS, a linear mixed-effects regression model, accounting for the limits of detection of the IRT assay (<3 ng/mL) by the statistical model (21), was used to estimate patient-specific IRT measurements at birth and rates of decline. IRT and age (days) were natural log (loge) transformed. Meconium ileus status was included in the regression model as a covariate since individuals with meconium ileus, on average, had IRT measurements taken at younger ages.
For the Colorado sample, we used linear mixed-effects modeling with loge transformations of IRT and age, accounting for values >400 and <5 ng/mL beyond the limits of detection of the assay (21). Meconium ileus was not included in the model because individuals were ascertained at birth.
The patient-specific IRT birth and rates of decline estimates from these models were used as individual measures of prenatal and early postnatal PD.
To assess association between exocrine PD and CFRD, age at CFRD diagnosis (or age at last clinic visit without CFRD) was regressed on estimated values of loge(IRT) at birth and rate of decline using sex-adjusted Cox proportional hazard models for the Canadian and Colorado samples with the R software package (22).
To determine if exocrine PD contributes to the cause of CFRD, we estimated the causal effect using MR in the Canadian sample. We compared it to the (possibly confounded) direct estimate obtained by regressing age of CFRD diagnosis on estimated values of loge(IRT) at birth in sex-adjusted Cox models. MR requires one exposure variable per instrumental variable. In this study, we use estimated loge(IRT) at birth, as opposed to both birth and rate of decline measures, to reflect the exocrine PD exposure. The causal effect on CFRD risk was estimated using the two-sample MR method with rs7512462 coded additively as the instrumental variable (18,23). The two-sample method uses the full CGS-genotyped sample (n = 1,661) in the MR analysis (23). Dividing the association estimate from the Cox regression of CFRD on rs7512462 by the average increase in loge(IRT) at birth per additional rs7512462 risk allele gives the loge(hazard ratio [HR]) that represents the association of genetically determined loge(IRT) levels at birth with CFRD risk (18). SEs for the two-sample causal HR were estimated using the delta method (24).
Results
Is Early Exocrine PD Associated With CFRD?
IRT measurements decline with advancing age (Fig. 1 and Table 1). After adjustment for rate of loge(IRT) decline and sex, loge(IRT) at birth was associated with CFRD risk in the Canadian (HR 0.30; P = 0.001) and Colorado samples (HR 0.39; P = 0.012); we observed a 70 and 61% decrease, respectively, in the rate of CFRD for every one-unit increase in loge(IRT) at birth. Rate of decline of loge(IRT) was also associated with CFRD in the Canadian (P = 0.007) and Colorado (P = 0.01) samples; a sharper decline in IRT, reflective of a more rapid depletion of acinar cell mass, predicted greater CFRD risk (Table 2).
Figure 1.
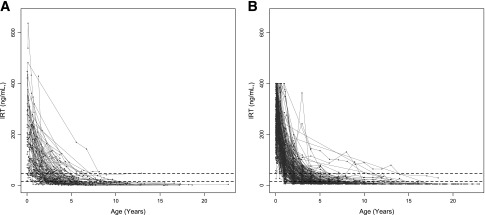
Longitudinal IRT trajectories for each participant in the Canadian (A) (n = 126) and Colorado (B) (n = 260) samples. Dotted lines represent the normal range of IRT measurement in the non-CF population (14.6–46.5 ng/mL).
Table 1.
Characteristics of study participants
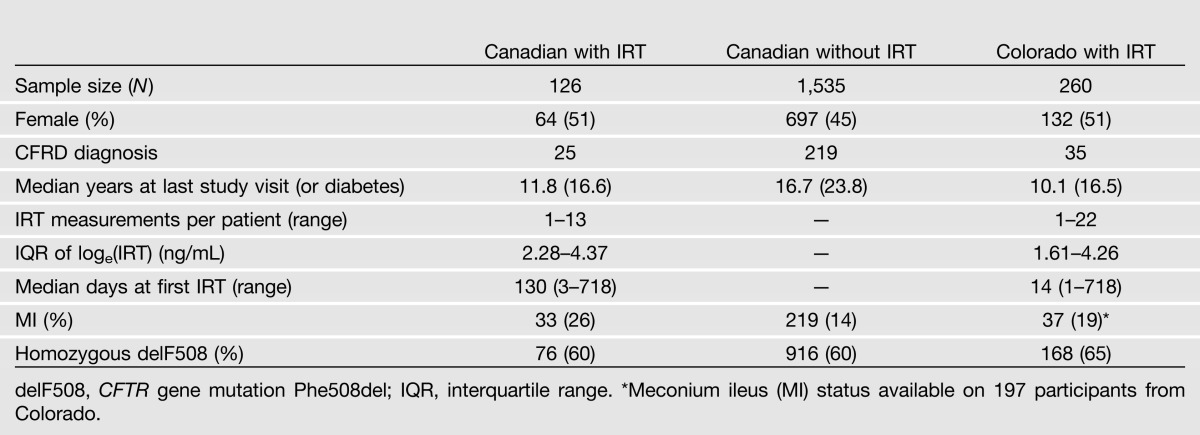
Table 2.
Cox proportional hazards model for effect of early exocrine PD [measured by loge(IRT) at birth and rate of decline] on CFRD risk adjusted for sex (other risk factors such as BMI and type 2 diabetes family history were not available) and the effect of prenatal exocrine PD [measured by loge(IRT) at birth] on CFRD risk (comparison between direct association by Cox proportional hazards modeling and two-sample causal estimation by MR; HR <1 implies greater CFRD risk with greater PD [lower IRT])

Is the Exocrine PD–CFRD Relationship Causal?
Using estimated loge(IRT) at birth (see Research Design and Methods), we assessed whether PD causally contributes to CFRD by applying MR with rs7512462 as an instrumental variable. We then compared this causal estimate to the risk estimate obtained directly from regressing CFRD on loge(IRT) at birth (Fig. 2 and Table 2).
Figure 2.

MR framework. MR estimate of causal effect of prenatal exocrine PD on CFRD risk was obtained by dividing the coefficient from the regression of CFRD diagnosis age on rs7512462 genotype, loge(HR 1.19), by the average increase in loge(IRT) at birth per additional SLC26A9 rs7512462 risk allele, β = −0.53, yielding: loge(HRcausal) = loge(1.19)/(−0.53) = −0.33. Exponentiation provided an HRcausal of 0.72. This causal effect estimate of the HR was equivalent to the estimate obtained by directly regressing CFRD diagnosis age on loge(IRT) at birth (HR 0.72). Application of MR required that the SLC26A9 instrument be robustly associated with the exposure and independent of confounding factors; these were satisfied by our F statistic of 8.99 and Mendel’s law of independent assortment, respectively. Additionally, it was assumed that the relationship between rs7512462 and CFRD was mediated by prenatal exocrine PD (see Discussion).
In the Canadian sample, rs7512462 was associated with estimated loge(IRT) at birth (β = −0.53; P = 0.003; F1,124 = 8.99) in a linear regression and explained 7% of its variance; individuals with the T allele have lower levels of IRT at birth. This coefficient was the denominator for the MR estimate.
In the larger genotyped CGS sample, the rs7512462 T allele conferred increased CFRD risk (sex-adjusted HR 1.19; P = 0.04). The loge(HR) from this analysis was the numerator in the MR. These 1,661 individuals are a subset from a previously reported SLC26A9–CFRD association (11).
The two-sample causal estimate of the HR for CFRD was 0.72 (95% CI 0.49–1.05) (Fig. 2 and Table 2), corresponding to a 28% decrease in the rate of CFRD for every one-unit increase in loge(IRT) at birth. Although the MR–HR CI slightly overlaps 1, its width is overestimated (18,24). This MR estimate is equivalent to the HR estimated directly from a regression of CFRD on loge(IRT) at birth, without adjustment for rate of loge(IRT) decline (sex-adjusted HR 0.72; P = 0.017). This agreement between the causal and the direct HR estimate, along with an absence of association between rs7512462 and other CFRD correlates (12), suggests that the direct measure of association between exocrine PD and CFRD is largely free of confounding.
Discussion
We demonstrate a robust, replicated association between exocrine PD and CFRD among CF patients with severe CFTR genotypes, providing evidence that lower early IRT is a biomarker for CFRD risk. Using the SLC26A9 CF modifier (12,20) in an MR framework, we provide evidence that this relationship is causal. CFRD is also associated with type 2 diabetes family history and susceptibility genes (11). Therefore, severe injury to the pancreatic islets by exocrine pancreatic inflammation and fibrosis, perhaps acting in concert with a genetic background that confers increased susceptibility to insulin resistance, may contribute to greater risk of CFRD.
Among other assumptions (Fig. 2), MR requires that rs7512462 is unrelated to CFRD other than through the effect of exocrine PD. SLC26A9 is expressed in the pancreas, lungs, and prostate (16), but replicated evidence suggests SLC26A9 is not associated with CF lung disease (12,13). This assumption further requires that rs7512462 is not pleiotropic for CFRD, but the relationship is mediated by proximity of the endocrine compartment [only 15% of the pancreas in neonates (25)] to damaged exocrine pancreas. The co-occurrence of SLC26A9 and CFTR in exocrine ducts is consistent with recognized direct (14) and functional (15) interactions and with the early evolving CF exocrine disease (26). As both SLC26A9 and CFTR proteins provide ion transport (chloride and bicarbonate), a straightforward interpretation is that SLC26A9 may augment ion transport when CFTR function is impaired, explaining its modifying feature. However, epithelial solute carriers such as SLC26A9 would not appear to have a direct role in endocrine compartments. Animal models can be insightful, provided cross-species distinctions in disease processes are known. A chronological picture of disease in the CF pig model is consistent with human observations, with exocrine disease initiating prior to birth (27). The pig pancreas is noted for similarity in a number of respects to the human pancreas and provides the source of digestive enzyme-replacement therapy for CF patients. In the less-studied CF ferret model, there are indications of abnormalities in blood glucose and insulin regulation shortly after birth, but dilated pancreatic ducts, intraluminal material, and apoptotic cells of exocrine tissue are present, also supporting exocrine disease prior to birth (28).
Untreated CFRD negatively affects nutritional status and pulmonary function and is associated with increased mortality (29). Our findings highlight the possibility of predicting CFRD susceptibility in early life using, among other factors, the widely available NBS IRT measurement. The added value of including IRT rate of decline (i.e., decline in pancreatic reserve) in assessing CFRD risk (R2 = 10.4 vs. 4.6%) suggests that CFRD is not determined by prenatal damage alone, and collecting additional measurements would improve prediction. The causal evidence between exocrine and endocrine pancreatic disease indicates that therapeutic interventions to promote ductal fluid flow in the exocrine pancreas could slow the progression of exocrine PD and perhaps delay onset of CFRD.
Article Information
Acknowledgments. The authors thank Drs. Andrew Paterson and Christine Bear for helpful discussions that contributed to this work and Dr. Mary Corey and Lynda Ellis for their contributions to the data resources.
Funding. This work was funded by the Canadian Institutes of Health Research (MOP-258916), Cystic Fibrosis Canada, and the United States Cystic Fibrosis Foundation (SONTAG07AO) and supported in part by the National Institutes of Health/National Center for Advancing Translational Sciences Colorado Clinical and Translational Sciences Institute (UL1-TR-000154).
Duality of Interest. No potential conflicts of interest relevant to this article were reported.
Author Contributions. D.S., J.M.R., P.R.D., and L.J.S. researched data; contributed to discussion; and wrote, reviewed, and edited the manuscript. M.R.M., W.L., J.G., L.S., and M.S. researched data and reviewed and edited the manuscript. K.K. and W.I. researched data and provided administrative and technical support. F.A. reviewed and edited the manuscript and provided administrative and technical support. L.J.S. is the guarantor of this work and, as such, had full access to all the data in the study and takes responsibility for the integrity of the data and the accuracy of the data analysis.
Footnotes
The contents of this article are the authors’ sole responsibility and do not necessarily represent official National Institutes of Health views.
References
- 1.Waters DL, Dorney SF, Gaskin KJ, Gruca MA, O’Halloran M, Wilcken B. Pancreatic function in infants identified as having cystic fibrosis in a neonatal screening program. N Engl J Med 1990;322:303–308 [DOI] [PubMed] [Google Scholar]
- 2.Durie PR, Forstner GG, Gaskin KJ, et al. Age-related alterations of immunoreactive pancreatic cationic trypsinogen in sera from cystic fibrosis patients with and without pancreatic insufficiency. Pediatr Res 1986;20:209–213 [DOI] [PubMed] [Google Scholar]
- 3.Ahmed N, Corey M, Forstner G, et al. Molecular consequences of cystic fibrosis transmembrane regulator (CFTR) gene mutations in the exocrine pancreas. Gut 2003;52:1159–1164 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lebenthal E, Lee PC. Development of functional responses in human exocrine pancreas. Pediatrics 1980;66:556–560 [PubMed] [Google Scholar]
- 5.Imrie JR, Fagan DG, Sturgess JM. Quantitative evaluation of the development of the exocrine pancreas in cystic fibrosis and control infants. Am J Pathol 1979;95:697–708 [PMC free article] [PubMed] [Google Scholar]
- 6.Oppenhelmer EH, Esterley JR. Pathology of cystic fibrosis: review of the literature and comparison with 146 autopsied cases. In Perspectives in Pediatric Pathology. 2nd ed. Rosenberg HS, Bolande RP, Eds. Chicago, Year Book Medical Publishers, 1976, p. 241–278 [PubMed] [Google Scholar]
- 7.Iannucci A, Mukai K, Johnson D, Burke B. Endocrine pancreas in cystic fibrosis: an immunohistochemical study. Hum Pathol 1984;15:278–284 [DOI] [PubMed] [Google Scholar]
- 8.Sontag MK, Corey M, Hokanson JE, et al. Genetic and physiologic correlates of longitudinal immunoreactive trypsinogen decline in infants with cystic fibrosis identified through newborn screening. J Pediatr 2006;149:650–657 [DOI] [PubMed] [Google Scholar]
- 9.Moran A, Dunitz J, Nathan B, Saeed A, Holme B, Thomas W. Cystic fibrosis-related diabetes: current trends in prevalence, incidence, and mortality. Diabetes Care 2009;32:1626–1631 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Marshall BC, Butler SM, Stoddard M, Moran AM, Liou TG, Morgan WJ. Epidemiology of cystic fibrosis-related diabetes. J Pediatr 2005;146:681–687 [DOI] [PubMed] [Google Scholar]
- 11.Blackman SM, Commander CW, Watson C, et al. Genetic modifiers of cystic fibrosis-related diabetes. Diabetes 2013;62:3627–3635 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Li W, Soave D, Miller MR, et al. Unraveling the complex genetic model for cystic fibrosis: pleiotropic effects of modifier genes on early cystic fibrosis-related morbidities. Hum Genet 2014;133;151–161 [DOI] [PubMed] [Google Scholar]
- 13.Wright FA, Strug LJ, Doshi VK, et al. Genome-wide association and linkage identify modifier loci of lung disease severity in cystic fibrosis at 11p13 and 20q13.2. Nat Genet 2011;43:539–546 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chang MH, Plata C, Sindic A, et al. Slc26a9 is inhibited by the R-region of the cystic fibrosis transmembrane conductance regulator via the STAS domain. J Biol Chem 2009;284:28306–28318 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bertrand CA, Zhang R, Pilewski JM, Frizzell RA. SLC26A9 is a constitutively active, CFTR-regulated anion conductance in human bronchial epithelia. J Gen Physiol 2009;133:421–438 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lohi H, Kujala M, Makela S, et al. Functional characterization of three novel tissue-specific anion exchangers SLC26A7, -A8, and -A9. J Biol Chem 2002;277:14246–14254 [DOI] [PubMed] [Google Scholar]
- 17.Chang MH, Plata C, Zandi-Nejad K, et al. Slc26a9—anion exchanger, channel and Na+ transporter. J Membr Biol 2009;228:125–140 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Didelez V, Meng S, Sheehan NA. Assumptions of IV Methods for Observational Epidemiology. Stat Sci 2010;25:22–40 [Google Scholar]
- 19.Couper RT, Corey M, Durie PR, Forstner GG, Moore DJ. Longitudinal evaluation of serum trypsinogen measurement in pancreatic-insufficient and pancreatic-sufficient patients with cystic fibrosis. J Pediatr 1995;127:408–413 [DOI] [PubMed] [Google Scholar]
- 20.Sun L, Rommens JM, Corvol H, et al. Multiple apical plasma membrane constituents are associated with susceptibility to meconium ileus in individuals with cystic fibrosis. Nat Genet 2012;44:562–569 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Vaida F, Liu L. Fast Implementation for Normal Mixed Effects Models With Censored Response. J Comput Graph Stat 2009;18:797–817 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.R Core Development Team. R: A Language and Environment for Statistical Computing. R Foundation for Statistical Computing, Vienna, Austria, 2013. Available from http://www.R-project.org/ Accessed 21 February 2014
- 23.Angrist JD, Krueger AB. The Effect of Age at School Entry on Educational Attainment: An Application of Instrumental Variables with Moments from Two Samples. J Am Stat Assoc 1992;87:328–336 [Google Scholar]
- 24.Thomas DC, Lawlor DA, Thompson JR. Re: Estimation of bias in nongenetic observational studies using “Mendelian triangulation” by Bautista et al. Ann Epidemiol 2007;17:511–513 [DOI] [PubMed] [Google Scholar]
- 25.Rahier J, Wallon J, Henquin JC. Cell populations in the endocrine pancreas of human neonates and infants. Diabetologia 1981;20:540–546 [DOI] [PubMed] [Google Scholar]
- 26.Walkowiak J, Sands D, Nowakowska A, et al. Early decline of pancreatic function in cystic fibrosis patients with class 1 or 2 CFTR mutations. J Pediatr Gastroenterol Nutr 2005;40:199–201 [DOI] [PubMed] [Google Scholar]
- 27.Abu-El-Haija M, Ramachandran S, Meyerholz DK, et al. Pancreatic damage in fetal and newborn cystic fibrosis pigs involves the activation of inflammatory and remodeling pathways. Am J Pathol 2012;181:499–507 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Olivier AK, Yi Y, Sun X, et al. Abnormal endocrine pancreas function at birth in cystic fibrosis ferrets. J Clin Invest 2012;122:3755–3768 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Koch C, Rainisio M, Madessani U, et al. Investigators of the European Epidemiologic Registry of Cystic Fibrosis Presence of cystic fibrosis-related diabetes mellitus is tightly linked to poor lung function in patients with cystic fibrosis: data from the European Epidemiologic Registry of Cystic Fibrosis. Pediatr Pulmonol 2001;32:343–350 [DOI] [PubMed] [Google Scholar]