

Abstract
Generation of mitochondrial reactive oxygen species (ROS) can be perturbed following exposure to environmental chemicals such as 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD). Reports indicate that the aryl hydrocarbon receptor (AhR) mediates TCDD-induced sustained hepatic oxidative stress by decreasing hepatic ATP levels and through hyperpolarization of the inner mitochondrial membrane. To further elucidate the effects of TCDD on the mitochondria, high-throughput quantitative real-time PCR (HTP-QRTPCR) was used to evaluate the expression of 90 genes encoding mitochondrial proteins involved in electron transport, oxidative phosphorylation, uncoupling, and associated chaperones. HTP-QRTPCR analysis of time course (30 μg/kg TCDD at 2, 4, 8, 12, 18, 24, 72, and 168 hrs) liver samples obtained from orally gavaged immature, ovariectomized C57BL/6 mice identified 54 differentially expressed genes (|fold change|>1.5 and P-value <0.1). Of these, 8 exhibited a dose response (0.03 to 300 μg/kg TCDD) at 4, 24 or 72 hrs. Dose responsive genes encoded proteins associated with electron transport chain (ETC) complex I (NADH dehydrogenase), III (cytochrome c reductase), IV (cytochrome c oxidase), and V (ATP synthase) and could be generally categorized as having proton gradient, ATP synthesis, and chaperone activities. In contrast, transcript levels of ETC complex II, succinate dehydrogenase, remained unchanged. Putative dioxin response elements were computationally found in the promoter regions of the 8 dose-responsive genes. This high-throughput approach suggests that TCDD alters the expression of genes associated with mitochondrial function which may contribute to TCDD-elicited mitochondrial toxicity.
Keywords: TCDD, mitochondria, transcription, liver, mouse, QRTPCR
INTRODUCTION
2,3,7,8-Tetrachlorodibenzo-p-dioxin (TCDD) is a persistent environmental contaminant that elicits a wide range of toxic and biochemical responses including hepatotoxicity, enzyme induction, immunotoxicity, and lethality (Nebert et al., 2000; Poland and Knutson, 1982; Senft et al., 2002a). In addition, TCDD induces dose- and time-dependent hepatic oxidative stress characterized by increased levels of reactive oxygen species (ROS), DNA damage, lipid peroxidation, changes in oxidative enzyme activities and glutathione redox state (Bagchi et al., 2002; Kern et al., 2002; Shen et al., 2005; Stohs, 1990; Stohs et al., 1986). In C57BL/6 mice specifically, sustained hepatic oxidative stress elicited by TCDD results in decreased hepatic ATP levels, increases in mitochondrial respiration-dependent ROS production, mitochondrial aconitase, and glutathione levels, as well as hyperpolarization of the mitochondrial inner membrane (Shen et al., 2005; Shertzer et al., 2006; Shertzer et al., 1998).
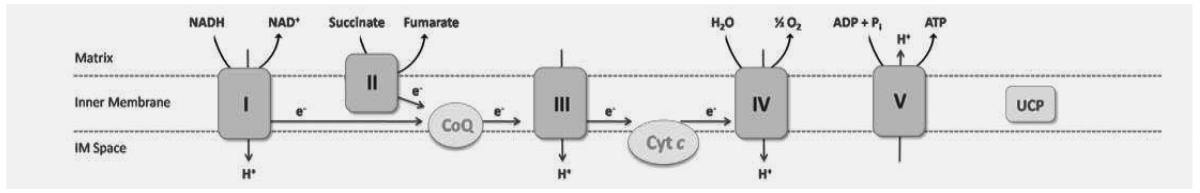
Although mitochondrial dysfunction is believed to contribute to TCDD-induced oxidative stress, the mechanisms involved are poorly understood. Collectively, the observed phenotypes suggest that the mitochondrial electron transport chain (ETC), may be a potential target. ROS byproducts from oxidative phosphorylation may also be a contributing factor as ETC complexes I (NADH dehydrogenase) and III (cytochrome c reductase) are major sites of ROS generation (McLennan and Degli Esposti, 2000). In total, complexes I and III consist of 40 and 10 genomic DNA encoded subunits, respectively. Moreover, five mitochondrial uncoupling proteins (UCPs) that can dissipate the proton gradient across the inner membrane to maintain redox balance, have also been implicated in reducing ROS production (Ledesma et al., 2002).
TCDD may also elicit changes in gene expression that disrupt electron flow through ETC complex III and cytochrome c oxidase (ETC complex IV) (Senft et al., 2002b). TCDD reportedly interferes with electron transfer at the oxidant side of cytochrome b which shuttles electrons to cytochrome c (Nohl et al., 1989). This would increase reduced cytochrome b and oxidized cytochrome c levels within complex III, increasing ROS generation. Furthermore, TCDD-induced increases in hydrogen peroxide may contribute to oxidative stress as a result of ETC complex IV inhibition and restrict electron flow leading to increases in reduced ETC complexes II (succinate dehydrogenase) and III (Senft et al., 2002a). However, decreases in complex IV activity do not account for the overall ROS production (Senft et al., 2002a). Alternatively, TCDD may produce highly reducing conditions that increase reduced glutathione levels and membrane hyperpolarization, resulting in increased mitochondrial ROS production (Shen et al., 2005).
The aryl hydrocarbon receptor (AhR) mediates the effects of TCDD by heterodimerizing with the aryl hydrocarbon receptor nuclear translocator (ARNT) upon ligand binding and nuclear translocation. This complex binds dioxin response elements (DREs) in regulatory regions to alter the expression of diverse genes including the well characterized “AhR gene battery” (Nebert et al., 2000). Despite the induction of Cyp1a1 and Cyp1a2 activity, ROS from these sources has not been implicated in mitochondrial damage (Senft et al., 2002b). Instead, mitochondrial toxicity is associated with respiration-dependent ROS production mediated by the AhR (Alsharif et al., 1994). C57BL/6 mice possessing high affinity AhR produce more ROS than the lower-affinity AhR containing DBA/2 mice, furthermore, AhR null mice exhibit less acute toxicity than wild-type mice (Alsharif et al., 1994; Senft et al., 2002b). TCDD-induced changes in glutathione levels, redox state, and DNA damage have also been shown to be AhR dependent (Burgoon and Zacharewski, 2008; Chan et al., 2004; Shen et al., 2005).
AhR dependency suggests that perturbations in gene expression may contribute to TCDD elicited mitochondrial toxicity. In this study, the expression of 90 genes associated with electron transport, oxidative phosphorylation, proton gradient uncoupling, and chaperone activities were examined. HTP-QRTPCR and dose-response studies identified several genes encoding mitochondrial proteins that were differentially regulated and may have a role in TCDD elicited mitochondrial toxicity.
MATERIALS AND METHODS
Animal husbandry
Female C57BL/6 mice, ovariectomized by the vendor on postnatal day (PND) 20, were acquired from Charles River Laboratories (Raleigh, NC) on PND 25. Animals were housed in polycarbonate cages containing cellulose fiber chips (Aspen Chip Laboratory Bedding, Northeastern Products, Warrensberg, NY) at 30-40% humidity in a 12 hrs (7AM – 7PM) light/dark cycle with free access to deionized water and Harlan Teklad 22/5 Rodent Diet 8640 (Madison, WI). Mice were allowed to acclimatize for 4 days prior to dosing. Animals were weighed and dosed with either TCDD (provided by S. Safe, Texas A&M University, College Station, TX) or sesame oil vehicle (Sigma, St. Louis, MO). For consistency this animal model was used for all the presented studies, and is typically used in our lab to facilitate comparisons with other studies (Boverhof et al., 2005). All procedures were performed with the approval of the Michigan State University Committee on Animals Use and Care.
Time course and dose-response studies
For the time course study, animals (n=5 per treatment group) were orally gavaged with 0.1 ml of sesame oil vehicle or 30 μg/kg body weight of TCDD. Mice were sacrificed at 2, 4, 8, 12, 18, 24, 72, or 168 hrs post dose. The dose was selected to elicit moderate hepatic effects while avoiding overt toxicity (Boverhof et al., 2005). Three separate dose-response studies (n=5 per treatment group) at 4, 24 and 72 h were independently conducted. Each animal was gavaged with 0.1 ml of sesame vehicle or 0.001 – 300 μg/kg TCDD and sacrificed by cervical dislocation. Tissue samples were removed, weighed, flash-frozen in liquid nitrogen, and stored at −80°C.
RNA isolation
Frozen liver samples (~100 mg of left lobe) were removed from −80°C storage and immediately placed into 1.0 ml of TRIzol (Invitrogen, Carlsbad, CA) and homogenized using a Mixer Mill 300 tissue homogenizer (Retsch, Germany). RNA was isolated according to the manufacturer’s protocol with an additional phenol:chloroform extraction, and re-suspended in RNA storage solution (Ambion Inc., Austin, TX). RNA was spectrophotometrically (A260) quantified and purity was assessed using the A260/A280 ratio and denaturing gel electrophoresis.
Quantitative Real-Time PCR (QRTPCR)
Total RNA (1 μg) was reverse transcribed by SuperScript II using an anchored oligo-dT primer as described by the manufacturer (Invitrogen, Carlsbad, CA). The resulting cDNA was used as template for HTP-QRTPCR (time course) and manual QRTPCR (dose response).
Primer pairs were designed to amplify 120-140bp fragments that cross intron/exon borders using Primer3 (http://frodo.wi.mit.edu/primer3/)(see Supplementary Table 1). Primer pairs were validated using in-silico PCR (http://genome.ucsc.edu/cgi-bin/hgPcr) and BLAST (http://blast.ncbi.nlm.nih.gov/Blast.cgi). In addition, primer pairs were further evaluated by PCR of cDNA where dissociation curve analysis to verify single product formation. Primers that failed were re-designed or the gene was not analyzed. In total, 96 genes encoding specific mitochondrial proteins (i.e., 40 ETC complex I, 4 ETC complex II, 10 ETC complex III, 15 ETC complex IV and 19 ETC complex V, 5 uncoupled protein (UCP) and 3 chaperone genes) were assayed. Additionally, 4 control genes (housekeeping and TCDD battery genes) were also examined (Table 1). Low fidelity primers resulted in multiple products for Ndufs6, Ndufv1, Atp5e, Atp5k, AtpSuGL, and Ucp3, thus these genes were not evaluated and only 90 genes are discussed.
Table 1.
Electron transport chain complexes and regulated genes
| |||||||
|---|---|---|---|---|---|---|---|
| Complex I NADH Dehydrogenase |
Complex II Succinate Dehydrogenase |
Complex III Cytochrome c Reductase |
Complex IV Cytochrome c Oxidase |
Complex V F0F1 ATPase |
UCP Uncoupling Protein |
Chaperone | |
|
| |||||||
| Mitochondrial genome encoded subunits |
7 | 0 | 1 | 3 | 2 | 0 | 0 |
| Nuclear genome encoded subunits |
40 | 4 | 10 | 15 | 19 | 5 | 3 |
|
| |||||||
| Number of differentially regulated genesa |
26 | - | 8 | 8 | 9 | 2 | 1 |
|
| |||||||
| Most temporally regulatedb |
Ndufb6 | - | Uqcrc1 | Cox4i1 | Atpaf1 | Ucp2 | Sco1 |
| (Fold changed)c | ▼ 9.1 | ▲ 3.2 | ▲ 4.0 | ▲ 5.3 | ▲ 3.1 | ▼ 1.64 | |
|
| |||||||
| Dose-responsived | Ndufa10 | - | Cyc1 | Cox7b | Atp5g3 | Ucp2 | Sco1 |
| Atp5l | Ucp5 | ||||||
|
| |||||||
Where |fold change| > 1.5 and P-value < 0.1 in the time course experiment
Gene exhibiting maximum temporal fold change across the time course experiment with P-value < 0.1 in the given ETC complex
Maximum temporal fold change across the time course experiment with P-value < 0.1
Gene exhibiting dose-response to TCDD in the given ETC complex
For HTP-QRTPCR time course samples (n=3), 10 μl reactions consisting of 3 μl diluted cDNA and 0.3 μM of forward and reverse gene-specific primers combined with 2× Power SYBR Green PCR Master Mix (Applied Biosystems, Foster City, CA) were aliquoted into 384-well plates using a Biomeck® 2000 Laboratory Automation Workstation (Beckman Coulter Inc., Fullerton, CA). Amplification was conducted on an Applied Biosystems PRISM 7900HT Sequence Detection System consisting of a 95°C denaturation stage for 10 min, then 40 repetitions of 95°C for 15 sec followed by 60°C for 1 min. Quantification was determined using the comparative CT method (ΔΔCT) (Applied Biosystems, Foster City, CA). The geometric mean of housekeeping gene (ActB, Gapdh, Hprt) expression was used to control for differences in RNA loading, quality and cDNA synthesis (Vandesompele et al., 2002). Relative expression levels were scaled such that fold change levels of time-matched vehicle control groups equaled one for graphing purposes.
For dose-response studies, QRTPCR was carried out manually rather than by high-throughput, and quantification was carried out using standards rather than ΔΔCT extrapolation. Briefly, genes identified using HTP-QRTPCR at 4, 24, and 72 hrs were evaluated in 30 μl QRTPCR reactions manually aliquoted into 96-well plates (manual QRTPCR), respectively. Reactions were carried out with master mix formulated to be as close as possible to that used for HTP-QRTPCR. Each reaction contained 1 μl of cDNA template, 0.1 μM forward and reverse gene-specific primers, 3 mM MgCl2, 1.0 mM dNTPs, 0.025 IU AmpliTaq Gold, and 1 × SYBR Green PCR Buffer (Applied Biosystems, Foster City, CA) and amplified using an Applied Biosystems PRISM 7500 Sequence Detection System. Results were quantified using a standard curve approach. Briefly, a standard curve was generated by using purified PCR product cDNA template specific for each gene/primer pair (serial 10× dilutions from 108 to 10 copies) as template for the QRTPCR reaction on the same 96well plate as the samples. The slope of the standard curve was used to assess amplification efficiency, as described by the manufacturer. All amplification efficiencies were 80-100%. Quantification of gene copy number for each sample was extrapolated from the generated standard curve, and fold changes were calculated. Sample standardization to the geometric mean of three housekeeping genes (ActB, Gapdh, Hprt) was used to control for differences in RNA loading, quality and cDNA synthesis (Vandesompele et al., 2002). Relative expression levels were scaled such that the expression level of the time-matched vehicle control group equaled one for graphing purposes. Select UCPs, chaperones and complex V genes were additionally evaluated beyond those identified by HTP-QRTPCR at 72 hrs.
Dose-response modeling
Our automated dose-response modeler was used to identify the best-fit dose-response model (Burgoon and Zacharewski, 2008). The algorithm uses particle swarm optimization to identify the best-fit model across five model classes (exponential, sigmoidal, Gaussian, linear, quadratic). A gene was considered dose responsive if its expression followed a sigmoidal or exponential curve. The best-fit model was used to calculate ED50 values.
Identification of Putative DREs
Putative DREs were computationally identified within the mouse genome (Build 37; obtained from the University of California, Santa Cruz Genome Browser) using an updated position weight matrix (Burgoon, 2009, in preparation; (Sun et al., 2004)). Briefly, select genes were scanned from 10,000 bp upstream of the transcriptional start site (TSS) to the end of the 3′-UTR to evaluate similarity to the position weight matrix. A similarity score of >0.80 was used to tentatively assign functionality, as previously described (Sun et al., 2004).
Statistical Analysis
HTP-QRTPCR data was analyzed using the Wilcoxon rank-sum test (R v2.6.2). Specifically, data were ranked and prioritized, rather than treated as hypothesis tests, due to Type I error rate inflation concerns from multiple tests. Differences with treatment were considered significant at time points where P-value < 0.1. Conversely, dose-response manual QRTPCR analysis was performed with SAS v9.1 (SAS Institute, Cary, NC) using analysis of variance followed by Dunnett’s post hoc test. Differences between treatment groups were considered significant when p < 0.05.
RESULTS
Temporal Gene Expression Profiling
Temporal expression profiles of 90 (as well as 6 controls for a total of 96) nuclear genes encoding mitochondrial proteins were analyzed by HTP-QRTPCR (Table 2, see Supplementary Table 2). Time course studies identified 54 differentially expressed genes (|fold change|>1.5 and P-value <0.1) following exposure to 30 μg/kg TCDD at one or more time points (Table 1, Table 3). Of these differentially expressed genes, 43 were induced and 11 were repressed. Twenty-six of the differentially regulated genes were associated with electron transport chain (ETC) complex I, NADH dehydrogenase, which facilitates the transfer of electrons from NADH to coenzyme Q and directly contributes to the formation of a proton gradient. The bias towards differentially regulated complex I genes is likely due to the large number of unique proteins that comprise complex I (~40% of all genes assayed). However, only 12 of the 54 differentially expressed genes exhibited AhR enrichment within −10 kb of their transcription start site (TSS) to the end of their 3′ untranslated region (UTR) in genome-wide chromatin immunoprecipitation (ChIP-chip) assays carried out at 24 hrs (J.B. Matthews et al., manuscript in preparation).
Table 2.
Time course identified differentially regulated genes elicited by 30μg/kg TCDD using HTP-QRTPCR
| RefSeq Accession |
Entrez Gene ID |
Complex | Gene Name | Gene Symbol |
Fold Changea | Time Pointb |
|---|---|---|---|---|---|---|
| NM_024197 | 67273 | Complex I | NADH dehydrogenase (ubiquinone) 1 alpha subcomplex 10 | Ndufa10 | ▲ 2.3 | 24, 168 |
| NM_027244 | 69875 | Complex I | NADH dehydrogenase (ubiquinone) 1 alpha subcomplex 11 | Ndufa11 | ▲ 2.4 | 2, 24 |
| NM_025551 | 66414 | Complex I | NADH dehydrogenase (ubiquinone) 1 alpha subcomplex 12 | Ndufa12 | ▲ 1.9 | 18, 24 |
| NM_001127346 | 75597 | Complex I | NADH dehydrogenase (ubiquinone) 1 alpha subcomplex, assembly factor 2 | Ndufa12l | ▲ 1.9 | 168 |
| NM_010886 | 17991 | Complex I | NADH dehydrogenase (ubiquinone) 1 alpha subcomplex 2 | Ndufa2 | ▲ 1.6 | 168 |
| NM_025348 | 66091 | Complex I | NADH dehydrogenase (ubiquinone) 1 alpha subcomplex 3 | Ndufa3 | ▲ 2.4 | 2, 12, 24 |
| NM_010886 | 17992 | Complex I | NADH dehydrogenase (ubiquinone) 1 alpha subcomplex 4 | Ndufa4 | ▲ 2.9 | 2, 4. 24 |
| NM_001098789 | 407790 | Complex I | NADH dehydrogenase (ubiquinone) 1 alpha subcomplex, 4-like 2 | Ndufa4l2 | ▲ 2.2 | 168 |
| NM_026614 | 68202 | Complex I | NADH dehydrogenase (ubiquinone) 1 alpha subcomplex 5 | Ndufa5 | ▼ 1.6 | 18 |
| NM_025987 | 67130 | Complex I | NADH dehydrogenase (ubiquinone) 1 alpha subcomplex 6 (B14) | Ndufa6 | ▼ 3.1 | 18 |
| NM_023202 | 66416 | Complex I | NADH dehydrogenase (ubiquinone) 1 alpha subcomplex 7 (B14.5a) | Ndufa7 | ▲ 1.8 | 72 |
| NM_026703 | 68375 | Complex I | NADH dehydrogenase (ubiquinone) 1 alpha subcomplex 8 | Ndufa8 | ▲ 4.8 | 8, 168 |
| NM_027175 | 69702 | Complex I | NADH dehydrogenase (ubiquinone) 1 alpha subcomplex, assembly factor 1 | Ndufaf1 | ▲ 2.0 | 2 |
| NM_026684 | 68342 | Complex I | NADH dehydrogenase (ubiquinone) 1 beta subcomplex 10 | Ndufb10 | ▲ 2.2 | 4 |
| NM_019435 | 104310 | Complex I | NADH dehydrogenase (ubiquinone) 1 beta subcomplex 11 | Ndufb11 | ▲ 2.1 | 168 |
| NM_026612 | 68198 | Complex I | NADH dehydrogenase (ubiquinone) 1 beta subcomplex 2 | Ndufb2 | ▲ 2.1 | 168 |
| NM_025597 | 66495 | Complex I | NADH dehydrogenase (ubiquinone) 1 beta subcomplex 3 | Ndufb3 | ▲ 2.7 | 24 |
| NM_001033305 | 230075 | Complex I | NADH dehydrogenase (ubiquinone) 1 beta subcomplex 6 | Ndufb6 | ▼ 9.1 | 2, 8, 24 |
| NM_025843 | 66916 | Complex I | NADH dehydrogenase (ubiquinone) 1 beta subcomplex 7 | Ndufb7 | ▲ 4.8 | 4 |
| NM_024220 | 68197 | Complex I | NADH dehydrogenase (ubiquinone) 1 subcomplex unknown 2 | Ndufc2 | ▲ 2.1 | 4 |
| NM_145518 | 227197 | Complex I | NADH dehydrogenase (ubiquinone) Fe-S protein 1 | Ndufs1 | ▲ 2.8 | 4, 168 |
| NM_153064 | 226646 | Complex I | NADH dehydrogenase (ubiquinone) Fe-S protein 2 | Ndufs2 | ▲ 2.1 | 4, 8 |
| M_026688 | 68349 | Complex I | NADH dehydrogenase (ubiquinone) Fe-S protein 3 | Ndufs3 | ▲ 2.5 | 2, 18, 168 |
| NM_144870 | 225887 | Complex I | NADH dehydrogenase (ubiquinone) Fe-S protein 8 | Ndufs8 | ▲ 2.7 | 72 |
| NM_028388 | 72900 | Complex I | NADH dehydrogenase (ubiquinone) flavoprotein 2 | Ndufv2 | ▼ 2.4 | 12, 18 |
| NM_030087 | 78330 | Complex I | NADH dehydrogenase (ubiquinone) flavoprotein 3 | Ndufv3 | ▲ 3.5 | 4, 18 |
| NM_025567 | 66445 | Complex III | Cytochrome c-1 | Cyc1 | ▲ 1.7 | 72 |
| NM_025710 | 66694 | Complex III | Ubiquinol-cytochrome c reductase, Rieske iron-sulfur polypeptide 1 | Rieske | ▲ 1.7 | 12 |
| NM_197979 | 66152 | Complex III | Ubiquinol-cytochrome c reductase (7.2 kD) subunit | Ucrc | ▼ 1.8 | 18 |
| NM_025650 | 66594 | Complex III | Ubiquinol-cytochrome c reductase (6.4kD) subunit | Uqcr | ▲ 1.9 | 72 |
| XM_484346 | 432822 | Complex III | Ubiquinol-cytochrome c reductase binding protein | Uqcrb | ▼ 1.6 | 18 |
| NM_025407 | 22273 | Complex III | Ubiquinol-cytochrome c reductase core protein 1 | Uqcrc1 | ▲ 3.2 | 168 |
| NM_025899 | 67003 | Complex III | Ubiquinol-cytochrome c reductase core protein 2 | Uqcrc2 | ▲ 2.7 | 4, 24, 72 |
| NM_025352 | 22272 | Complex III | Ubiquinol-cytochrome c reductase, complex III subunit VII | Uqcrq | ▼ ▲1.7 | 4, 18 |
| NM_009941 | 12857 | Complex IV | Cytochrome c oxidase subunit IV isoform 1 | Cox4i1 | ▲4.0 | 72 |
| NM_053091 | 84682 | Complex IV | Cytochrome c oxidase subunit IV isoform 2 | Cox4i2 | ▲ 1.7 | 168 |
| NM_009942 | 12859 | Complex IV | Cytochrome c oxidase, subunit Vb | Cox5b | ▲ 1.6 | 2 |
| NM_009943 | 12862 | Complex IV | Cytochrome c oxidase, subunit Via, polypeptide 2 | Cox6a2 | ▼ 2.3 | 8, 18 |
| NM_025628 | 110323 | Complex IV | Cytochrome c oxidase, subunit Vib polypeptide 1 | Cox6b1 | ▲ 3.1 | 2, 72, 168 |
| NM_053071 | 12864 | Complex IV | Cytochrome c oxidase, subunit Vic | Cox6c | ▲ 2.7 | 4, 168 |
| NM_009945 | 12866 | Complex IV | Cytochrome c oxidase, subunit VIIa 2 | Cox7a2 | ▲ 2.1 | 72 |
| NM_025379 | 66142 | Complex IV | Cytochrome c oxidase, subunit VIIb | Cox7b | ▲ 2.1 | 72 |
| NM_181040 | 230649 | Complex V | ATP synthase mitochondrial F1 complex assembly factor 1 | Atpaf1 | ▲ 5.3 | 2, 24 |
| NM_145427 | 246782 | Complex V | ATP synthase mitochondrial F1 complex assembly factor 2 | Atpaf2 | ▼ 2.3 | 12 |
| NM_016774 | 11947 | Complex V | ATP synthase, H+ transporting, mitochondrial F1 complex, beta subunit | Atp5b | ▼ 2.1 | 8, 168 |
| NM_025313 | 66043 | Complex V | ATP synthase, H+ transporting, mitochondrial F1 complex, delta subunit | Atp5d | ▲ 2.0 | 2 |
| NM_009725 | 11950 | Complex V | ATP synthase, H+ transporting, mitochondrial F0 complex, subunit b, isoform 1 | Atp5f1 | ▲ 2.5 | 8, 168 |
| NM_026468 | 67942 | Complex V | ATP synthase, H+ transporting, mitochondrial F0 complex, subunit c (subunit 9), isoform 2 | Atp5g2 | ▲ 1.6 | 72 |
| NM_175015 | 228033 | Complex V | ATP synthase, H+ transporting, mitochondrial F0 complex, subunit c (subunit 9), isoform 3 | Atp5g3 | ▲ 2.0 | 72 |
| NM_016755 | 11957 | Complex V | ATP synthase, H+ transporting, mitochondrial F0 complex, subunit f | Atp5j | ▼ 1.7 | 8 |
| NM_013795 | 27425 | Complex V | ATP synthase, H+ transporting, mitochondrial F0 complex, subunit g | Atp5l | ▼ 1.8 | 18, 168 |
| NM_011671 | 22228 | UCP | Uncoupling protein 2 (mitochondrial, proton carrier) | Ucp2 | ▲ 3.1 | 4 |
| NM_028711 | 74011 | UCP | Uncoupling protein 4 (mitochondrial solute carrier) | Ucp4 | ▲ 2.2 | 168 |
| NM_001040026 | 52892 | Chaperone | SCO cytochrome oxidase deficient homolog 1 (respiratory chain complex IV assembly) | Sco1 | ▲ 1.7 | 4 |
Time points with maximum fold change and P-value < 0.1
| fold changej > 1.5 and P-value < 0.1
Table 3.
Temporal distribution of differentially regulated genes after 3 μg/kg TCDD exposure
| Time point (hrs) |
Number of Differentially Regulated Genes |
Primary Complex Altered | Genes Meeting Cut-offs |
|---|---|---|---|
| 2 | 10 | Complex I | Ndufal 1, Ndufa3, Ndufa4, Ndufafl, Ndufb6, Ndufs3, Cox5b, Cox6b1, Atpaf1, Atp5d |
| 4 | 10 | Complex I | Ndufa4, Ndufb10, Ndufb7, Ndufs1, Ndufs2, Ndufv3, Uqcrc2, Uqcrq, Cox6c, Ucp2 |
| 8 | 7 | Complex I and V | Ndufa8, Ndufb6, Ndufs2, Cox6a2, Atp5b, Atp5f1, Atp5j |
| 12 | 4 | Complex I | Ndufa3, Ndufv2, Rieske, Atpaf2 |
| 18 | 12 | Complex I | Ndufa12, Ndufa5, Ndufa6, Ndufs3, Ndufv2, Ndufv3, Ucrc, Uqcrb, Uqcrq, Cox6a2, Atp5l, Sco1 |
| 24 | 10 | Complex I | Ndufa10, Ndufa11, Ndufa12, Ndufa3, Ndufa4, Ndufb3, Ndufb6, Ndufc2, Uqcrc2, Atpaf1 |
| 72 | 11 | Complex IV | Ndufa7, Ndufs8, Cyc1, Uqcr, Uqcrc2, Cox4i1, Cox6b1, Cox7a2, Cox7b, Atp5g2, Atp5g3 |
| 168 | 17 | Complex I | Ndufa10, Ndufa12l, Ndufa2, Ndufa4l2, Ndufa8, Ndufb11, Ndufb2, Ndufs1, Ndufs3, Uqcrc1, Cox4i2, Cox6b1, Cox6c, Atp5b, Atp5f1, Atp5l, Ucp4 |
Note: genes included meet statistical (P-value < 0.1) and fold change (fold change| > 1.5) cut-offs. Primary complex altered indicates the complex most represented at that time point.
Dose-Response Modeling
Time points for subsequent dose response studies were selected based on a review of the time course data (Table 2, Table 3). Three independent dose-response studies were conducted at 4, 24, and 72 hrs using the same animal model and experimental design as the time course study. Cyp1a1 was examined in all studies as a positive control, and exhibited dose-dependent induction at each time point (Figure 1). However, dose-dependent induction at 24 hrs exhibited a sigmoidal response while the curves for 4 and 72 hrs were exponential. This may be partially due to differences in TCDD tissue levels that are still accumulating at 4 hrs and are beginning to decrease by 72 hrs (Boverhof et al., 2005). Manual QRTPCR data for dose-response evaluation at 4, 24, and 72 hr time points are available in supplementary tables 3, 4 and 5, respectively.
Figure 1.
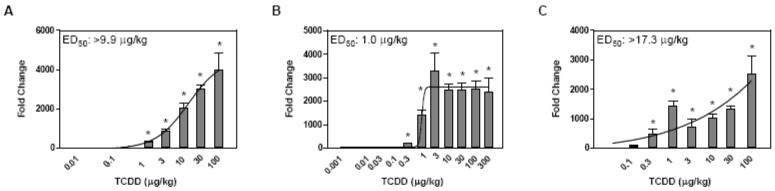
Manual QRTPCR evaluation of Cyp1a1 dose-responsiveness at 4 (A), 24 (B) and 72 (C) hrs after exposure to TCDD. The x-axis indicates the dose of TCDD, the y-axis represents the fold change calculated relative to vehicle controls. Curves were generated in GraphPad Prism 4.0 using non-linear regression; ED50 values were calculated using an automated dose-response modeler (Burgoon and Zacharewski, 2008). The asterisk (*) indicates significant fold induction by TCDD relative to vehicle controls at p < 0.05
Temporally responsive genes were classified as ‘confirmed’ when |fold change|>1.5 and statistical p-value < 0.05 in the dose-response study conducted using manual QRTPCR. Although the 10 dysregulated genes at 4 hrs did not exhibit a sigmoidal or exponential dose response, all were confirmed with induced expression between 2- to 6-fold. Ucp2 and Ndufa10 (complex I) exhibited dose-responsive induction at 24 hrs with ED50 values of 2.6 and 38.2 μg/kg, respectively (Figure 2). Of the remaining genes identified at 24 hrs, all were confirmed as differentially regulated but not dose responsive with the exception of Ndufb3 and Ndufc2 which did not meet the statistical cut-off. Ndufa11 is a gene confirmed as differentially regulated but not as dose responsive, it was significantly induced at 24 hrs in the HTP-QRTPCR time course study but did not exhibit a dose response at 24 hrs that could be modeled (Figure 3). Of the 11 differentially regulated genes identified at 72 hrs using HTP-QRTPCR, all were confirmed using manual QRTPCR and 3 (Cyc1, Cox7b and Atp5g3) exhibited sigmoidal dose response relationships (Figure 4). Sco1, Atp5l and Ucp5 were also dose-responsive at 72 hrs, but were not among the 11 differentially regulated genes identified by HTP-QRTPCR. Automated dose-response modeling yielded ED50 values for all dose response genes ranging from 3.2 μg/kg for Atp5l to >46.1 μg/kg for Cyc1 which exhibited an exponential dose response relationship.
Figure 2.

Dose response evaluation of genes at 24 hrs by manual QRTPCR. The x-axis indicates the dose of TCDD, the y-axis represents the fold change calculated relative to time-matched vehicle controls (n=4). Curves were generated in GraphPad Prism 4.0 using non-linear regression; ED50 values were determined using an automated dose-response modeler (Burgoon and Zacharewski, 2008). The asterisk (*) indicates significant fold induction by TCDD relative to vehicle controls at p < 0.05
Figure 3.

Evaluation of Ndufa11, an ETC complex I gene. HTP-QRTPCR temporal expression profile (A) and dose-response (B) evaluation at 24 hrs by manual QRTPCR. The x-axis indicates the time or dose of TCDD, respectively, while the y-axis represents the fold change calculated relative to time-matched vehicle controls (n=4). The asterisk (*) indicates significant fold induction by TCDD relative to vehicle controls at p < 0.05
Figure 4.

72 hr dose response evaluation using manual QRTPCR. The x-axis indicates the dose of TCDD, the y-axis represents the fold change calculated relative to time-matched vehicle controls (n=5). Curves were generated in GraphPad Prism 4.0 using non-linear regression; ED50 values were obtained using an automated dose-response modeler (Burgoon and Zacharewski, 2008). The asterisk (*) indicates significant fold induction by TCDD relative to vehicle controls at p < 0.05
Putative Dioxin Response Element (DRE) Identification
The genomic sequences of the 8 dose-responsive genes (Ndufa10, Cyc1, Cox7b, Atp5g3, Atp5l, Ucp2, Ucp5, Sco2) were computationally scanned to identify putative DREs located within 10,000 bp upstream of the transcriptional start site (TSS) to the end of the 3′-UTR. A matrix similarity score of >0.80 was used to tentatively assign functionality, as previously described (Sun et al., 2004). Each dose-responsive gene possessed several putative DREs. However, only 3 of the 8 confirmed dose-responsive genes exhibited AhR enrichment within −10 kb of their TSS to the end of their 3′ UTR in genome-wide chromatin immunoprecipitation (ChIP-chip) assays (J.B. Matthews et al., manuscript in preparation). The highest incidence of DREs was located within 10kb upstream of the TSS (Table 4). In contrast, the incidence of DREs within the open reading frames and the untranslated regions were much lower, consistent with functional DREs tending to reside within −1,500 bp of the TSS (Sun et al., 2004).
Table 4.
DRE distribution within the genomic sequence of dose-responsive genes
| Gene | ETC Complex | 5-10kb upstream of TSS |
Within 5Kb of TSS |
In Introns |
In Exons |
In 5′ UTR |
In 3′ UTR |
|---|---|---|---|---|---|---|---|
| Ndufa10 | Complex I | 1 | 1 | 6 | 0 | 0 | 0 |
| Cyc1 | Complex III | 3 | 5 | 2 | 0 | 1 | 0 |
| Cox7b | Complex IV | 2 | 6 | 0 | 1 | 0 | 0 |
| Atp5g3 | Complex V | 1 | 1 | 1 | 0 | 0 | 0 |
| Atp5l | Complex V | 5 | 1 | 0 | 0 | 0 | 0 |
| Ucp2 | Uncoupled Protein | 4 | 2 | 3 | 0 | 0 | 0 |
| Ucp5 | Uncoupled Protein | 0 | 2 | 4 | 1 | 0 | 1 |
| Sco1 | Chaperone | 6 | 3 | 8 | 0 | 0 | 0 |
Note: Identified DREs must have a similarity score >0.8 as identified in UCSC mouse build 37
DISCUSSION
This study is the first targeted comprehensive evaluation of the effect of TCDD on the expression of nuclear encoded mitochondrial electron transport and oxidative phosphorylation associated genes by HTP-QRTPCR. Responses exhibited both time- and dose-dependent expression patterns, in a gene-specific manner. This is consistent with TCDD eliciting a distinct hepatic gene expression profile that included induction, repression, early, mid, and late responses, many of which could be directly associated with the resulting phenotypes (e.g. fatty accumulation, immune cell infiltration) (Boverhof et al., 2005). In all, the temporal expression of 90 mitochondrial associated genes was examined using HTP-QRTPCR. Fifty four (54) mitochondrial genes were differentially expressed in response to TCDD, of which 8 exhibited sigmoidal or exponential dose-dependent pattern of expression at 4, 24 or 72 hrs. The ED50 values obtained for the 8 identified genes ranged from 0.01 – 100 μg/kg. Meanwhile the ED50 for Cyp1a1 mRNA induction was comparable to previous reports using manual QRTPCR (Boverhof et al., 2005).
Although HTP-QRTPCR was used to rapidly examine gene expression, differences in assay conditions and sensitivity as well as hepatic TCDD levels likely contributed to the identification of the limited number of dose responsive genes using manual QRTPCR. Moreover, ChIP-chip data suggests that these may be secondary responses that do not involve direct interactions between AhR and DREs (J.B. Matthews et al, manuscript in preparation). Although, TCDD altered the expression of ETC subunit mRNA levels in complexes I, III, IV and V as well as UCPs and chaperone, no complex II subunit mRNA levels were altered, consistent with TCDD having no impact on its activity (Shertzer et al., 2006).
Previous studies suggest that membrane potential may regulate mitochondrial ROS production (McLennan and Degli Esposti, 2000). This is highly influenced by UCPs which mediate proton leakage to reduce the inner membrane potential (Ledesma et al., 2002). TCDD elicited temporal and dose-dependent differential expression of UCP2 and UCP5. UCP2 is highly expressed in Kupffer cells, but not in healthy hepatocytes (Fulop et al., 2006). However, UCP2 is highly expressed in fatty liver hepatocytes (Fulop et al., 2006; Grav et al., 2003). TCDD (30 μg/kg) induces fatty accumulation in C57BL/6 mouse liver (Boverhof et al., 2005). Fatty liver, as well as increased ROS production and lipid peroxidation are all manifestations of TCDD-induced hepatotoxicity (Alsharif et al., 1990; Boverhof et al., 2005; Burgoon and Zacharewski, 2008; Stohs, 1990). UCP2 has also been implicated in moderating hydrogen peroxide release, as well as lipid peroxidation, while regulating oxidative stress, all of which further correlate with TCDD-induced hepatotoxicity (Demori et al., 2008; Negre-Salvayre et al., 1997). Although no effect on hepatic UCP2 protein levels were reported following 5 μg/kg TCDD treatment in C57BL/6J mice (Shertzer et al., 2006), our results suggest UCP2 mRNA expression increases over time and is dose-responsive. It is possible that in TCDD-induced fatty liver, UCP2 is activated to reduce the hyperpolarization of the inner mitochondrial membrane to modulate hydrogen peroxide generation (Fulop et al., 2006; Negre-Salvayre et al., 1997). In fact, TCDD depolarizes the inner mitochondrial membrane in primary rat hepatocytes, treated with 15nM TCDD for 72 hrs (Aly and Domenech, 2009). Ultimately this could attenuate membrane potential and alter ATP synthase (ETC complex V) activity resulting in reduced ATP production (Shertzer et al., 2006). Interestingly, the ATP synthase genes, Atp5g3 and Atp5l, also exhibited dose-dependent induction by TCDD, further suggesting dysregulation of complex V. Together, the induction of UCP2 as well as ATP synthase components suggest potential mechanisms contributing to TCDD-induced mitochondrial toxicity.
TCDD also altered the expression of subunits of complexes I, III and IV which may further disrupt mitochondrial function. Complex I (NADH dehydrogenase) has been implicated in ROS production, more specifically in superoxide production and lipid peroxidation (Lenaz, 2001; McLennan and Degli Esposti, 2000). Ndufa10, a component of complex I, exhibited dose-dependent induction by TCDD at 24 hrs, and could affect complex I activity. Increased respiration in response to TCDD could alter membrane potential as well as account for the observed increase in ROS production (Shertzer et al., 2006). However, complex III is the rate limiting step in electron flow, though membrane fluidity and complex IV and V activity may also affect respiration (Shertzer et al., 2006). Cytochrome c-1 (Cyc1), a soluble electron carrier that is reduced at complex III and is oxidized by complex IV, exhibited dose-dependent induction. Increases in Cyc1 could contribute to excessive electron transfer to complex IV, and increase mitochondrial respiration. Consequently, both complex III and IV associated transcripts and their activities could be differentially regulated by TCDD. In addition to Cyc1 (categorized as complex III in this study), the chaperone Sco1, which is involved in complex IV assembly, as well as Cox7b, a complex IV component, were both dose-responsive. Collectively, these results suggest that TCDD may affect mitochondrial respiration by altering subunit expression and subsequent complex activity.
Finally, putative DREs (pDREs) were identified for all genes showing dose-dependent induction by TCDD. While the expected occurrence of the DRE core consensus sequence (GCGTG) would be every 512 bp within both strands of the genome if randomly distributed, pDREs are typically located proximal (within −1,500 bp) to the TSS in TCDD-inducible genes such as the members of the “AhR gene battery” (Nebert et al., 2000; Sun et al., 2004). Moreover, pDREs occur less frequently in coding and distal regions of the known TCDD-regulated genes (Sun et al., 2004). The pDREs computationally identified for Cyc1, Cox7b, Atp5g3, Atp5l and Ucp2 (Table 4) were, in general, proximal to the TSS, suggesting potential regulation by AhR.
In summary, 8 nuclear encoded mitochondrial genes involved in electron transport and oxidative phosphorylation exhibited TCDD-dependent differential regulation. The dysregulation of these genes may directly contribute to oxidative stress and subsequent TCDD elicited mitochondrial toxicity. Temporal and dose-dependent expression patterns as well as the computational identification of putative DREs proximal to the TSS suggest AhR-mediated regulation. However, it is possible that the differential expression of some identified genes may be secondary responses to oxidative stress. Our data suggest that the oxidative stress and subsequent mitochondrial toxicity may involve multiple interactions that affect several respiratory pathway targets. Additional studies are warranted to assess the effects of TCDD on protein expression and activity in order to further elucidate the role of these targets in TCDD-elicited oxidative stress and mitochondrial toxicity.
Supplementary Material
ACKNOWLEDGEMENTS
Special thanks to Anna Kopec and Edward Dere for the critical reading of the manuscript. This work was funded by Superfund grant NIEHS SBRP P42ES04911.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- Alsharif NZ, Grandjean CJ, Murray WJ, Stohs SJ. 2,3,7,8-Tetrachlorodibenzo-p-dioxin (TCDD)-induced decrease in the fluidity of rat liver membranes. Xenobiotica. 1990;20:979–988. doi: 10.3109/00498259009046913. [DOI] [PubMed] [Google Scholar]
- Alsharif NZ, Lawson T, Stohs SJ. Oxidative stress induced by 2,3,7,8-tetrachlorodibenzo-p-dioxin is mediated by the aryl hydrocarbon (Ah) receptor complex. Toxicology. 1994;92:39–51. doi: 10.1016/0300-483x(94)90166-x. [DOI] [PubMed] [Google Scholar]
- Aly HA, Domenech O. Cytotoxicity and mitochondrial dysfunction of 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD) in isolated rat hepatocytes. Toxicol Lett. 2009 doi: 10.1016/j.toxlet.2009.08.008. [DOI] [PubMed] [Google Scholar]
- Bagchi D, Balmoori J, Bagchi M, Ye X, Williams CB, Stohs SJ. Comparative effects of TCDD, endrin, naphthalene and chromium (VI) on oxidative stress and tissue damage in the liver and brain tissues of mice. Toxicology. 2002;175:73–82. doi: 10.1016/s0300-483x(02)00062-8. [DOI] [PubMed] [Google Scholar]
- Boverhof DR, Burgoon LD, Tashiro C, Chittim B, Harkema JR, Jump DB, Zacharewski TR. Temporal and dose-dependent hepatic gene expression patterns in mice provide new insights into TCDD-Mediated hepatotoxicity. Toxicol Sci. 2005;85:1048–1063. doi: 10.1093/toxsci/kfi162. [DOI] [PubMed] [Google Scholar]
- Burgoon LD, Zacharewski TR. Automated quantitative dose-response modeling and point of departure determination for large toxicogenomic and high-throughput screening data sets. Toxicol Sci. 2008;104:412–418. doi: 10.1093/toxsci/kfn083. [DOI] [PubMed] [Google Scholar]
- Chan CY, Kim PM, Winn LM. TCDD-induced homologous recombination: the role of the Ah receptor versus oxidative DNA damage. Mutat Res. 2004;563:71–79. doi: 10.1016/j.mrgentox.2004.05.015. [DOI] [PubMed] [Google Scholar]
- Demori I, Burlando B, Gerdoni E, Lanni A, Fugassa E, Voci A. Uncoupling protein-2 induction in rat hepatocytes after acute carbon tetrachloride liver injury. J Cell Physiol. 2008;216:413–418. doi: 10.1002/jcp.21415. [DOI] [PubMed] [Google Scholar]
- Fulop P, Derdak Z, Sheets A, Sabo E, Berthiaume EP, Resnick MB, Wands JR, Paragh G, Baffy G. Lack of UCP2 reduces Fas-mediated liver injury in ob/ob mice and reveals importance of cell-specific UCP2 expression. Hepatology. 2006;44:592–601. doi: 10.1002/hep.21310. [DOI] [PubMed] [Google Scholar]
- Grav HJ, Tronstad KJ, Gudbrandsen OA, Berge K, Fladmark KE, Martinsen TC, Waldum H, Wergedahl H, Berge RK. Changed energy state and increased mitochondrial beta-oxidation rate in liver of rats associated with lowered proton electrochemical potential and stimulated uncoupling protein 2 (UCP-2) expression: evidence for peroxisome proliferator-activated receptor-alpha independent induction of UCP-2 expression. J Biol Chem. 2003;278:30525–30533. doi: 10.1074/jbc.M303382200. [DOI] [PubMed] [Google Scholar]
- Kern PA, Fishman RB, Song W, Brown AD, Fonseca V. The effect of 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD) on oxidative enzymes in adipocytes and liver. Toxicology. 2002;171:117–125. doi: 10.1016/s0300-483x(01)00564-9. [DOI] [PubMed] [Google Scholar]
- Ledesma A, de Lacoba MG, Rial E. The mitochondrial uncoupling proteins. Genome Biol. 2002:3. doi: 10.1186/gb-2002-3-12-reviews3015. REVIEWS3015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lenaz G. The mitochondrial production of reactive oxygen species: mechanisms and implications in human pathology. IUBMB Life. 2001;52:159–164. doi: 10.1080/15216540152845957. [DOI] [PubMed] [Google Scholar]
- McLennan HR, Degli Esposti M. The contribution of mitochondrial respiratory complexes to the production of reactive oxygen species. J Bioenerg Biomembr. 2000;32:153–162. doi: 10.1023/a:1005507913372. [DOI] [PubMed] [Google Scholar]
- Nebert DW, Roe AL, Dieter MZ, Solis WA, Yang Y, Dalton TP. Role of the aromatic hydrocarbon receptor and [Ah] gene battery in the oxidative stress response, cell cycle control, and apoptosis. Biochem Pharmacol. 2000;59:65–85. doi: 10.1016/s0006-2952(99)00310-x. [DOI] [PubMed] [Google Scholar]
- Negre-Salvayre A, Hirtz C, Carrera G, Cazenave R, Troly M, Salvayre R, Penicaud L, Casteilla L. A role for uncoupling protein-2 as a regulator of mitochondrial hydrogen peroxide generation. FASEB J. 1997;11:809–815. [PubMed] [Google Scholar]
- Nohl H, de Silva D, Summer KH. 2,3,7,8, tetrachlorodibenzo-p-dioxin induces oxygen activation associated with cell respiration. Free Radic Biol Med. 1989;6:369–374. doi: 10.1016/0891-5849(89)90081-6. [DOI] [PubMed] [Google Scholar]
- Poland A, Knutson JC. 2,3,7,8-tetrachlorodibenzo-p-dioxin and related halogenated aromatic hydrocarbons: examination of the mechanism of toxicity. Annu Rev Pharmacol Toxicol. 1982;22:517–554. doi: 10.1146/annurev.pa.22.040182.002505. [DOI] [PubMed] [Google Scholar]
- Senft AP, Dalton TP, Nebert DW, Genter MB, Hutchinson RJ, Shertzer HG. Dioxin increases reactive oxygen production in mouse liver mitochondria. Toxicol Appl Pharmacol. 2002a;178:15–21. doi: 10.1006/taap.2001.9314. [DOI] [PubMed] [Google Scholar]
- Senft AP, Dalton TP, Nebert DW, Genter MB, Puga A, Hutchinson RJ, Kerzee JK, Uno S, Shertzer HG. Mitochondrial reactive oxygen production is dependent on the aromatic hydrocarbon receptor. Free Radic Biol Med. 2002b;33:1268–1278. doi: 10.1016/s0891-5849(02)01014-6. [DOI] [PubMed] [Google Scholar]
- Shen D, Dalton TP, Nebert DW, Shertzer HG. Glutathione redox state regulates mitochondrial reactive oxygen production. J Biol Chem. 2005;280:25305–25312. doi: 10.1074/jbc.M500095200. [DOI] [PubMed] [Google Scholar]
- Shertzer HG, Genter MB, Shen D, Nebert DW, Chen Y, Dalton TP. TCDD decreases ATP levels and increases reactive oxygen production through changes in mitochondrial F(0)F(1)-ATP synthase and ubiquinone. Toxicol Appl Pharmacol. 2006;217:363–374. doi: 10.1016/j.taap.2006.09.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shertzer HG, Nebert DW, Puga A, Ary M, Sonntag D, Dixon K, Robinson LJ, Cianciolo E, Dalton TP. Dioxin causes a sustained oxidative stress response in the mouse. Biochem Biophys Res Commun. 1998;253:44–48. doi: 10.1006/bbrc.1998.9753. [DOI] [PubMed] [Google Scholar]
- Stohs SJ. Oxidative stress induced by 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD) Free Radic Biol Med. 1990;9:79–90. doi: 10.1016/0891-5849(90)90052-k. [DOI] [PubMed] [Google Scholar]
- Stohs SJ, Al-Bayati ZF, Hassan MQ, Murray WJ, Mohammadpour HA. Glutathione peroxidase and reactive oxygen species in TCDD-induced lipid peroxidation. Adv Exp Med Biol. 1986;197:357–365. doi: 10.1007/978-1-4684-5134-4_33. [DOI] [PubMed] [Google Scholar]
- Sun YV, Boverhof DR, Burgoon LD, Fielden MR, Zacharewski TR. Comparative analysis of dioxin response elements in human, mouse and rat genomic sequences. Nucleic Acids Res. 2004;32:4512–4523. doi: 10.1093/nar/gkh782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vandesompele J, De Preter K, Pattyn F, Poppe B, Van Roy N, De Paepe A, Speleman F. Accurate normalization of real-time quantitative RT-PCR data by geometric averaging of multiple internal control genes. Genome Biol. 2002:3. doi: 10.1186/gb-2002-3-7-research0034. RESEARCH0034. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.