

Abstract
Catalysis and synthesis are intimately linked in modern organic chemistry. The synthesis of complex molecules is an ever evolving area of science. In many regards, the inherent beauty associated with a synthetic sequence can be linked to a certain combination of the creativity with which a sequence is designed and the overall efficiency with which the ultimate process is performed. In synthesis, as in other endeavors, beauty is very much in the eyes of the beholder.[**] It is with this in mind that we will attempt to review an area of synthesis that has fascinated us and that we find extraordinarily beautiful, namely the combination of catalysis and sigmatropic rearrangements in consecutive and cascade sequences.
Keywords: homogeneous catalysis, sigmatropic reactions, tandem reactions
1. Introduction
The development of tandem processes has had a profound impact on organic synthesis and synthetic planning. Combining multiple reactions in a series affords highly functionalized products with minimal handling. Likewise, with the growing body of catalytic reactions and their importance for the synthesis of asymmetric intermediates, it is understandable that catalysis is a broad area of focus in the 21st century. Although the coupling of highly efficient catalytic transformations to other processes, particularly those that are thermally mediated, could be expected to be a key motif for accessing highly complicated substances with exquisite control and efficiency, this line of research is in its relative infancy. In this review, we cover cascade reactivity in the context of catalytic transformations that are coupled to bond rearrangements, specifically sigmatropic processes. We will discuss ensembles of reactions involving the combination of catalysis and sigmatropic rearrangements, including Claisen, Cope, [2,3], [1,2], [1,3], and [1,5] rearrangements. It is our aim that this analysis will afford future researchers the opportunity to build on such cascades for further development of more selective and powerful transformations.
1.1. Defining and Classifying Cascades
1.1.1. Defining Tandem Reactions and Reaction Ensembles
Chemical reactivity is difficult to define and classify. Classifications of tandem reactions become even more cumbersome because of the fact that nearly every chemical reaction comprises multiple elementary steps. Such combinations of elementary steps are generally not categorized as tandem or cascade reactions, and are simply referred to as the mechanism of a particular process. Alternatively, combining multiple reactions leads to the concept of tandem reactivity. For our purposes, we will adhere to the definitions put forth by Tietze, such as the overarching “sequential” or “one-pot” reaction.[1] This includes “domino” reactions (also known as tandem or cascade) whereby “subsequent reactions result as a consequence of the functionality formed by bond formation or fragmentation in the previous step.” “Consecutive” processes would be ones where additional reagents are added without isolation of the first formed product. One caveat that we would like to add to the literature involves more precisely defining these processes based on the number of discrete reactions involved. In fact, the need for such a classification system arose in one of our own publications involving a cascade of three reactions. We initially named this a triple tandem reaction. This descriptor unfortunately implies three sequential tandem events, or six reactions in total, which is incorrect.
After a number of discussions with one of our esteemed colleagues, the now late Professor Nelson Leonard, we developed a more precise terminology. We propose to use the descriptors duet, trio, quartet, quintet, etc. for defining tandem events involving 2, 3, 4, 5, etc. reactions in a reaction ensemble. Ultimately, this naming system leads one to envision entire symphonies of reactions occurring in con- trolled, multi-reaction cascades. Moreover, the beauty and creativity associated with music allows one to draw a more subtle analogy between music and synthesis as well as art and science. The classification herein is in contrast to “concerto catalysis,” which seeks to improve traditional metallic catalysts by uniting them with all scales of catalytic assemblies, from large biological macromolecules to small molecules, an endeavor which would require and encourage cross-disciplinary thinking.[2] Both concepts appeal to the musical sense of orchestration in chemical synthesis, and illustrate the goal of chemists to attain more exquisite control over chemical processes.
1.1.2. Defining Catalysis
Chemical catalysis can be defined as the utilization of a reagent for the purpose of enhancing the rate of a reaction without altering the makeup of the reagent. This definition is sufficiently broad to encompass catalysts used in both small (substoichiometric) and large (stoichiometric and superstoichiometric) quantities as well as relatively complicated ([Ir-(PCy3)3]+BPh4-) and simple (H+) catalysts. Because of the potentially huge numbers of reactions that could fall under this definition, we have necessarily been somewhat selective in terms of what is included in this review. This is not a comprehensive review; rather it is our intention to provide illustrative examples that may ultimately provide the reader with not only breadth in the area, but enough depth to push the science beyond what is currently possible.
1.1.3. Defining Sigmatropic Rearrangements
Woodward and Hoffmann defined a sigmatropic rearrangement of order [i,j] to be “the migration of a s bond, flanked by one or more π-electron systems, to a new position whose termini are i–1 and j–1 atoms removed from the original bonded loci, in an uncatalyzed, intramolecular process.”[3] For the purposes of this review, we will use a somewhat more loose definition based solely on the transformation and the bonds formed and broken. The strict uncatalyzed, unimolecular, and concerted definition of Woodward and Hoffmann are simply too constricting for this topic. Moreover, we have deliberately limited this review to sigmatropic processes and not attempted to review all pericyclic processes involved in catalyzed tandem reactions. There are excellent reviews written specifically on topics dealing broadly with tandem and/or pericyclic reactions.[4] It was our belief that including such areas in the review would be an unnecessary repetition of these topics, and the examples involving sigmatropic rearrangements (of which there are significantly fewer) would not stand out. By incorporating these two relatively simple strategies (catalysis and sigma-tropic rearrangements) in cascade sequences, an incredible diversity of transformations is possible. As a result, the material described herein does not always fall neatly into subcategories, and multiple transformations presented in one section are connected to transformations presented elsewhere. Figure 1 provides a visual overview to our organization; reactions are grouped according to major “themes” to the extent that we could find them. In particular, incorporation of the Claisen rearrangement into tandem sequences showed the broadest versatility and diversity, a clear outcome of the many known variants of the Claisen rearrangement. Rather than being a drawback, we hope the broad coverage will inspire further discovery by highlighting the multiple connections possible between various methodologies, and providing a framework for thinking about new ways to combine multiple processes into one.
Figure 1.

Graphical overview.
1.1.4. Classification: Duets, Trios, Quartets, and Higher
The review is primarily classified by the type of sigma-tropic process. Within each group, the examples are subdivided by the first sigmatropic rearrangement that occurs in that cascade. Although it would be illustrative to organize according to increasing complexity, we found the discussions to be too fragmented. Within each section, reactions are thus named according to the number of discrete events occurring in a cascade sequence (i.e. duets (2), trios (3), quartets (4), etc.). The duet constitutes the simplest musical group beyond a soloist, but allows for nearly unlimited binary combinations of musicians. Likewise a chemical duet is the simplest, and by far the most common, type of tandem reaction. As in music, the chemical interplay of the two voices of reactivity leads to a combination that is greater than the sum of the individuals. Even more impressive is the choreographing of additional chemical processes, each with their own selectivity, into trios and quartets.
In many cases, particularly when mechanistic information is deficient, it can be difficult to quantify the discrete number of steps. A number of judgements are required. For example, do tautomerizations count as discrete steps? Do reversible processes to form intermediates at low concentration merit tallying? We have attempted to make those judgments according to context and have used our best discretion when selecting the most noteworthy examples. The inclusion of reactions that are chemically catalytic without being operationally catalytic, or those that are consecutive (multiple reagent additions) rather than the more elegant and advantageous tandem, is intended to reveal both the advances and shortcomings of attempts to incorporate multiple (catalytic) transformations under one given set of conditions (often not yet possible). The true testiment to our ability to “orchestrate” will be to have all musical partners present at the outset – with all the requirements for reagent selectivity and compatability that would entail.[*]
2. [3,3]-Sigmatropic Rearrangements
2.1. Cope Rearrangement
Although it is the prototypical example of a [3,3]-sigma-tropic rearrangement, the all-carbon Cope rearrangement was discovered after its oxygenated counterpart, the Claisen rearrangement. Found by Cope in 1940[5] and subsequently developed further, the simplest example (the rearrangement of 1,5-cyclohexadiene, 1) is a degenerate reversible process that can be studied only by labelling and/or substitution experiments. The reversibility of the Cope rearrangement presents a challenge for its use in a synthetic or practical sense, as well as for the development of asymmetric variants.[6] Different strategies have been employed to drive the rearrangement toward a desired product. For example, the thermal rearrangement disclosed in Cope s original report (2→3, Scheme 1), involves a significant thermodynamic driving force from the introduction of conjugative stabilization. Introduction of aromaticity either directly or through tautomerization is another way to favor Cope-rearrangement products.[7] Two other widely used strategies are the strain-release Cope and oxy-Cope rearrangements, each of which are discussed further below. A number of good reviews of catalysis in the Cope and Claisen rearrangements have been published.[8] Most of the examples included herein do not necessarily involve catalysis of the Cope rearrangement step.
Scheme 1.
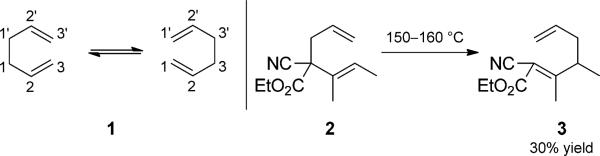
Cope rearrangement. [Cope, 1940, Ref. [5]]
2.1.1. Strain-Release Cope Rearrangement
Introduction of a cyclopropane or cyclobutane is a very commonly used strategy for rendering a Cope rearrangement irreversible. Cis-divinyl cyclopropranes cannot often be isolated because they rearrange too quickly, although mechanistic studies by Brown et al. were successful in measuring the free energy of activation for the rearrangement of the parent divinyl cyclopropane in CFCl3 (ΔG† = 20.6 kcalmol 1, for example, t½ = 90 s at 35°C).[9] The facility with which the Cope rearrangement occurs in these systems makes it a prime candidate for incorporation into tandem reaction sequences.
Ever since Davies and co-workers[10] first reported the rhodium-catalyzed cyclopropanation/Cope rearrangement duet, the strategy has been used to great effect in a number of total syntheses to generate seven-membered rings. The use of enantiomerically pure rhodium catalysts that render the reaction asymmetric has been of particular impact. To date, these are the most prevalent examples of catalyst control in asymmetric all-carbon Cope rearrangements.[6b,11] A recent example from the Davies group in the context of natural product total synthesis is shown in Scheme 2. Asymmetric cyclopropanation of triene 4 followed by Cope rearrangement, with transfer of chirality generated cycloheptadiene 7 in good yield and high enantiomeric excess. This intermediate was further elaborated to the natural product (–)-5-epivibsanin, 8.[10c]
Scheme 2.

Rhodium-catalyzed asymmetric cyclopropanation/Cope rearrangement duet. [Davies, 2009, Ref. [10c]]
One of the authors of this review employed an enantio-selective cyclopropanation/Cope rearrangement duet in a “parallel kinetic resolution” to prepare enantiomerically enriched diastereomers from racemic starting material (Scheme 3).[12] When a racemic mixture of diene 10 was treated with a vinyl diazoester and an enantiomerically pure dirhodium tetraprolinate catalyst, diastereomeric cycloheptadienes 13 and 14 were isolated as a 1:1 mixture, each with enantiomeric ratios of 88:12. The moderate e.r. values indicate imperfect facial selectivity of the catalyst. The resolution of racemic 10 could thus be used to prepare tricylic cores of the cyanthiwigin and the cyathane diterpenes, each enriched in the naturally occurring enantiomer.
Scheme 3.

Parallel kinetic resolution using a cyclopropanation/Cope rearrangement duet. Dosp=N-[(4-dodecylphenyl)sulfonyl] prolinate. [Sarpong, 2009, Ref. [12]]
Padwa and co-workers explored extensively the isomerization of α-diazo ketones that bear tethered alkyne units (Scheme 4).[13] Thus, diazoketone 17 isomerized to rhodium carbenoid 18 upon treatment with catalytic rhodium(II) mandelate. A cyclopropanation occurs with the closer of two double bonds. The E configuration is necessary, as it alone can furnish the reactive cis-divinyl cyclopropane 19. Cope rearrangement terminated the reaction trio to generate cycloheptadiene 20.
Scheme 4.

Rhodium-catalyzed alkyne metathesis/cyclopropanation/ Cope rearrangement trio. [Padwa, 1991, Ref. [13a]]
Montgomery and Ni showed that nickel can catalyze the coupling of enynes with diazocompounds in a related enyne cylization reaction (Scheme 5).[14] The details of the mechanism are not known, but model and mechanistic studies support the intermediacy of cyclopropanes such as 24. Without a clear mechanistic understanding of the process it is difficult to characterize according to the number of reactions, however, prior experimental observation and understanding is most consistent with an alkyne metathesis/ cyclopropanation/Cope rearrangement trio.
Scheme 5.

Nickel-catalyzed diazo/alkyne coupling and cyclopropanation/Cope rearrangement trio. [Montgomery, 2006, Ref. [14]]
Tandem cyclopropanation/Cope rearrangement sequences have relied on other transformations beyond [2+1] addition of carbenes to alkenes. Enyne isomerizations with electrophilic metal catalysts can also be used for the in situ generation of divinyl cylclopropanes (Scheme 6).[15] For example, treatment of dienyne 26 with catalytic PtCl2 led to isolation of the bicylic cycloheptadiene 28,[15a] while treatment of trienyne 29 with catalytic [W(CO)6] led to isolation of bicyclic cycloheptadiene 31.[15b]
Scheme 6.

Enyne cycloisomerization/Cope rearrangement duets postulated to involve divinylcyclopropane intermediates. (a) [Chung, 2010, Ref. [15a]] (b) [Iwasawa, 2006, Ref. [15b]]
The mechanistic details of the two reactions may diverge slightly, but according to current thinking, each process is initiated by electrophilic activation of the alkyne followed by nucleophilic attack of the pendant alkene (A, Scheme 7).[16] Nucleophilic closure by vinyl metal species B can then be envisioned to generate the metal carbene C. Hydride shift and loss of metal would then lead to cyclopropane D. Cope rearrangement can feasibly occur at this stage. In the case of platinum catalysis, divinyl cylopropane intermediates can be isolated and have been shown to undergo the postulated Cope rearrangement under thermal conditions, suggesting that metal loss is fast in C.[15a] Intermediates were not isolated in the tungsten-mediated reaction, thus preventing conlusions about when demetallation occurs. The nature of the intermediates shown in Scheme 7 has been the subject of a fascinating discussion along the lines of the historical debate regarding non-classical ion intermediates.[17]
Scheme 7.

Proposed mechanism for cyclopropane generation from enynes.
Two authors of this review expanded the strain-release Cope rearrangement to ketene substrates and developed a Wolff/Cope rearrangement duet.[18] Sonication of diazo ketone 32 with a catalytic amount of AgOBz and a stoichio-metric amount of Et3N at 45°C led to the desired cycloheptadienone 34 in excellent yield (Scheme 8). Under photo-lytic conditions, the success of the reaction was mixed. Prolonged exposure to light ultimately led to vinyl cyclopentenone 35. The intermediacy of 34 was confirmed and suggests a subsequent Norrish type I fragmentation/recombination as the final step in a Wolff/Cope/1,3-acyl-shift rearrangement trio.
Scheme 8.

Wolff/Cope rearrangement duet and isomerization trio. [Stoltz, 2003, Ref. [18]]
The diminished ring strain in cyclobutanes, compared to cyclopropanes, makes them less reactive but competent substrates for strain-release Cope rearrangements. In the synthesis of the sesquiterpene lactone (+)-asteriscanolide, Snapper and co-workers utilized a cyclobutene ring-opening metathesis/Cope rearrangement duet (Scheme 9).[19] Although precedent was poor for selective ring-opening metathesis of a strained trisubstituted olefin, treatment of olefin 36 with Grubbs II catalyst under optimized conditions produced cyclooctadiene 38 in excellent yield. In an earlier report, related divinyl cyclobutanes underwent Cope rearrangement only at significantly higher temperatures (200°C). The lower propensity of those substrates to undergo a Cope rearrangement was attributed to substitution at C8, which would disfavor the required parallel arrangement of the two vinyl groups, as in 37.
Scheme 9.

Ruthenium-catalyzed metathesis/Cope rearrangement duet. [Snapper, 2000, Ref. [19]]
Kanematsu and co-workers reported a [2+2] cycloaddition/[3,3] rearrangement sequence of an allenyl ether, which constituted a unique example of the cyclobutane strain-release Cope rearrangement (Scheme 10).[20] The transformation was initiated by base-catalyzed isomerization of enantioenriched propargyl ether 40 to allenyl ether 41, which could then engage in a [2+2] cycloaddition. The resultant methylene cyclobutane 42 subsequently underwent Cope rearrangement to generate cyclic ether 43, a potential precursor to taxane derivatives, in quantitative yield.
Scheme 10.

[2+2] Cycloaddition/[3,3] rearrangement trio initiated by base-catalyzed isomerization of a propargyl ether to an allenyl ether. [Kanematsu, 1995, Ref. [20]]
2.1.2. Oxy-Cope Rearrangements
In 1964, Jones and Berson demonstrated that particular Cope rearrangement products could be favored by incorporating an oxygen substituent at position 1 (Scheme 1).[21] Such substrates did not only rearrange “irreversibly” as a result of keto–enol tautomerization, but a kinetic effect was also observed.[21b] Furthermore, Evans found that rate enhancements were even more significant for anionic alkoxides.[21c]
Under thermal conditions, tandem pericyclic processes were combined with oxy-Cope rearrangements in complex cascade sequences to great success.[22] Although a deprotonation would fit within the concept of catalysis, even if the base were used stoichiometrically, we have excluded clearly irreversible examples that utilize bases (e.g. NaH, nBuLi), unless such bases are used substoichiometrically. We use a similar constraint to limit the number of examples discussed throughout this review.
The anionic oxy-Cope reaction was first invoked to explain a base-catalyzed dienol rearrangement that could also be explained by a fragmentation–recombination process, specifically a retro-aldol/Michael addition.[8] For example, Tice and Heathcock reported divergent base-mediated rearrangements of enone 44 (Scheme 11).[23] With KOH in aqueous methanol, dicarbonyl compound 47 was isolated. With excess KH in THF, dicarbonyl compound 51 was isolated. If a catalytic amount of KH was used, both compounds were isolated in a 1:1 ratio. Both rearrangements were proposed to proceed by retro-aldol reaction to provide enolate 45. Cyclization could then produce epimeric alkoxide 46 and isomeric alkoxide 48, which are both prone to undergo oxy-Cope rearrangement. In the case of 46, enolate protonation terminated the base-catalyzed rearrangement trio to give bicycle 47. In the case of 48, the resultant enolate underwent isomerization and Michael addition to form 51, thus terminating a rearrangement quartet. As mentioned above, such rearrangements need not necessarily invoke an anionic oxy-Cope reaction; direct Michael reactions of 45 constitute a similarly plausible mechanism. We include these reactions in the current review with that caveat.
Scheme 11.

Base-catalyzed oxy-Cope sequences; a retro-aldol/aldol/ oxy-Cope trio and oxy-Cope/Michael quartet. [Heathcock, 1981, Ref. [23]]
Another example of a similar catalyzed tandem anionic oxy-Cope rearrangement was reported by Rajagopalan et al. and involved allenic system 52, a previously unexplored type of substrate for the anionic oxy-Cope reaction (Scheme 12).[24] Treatment with s substoichiometric amount of sodium ethoxide gave solely the product of anionic oxy-Cope rearrangement, dienone 53, which formed aldol product 54 upon purification on alumina. Use of a stoichiometric amount of base effected the tandem sequence, oxy-Cope rearrangement and transannular cyclization, to give the bicyclic exo-methylene compound 54. The allenic oxy-Cope systems were shown to react faster than simple vinyl counterparts; this was attributed to allenic strain, which reduces the strength of the π bond involved in the rearrangement.
Scheme 12.

Base-mediated anionic oxy-Cope/aldol reaction duet. [Rajagopalan, 1993, Ref. [24]]
In the course of synthetic studies toward the sesquiterpene gnididione, Jacobi and co-workers observed a duet rearrangement/spirocyclization of diynyl carbinols (Scheme 13).[25] The sequence was initiated by thermal oxy-Cope rearrangement to enynone 55. tert-Butyl catechol (TBC) then catalyzed a radical cyclization to form cyclopentenone 57. Mechanistic studies supported cyclization following single-electron reduction of the enol tautomer of 56.
Scheme 13.
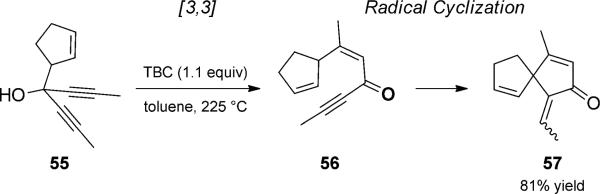
Phenol-catalyzed electrocylization preceded by oxy-Cope rearrangement. [Jacobi, 1994, Ref. [25]]
In their work on the preparation of polycylic structures, Huq and co-workers observed a unique dianionic aromatic oxy-Cope rearrangement trio (Scheme 14).[26] This was the first report of a dianionic oxy-Cope rearrangement engaging two benzene π bonds as part of a 1,5-hexadiene system.
Scheme 14.

Base-catalyzed dianionic oxy-Cope rearrangement/aldol/ oxidation trio. [Huq, 2006, Ref. [26]]
Although pentacyclic diols, such as 61, were the expected products, in the case of 58 an unexpected rearrangement trio also occurred. The dianionic oxy-Cope rearrangement led to dienolate 60, which then underwent single tautomerization, partial proton quenching, and aldol cyclization to give hexacycle 62. Air oxidation rearomatized one of the phenyl rings to provide 63 in good yield.
In a very elegant use of strain release in a tandem oxy-Cope sequence, Leighton and co-workers developed a method that allows the rapid preparation of the core of phomoidrides, a series of compounds that are squalene synthase and Ras farnesyl transferase inhibitors.[27] Originally discovered by Pfizer, they are also known as CP molecules. Although there was good precedent for an originally proposed simple anionic oxy-Cope approach, the harsh reaction conditions indicated an unfavorable process that was highly sensitive to even small changes in substrate structure. In order to promote the rearrangement, they decided to introduce an additional strain-inducing feature.
Specifically, an oxy-Cope precursor that already contained the pseudoester ring system of 67 was proposed to have a strained and twisted exo-methylene group more prone to engage in a [3,3] rearrangement (Scheme 15). To generate such a strained tricyclic system, they envisioned a novel carbonylation reaction starting from enol triflate 64. In practice, carbonylation with [Pd(PPh3)4] gave lactone 67 in good yield. The reaction was proposed to proceed by oxidative addition and CO insertion followed by intramolecular trapping of putative palladium-acyl 65 by the pendant alcohol. Studies on a model system showed that either triflate stereoisomer could be used, implicating a palladium-mediated isomerization. The high complexity of this process was thus more remarkable by potentially being stereoconvergent.
Scheme 15.

Novel carbonylation strain-release Cope rearrangement duet in the synthesis of phomoidrides. [Leighton, 2003, Ref. [27a]]
Bode and co-workers reported an asymmetric annulation that was catalyzed by an N-heterocyclic carbene (NHC) and proposed to proceed through a unique oxy-Cope rearrangement (Scheme 16).[28a] The proposed mechanism involves a crossed benzoin reaction between cinnamaldehyde (70) and conjugated ester 69 to generate alkoxide 71, which is poised for a [3,3] rearrangement. Tautomerization of enol 72 followed by aldol addition would then generate isomeric alkoxide 73. Acylation and elimination of the NHC catalyst led to a β-lactone, which then underwent a [2+2] cyclo-reversion to form CO2 and the observed cyclopentene 74. They also reported a related reaction involving enals and unsaturated N-sulfonyl ketimines to form cyclopentane-fused β-lactams.[28b]
Scheme 16.

Asymmetric Benzoin-type coupling/cyclization/decarboxylation quartet through oxy-Cope rearrangement catalyzed by an N-heterocyclic carbene. [Bode, 2007, Ref. [28a]]
2.1.3. Miscellaneous Cope Rearrangements
In an elegant total synthesis of strychnine, Kuehne and coworkers developed a complex Pictet-Spengler/Cope rearrangement trio to generate key tetracyclic intermediate 81 (Scheme 17).[29] Studies using p-anisaldehyde, with which Cope rearrangement is not possible, indicate that although spiroindolene 78 can and likely does form as a mixture of four diastereomers, only one adopts a conformation favorable for Cope rearrangement. Thus, prolonged heating funnelled the reaction through a single reactive diastereomer. Use of tryptophan-derived 75 resulted in formation of tetracycle 81 as a single enantiomer. This is a fine example of internal asymmetric induction.[6b]
Scheme 17.

Diastereoselective Pictet–Spengler/Cope rearrangement trio used in a total synthesis of strychnine. [Kuehne, 1997, Ref. [29]]
Tunge et al. reported a palladium-catalyzed decarboxylative C(sp3)—C(sp3) coupling reaction followed by palladium-catalyzed Cope rearrangement (Scheme 18).[30] To effect decarboxylative coupling, β-ketoesters or alkylidene malononitriles, such as 83, were treated with catalytic [Pd(PPh3)4]. In all cases examined, the predominant products were the aallylated products (e.g. 84) rather than the conjugated gallylated products (e.g. 85), indicating kinetic control in the reaction. When the conditions were altered to include higher temperatures, a decarboxylative coupling/Cope rearrangement duet was observed that allowed access to the thermodynamic γ-allylation products. In the presence of the Trost ligand 82, high diastereoselectivity and moderate enantioselectivity were enforced.
Scheme 18.

Palladium-catalyzed decarboxylative coupling/Cope rearrangement duet. [Tunge, 2006, Ref. [30]]
In an uncommon example of Brønsted acid catalysis in a tandem Cope sequence, Dauben and Chollet observed an enol hydrolysis/Cope rearrangement duet (Scheme 19).[31]
Scheme 19.

Acid-catalyzed Cope rearrangement duet. [Dauben, 1981, Ref. [31]]
Seeking enone 87, they were surprised to isolate instead enone 88. The facile nature of the rearrangement suggested that acid catalysis was operational. Comparative studies on related 2-acyl-1,5-dienes gave credence to that proposal, as Cope rearrangement in the absence of acid occurred only upon heating.
In another unexpected Cope sequence, Krohn and Bernhard sought to synthesize α-hydroxy ketones en route to angucycline antibiotics (Scheme 20).[32] Instead, treatment of allyl naphthols, such as 89, with catalytic [Zr(acac)4] and excess tBuOOH led to the isolation of o-quinones such as 92. Initial hydroxylation was followed by oxy-Cope rearrangement and subsequent oxidation. Substrates with simple alkyl substituents that cannot rearrange resulted in the desired ahydroxy ketones.
Scheme 20.

Zirconium-catalyzed oxidation oxy-Cope rearrangement trio. [Krohn, 1996, Ref. [32]]
2.2. Claisen Rearrangement
The Claisen rearrangement, a heteroatom variant of the Cope rearrangement, was first discovered by Ludwig Claisen in 1912 (Scheme 21).[33] The heteroatom of the Claisen rearrangement provides a much broader array of reactions
Scheme 21.
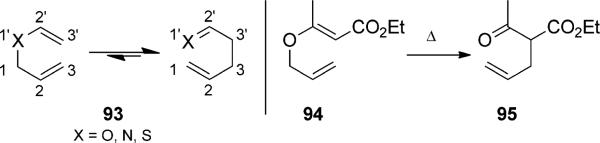
Claisen rearrangement. [Claisen, 1912, Ref. [33]]
that can be combined into a tandem sequence. It also presents a functional-group handle that makes catalytic and asymmetric variants more feasible. Although the Claisen rearrangement is not strictly irreversible, reaction equilibria tend toward carbonyl-type products, thus also increasing its practicality in synthesis. A number of excellent reviews on the Claisen rearrangement have appeared.[34] Catalytic tandem processes involving Claisen rearrangement can be organized loosely into the following types: tandem pericylic processes, coupling and isomerization, and cyclization reactions.
2.2.1. Tandem Pericyclic Processes
The most conceptually simple tandem Claisen rearrangement is a duet involving two sequential [3,3] rearrangements. A powerful example of this on an industrial scale is exemplified by the BASF Citral process, in which acetal 96 reacts with an acid catalyst to generate diene 97 (Scheme 22).[35] Subsequent Claisen and Cope rearrangement efficiently give the important fragrant molecule 99.
Scheme 22.

BASF continuous Citral process including a Claisen/Cope rearrangement duet. [1981, Ref. [35]]
A europium(III)-catalyzed tandem Claisen/Cope rearrangement was reported by Metz and co-workers (Scheme 23).[36] In their synthesis of the flavanoid 8-prenylnaringenin (103), they found that the europium-catalyzed tandem sequence presented a much more efficient route to preparing material needed for biological testing than the corresponding thermal process. Additionally, the Claisen/ Cope rearrangement duet was a more selective alternative to previous preparative methods, which suffered from low yields and/or relied on C prenylation. They found that 103 as well as the de-acetylated version of 101 both displayed strong estrogenic activities. Related thermal aromatic Claisen/ Diels–Alder cascades have been used by Nicolaou and Theodorakis to prepare complex polycyclic scaffolds in the syntheses of Garcinia natural products.[37]
Scheme 23.

Claisen/Cope rearrangement duet. [Metz, 1991, Ref. [36]]
Tandem Claisen rearrangements have found great utility in the preparation of butenylidene-bridged aromatic compounds such as 107 for further use in the synthesis of rotaxanes, helicates, and other compounds that are structurally relevant to supramolecular chemistry (Scheme 24). Most commonly, the tandem rearrangement is performed under thermal conditions; however, Hiratani and co-workers reported a Lewis acid catalyzed variant that proceeded under milder conditions.[38] Thus, treatment of allyl phenyl ether 104 with EtAlCl2 at room temperature in the presence of 2-methyl-2-butene resulted in iterative tandem Claisen rearrangements. Intermediates 105 and 106 are depicted to highlight the process, even though the two sequences operate independently of each other.
Scheme 24.

Iterative Claisen rearrangement duets. [Hiratani, 1998, Ref. [38]]
In their formal synthesis of (–)-morphine, Chida and co-workers used iterative Claisen rearrangements to set the stereochemistry of the vicinal tertiary and quaternary carbon atoms of morphine in a single transformation (Scheme 25).[39] Their strategy utilized the chiral pool by synthesizing enantiomerically pure trans-diol 109 from d-glucal. Treatment with catalytic 2-nitrophenol and ethyl orthoacetate at 140°C results in sequential Claisen rearrangements with good chirality transfer in moderate yield.
Scheme 25.

Iterative acid-catalyzed Johnson–Claisen duets. [Chida, 2008, Ref. [39]]
In some cases, cyclic enol ethers, presumed to arise from inverse hetero-Diels–Alder (IHDA) cycloadditions, have been shown to arise from a Diels–Alder/retro-Claisen rear- rangement duet (Scheme 26). In their studies of unsaturated phosphonate reactivity, Hanessian and Compain observed Lewis acid sensitive ratios of the classic Diels–Alder adducts (analogous to 116) and the IHDA product 114.[40] In work reported by Davies and co-workers,[41] treatment of a,bunsaturated aldehydes such as 115 resulted in isolation of a minor amount of Diels–Alder cycloaddition product 116 and a significant amount of the tandem DA/Claisen rearrangement product 117. They determined, however, that direct IHDA competes under certain conditions. In both cases, isolation of the initial adduct and treatment under Lewis acidic conditions were used to support the tandem mechanism.
Scheme 26.

Lewis acid catalyzed Diels–Alder/retro-Claisen rearrangement duets appear as inverse hetero-Diels–Alder reaction. a) [Hanes-sian, 2002, Ref. [40]]; b) [Davies, 2005, Ref. [41]]
In their efforts to rapidly and efficiently access skeletons of iboga-type alkaloids, Neier and co-workers developed a Lewis acid catalyzed tandem Diels–Alder reaction involving thiocyanic acid ester 118 (Scheme 27).[42] Following a methanol/triethylamine quench, isothiocyanate 121 was isolated, which results from a [3,3]-sigmatropic rearrangement of the cyclohexadiene adduct.
Scheme 27.

Lewis acid catalyzed Diels–Alder “hetero-Claisen” rearrangement duet. [Neier, 1994, Ref. [42]]
The first report of a tandem Johnson–Claisen rearrangement/Diels–Alder reaction was by Mulzer and co-workers (Scheme 28).[43] The distribution of products was sensitive to olefin substitution and the ring size of the product. The most selective transformation was that of allylic alcohol 122, which upon treatment with ethyl orthoacetate and catalytic propionic acid underwent a reaction trio.
Scheme 28.

Acid-catalyzed Johnson–Claisen/Diels–Alder cycloaddition trio. [Mulzer, 1991, Ref. [43]]
Indeed, methods combining the Claisen rearrangement in tandem with one or more other pericyclic reactions are very powerful, as they are often stereoselective at each stage. Drawing on reports that enantiomerically pure copper(II) bis(oxazolines) asymmetrically catalyze Claisen rearrangements and intermolecular carbonyl-ene reactions independently, Hiersemann and co-workers successfully applied these catalysts in a tandem process (Scheme 29).[44] Treatment of allyl vinyl ether 126 with catalytic [Cu{(S,S)-Ph-Box}](OTf)2 resulted in isolation of cyclohexanol 128 in high yield, good diastereoselectivity, and outstanding enantioselectivity. Based on their previous work on the catalyzed Claisen rearrangement, they were able to conclude that the ene reaction proceeds with almost complete enantioselectivity and that the low diastereoselectivity observed is a result of incomplete enantioselectivity on the part of the Claisen rearrangement. The selectivity in the ene reaction is thus completely catalyst controlled.
Scheme 29.

Copper-catalyzed Claisen rearrangement/ene reaction duet. [Hiersemann, 2002, Ref. [44]]
2.2.2. Metal-Catalyzed Coupling/Claisen Rearrangement and Isomerization/Claisen Rearrangement
Exploration of various strategies to synthesize vinyl ethers have revealed many challenges, thus their preparation as intermediates in tandem processes presents a great improvement. The Johnson–Claisen rearrangement (see above Scheme 25 and 28) is itself a tandem reaction; it involves initial acid-catalyzed condensation of orthoformates with allyl alcohols to form reactive ketene acetals.[45] Metal-catalyzed couplings or isomerization reactions are attractive strategies to generate vinyl ethers in situ, and the following examples represent advances in this area.
Buchwald and Nordmann reported a copper-catalyzed coupling reaction of allylic alcohols and viny iodides to generate Claisen substrates (Scheme 30a).[46] For example, 2-hexen-1-ol and vinyl iodide 129 reacted in the presence of CuI to generate diene 131, which underwent further rearrange ment to aldehyde 132. The observed relative stereochemistry was consistent with a chair-like transition state for the Claisen rearrangement. Palladium-catalyzed coupling of allylic alcohols with triethylene glycol divinyl ether, as reported by Wei and co-workers, also gave allylic vinyl ether substrates for Claisen rearrangement (Scheme 30b).[47] Their method could be performed on a kilogram scale. Copper and palladium catalysts provide good alternatives to the use of toxic mercury(II) catalysts commonly used for the preparation of vinyl enol ethers.
Scheme 30.

Copper- and palladium-catalyzed alcohol coupling/Claisen rearrangement duets. a) [Buchwald, 2003, Ref. [46]]; b) [Wei, 2007, Ref. [47]]
Wood and co-workers disclosed a rhodium-catalyzed carbene insertion coupling methodology in tandem with a Claisen rearrangement (Scheme 31).[48a] In these duets, allylic alcohols inserted into rhodium carbenoids with proton transfer to generate allyloxy enols (many of which could be observed by 1H NMR spectroscopy), such as 137. Mechanistic studies highlighted the inherent propensity of allyloxy enols to undergo Claisen rearrangement. The facile nature of the rearrangement is what allowed the tandem sequence to proceed beyond the formation of simple O–H insertion products. When enantiomerically enriched allylic alcohols were used, a number of a-hydroxyketones could be isolated in good yield with transfer of chirality. The methodology was used in the total synthesis of (+)-latifolic acid[48b] as well as the synthesis of the welwitindolinone skeleton.[48c]
Scheme 31.

Rhodium-mediated OH insertion/Claisen rearrangement duet. [Wood, 1999, Ref. [48a]]
During their studies on welwitindolinone, attempts to isolate the key a-hydroxyketone 141 were accompanied by aketol rearrangement to 142 (Scheme 32).[48c] In the total synthesis of furanosylated indolocarbazoles (+)- and (–)-K252a, the hydroxyketones were deliberately treated with BF3·OEt2 to effect the rearrangements (not shown), thus efficiently promoting such a Claisen rearrangement trio.[48d]
Scheme 32.

Rhodium-mediated OH insertion/Claisen/ketol rearrangement trio. [Wood, 1999, Ref. [48c]]
In a related carbene trapping reaction, one that could also be called a “metallo-Claisen” rearrangement, Trost and coworkers utilized the propensity of alkynes to isomerize to metal vinylidene complexes and generate carbenoid intermediates by trapping with allyl alcohols (143 to 145, Scheme 33).[49] [3,3] Rearrangement to a ruthenium–acyl complex and reductive elimination terminated the reaction trio to generate a g,d-unsaturated ketone 148. An allyl isomerization was invoked to account for the observed selectivity.
Scheme 33.

Ruthenium-catalyzed metallo-Claisen rearrangement/coupling duet. [Trost, 1990, Ref. [49]]
Two authors of this review used rhodium carbenoids to effect an isomerization reaction that allowed alternative in situ preparation of vinyl enol ethers (Scheme 34).[50] This method addressed the difficulty associated with stereoselectively preparing Z-enol ethers. Additionally, their transformation, which relied on the in situ generation of a diazo compound, expanded the arena of utility to non-carbonyl-stabilized carbene precursors. Thus, N-aziridinyl imines, such as 149 (i.e. Eschenmoser hydrazones), thermally decomposed by chelotropic extrusion in the presence of rhodium(II) catalysts to produce rhodium–carbenoid intermediates, which isomerize controllably to form Z-enol ethers. In the specific case of enol ether 150, subsequent addition of Me2AlCl led to aldehyde 151 by a Claisen rearrangement trio. If DIBAL was used, a reductive rearrangement generated alcohol 152.
Scheme 34.

Rhodium-catalyzed Bamford–Stevens/Claisen rearrangement trio and reduction quartet. [Stoltz, 2002, Ref. [50]]
Conversely, the presence of an additional double bond resulted in tandem ene or Cope reactions (Scheme 35). Treatment of neryl ether 153 with rhodium acetate followed by a Lewis acid induced cascade terminated in a carbonyl-ene reaction to produce cyclohexenol 155 with excellent diaste reoselectivity, while a,b-unsaturated hydrazone 156 underwent a Bamford–Stevens/Claisen/Cope rearrangement cascade to produce aldehyde 158. Both sequences proceed by relatively rare quartet ensembles.
Scheme 35.

Bamford–Stevens/Claisen rearrangement quartets. [Stoltz, 2002, Ref. [50]]
Metal-catalyzed isomerization of bis(allyl) ethers has also been a fruitful strategy for the in situ generation of Claisen substrates. The strategy was originally reported by Reuter and Salomon using ruthenium catalysis.[51] During explorations in the realm of ruthenium-catalyzed alkene metathesis, Dixneuf and co-workers discovered an alternate reactivity pattern involving tandem isomerization/Claisen rearrangement (Scheme 36).[52] They used a three-component catalyst system (ruthenium, NHC, base) to catalyze the isomerization of 1,6-diene 159 to an intermediate vinyl ether, which underwent Claisen rearrangement to g,d-unsaturated alde- hyde 160 in good yield. Double isomerization of 1,7-diene (allyl homoallyl) systems could also be accomplished.
Scheme 36.

Ruthenium-catalyzed isomerization/Claisen rearrangement duet. [Dixneuf, 2002, Ref. [52a]]
Schmidt explored the competition between isomerization/ Claisen rearrangement and alkene metathesis (Scheme 37). The ability to control the pathway by judicious choice of reaction conditions in any given substrate system increases the diversity of products that can be generated using a single catalyst.[53] Whereas Grubbs I catalyst, [Cl2(PCy3)2Ru=CHPh], rapidly catalyzed conversion of diene 162 to the dihydrofuran 163, it was reasoned that the isomerization reactivity of the ruthenium carbene could be enhanced by formation of a ruthenium–hydride species. Schmidt and coworkers exploited Grubbs observation that [Cl2(PCy3)2Ru=CHOEt] (formed by the reaction of [Cl2(PCy3)2Ru=CHPh] with electron-rich olefins) decomposes to the ruthenium–hydride complex [RuHCl(CO)(PCy3)2] upon heating. Thus, by altering the alkene metathesis conditions to include ethyl vinyl ether, they were able to switch the reactivity almost entirely to the isomerization/Claisen pathway to preferentially generate aldehyde 164.
Scheme 37.

Orthogonal reactivity using Grubbs catalyst; metathesis versus isomerization/Claisen duet. [Schmidt, 2000/2004, Ref. [53]]
Iridium catalysts are also effective for the isomerization duet.[54,55] In their work on quinone monoketals, Swenton and co-workers observed that quinol vinyl ethers 165 could be converted thermally and photochemically to spiro-fused 2,5-cyclohexadienones 166 through [1,3] rearrangement (Scheme 38). Although [3,3] rearrangement is not feasible in such substrates, they were interested in understanding the competition in substrates where [1,3] and [3,3] rearrangements were both sterically and electronically possible. As a result, they reported an iridium-catalyzed isomerization/ Claisen rearrangement/cylization trio (167 → 170).
Scheme 38.

Iridium-catalyzed isomerization/Claisen rearrangement trio terminated by lactol formation. [Swenton, 1991, Ref. [55]]
Nelson[56] and co-workers reported a strategy that combined the addition of dialkyl zinc reagents to aldehydes catalyzed by asymmetric aminoalcohols with the iridiumcatalyzed isomerization/Claisen rearrangement (Scheme 39). Of paramount importance was finding a catalyst to achieve selective isomerization without epimerizing the aldehyde product. Thus, consecutive nucleophilic addition and palladium-catalyzed etherification of cinnamaldehyde with diethyl zinc and allyl acetate generates bis(allyl) ether 172 in high yield and enantioselectivity. Iridium-catalyzed isomerization of the terminal allyl group followed by rearrangement afforded aldehyde 173 with negligible loss of enantioselectivity. The duet could be expanded to a reduction trio by quenching with NaBH4.
Scheme 39.

A consecutive quartet ensemble: asymmetric alkyl zinc addition/palladium etherification duet, followed by isomerization/ Claisen rearrangement duet. [Nelson, 2005, Ref. [56b]]
This method was also applied to 1,6-enynes in a report by Tanaka and co-workers (Scheme 40).[57] When enyne 174 was treated with a rhodium(I) catalyst, allenic aldehyde 175 was generated. At high temperatures, the allenes isomerized further to generate dienals such as 176. The mechanism of this carbonyl migration was proposed to proceed by initial insertion in the aldehyde C–H bond.
Scheme 40.

Rhodium-catalyzed isomerization/Claisen rearrangement duet (to 175) and migration trio (to 176). [Tanaka, 2009, Ref. [57]]
Eilbracht and co-workers developed a rhodium(I)-catalyzed tandem Claisen rearrangement/hydroacylation reaction duet of allyl vinyl ethers to form cyclopentanones.[58] They then expanded the reaction to include an isomerization to generate the allyl vinyl ethers in situ (Scheme 41). In the rearrangement trio, bis(allyl) ethers such as 178 were treated with a rhodium(I) or ruthenium(II) catalyst. Isomerization to vinyl enol ethers such as 179 was followed by Claisen rearrangement to aldehyde 180. The quaternary center at C3 was necessary to block double bond isomerization to generate a,b-unsaturated aldeydes, a common side reaction under these conditions. Intramolecular olefin hydroacylation then proceeded to generate cyclopentanone 181. They have demonstrated the utility of the Claisen/hydroacylation in the preparation of spirocylic precursors to sesquiterpene natural products, and a polystyrene-supported catalyst has also been shown to be effective for the duet.
Scheme 41.

Rhodium-catalyzed isomerization/Claisen/hydroacylation trio. [Eilbracht, 1995, Ref. [58]]
Ovaska and co-workers made extensive use of an anionic cylization/Claisen rearrangement duet in the rapid synthesis of relatively complex polycylic structures (Scheme 42).[59] For example, in the formal total synthesis of frondosin A, treatment of allylic alcohol 182 with a substoichiometric amount of methyllithium proceeded to cycloheptene 184 as a mixture of diastereomers. Epimerization in basic methanol increased the ratio to favor the desired isomer. The versatility of the method was shown by the ability to use structurally isomeric 185 to generate 186, also a viable precursor to frondosin A. A gold-catalyzed cationic variant of this reaction is presented in Section 2.2.5, Scheme 63.
Scheme 42.

Base-initiated 5-exo-dig cyclization/Claisen rearrangement duets. [Ovaska, 2008, Ref. [59]]
Scheme 63.

Gold-catalyzed cyclization/[3,3] rearrangement duet. [Rhee, 2008, Ref. [96]]
A related rearrangement that involves p-alkyne activation is the platinum-catalyzed carboalkoxylation/Claisen rearrangement duet (Scheme 43).[60] Yamamoto and co-workers reported the platinum(II)-catalyzed cycloisomerization of saturated cylic acetals to 1,2-dialkoxyindenes, related to structure 188. When the acetal contained a double bond, as in 187, the carboalkoxylation was followed by Claisen rearrangement to give bridged bicycle 189.
Scheme 43.

Platinum-catalyzed cycloisomerization/Claisen rearrangement duet. [Yamamoto, 2005, Ref. [60]]
In an elegant demonstration of the importance of understanding catalyst structure and reactivity when designing a new tandem reaction, Chatani and co-workers reported a rhodium(I)-catalyzed [2+2+1] carbonylative cycloaddition/Claisen rearrangement duet of phenoxide-substituted diynes 190 (Scheme 44).[61] To that point, the carbonylative cyclo-addition of diynes had rarely been studied, and the strong binding of product cyclopentadienes to the rhodium catalyst was recognized early as a barrier to developing a catalytic process. Having isolated and characterized cyclopentadiene-bound rhodium complex 193 by X-ray crystallography, they considered ways to release the catalyst for turnover. Introduction of an aryl ether substituent resulted in [3,3]-sigma-tropic rearrangement to 192, a more weakly coordinating product. In fact, the rearrangement benefits from coordination by rhodium; this aromatic Claisen rearrangement proceeds at 120°C while much higher temperatures (> 200°C) are typically required. Thus, treatment of diyne 190 with catalytic rhodium(I) under CO atmosphere and with heating produced phenol 192 in moderate yield.
Scheme 44.

Rhodium-catalyzed Pauson–Khand/Claisen rearrangement duet. [Chatani, 2010, Ref. [61]]
2.2.3. Claisen Rearrangement/Cyclization
A variety of N and O heterocycles can be synthesized using tandem Claisen reactions. For example, aromatic ether 194 in the presence of catalytic AlCl3 at -70°C rearranges to cyclic ether 195, presumably via a phenol intermediate (Scheme 45a).[62a] Similar transformations were catalyzed by gold, iridium, molybdenum, zinc, titanium, and zeolites.[62b–h] The sequence could be taken one step further, as in the copper-catalyzed rearrangement trio reported by Adio and co-workers (Scheme 45b).[63a] The transformation proceeded by initial hydroalkoxylation to form allyl aryl ethers (e.g. 198), which then underwent a Claisen rearrangement to allyl aryl compounds (e.g. 199). In contrast to the preceding duets, a 6-endo-trig cyclization terminates the sequence. Annulation strategies such as these have been reported previously using other Brønsted and Lewis acid catalysts. However, a caveat should be mentioned: an intermediate Claisen rearrangement has not always been suggested to explain the formation of the aryl–bond, nor has it been confirmed in the aforementioned case.[63b–f]
Scheme 45.

Lewis acid catalyzed Claisen rearrangement/cyclization (a) duet [Ryu, 1993, Ref. [62a]] and (b) trio. [Hii, 2008, Ref. [63a]]
Binder and Kirsch combined Claisen rearrangement with in situ amine condensation for use in the synthesis of pyrroles (Scheme 46). Their rearrangement trio commenced with a silver-catalyzed Claisen rearrangement. An aryl amine and a gold catalyst were subsequently added to the presumed allenyl ketone intermediates. Condensation and cyclization thus provided methylpyrolles such as 203. Although gold catalysts should be able to effect the initial Claisen rearrangement, yields were unfortunately lower when all reagents were initially present.[64] Gold-catalyzed rearrangements of prop-argyl enol ethers have also been reported by Toste.[65]
Scheme 46.

Silver- and gold-catalyzed Claisen trio. [Kirsch, 2006, Ref. [64]]
Methylpyrroles could also be obtained from propargylenamines through a related amino-Claisen/cyclization duet reported by Saito and co-workers (Scheme 47).[66] The same
Scheme 47.

Rhodium-catalyzed one-pot alkylation/amino-Claisen/cyclization trio. HFIP= hexafluoroisopropyl alcohol. [Saito and Hanzawa, 2009, Ref. [67]]
authors developed a rhodium-catalyzed amino-Claisen cyclization duet to generate indoles. The reaction could be combined with in situ synthesis of propargyl amines, extending the sequence to a trio.[67] For example, treatment of a pre-mixed solution of bromobutyne and aniline with rhodium catalyst in the presence of base and phosphine at reflux in hexafluoroisopropyl alcohol (HFIP) produced indole 208 in good yield.
In a report by Liu and co-workers,[68] gold(III) and silver(I) salts were combined to catalyze the Claisen rearrangement of (aryloxymethyl)alkenoates. Subsequent lacto- nization terminated the duet (Scheme 48). Thus, treatment of naphthyl ether 209 with AuCl3 and AgOTf in DCE or DCB at 120°C led to isolation of dihydrocoumarin 211 in good yield. At low temperatures and in cases where substituents disfavored the [3,3] rearrangement, a competing [1,3] rearrangement proceeded via an allyl cation/phenoxide ion pair to generate isomeric dihydrocoumarins.
Scheme 48.

Gold-/silver-catalyzed Claisen rearrangement/lactonization duet. [Liu and Xu, 2009, Ref. [68]]
Propargyl esters are increasingly studied substrates in the context of enyne cycloisomerization reactions because of their propensity to undergo gold- or platinum-catalyzed 1,2-and/or 1,3-acyl migration reactions.[69] Rhodium catalysts have also been shown to be effective for the process.[70] The 1,3-acyl migration is a formal [3,3]-sigmatropic rearrangement that generates allenyl acetates, which under catalyzed conditions are themselves activated by the metal to undergo further rearrangements (Scheme 49). Toste and co-workers showed that the process was reversible, so tandem chemistry is again required to complete the reaction and achieve selectivity.[71] The allenyl acetate formed could be captured in many ways, such as by a pendant nucleophile (Scheme 49a)[72] or Nazarov cyclization (Scheme 49b).[73a] In one case, trapping with a pendant indole resulted in a formal [2+2] reaction duet (Scheme 49c).[73b] The intermediate allenyl acetates could also undergo further isomerization reactions[74] or trapping with an external nucleophile.[75] Only a few of the multitude of possible reactions are shown here; for further information we direct the reader to reviews by Tang,[70] Zhang,[76] and Marion.[69] The propargyl esters could also participate in a 1,2-acyl shift leading to metal intermediates that display carbene-like reactivity;[77] for combination with a [2,3] rearrangement, see Section 3.1 (Scheme 87 and 88).
Scheme 49.

Reaction duets initiated by Claisen rearrangement of propargyl esters. a) [Gagosz, 2006 and related, Shin, 2007, Ref. [72]], b) and c) [Zhang, 2006/2005, Ref. [73a,b]]]
Scheme 87.

Gold-catalyzed carbenoid generation/ylide formation/rearrangement quartet. [Davies, 2008, Ref. [144]]
Scheme 88.

Alternate gold-catalyzed carbenoid generation/ylide formation/rearrangement quartet. [Davies, 2008, Ref. [144]]
Gevorgyan and co-workers reported a copper-catalyzed [3,3] rearrangement/cycloisomerization duet involving phosphatyloxy alkynyl ketones such as 218 (Scheme 50).[78] This reaction involved the rearrangement of a phosphate ester (see analogous propargyl ester Claisen; Scheme 49) and was followed by cylization to form furans. Bicyclic products could also be obtained when alkynyl pyridines such as 221 were used. Oxygen isotope labelling provided support for the [3,3] rearrangement pathway; interestingly, under these conditions the standard carboxylic ester substrates did not appear to undergo the Claisen-type rearrangement, but instead formed similar products by alternate rearrangement mechanisms.
Scheme 50.

Cascade [3,3] rearrangement/cycloisomerization duet. [Gevorgyan, 2007, Ref. [78]]
2.2.4. Claisen Rearrangement/Coupling
The following reactions are unified in that they combine Claisen reaction with a C–C bond-forming event. During studies of 2-allyloxypyridines as substrates in the chelation-controlled Heck reaction, Itami and co-workers discovered a Claisen rearrangement/Heck reaction duet (Scheme 51).[79] Treatment of allyloxypyridines such as 223 with palladium, followed by addition of aryl iodides and base, resulted in formation of N-substituted 2-pyridones in good yield and selectivity for E olefins.
Scheme 51.
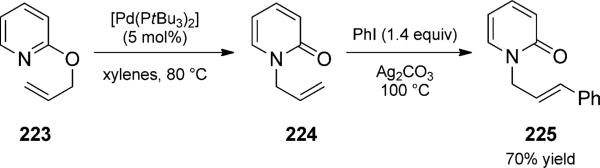
Palladium-catalyzed aza-Claisen/Heck duet. [Itami and Yoshida, 2003, Ref. [79]]
As part of their substance P antagonist programme, Watson and co-workers sought a route to spiroindane 229 (Scheme 52).[80] Since α-allyl ketone 228 could not be synthesized by direct C alkylation, they instead explored a palladium-catalyzed Claisen rearrangement. When allyl enol ether 226 was heated in the presence of [Pd(PPh3)4], the rearrangement terminated at amide 228. When the reaction was performed in the presence of base, subsequent intra-molecular Heck reaction allowed access to amide 229. Use of a crotyl analogue of 226 indicated that the reaction proceeds via a π-allyl/enolate ion pair, rather than a palladium(II)-accelerated cyclic sigmatropic transition state.
Scheme 52.

Palladium-catalyzed formal Claisen/Heck duet. [Watson, 1994, Ref. [80]]
Wipf and co-workers developed an asymmetric zirconocene-mediated Claisen rearrangement/carboalumination duet.[81b] They have since expanded on that tandem sequence by developing a zirconocene-catalyzed carboalumination of alkynes in the presence of enol ethers.[81a] The resultant trio led to the isolation of 1,6-dienes (Scheme 53). Zirconiumcatalyzed carboalumination of alkynes led initially to vinyl alanes such as 231. The vinyl alane itself could then serve as a Lewis acid for the catalysis of Claisen rearrangement of vinyl enol ether 232. Vinyl transfer from the alane to the resultant aldehyde 233 generated allylic alcohol 234 with good selectivity. Water is a key accelerating additive for these sequences.
Scheme 53.

Zirconium-catalyzed carboalumination coupled with Claisen rearrangement and carbonyl addition, a trio ensemble. [Wipf, 2005, Ref. [81a]]
The Claisen rearrangement has also been coupled with alkene metathesis. Specifically, Sutherland and co-workers combined the asymmetric Overman rearrangement, a hetero-Claisen variant involving trichloroacetimidate 237, with a ring-closing metathesis to synthesize the cyclic allylic trichloroacetamide 239 (Scheme 54).[82,104] The Overman rearrangement has wide appeal since the trichloroacetamide handle has utility for a number of subsequent transforma tions, and use of the enantiopure palladium complex (S)-COP-Cl makes it catalytic and asymmetric.[83] Trichloracet-amide 239 was used in a synthesis of the tropane alkaloid (+)-physoperuvine.[82a]
Scheme 54.

One-pot Overman rearrangement/alkene metathesis duet. [Sutherland, 2009, Ref. [82a]]
Borrelly and Paquette used a Claisen rearrangement/ Sakurai cyclization duet to prepare vinyl bromide 245, a cyclopentenyllithium reagent precursor that is important in their efforts toward the total synthesis of natural products (Scheme 55).[84] Treatment of vinyl enol ether 242 with Et2AlCl, followed by HCl and TBAF in the workup, resulted in facile formation at room temperature of the vinyl bromide 245, presumably via intermediate aldehyde 243. Thermal conditions were unsuccessful.
Scheme 55.

Lewis acid catalyzed Claisen rearrangement/Sakurai allylation duet. [Paquette, 1993, Ref. [84]]
Another set of atypical Claisen rearrangements are the transpositions invoked in oxidative processes mediated by classic chromium reagents, that is, metallo-Claisen rearrangements (see also Scheme 33).[85] A catalytic tandem varient of this process was developed by Trost and co-workers using vanadium (Scheme 56).[86] The transposition of allenic alco hols had previously been unexplored and it was anticipated that the products would be reactive enolates. Thus, treatment of allenic alcohols such as 246 with an oxo-vanadium catalyst resulted in rearrangement to enolate 248. Subsequent capture by benzaldehyde terminated the aldol reaction duet.
Scheme 56.

Vanadium-catalyzed allenic alcohol transposition/aldol-type addition duet. [Trost, 2001, Ref. [86]]
In an impressive combination of four elementary events, M ller and co-workers reported Claisen rearrangement quartets initiated by palladium-catalyzed alkyne coupling.[87] Treatment of aryl or aroyl halides (“R-X”) with alkynes in the presence of palladium(II) generated aryl or aroyl alkynes, such as 252 (Scheme 57). Isomerization of the resultant propargyl ether led to allenyl ethers that could undergo subsequent Claisen rearrangment to a key diene intermediate 254. Intermediate 254 underwent an array of subsequent pericyclic reactions, depending on the electronic nature of the substituents (Scheme 58).
Scheme 57.

Palladium-catalyzed coupling/Claisen rearrangement. [M ller, 2006, Ref. [87]]
Scheme 58.

Pericylic reaction terminated quartets. [Müller, 2006, Ref. [87]]
2.2.5. Charge-Accelerated Claisen Rearrangement
Allylic ethers, amines, and sulfides can add to ketenes to generate zwitterionic intermediates (e.g. 263, Scheme 59), which undergo facile [3,3] rearrangement, a variant known as the Belluš–Claisen rearrangement.[88] Such duets have been used extensively for the formation of building blocks for natural product heterocycle synthesis, and benefit from exceptional asymmetric induction when enantiopure substrates are used.[89] Although stoichiometric amounts of Lewis acids are typically required, MacMillan and co-workers[90] made a significant advance in showing that asymmetric induction could be externally controlled by the Lewis acid.[90a] The reaction could be extended a step further when allyl diamines were used (Scheme 59).[90c] The initial [3,3]rearrangement was proposed to proceed through the (E)-ammonium intermediate 261, wherein pseudo 1,3-diaxial interactions are minimized. Minimization of (1,2) allylic strain was then proposed as the driving force for the subsequent [3,3] rearrangement of ammonium 263 to selectively generate the 2,3-syn-3,6-anti isomer 264. The preparation of said 1,7-diamidoheptane represents a rapid method for preparing a stereochemical motif commonly found in macro-lide antibiotics. The ability to selectively cleave the α,β disubstituted amide using an iodolactonization/ring-opening sequence demonstrates further the utility of this process.
Scheme 59.

Diastereoselective Bellus–Claisen rearrangement trio. [Macmillan, 2001, Ref. [90c]]
A number of related charge-accelerated Claisen rearrangements have been reported. For example, Brønsted or Lewis acid catalyzed amine addition to alkynes[91] or alle nes[90b] results in vinyl ammonium intermediates poised for [3,3] rearrangement (Scheme 60a,b). A base-catalyzed isomerization/rearrangement trio of propargylammonium salt 273 was reported by Honda and co-workers (Scheme 60c).[92] Previously, ammonium salts under basic conditions generated ylides that underwent [2,3] rearrangement. With protic solvents, base deprotonation (catalytic in principle) led to alkyne–allene isomerization, which was then followed by [3,3] rearrangement and in situ reduction.
Scheme 60.

Acid-catalyzed ammonium-Claisen rearrangement duets and a base-mediated isomerization/aza-Claisen/reduction trio. a) [Vedejs, 1994, Ref. [91]], b) [MacMillan, 2002, Ref. [90b]], c) [Honda, 2003, Ref. [92]]
In synthetic efforts toward analogues of naturally occurring prostaglandins, Florent and co-workers developed a related ionic aza-Claisen/Mannich trio (Scheme 61).[93] Treatment of either diastereomer of aldehyde 277 with pyrrolidine resulted in a crude mixture of enamine isomers 278. Alkylation with an allylic iodide generated the ammonium 279, which underwent facile [3,3] rearrangement to iminium 280. They expected to isolate the aldehyde derived from 280, but were pleased to isolate instead cyclopentenone 282. Thus, an immediate Mannich cyclization occured through nucleophilic attack of the pendant MOM-protected enol ether. Finally, pyrrolidine β-elimination terminated the alkylation/Claisen/Mannich quartet. The poor overall diastereo-selectivity is likely linked to the mixed enamine stereochemistry of 278.
Scheme 61.

Pyrrolidine-mediated alkylation/cationic aza-Claisen/Mannich rearrangement/elimination quartet. [Florent, 2003, Ref. [93]]
Rainier and co-workers reported a related tandem reaction that involved formation of a 1,3-dipole from a thioether and a rhodium carbenoid (Scheme 62).[94] In their case, thioether addition to the carbene led to the formation of the intermediate sulfur ylide, 285. They proposed that proton transfer precedes the [3,3]-sigmatropic rearrangement to give indoline 286. The reaction displayed high diastereoselectivity for certain substrates. The methodology provided an attractive route to substituted indolines with a quaternary carbon center and could also be used to prepare indolines with vicinal quaternary carbon centers. The products formed have significant potential for use in the total synthesis of bioactive indoline natural products.
Scheme 62.

Rhodium-catalyzed ylide formation/thio-Claisen rearrangement duet. [Rainier, 2008, Ref. [94a]]
While Ovaska s alkoxide cyclization conditions may be considered relatively harsh (see above, Scheme 42), the Lewis acid catalyzed Claisen rearrangement of cyclic enol ethers to form cycloheptenones provides an alternate method that fits within the context of charge-accelerated Claisen rearrangements. Although the strong Lewis acids used therein demand the consideration of functional-group compatibility,[95] Rhee and co-workers reported an extremely facile sequence that formally corresponds to the same type of duet as in Ovaska s work (Scheme 63).[96] Treatment of 3-alkoxy-1,6-enynes with a gold(I) catalyst (and AgSbF6 to favor the formation of cationic gold) resulted in rapid isomerization initially to the isolable methyl enol ether 290. The transfer of stereochemical information from the trans double bond of 288 strongly supported the concerted nature of the rearrangement.
2.3. Cationic Hetero-Cope Rearrangements
The “aza-Cope rearrangement” most commonly refers to the “2-aza-Cope rearrangement” of iminium ions (293 → 294, Scheme 64). While the “oxy-Cope rearrangement” generally refers to rearranging p systems with appending oxygen substituents, the “oxonia-Cope rearrangement” is the term given to the oxocarbenium analogue of the aza-Cope rearrangement of iminium ions. This section will focus on the incorporation of the cationic aza-Cope and cationic oxonia-Cope rearrangement within catalytic tandem sequences.
Scheme 64.
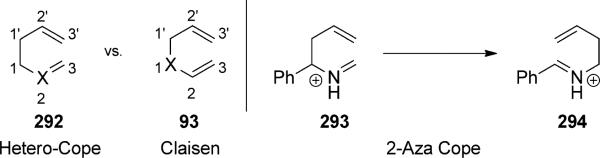
Distinction between hetero-Cope (such as the 2-aza-Cope) and Claisen rearrangement substrates. [Horowitz and Geissman, 1950, Ref. [97]]
2.3.1. Aza-Cope Rearrangements
In many instances, the aza-Cope rearrangement is itself a reaction duet, initiated by acid-catalyzed condensation between an amine and carbonyl group to form a reactive iminium ion. The aza-Cope rearrangement was first invoked to explain a C–C cleavage reaction in the reductive alkylation of homoallylic amine 295 (Scheme 65).[97] In this case,
Scheme 65.
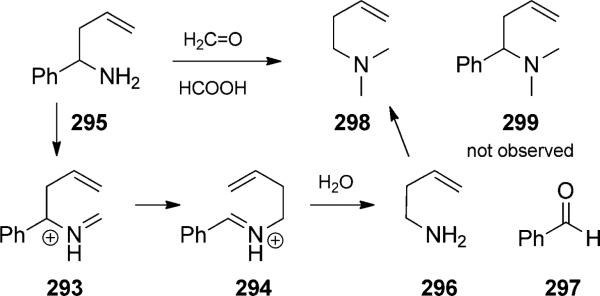
First aza-Cope rearrangement duet. [Horowitz and Geissman, 1950, Ref. [97]]
condensation between formaldehyde and the benzylamine generates iminium 293, which rearranges to iminium 294. Hydrolysis leads to formation of homoallylic amine 296, and it is this amine rather than the starting material 295 that is alkylated through reductive amination to the tertiary amine 298. Formic acid is the reductant.
Recently, Rueping and Antonchick reported a catalytic enantioselective variant that utilized the enantiopure binol phosphoric acid derivative 303 (Scheme 66).[98] Enantioselectivity was proposed to arise from a strong ion-pair interaction (E) between the protonated imine and the anionic phosphate. This method expands the available routes for preparing optically active homoallylic amines, which are important building blocks in natural product synthesis.
Scheme 66.

Catalytic asymmetric aza-Cope rearrangement duet. MTBE= methyl tert-butyl ether [Rueping, 2008, Ref. [98]]
As with the parent Cope and Claisen rearrangements, the aza-Cope rearrangement displays good stereocontrol through a chair-like transition state. Use of a judiciously placed substituent led to the observation by Overman and coworkers of a dynamic kinetic resolution in the aza-Cope rearrangement (Scheme 67).[99] The possibility of resolution was deduced from the observation that iminium ions such as 305 can epimerize rapidly by a fast equilibrium with enammonium ions 306. Thus, treatment of amine 304 with trifluoroacetic acid, dimedone, and morpholine, followed by quenching with benzyl chloroformate, leads to isolation of carbamate 308 as a single diastereomer in high enantiomeric excess. The full transformation constitutes an iminium ion aza-Cope/acylation quartet if you include initial acetal hydrolysis. The dynamic kinetic resolution occurs, because aza-Cope rearrangement proceeds slowly, but selectively, via diastereomer 305a. Trapping of the thermodynamically less stable iminium 307 by dimedone is fast, so that the aza-Cope rearrangement is rendered effectively irreversible. Rearrangement of 305a across the concave face by a chair-like transition state is more facile, as it places the phenyl substituent in a pseudo-equatorial orientation, whereas in 305b that substituent would be placed in a pseudo-axial position.
Scheme 67.

Dynamic kinetic resolution using an aza-Cope rearrangement quartet. [Overman, 2010, Ref. [99]]
Tehrani and co-workers combined the Lewis acid catalyzed aza-Cope rearrangement with a Petasis reaction (Scheme 68).[100] Addition of an alkynylborate salt generated not the product of direct addition (311), but instead the rearranged amine 314. BF3 coordination catalyzes the aza-Cope rearrangement to isomeric iminium 313. Acetylide addition to this more reactive iminium compound leads to amine 314 upon workup. The Petasis reaction shares some mechanistic steps with the Mannich reaction, and tandem aza-Cope/Mannich reactions have long been known to have excellent utility in the preparation of nitrogen-containing heterocycles. They are discussed further below.
Scheme 68.

Lewis acid mediated aza-Cope/Petasis reaction duet. [Tehrani, 2008, Ref. [100]]
In their research aimed at the design of methods to prepare α-amino acids, Agami and co-workers made use of glyoxyl condensation/aza-Cope rearrangement trios (Scheme 69).[101] Two possible pathways could be observed, depending on the nucleophile present (water or formic acid) to trap the reactive iminium ion. The reactions were initiated by spontaneous condensation between glyoxal- and phenylglycinol-derived amino alcohol 315 to generate iminium 316. Aza-Cope rearrangement generated iminium ion 317. In the presence of water, the iminium compound was cleaved to form amine 318 in good yield through an uncatalyzed condensation/aza-Cope/hydrolysis trio. On the other hand, in the presence of the weaker formic acid nucleophile, iminium ion 317 underwent a further iminium-ene cyclization, the product of which was trapped to form bicycle 319, thus concluding a condensation/aza-Cope/iminium-ene cyclization quartet. Waters and co-workers combined a similar condensation/aza-Cope reaction with silver-mediated [3+2] cyclo-addition in a consecutive reaction trio.[102]
Scheme 69.

Aza-Cope rearrangements. [Agami, 1992, Ref. [101]]
The aza-Cope/Mannich sequence represents by far the most developed tandem sequence involving an aza-Cope rearrangement (Scheme 70). It was developed by Overman in the late 1970s and was specifically designed to capitalize on both the acceleration of rearrangements involving charged species and the tandem intramolecular trapping of a reactive iminium ion to render the aza-Cope rearrangement effectively irreversible.[103] When the iminiumion is formed by an intramolecular reaction involving a pendant carbonyl group, complex polycyclic structures result. The method has been used to great success to construct complex natural products, including Overman s own elegant synthesis of actinophyllic acid.[103c] For a personal account of Overman's methods development in the broad area of Claisen rearrangements, see reference [104].
Scheme 70.
Overman's aza-Cope/Mannich reaction duet.
Brummond and co-workers used the condensation/aza-Cope/Mannich reaction trio in the formal synthesis of a tricyclic immunosuppressant.[105] Condensation of amino ketone 323 to form iminium 324 demonstrated the feasibility of proceeding through a bridgehead iminium ion (Scheme 71). Rearrangement and enol ether attack on iminium 325 led to the isolation of tricycle 326 as a mixture of diastereomers. Because of the instability of the aldehyde, it was protected as a ketal (72% yield after two steps). A more functionalized substrate was also competent in the reaction and used for the preparation of an advanced intermediate toward immunosuppressant FR901483.
Scheme 71.

Aza-Cope/Mannich trio via a bridgehead iminium ion intermediate. [Brummond, 2001, Ref. [105b]]
The aza-Cope/Mannich sequence has been utilized to great extent in synthesis and medicinal chemistry,[106] and solid-supported acid resins[107] have been developed to effect the transformation.
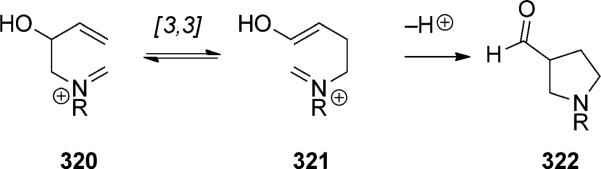
2.3.2. Oxonia-Cope Rearrangement
The oxygen analogue of the cationic aza-Cope rearrangement is typically referred to as the oxonia-Cope rearrangement (not to be confused with oxy-Cope rearrangements, see Section 2.1.2). The term oxa-Cope rearrangement is sometimes used, but this term has also been applied to the Claisen rearrangement. Historically, the oxonia-Cope rearrangement has been considered mainly as a competing background process in the ene cyclization of oxocarbenium ions (i.e. the Prins reaction; Scheme 72). Overman and co-workers had originally designed this reaction after the success of the Cope rearrangement of iminium ions, but found that the oxocarbenium analogues followed a different pathway. The isolated products instead implicated a Prins cyclization/pinacol rearrangement pathway.[103a,104]
Scheme 72.
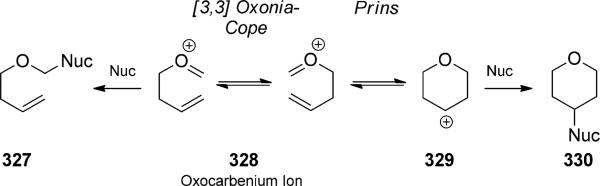
Competing oxocarbenium reactions.
The mechanism of the oxonia-Cope rearrangement was first invoked by Speckamp and co-workers in their reports on the acid-mediated π cyclizations of a-methoxycarbonyl oxycarbenium ions to form oxacyclic carboxylic esters.[108] Although such cyclization reactions fall outside the scope of this article, there is a significant body of work, particularly by Rychnovsky,[109] that suggests the oxonia-Cope rearrangement is a background process for many of them.[110] The background process can be detrimental to stereoselective processes, and can be minimized.[111]
The oxonia-Cope rearrangement product can sometimes be selectively intercepted. The cationic intermediates can be captured by a pendant or external nucleophile, lose a proton to form alkene products, or be hydrolyzed to release homo-allylic alcohols. For example, the oxonia-Cope rearrangement without a competing Prins cyclization has been developed by Nokami and co-workers as an improved method for a-selective allylation of aldehydes (Scheme 73).[112] While many allylic transition-metal reagents preferentially give g adducts, such as homoallylic alcohol 331, the a-selective allylation is synthetically more challenging. Rearrangement via oxocarbenium ions was seen as a way to isomerize undesired (but easily synthesized) g adducts. The reaction is driven toward products that are derived from more stable cations or those that contain less hindered alcohols and more stable olefins. While the original reaction was planned with a stoichiometric amount of aldehyde, it was shown that a catalytic amount of aldehyde is sufficient to promote the isomerization. Stereo-chemical fidelity is observed when enantiomerically enriched alcohols are used, confirming the predictive stereocontrol provided by the cyclic transition state. Loh and co-workers showed that indium(III) was a competent oxonia Cope/cyclization catalyst and observed two different reaction quartets (Scheme 74).[113] At the outset of their studies, they sought to expand Nokami s methodology, which was less effective with prenyl adducts. Loh proposed to thus explore the stronger Lewis acid In(OTf)3 in the allyl-transfer exchange reaction. Instead of the expected homo-allylic alcohol 341, they observed further reactivity depending upon the amount of excess aldehyde. With a catalytic amount of aldehyde, oxocarbenium 338 was hydrolyzed to generate the expected alcohol. Indium then catalyzed the cyclization to form tetrahydrofuran 335. With a full equivalent of aldehyde, an oxonium-ene cyclization proceeded presumably to tertiary cation 339. Oxygen [1,3] shift followed by proton transfer led to the formation of alkene 336. The intermediacy of alcohol 341 was verified by separate control experiments. Despite the unexpected reactivity in this case, the method was later successfully expanded to the preparation of a-prenylated aldehydes by careful selection of the prenylated donor alcohol. The oxonia-Cope rearrangement continues to be used in the preparation of a adducts in methodology development[114] and total synthesis.[115] Transfer of chirality is generally very good in these reactions, and stereodefined products are often obtained.
Scheme 73.

Isomerization of allylation adducts by oxonia-Cope rearrangement trio. [Nokami, 2000, Ref. [112a]]
Scheme 74.

Indium-catalyzed oxonia-Cope/cyclization quartet. [Loh, 2001, Ref. [113c]]
Ramachandran and co-workers observed adduct isomerization following crotyl borane addition to aldehydes in tandem oxonia-Cope rearrangements (Scheme 75).[116] For the case of vinyl esters such as 342, the rearrangement was terminated by lactonization. Lewis acid accelerated crotylbo-ration led initially to γ adduct 343. In the presence of a ytterbium(III) catalyst, lactonization without rearrange ment to form lactone 348 occured. Conversely, with an indium(III) catalyst, acetal formation and elimination resulted in the formation of oxonium ion 344. Oxonia-Cope rearrangement to 345 was followed by regeneration of the borate ester a adduct 346. Lactonization then provided rearranged 347 in good yield.
Scheme 75.

Indium-catalyzed crotylboration/oxonia-Cope/lactonization trio. [Ramachandran, 2007, Ref. [116a]]
In a Lewis acid mediated oxonia-Cope rearrangement trio Rychnovsky and co-workers utilized a Mukaiyama–Michael addition to generate a reactive oxocarbenium ion in situ (Scheme 76).[117] Treatment of enol ethers such as 349 with TiBr4 (catalytic in principle) and 350 resulted in a Michael addition to generate oxocarbenium 351. [3,3] Rearrangement to oxocarbenium 352 followed by cyclization generated tetrahydropyran 353 in good yield. The appropriate placement of a tethered nucleophile favors oxonia-Cope rearrangement without a Prins cyclization “interrupting”. Rychnovsky used this strategy to great success in other, non-catalytic sequences.[118]
Scheme 76.

Titanium-mediated Mukaiyama/oxonia-Cope rearrangement trio. DTBMP= 2,6-di-tert-butyl methyl pyridine. [Rychnovsky, 2005, Ref. [117]]
3. [2,3]-Sigmatropic Rearrangements
Although [2,3]-sigmatropic rearrangements are far less ubiquitous in organic synthesis, they present many features similar to the more utilized [3,3] counterparts that make them attractive strategies. Virtually all known [2,3] rearrangements involve heteroatoms,[119] commonly N, O, and S, and are generally divided into two types (Scheme 77). They often proceed with high stereoselectivity; the higher conformational flexibility of five-membered cyclic transition states suggests that they are more susceptible to the effects of stereochemical control by substituents. This has been summarized in detail by Hoffmann.[120]
Scheme 77.

Representative [2,3]-sigmatropic rearrangements. [type I representative: Hiersemann, 2001, Ref. [121]], type II: Clark, 1992, Ref. [133]]
Type I reactions encompass anionic rearrangements such as the Wittig rearrangement.[122] They typically involve the stoichiometric, irreversible generation of an anion, and so none are included in this review. Conversely, type II rearrangements encompass ylide reactions such as the carbon-oxonium ylide rearrangement. Because of the recent increase in the number of methods involving catalytic generation of ylides from diazo compounds,[123] several examples are discussed in the current review.
3.1. Carbon Ylides
There have been a number of reports on the metal-catalyzed ylide formation/[2,3]-sigmatropic rearrangement duet using α-diazocarbonyl compounds and sulfides,[124] ethers, and amines.[125] These methods have provided a much milder alternative to stoichiometric base-promoted methodologies. Competing carbenoid reactions (e.g. carbene dimerization, C H insertion, cyclopropanation, and Stevens [1,2] rearrangement), some of which are discussed in Section 4, and catalyst deactivation by the heteroatom have presented the main challenges to their development. Many asymmetric variants have been reported.[126]
The intermolecular coupling of diazo compounds with sulfides through [2,3] rearrangement is known as the Doyle– Kirmse reaction, itself a sulfur ylide duet (Scheme 78). Initial developments by Doyle and co-workers[127] provided homo-allyl sulfides (e.g. 356 or 359) in good yields from the reaction of allyl sulfides and ethyl diazoacetate, but relied on a high excess of diazoester and the slow addition of sulfide. Although rhodium is most commonly used to generate sulfonium ylides, Carter and Van Vranken showed that palladium was a suitable catalyst for the transformation and that iron was even better.[128] Use of iron offered two key improvements to the methodology: the slow addition of reagents was unnecessary and the excess of diazo compound could be reduced significantly.
Scheme 78.

Metal-catalyzed Doyle–Kirmse sulfur ylide duets. dppe =1,2-bis(diphenylphosphino)ethane. DTBP= 2,6-di-tert-butyl-4-methylpyridine. a) [Doyle, 1981, Ref. [127]], b) [Van Vranken, 2000/ 2001, Ref. [128]]
As the methodology evolved, asymmetric variants have been reported using enantiomerically pure phthalimido piperidinonate dirhodium(II) catalysts[129] as well as diaste reoselective variants using enantiopure allylic sulfides.[130] Intramolecular insertion/rearrangements via sulfonium ylides are also known.[131]
Related reactions of diazo compounds and ethers via oxygen ylides have also been developed. Johnson with Roskamp and Pirrung with Werner independently reported the first intramolecular carbenoid insertion/ylide rearrangement duets (Scheme 79).[132] Clark improved upon these rhodium-catalyzed methods by showing that [Cu(acac)2] led to higher yields and levels of diastereoselectivity, such as in the rearrangement via oxonium 361 to produce tetrahydrofuranone 362.[133] Intermolecular and asymmetric insertions of carbenoids into allylic ethers have been reported, but with variable results.[134] Recently, Davies and Li showed that oxonium ylide rearrangement proceeded with high enantioselectivity and could compete with O H insertion when donor/acceptor carbenoids and a highly substituted alcohol were used as substrates (Scheme 80).[135] Thus, treatment of allylic alcohol 364 and donor/acceptor diazo compound 363 with the enantiomerically pure [Rh2{(S)-dosp}4] catalyst gave allylic alcohol 366 with good diastereoselectivity and chemoselectivity over the O H insertion product 367. The yield and diastereoselectivity could be improved by using the enantiomeric form of the catalyst. The example shown demonstrates that even in a “mismatched” case, the catalyst is capable of dominating the control of the reaction stereochemistry.
Scheme 79.
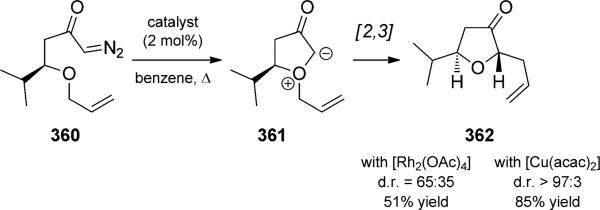
Metal-catalyzed oxonium [2,3] rearrangement duet. [Clark, 1992, Ref. [133]; for first reports see Pirrung, 1982 and Johnson, 1986, Ref. [132]]
Scheme 80.

Use of alcohols in ylide formation/[2,3]-sigmatropic rearrangement duet. [Davies, 2010, Ref. [135]]
Initial forays into catalytic ammonium ylide formation/ [2,3] rearrangement were reported by Doyle and involved intermolecular coupling of allylic amines and metal carbenoids.[127] These reactions also relied on high excess of the allylic amine as solvent, and metal carbenoid dimerization had to be suppressed by slow combination of reagents. An intramolecular reaction to generate cyclic amines was first reported by Clark and co-workers[136] and later explored by McMills and co-workers.[137] This method was used to prepare a bicyclic ring system relevant to the synthesis of manzamine alkaloids (Scheme 81).[136b] Transfer of chirality from the (S)-prolinol building block was quite efficient to generate bicyclic amine 372 in high enantiomeric excess. The stereochemistry and conformation of the ylide is inferred from the product observed.[138]
Scheme 81.

Diastereoselective ammonium ylide formation/[2,3]-sigma-tropic reaction duet. [Clark, 1995, Ref. [136b]]
Hodgson and co-workers demonstrated that ylide formation sequences could be combined in one-pot processes with alkene cross-metathesis reactions.[139] For example, they reported a consecutive reaction trio involving a cross-meta-thesis/oxonium ylide formation/[2,3]-sigmatropic rearrangement to access dihydrofuranones with high diastereoselectivity. Thus, α-diazo-β-ketoesters such as 373 were first treated with Grubbs II catalyst and a substituted alkene, followed by addition of a catalytic amount of rhodium(II) acetate (Scheme 82). Because of instability, the final aldehyde was immediately treated with sodium cyanoborohydride to give hemiketal 377 as an 80:13:4:3 mixture of diastereomers in favor of the desired isomer shown. The observation of allylic transposition indicates that a pericyclic [2,3]-sigmatropic rearrangement pathway is likely.
Scheme 82.

Ruthenium-catalyzed cross-metathesis and rhodium-catalyzed oxonium ylide formation/[2,3]-sigmatropic rearrangement with reduction; a consecutive reaction quartet. [Hodgson, 2008, Ref. [139a]]
In exploring ways to functionalize the C5 position of dpyrones, de March and co-workers utilized a [2,3]-sigmatropic rearrangement of sulfonium ylides in two separate reaction trios.[140] When the parent phenylthiomethyl pyrone (378a, X = H) was treated under rhodium catalysis in the presence of two equivalents of dimethyl diazomalonate, ylide formation was followed by [2,3]-sigmatropic rearrangement (Scheme 83). Under the reaction conditions, the exocylic double bond did not isomerize to form the fully conjugated pyrone, and so underwent a cyclopropanation with dimethyl diazomalonate. Approach of the carbenoid from the less hindered side led diastereoselctively to spirocycle 382. Cyclopropanation could be avoided by using a single equivalent of dimethyl diazomalonate. Conversely, if the bromine analogue was used (378b, X = Br), the exocyclic double bond participated in a [1,3] bromine allylic rearrangement that resulted in the formation of conjugated pyrone 383.
Scheme 83.

Pyrone reaction trios. [de March and Moreno-MaÇas, 1986/1988, Ref. [140]]
The groups of Hoye[141] and Padwa[13] both combined ylide
formation/[2,3]-sigmatropic rearrangement with rhodiumcatalyzed carbene/alkyne metathesis in a number of related reaction trios. Both groups showed that a-diazo ketones bearing tethered alkyne units isomerize under rhodium catalysis to vinyl metal carbenoids (e.g. 385, Scheme 84). These intermediates could be trapped and used in a number of tandem reactions to provide substituted cylopentenones. Oxygen and sulfur ylide representatives of the cylization/ylide formation/[2,3]-sigmatropic rearrangement trio are shown in Scheme 84. As a probe for the mechanism, Hoye used trapping with external sulfides to identify isomeric carbene intermediates resulting from alkyne insertion (not shown).
Scheme 84.

Rhodium-catalyzed alkyne metathesis/[2,3] rearrangement trios. [Padwa, 1995, Ref. [13]; see also Hoye, 1992, Ref. [141]]
A recent report demonstrated an alternative to the general practice of independently preparing diazo compounds for use in the tandem ylide formation/[2,3]-sigma-tropic rearrangement sequence. Murphy and West[142] reported that β-ketoesters with pendant allyl or benzyl ethers or sulfides could be converted in a one-step sequence to cyclic ethers and sulfides. Treatment of β-ketoesters 388a and 388b with phenyl iodonium acetate, cesium carbonate, and rhodium acetate catalyst provided 2-substituted hetero-cycles 391a and 391b, presumably via ylide 390 (Scheme 85). For many of the substrates examined, the yields of this reaction trio were competitive with those obtained by the corresponding two-step diazo preparation/carbene-transfer rearrangement sequence (68% and 55% yields respectively for 388a and 388b). For allyl substrates, the Stevens [1,2] shift was not ruled out.
Scheme 85.

Rhodium-catalyzed ylide formation/[2,3]-sigmatropic rearrangement trio utilizing in situ generation of an iodonium ylide. [West, 2006, Ref. [142]]
Ohe and co-workers showed that related vinyl metal carbenoids could be generated by the cyclization of carbonyl compounds onto tethered alkynes, in a process that precludes the need to independently prepare precursor diazo compounds. Enyne carbonyl compounds, such as 392, in the presence of catalytic rhodium(II) acetate, cyclized to generate an intermediate metal carbenoid (Scheme 86). For example, in the presence of alkenes, cyclopropane products were isolated that are consistent with such presumed carbenoids. In the presence of sulfides however, a Doyle–Kirmse-type reaction, or ylide formation/[2,3]-sigmatropic rearrangement, occured. When performed in the presence of a bisallylic sulfide, a reaction quartet was observed that terminated by an intramolecular Diels–Alder reaction, leading to significantly more complex products, such as 397.[143]
Scheme 86.

Rhodium-catalyzed sulfonium ylide quartet. [Ohe and Uemera, 2003, Ref. [143]]
Onium ylides can be generated using gold catalysts, as demonstrated by the rearrangement quartet reported by Davies and Albrecht.[144] The reaction is initiated by the gold-catalyzed [1,2] migration of propargyl esters, such as 398 (Scheme 87).[69] A postulated gold carbene is generated that can be captured by an allylic sulfide to generate free (or gold-bound) ylide 401. Following [2,3] rearrangement, diene 402 is then poised to undergo Cope rearrangement to produce enol acetate 403 in excellent yield.
If an allyl propargyl sulfide is used, an alternate rearrangement quartet occurs. The previously described ylide formation/[2,3] rearrangement proceeds to presumably generate enyne 406 (Scheme 88).[144] Gold catalyst activation of the alkyne then leads to a carbothiolation and [3,3] rearrangement, similar to that shown in Scheme 63, to generate dihydrothiophene 408. Mechanistic understanding of these types of cycloisomerizations is still evolving.
Tambar and co-workers sought to expand the chemistry beyond diazo compounds for the catalytic generation of ammonium ylides by exploring allylic substitution methods, and recently reported a palladium-catalyzed method involving alkylation of tertiary amines for the generation of ammonium ylides for [2,3] rearrangement.[145] In the first example of the use of tertiary amines as intermolecular nucleophiles in metal-catalyzed allylic substitutions, they showed that carbonates such as 409 could be treated with amines in the presence of [Pd2dba3], phosphine, and base to afford homoallylic amines such as 412 in good yields and high diastereselectivity (Scheme 89). Mechanistic studies suggest that formation of the unstable ammonium intermediate is unfavored, but followed by rapid deprotonation and rearrangement, thus explaining the challenges associated with previous attempts to invoke tertiary amines in allylic alkylation. Tambar also developed asymmetric [2,3] rearrangements of sulfimides and amine-N-oxides, as noted below.
Scheme 89.
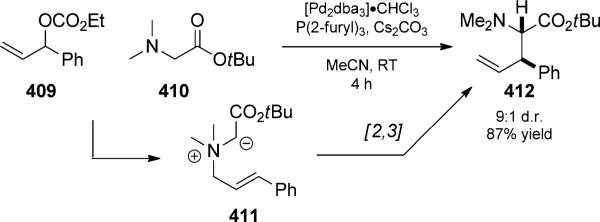
Palladium-catalyzed allylic amination/[2,3] rearrangement duet. [Tambar, 2011, Ref. [145]]
3.2. “Heteroatom Ylides”
In their program to develop new tandem cycloadditions utilizing 1,3 dipoles, Grigg and Markandu explored the in situ generation of nitrones by the [2,3]-sigmatropic rearrangement of O-allyl ethers of oximes.[146] However, thermal conditions afforded low yields of the products of [2,3]-sigmatropic rearrangement/dipolar cycloaddition duets (Scheme 90).
Scheme 90.

Palladium-catalyzed reaction duet: a [2,3]-sigmatropic rearrangement/1,3-dipolar cycloaddition. [Grigg, 1991, Ref. [146]]
The reaction was much more successful when catalyzed by palladium. Treatment of O-allyl ether oxime 413 and 417 with catalytic bis(acetonitrile) dichloropalladium provided bicycle 416 and tricycle 419 from trapping of intermediate nitrones. The lower yield of the latter reaction was attributed to the potential reactivity of aryl allyl ethers with palladium(II) to also form π-allyl species.
A similar reaction that does not involve the generation of reactive intermediate ylides is the [2,3]-sigmatropic rearrangement of sulfinate esters. This rearrangement was used by Mukai and co-workers in a reaction duet for the in situ preparation of a phenylsulfonyl allenyne Pauson–Khand substrate (Scheme 91).[147] Treatment of propargyl sulfinate ester 420 with a rhodium(I) catalyst gave cyclopentindenone 422, which was presumed to arise through carbonylative [2+2+1] ring-closing reaction of phenylsulfonyl allenyne 421. Interestingly, thermal [2,3] rearrangement was unsuccessful, suggesting that rhodium catalysis is in effect. These structures were targeted as possible intermediates in the synthesis of cyanosporasides.
Scheme 91.

Rhodium-catalyzed [2,3]-sigmatropic rearrangement Pauson–Khand reaction duet. [Mukai, 2009, Ref. [147a]]
The Mislow–Evans rearrangement is a commonly used strategy to prepare allylic alcohols (type II, Scheme 77). It involves the [2,3]-sigmatropic rearrangement of allylic sulf-oxides and is frequently performed as a single-pot reaction trio combining oxidation of a sulfide to a sulfoxide, followed by thermal rearrangement and cleavage of the resultant sulfenate. These sequences are rarely catalytic. However, in a selenium analogue of the Mislow–Evans rearrangement, Carter and Bourland showed that aryl selenides such as 424 could be oxidized to the corresponding selenate using catalytic vanadium(IV) acetylacetonate (Scheme 92).[148] Stoichiometric amounts of cumene hydroperoxide reoxidized the vanadium and a final tri n-butyl phosphine quench cleaved the O–Se bond to generate an allylic alcohol, thus completing this reaction trio sequence. Oxidation under Sharpless asymmetric conditions could also be used to asymmetrically generate selenoxides, ultimately leading to enantiomerically enriched allylic alcohols, however the latter reaction trio could not be accomplished catalytically.[149] Related two-pot stoichiometric oxidation/catalytic asymmetric [2,3] rearrangement of amine-N-oxides was recently reported by Tambar and co-workers.[150]
Scheme 92.

Vanadium-initiated selenoxide rearrangement reaction trio. [Carter, 2000, Ref. [148]]
Similarly, the [2,3] rearrangement of allyl sulfimides constitutes a nitrogen analogue of the Mislow–Evans rearrangement. Sulfimides can be generated catalytically to initiate tandem rearrangement sequences. In a report by Murakami and Katsuki, use of an enantiopure ruthenium(II) complex initiated an asymmetric rearrangement trio that was terminated by basic hydrolysis to generate enantiomerically enriched allylic amines.[151] Thus, treatment of allylic sulfide 357 with tosyl azide, molecular sieves, and [(CO)Ru(salen)] catalyst 431 led initially to sulfimide 428 (Scheme 93). Rearrangement gave sulfonamide 429. Basic cleavage of the nitrogen sulfer bond afforded allylic amines of type 430 in good yields and enantiomeric excess. This transformation is an improvement on a copper-catalyzed sulfimidation rearrangement duet.[152] Iron(II) is also a competent catalyst.[153] Two-pot sulfimide formation/[2,3] rearrangements were recently reported in a catalytic, enantioselective fashion by Tambar and co-workers.[154]
Scheme 93.

Ruthenium-catalyzed sulfimidation/[2,3]-sigmatropic reaction trio. [Katsuik, 2002, Ref. [151]]
A catalytically initiated tandem sequence that has been frequently utilized in total synthesis is the Knoevenagel reaction/Mislow–Evans rearrangement originally reported by Nokami.[155,156] Although the Knoevenagel reaction is typically induced with a stoichiometric amount of amine, the amine is a chemically catalytic reagent. A recent example of this reaction quartet utilized the solid support N,N-diethylaminopropylated silica gel (NDEAP) as a stable and recoverable catalyst (Scheme 94).[157] Knoevenagel reaction between citronellal and arylsulfinylacetonitrile 433 generated a vinyl sulfoxide that was in equilibrium with allyl sulfoxide 435. [2,3]-Sigmatropic Mislow–Evans rearrangement and sulfenate cleavage generated allylic alcohol 437 as a mixture of diastereomers. The Knoevenagal reaction can itself be considered a reaction duet: aldol addition followed by dehydration. Perhaps the aforementioned sequence would be more appropriately called a reaction quintet!
Scheme 94.

Knoevenagel/Mislow–Evans reaction quintet. [Hagiwara, 2006, Ref. [157]]
4. Miscellaneous [X,Y] Rearrangements
A number of other [X,Y]-sigmatropic rearrangements are thermally and photochemically allowed; however, their involvement can be more difficult to detect and distinguish from simple proton-transfer processes. Additionally, they often occur under similar conditions and in competition with other rearrangements, and rarely exist independently as distinct, selective processes. They do however represent an important area of exploration. In Scheme 38 and 48, the [1,3] rearrangement from oxygen to carbon was a competing process in Claisen rearrangements, and Rovis and Nasveshuk, for example, specifically explored optimizing it for use in stereoselective synthesis.[158] Only a few catalytic examples in tandem reactions are presented here.
With substrates related to those in Scheme 78–80, but lacking the pendant olefin, ammonium ylides can be generated from cyclic amines (439, Scheme 95). Spirocyclic inter mediates then undergo [1,2] shift, also known as the Stevens rearrangement, with concomitant ring expansion. The groups of Padwa, Saba, and West each utilized such a duet to prepare fused bicyclic frameworks.[159]
Scheme 95.

Padwa's ammonium ylide/[1,2] rearrangement duet. [Padwa, 2001, Ref. [159]]
In an effort to expand the chemistry of metallonitrenes into the broader realm of reactions such as those displayed by analogous metallocarbenes, Thornton and Blakey reported a metallonitrene/alkyne metathesis reaction quartet.[160] Thus, treatment of simple linear sulfamate ester (441) with phenyl iodonium acetate and a rhodium(II) catalyst followed by reductive workup provided good yields of bicyclic oxathiaze-panes (447, Scheme 96). The sequence is believed to proceed by alkyne attack on an electrophile rhodium nitrene leading potentially to a vinyl cation that isomerizes to an imine metallocarbene (443). The metallocarbene can then be captured by the pendant allyl ether to generate an ylide (444) that is poised for a sigmatropic rearrangement, likely a [1,2] rearrangement, since benzyl ethers are also competent. Although the resultant N-sulfonyl imines could be isolated, they were sensitive to hydrolysis upon purification. It was shown to be operationally convenient to reduce them in situ or capture them with an organometallic reagent in reactions that produce a single diastereomer.
Scheme 96.

Rhodium-catalyzed isomerization of metallonitrenes to metallocarbenes; a reaction quartet to efficiently convert simple alkynes to complex bicyclic structures. [Rh2(esp)2]=Rh2(a,a,a’,a'-tetramethyl-1,3-benzenedipropionate) [Blakey, 2008, Ref. [160]]
Under platinum catalysis, Liang and co-workers observed a [1,5] shift that followed [3,3] propargylic ester rearrangement (a process discussed in Section 2.2.3).[161] Specifically, propargylic ester 448 was proposed to first rearrange to allenyl ester 449 (two diastereomers possible, Scheme 97). A [1,5] shift leads to the formation of triene 450, which then undergoes 6p cyclization to give naphthyl acetate 451.
Scheme 97.

Platinum-catalyzed [1,5]-H-shift trio. [Liang, 2008, Ref. [161]]
In their work on the synthetic elaboration of N-protected bromomethylindoles, Mohanakrishnan and co-workers reported a ZnBr2-mediated arylation/[1,5]-sigmatropic rearrangement trio.[162] The domino sequence was initiated by the zinc(II)-mediated arylation of bromomethylindole 452 with anisole, a reaction that likely proceeds through a Friedel– Crafts-type mechanism, and so is included under our umbrella definition of catalysis (Scheme 98). A [1,5]-sigmatropic shift gave hexatriene 455, which then underwent a 6p electro-cyclization. Elimination of malonate with rearomatization provided the annulated product 457.
Scheme 98.

Zinc(II)-mediated arylation/sigmatropic rearrangement trio. [Mohanakrishnan, 2009, Ref. [162]]
Expanding on their work with rhodium carbenoids (see also Scheme 2 and Scheme 80), Davies and co-workers explored catalytic enantioselective C–H activations. Reactions involving dienes and vinyldiazoacetates resulted in nonconjugated products, revealing a new mechanistic process, the C–H activation/Cope rearrangement (for example 458 and 459 to 460, Scheme 99).[163] On its own, it is not a reaction duet, but rather a new reaction type that combines C–H activation with Cope rearrangement in a concerted process (see scheme for the proposed transition state). In the reaction of naphthalenes with phenyl-substituted vinyldiazoacetate 363, the reaction is followed by a retro-Cope rearrangement to form 462, the expected product of direct C–H insertion.[163b] The much higher diastereoselectivity observed compared to other direct C–H insertions provided evidence for this “hidden” reaction duet. Davies demonstrated the utility of the C–H activation/Cope rearrangement in total synthesis.[163c] The novel process is another hint of the exciting possibilities that still remain in synthetic chemistry for harnessing new types of reactivity.
Scheme 99.

Rhodium-catalyzed C H activation Cope rearrangement and C–H activation Cope/retro-Cope rearrangement duet. [Davies, 2004, Ref. [163b]]
5. Outlook
Although the reactions presented herein range from the very simple to the very complex, they were each made possible and became more versatile when chemists combined a strong mechanistic understanding of reactivity and selectivity with a consideration of possible influences in more complicated systems. Oftentimes, great successes result from an appreciation for and the pursuit of “unexpected” products. It is in such by-products that tandem processes are often revealed. As chemists strive for increasing levels of control, for each catalyst or reagent to accomplish more, advancement will surely depend upon both artistic creativity in substrate design and systematic mechanistic analysis. By using tandem reactions, we should be able to engineer contexts where multiple pathways are possible, but only the desired pathway proceeds. In some cases, this will involve a multitude of compatible reagents carrying out different processes; in others, the catalysts themselves will prove capable of mediating multiple different processes, as in “concurrent catalysis.”[164] Such total understanding of conformational elements, reaction selectivity, and catalyst compatibility would allow us to engage a single simple substrate and direct it to rearrange by a specific reaction sequence to the complex product of choice. Although outside the scope of our review, we take inspiration from the Heathcock syntheses in the daphniphyllum alkaloid series, where by judicious choice of starting material resulted in the molecule curling upon itself to assemble a structure mere steps away from the desired natural product.[165] As catalyst activity and control of selectivity in cascade processes becomes even better and more routine, synthetic chemists will have unprecedented success in creat ing intricate structures in only a few steps and with extreme efficiency.
Acknowledgments
We thank Dr. Wen-Bo Liu and Nathan Bennett for helpful comments and editing. We also thank NIH-NIGMS (R01GM080269), Amgen, Abbott, Boehringer Ingelheim, ACS Organic Division (fellowship for J.A.M), Bristol-Meyers Squibb (fellowship for J.A.M), Pfizer-UNCF (fellowship for R.S.), and Caltech for financial support. A.C.J. thanks Wake Forest University for start-up support and NIH-NRSA (F32GM082000) for support of a postdoctoral fellowship.
Biography
 Amanda C. Jones was born in Evanston, IL in 1979. She obtained an A.B. in chemistry from Princeton University in 2001 where she worked with Maitland Jones, Jr. In 2007, she obtained her Ph.D. from the University of Wisconsin under the direction of Hans J. Reich. She was an NIH postdoctoral scholar with Brian M. Stoltz from 2007–2010. In 2010, she started her independent career at Wake Forest University, where her research is focused on mechanistic organometallic chemistry and NMR spectroscopy.
Amanda C. Jones was born in Evanston, IL in 1979. She obtained an A.B. in chemistry from Princeton University in 2001 where she worked with Maitland Jones, Jr. In 2007, she obtained her Ph.D. from the University of Wisconsin under the direction of Hans J. Reich. She was an NIH postdoctoral scholar with Brian M. Stoltz from 2007–2010. In 2010, she started her independent career at Wake Forest University, where her research is focused on mechanistic organometallic chemistry and NMR spectroscopy.
 Jeremy A. May was born in Bozeman, MT in 1975. He obtained his B.S. in chemistry from the University of Utah in 2000 where he worked for Peter Beal. In 2006, he obtained his Ph.D. from Caltech under the direction of Brian M. Stoltz. He was an NIH postdoctoral fellow in the group of Samuel J. Danishefksy at the Memorial Sloan Kettering Cancer Center from 2005– 2008. In 2008, he began his independent career at the University of Houston, where his research is focused on natural product synthesis and methods development related to unsaturated and low-valent carbon.
Jeremy A. May was born in Bozeman, MT in 1975. He obtained his B.S. in chemistry from the University of Utah in 2000 where he worked for Peter Beal. In 2006, he obtained his Ph.D. from Caltech under the direction of Brian M. Stoltz. He was an NIH postdoctoral fellow in the group of Samuel J. Danishefksy at the Memorial Sloan Kettering Cancer Center from 2005– 2008. In 2008, he began his independent career at the University of Houston, where his research is focused on natural product synthesis and methods development related to unsaturated and low-valent carbon.
 Richmond Sarpong was born in 1974 in Bechem, Ghana. He obtained his B.A. from Macalester College in 1995, where he worked with Rebecca C. Hoye. In 2001, he obtained his Ph.D. from Princeton University, under the direction of Martin F. Semmelhack. He was a UNCF-Pfizer postdoctoral fellow in the group of Brian M. Stoltz at Caltech from 2001–2004. In 2004, he began his independent career at the University of California-Berkeley. His research is focused on the total synthesis of biologically active and complex natural products as a platform for the development of new synthetic methods and strategies.
Richmond Sarpong was born in 1974 in Bechem, Ghana. He obtained his B.A. from Macalester College in 1995, where he worked with Rebecca C. Hoye. In 2001, he obtained his Ph.D. from Princeton University, under the direction of Martin F. Semmelhack. He was a UNCF-Pfizer postdoctoral fellow in the group of Brian M. Stoltz at Caltech from 2001–2004. In 2004, he began his independent career at the University of California-Berkeley. His research is focused on the total synthesis of biologically active and complex natural products as a platform for the development of new synthetic methods and strategies.
 Brian M. Stoltz was born in Philadelphia, Pennsylvania, USA in 1970. He obtained his B.S. in Chemistry and B.A. in German from Indiana University of Pennsylvania in 1993. In 1997, he earned his Ph.D. under the direction of Professor John L. Wood at Yale University, specializing in synthetic organic chemistry. Following an NIH postdoctoral fellowship with Professor E. J. Corey at Harvard University (1998–2000), he joined the faculty at Caltech in 2000. His research focuses on the synthesis of complex molecules with important biological properties and the development of new synthetic methods including asymmetric catalysis and cascade processes.
Brian M. Stoltz was born in Philadelphia, Pennsylvania, USA in 1970. He obtained his B.S. in Chemistry and B.A. in German from Indiana University of Pennsylvania in 1993. In 1997, he earned his Ph.D. under the direction of Professor John L. Wood at Yale University, specializing in synthetic organic chemistry. Following an NIH postdoctoral fellowship with Professor E. J. Corey at Harvard University (1998–2000), he joined the faculty at Caltech in 2000. His research focuses on the synthesis of complex molecules with important biological properties and the development of new synthetic methods including asymmetric catalysis and cascade processes.
Footnotes
Sometimes the assessment of beauty is nearly unanimous. The first four notes of Beethoven's 5th Symphony (Symphony No.5 in C minor, Op.67) represent perhaps the most well-known and popular motif in classical music. The orchestral score is shown in the background of the cover graphic. Accessed March 20, 2013 from http://imslp.org/wiki/Symphony_No.5,_Op.67_%28Beethoven,_ Ludwig_van%29.
We thank one referee both for his or her close examination of our manuscript, but also for their careful consideration of our choice of language. We appreciate the insight that in musical ensembles, each “member usually contributes their sound at an identical moment in time, not one after the other. In contrast, a 1-pot reaction sequence is composed of a series of transformations, which occur consecutively.” We believe that part of the success of these reactions is the requisite compatibility and selectivity of multiple reagents and functionalities. Part of group music, for sure, is “waiting your turn.” At times an individual musician must remain silent; similarly, the success of these reactions relies on the minimization of other reaction pathways. Although our duet, trio, etc. terminology counts reaction steps as ensemble members (rather than the acting reagents themselves), we have chosen to stay faithful to our original conversation with Nelson Leonard. We invite the community to continue discussing the best way to characterize these reaction cascades.
Contributor Information
Amanda C. Jones, Chemistry Department, Wake Forest University Box 7486, Winston Salem, NC 27109 (USA).
Jeremy A. May, Department of Chemistry, University of Houston Houston, TX 77204-5003 (USA)
Richmond Sarpong, Department of Chemistry, University of California Berkeley, CA 94720 (USA).
Brian M. Stoltz, Department of Chemistry and Chemical Engineering, California Institute of Technology MC 101-20, Pasadena CA 91125 (USA) stoltz@caltech.edu
References
- 1.Tietze LF, Beifuss U. Angew. Chem. 1993;105:137–170. [Google Scholar]; Angew. Chem. Int. Ed. Engl. 1993;32:131–163. [Google Scholar]
- 2.a Ichikawa K, Nonaka K, Matsumoto T, Kure B, Yoon K, Higuchi Y, Yagi T, Ogo S. Dalton Trans. 2010;39:2993–2994. doi: 10.1039/b926061g. [DOI] [PubMed] [Google Scholar]; b Ikariya T. Bull. Chem. Soc. Jpn. 2011;84:1–16. [Google Scholar]
- 3.Hoffmann R, Woodward RB. Acc. Chem. Res. 1968;1:17–22. [Google Scholar]
- 4.For general reviews of tandem chemistry, see: Anderson EA. Org. Biomol. Chem. 2011;9:3997–4006. doi: 10.1039/c1ob05212h. Chapman CJ, Frost CG. Synthesis. 2007:1–21. Nicolaou KC, Edmonds DJ, Bulger PG. Angew. Chem. 2006;118:7292–7344. doi: 10.1002/anie.200601872. Angew. Chem. Int. Ed. 2006;45:7134–7186. doi: 10.1002/anie.200601872. Pellissier H. Tetrahedron. 2006;62:2143–2173. Fogg DE, dos Santos EN. Coord. Chem. Rev. 2004;248:2365–2379. Tietze LF. Chem. Rev. 1996;96:115–136. doi: 10.1021/cr950027e. Bunce RA. Tetrahedron. 1995;51:13103–13159. Hoffmann HMR. Angew. Chem. 1992;104:1361–1363. Angew. Chem. Int. Ed. Engl. 1992;31:1332–1334. for additional reviews involving pericyclic reactions, see: Neuschütz K, Velker J, Neier R. Synthesis. 1998:227–255. Denmark SE, Thorarensen A. Chem. Rev. 1996;96:137–165. doi: 10.1021/cr940277f.
- 5.Cope AC, Hardy EM. J. Am. Chem. Soc. 1940;62:441–444. [Google Scholar]
- 6.a Enders D, Knopp M, Schiffers R. Tetrahedron: Asymmetry. 1996;7:1847–1882. [Google Scholar]; b Nubbemeyer U. Synthesis. 2003:961–1008. [Google Scholar]
- 7.a Bramley RK, Grigg R. J. Chem. Soc. D. 1969:99b–100. [Google Scholar]; b Kawasaki T, Nonaka Y, Watanabe K, Ogawa A, Higuchi K, Terashima R, Masuda K, Sakamoto M. J. Org. Chem. 2001;66:1200–1204. doi: 10.1021/jo0014921. [DOI] [PubMed] [Google Scholar]; c Miller B. J. Am. Chem. Soc. 1965;87:5115–5120. [Google Scholar]
- 8.a Ganem B. Angew. Chem. 1996;108:1014–1023. [Google Scholar]; Angew. Chem. Int. Ed. Engl. 1996;35:936–945. [Google Scholar]; b Overman LE. Angew. Chem. Angew Chem. 1984;96:563–573. [Google Scholar]; Angew. Chem. 1984;96:565–573. [Google Scholar]; Angew. Chem. Int. Ed. Engl. 1984;23:579–586. [Google Scholar]; c Schenck TG, Bosnich B. J. Am. Chem. Soc. 1985;107:2058–2066. [Google Scholar]; d Lutz RP. Chem. Rev. 1984;84:205–247. [Google Scholar]
- 9.Brown JM, Golding BT, Stofko JJ. Chem. Commun. 1973:319–320. [Google Scholar]
- 10.a Davies HML, Clark DM, Smith TK. Tetrahedron Lett. 1985;26:5659–5662. [Google Scholar]; b Davies HML, Smith HD, Korkor O. Tetrahedron Lett. 1987;28:1853–1856. [Google Scholar]; c Schwartz BD, Denton JR, Lian Y, Davies HML, Williams CM. J. Am. Chem. Soc. 2009;131:8329–8332. doi: 10.1021/ja9019484. and references therein. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Felix RJ, Weber D, Gutierrez O, Tantillo DJ, Gagn MR. Nat. Chem. 2012;4:405–409. doi: 10.1038/nchem.1327. During final preparations of this manuscript, a gold-catalyzed enantioselective Cope rearrangement was reported. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Miller LC, Ndugnu JM, Sarpong R. Angew. Chem. 2009;121:2434–2438. [Google Scholar]; Angew. Chem. Int. Ed. 2009;48:2398–2402. doi: 10.1002/anie.200806154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.a Padwa A, Kassir JM, Semones MA, Weingarten MD. J. Org. Chem. 1995;60:53–62. [Google Scholar]; b Padwa A. J. Organomet. Chem. 2001;617:3–16. and references therein. [Google Scholar]; c Padwa A, Krumpe KE, Gareau Y, Chiacchio U. J. Org. Chem. 1991;56:2523–2530. [Google Scholar]
- 14.Ni Y, Montgomery J. J. Am. Chem. Soc. 2006;128:2609–2614. doi: 10.1021/ja057741q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.a Kim SY, Park Y, Chung YK. Angew. Chem. 2010;122:425–428. doi: 10.1002/anie.200905361. [DOI] [PubMed] [Google Scholar]; Angew. Chem. Int. Ed. 2010;49:415–418. doi: 10.1002/anie.200905361. [DOI] [PubMed] [Google Scholar]; b Kusama H, Onizawa Y, Iwasawa N. J. Am. Chem. Soc. 2006;128:16500–16501. doi: 10.1021/ja0671924. [DOI] [PubMed] [Google Scholar]
- 16.F rstner A, Davies PW. Angew. Chem. 2007;119:3478–3519. [Google Scholar]; Angew. Chem. Int. Ed. 2007;46:3410–3449. doi: 10.1002/anie.200604335. [DOI] [PubMed] [Google Scholar]
- 17.Hashmi ASK. Angew. Chem. 2008;120:6856–6858. [Google Scholar]; Angew. Chem. Int. Ed. 2008;47:6754–6756. doi: 10.1002/anie.200802517. [DOI] [PubMed] [Google Scholar]
- 18.a Sarpong R, Su JT, Stoltz BM. J. Am. Chem. Soc. 2003;125:13624–13625. doi: 10.1021/ja037587c. [DOI] [PubMed] [Google Scholar]; b Su JT, Sarpong R, Stoltz BM, Goddard WA., III J. Am. Chem. Soc. 2004;126:24–25. doi: 10.1021/ja037716p. [DOI] [PubMed] [Google Scholar]
- 19.a Limanto J, Snapper ML. J. Am. Chem. Soc. 2000;122:8071–8072. [Google Scholar]; b Snapper ML, Tallarico JA, Randall ML. J. Am. Chem. Soc. 1997;119:1478–1479. [Google Scholar]
- 20.a Yeo S, Hatae N, Seki M, Kanematsu K. Tetrahedron. 1995;51:3499–3506. [Google Scholar]; b Hayakawa K, Aso K, Shiro M, Kanematsu K. J. Am. Chem. Soc. 1989;111:5312–5320. [Google Scholar]
- 21.a Berson JA, Jones M., Jr. J. Am. Chem. Soc. 1964;86:5019–5020. [Google Scholar]; b Berson JA, Walsh EJ., Jr. J. Am. Chem. Soc. 1968;90:4730–4732. [Google Scholar]; c Evans DA, Golob AM. J. Am. Chem. Soc. 1975;97:4765–4766. [Google Scholar]
- 22.Arns S, Barriault L. Chem. Commun. 2007:2211–2221. doi: 10.1039/b700054p. [DOI] [PubMed] [Google Scholar]
- 23.Tice CM, Heathcock CH. J. Org. Chem. 1981;46:9–13. [Google Scholar]
- 24.Balakumar A, Seelvasekaran J, Rajagopalan K. J. Org. Chem. 1993;58:5482–5486. [Google Scholar]
- 25.Jacobi PA, Armacost LM, Brielmann HL, Cann RO, Kravitz JI, Martinelli MJ. J. Org. Chem. 1994;59:5292–5304. [Google Scholar]
- 26.Hussaini SS, Raj ARN, Huq CAMA. Tetrahedron Lett. 2007;48:775–778. [Google Scholar]
- 27.a Bio MM, Leighton JL. J. Org. Chem. 2003;68:1693–1700. doi: 10.1021/jo026478y. and references therein. [DOI] [PubMed] [Google Scholar]; b Bio MM, Leighton JL. Org. Lett. 2000;2:2905–2907. doi: 10.1021/ol0063609. [DOI] [PubMed] [Google Scholar]
- 28.a Chiang P-C, Kaeobamrung J, Bode JW. J. Am. Chem. Soc. 2007;129:3520–3521. doi: 10.1021/ja0705543. [DOI] [PubMed] [Google Scholar]; b He M, Bode JW. J. Am. Chem. Soc. 2008;130:418–419. doi: 10.1021/ja0778592. [DOI] [PubMed] [Google Scholar]
- 29.a Kuehne ME, Xu F. J. Org. Chem. 1998;63:9427–9433. [Google Scholar]; b Kuehne ME, Xu F. J. Org. Chem. 1997;62:7950–7960. doi: 10.1021/jo970207j. [DOI] [PubMed] [Google Scholar]
- 30.Waetzig SR, Rayabarapu DK, Weaver JD, Tunge JA. Angew. Chem. 2006;118:5099–5102. doi: 10.1002/anie.200600721. [DOI] [PubMed] [Google Scholar]; Angew. Chem. Int. Ed. 2006;45:4977–4980. doi: 10.1002/anie.200600721. [DOI] [PubMed] [Google Scholar]
- 31.Dauben WG, Chollet A. Tetrahedron Lett. 1981;22:1583–1586. [Google Scholar]
- 32.Krohn K, Bernhard S. Synthesis. 1996:699–701. [Google Scholar]
- 33.Claisen L. Chem. Ber. 1912;45:6157–3166. [Google Scholar]
- 34.a Majumdar KC, Safiul A, Chattopadhyay B. Tetrahedron. 2008;64:597–643. [Google Scholar]; b Martín Castro AM. Chem. Rev. 2004;104:2939–3002. doi: 10.1021/cr020703u. [DOI] [PubMed] [Google Scholar]; c Hiersemann M, Abraham L. Eur. J. Org. Chem. 2002:1461–1471. [Google Scholar]
- 35.Nissen A, Rebafka W, Aquila W. United States Patent. :4288636. 09/08/, 1981. [Google Scholar]
- 36.Gester S, Metz P, Zierau O, Vollmer G. Tetrahedron. 2001;57:1015–1018. [Google Scholar]
- 37.a Nicolaou KC, Li J. Angew. Chem. 2001;113:4394–4398. [Google Scholar]; Angew. Chem. Int. Ed. 2001;40:4264–4268. doi: 10.1002/1521-3773(20011119)40:22<4264::AID-ANIE4264>3.0.CO;2-1. [DOI] [PubMed] [Google Scholar]; b Tisdale EJ, Slobodov I, Theodorakis EA. Org. Biomol. Chem. 2003;1:4418–4422. doi: 10.1039/b311833a. [DOI] [PubMed] [Google Scholar]
- 38.a Uzawa H, Hiratani K, Minoura N, Takahashi T. Chem. Lett. 1998:307–308. [Google Scholar]; b Hiratani K, Albrecht M. Chem. Soc. Rev. 2008;37:2413–2421. doi: 10.1039/b719548f. For a review on the thermal tandem Claisen rearrangement in supramolecular chemistry, see. [DOI] [PubMed] [Google Scholar]
- 39.Tanimoto H, Saito R, Chida N. Tetrahedron Lett. 2008;49:358–362. [Google Scholar]
- 40.Hanessian S, Compain P. Tetrahedron. 2002;58:6521–6529. [Google Scholar]
- 41.Davies HML, Dai X. J. Org. Chem. 2005;70:6680–6684. doi: 10.1021/jo050821s. [DOI] [PubMed] [Google Scholar]
- 42.Schoepfer J, Marquis C, Pasquier C, Neier R. Chem. Commun. 1994:1001–1002. [Google Scholar]
- 43.Mulzer J, Bock H, Eck W, Buschmann J, Luger P. Angew. Chem. 1991;103:450–452. [Google Scholar]; Angew. Chem. Int. Ed. Engl. 1991;30:414–416. [Google Scholar]
- 44.Kaden S, Hiersemann M. Synlett. 2002:1999–2002. [Google Scholar]
- 45.Johnson WS, Werthemann L, Bartlett WR, Brocksom TJ, Li T-T, Faulkner DJ, Petersen MR. J. Am. Chem. Soc. 1970;92:741–743. [Google Scholar]
- 46.Nordmann G, Buchwald SL. J. Am. Chem. Soc. 2003;125:4978–4979. doi: 10.1021/ja034809y. [DOI] [PubMed] [Google Scholar]
- 47.Wei X, Lorenz JC, Kapadia S, Saha A, Haddad N, Busacca CA, Senanayake CH. J. Org. Chem. 2007;72:4250–4253. doi: 10.1021/jo062548f. [DOI] [PubMed] [Google Scholar]
- 48.a Wood JL, Moniz GA. Org. Lett. 1999;1:371–374. doi: 10.1021/ol990697x. [DOI] [PubMed] [Google Scholar]; b Drutu I, Krygowski ES, Wood JL. J. Org. Chem. 2001;66:7025–7029. doi: 10.1021/jo015741c. [DOI] [PubMed] [Google Scholar]; c Wood JL, Holubec AA, Stoltz BM, Weiss MM, Dixon JA, Doan BD, Shamji MF, Chen JM, Heffron TP. J. Am. Chem. Soc. 1999;121:6326–6327. [Google Scholar]; d Wood JL, Stoltz BM, Dietrich H-J, Pflum DA, Petsch DT. J. Am. Chem. Soc. 1997;119:9641–9651. [Google Scholar]
- 49.Trost BM, Dyker G, Kulawiec RJ. J. Am. Chem. Soc. 1990;112:7809–7811. [Google Scholar]
- 50.May JA, Stoltz BM. J. Am. Chem. Soc. 2002;124:12426–12427. doi: 10.1021/ja028020j. [DOI] [PubMed] [Google Scholar]
- 51.Reuter JM, Salomon RG. J. Org. Chem. 1977;42:3360–3364. [Google Scholar]
- 52.a Le Nôtre J, Brissieux L, Sémeril D, Bruneau C, Dixneuf PH. Chem. Commun. 2002:1772–1773. doi: 10.1039/b205805g. [DOI] [PubMed] [Google Scholar]; b Ammar HB, Le Nôtre J, Salem M, Kaddachi MT, Toupet L, Renaud J-L, Bruneau C, Dixneuf PH. Eur. J. Inorg. Chem. 2003;22:4055–4064. [Google Scholar]; c Ammar HN, Le Nôtre J, Salem M, Kaddachi MT, Dixneuf PH. J. Organomet. Chem. 2002;662:63–69. [Google Scholar]; d Le Nôtre J, Touzani R, Lavastre O, Bruneau C, Dixneuf PH. Adv. Synth. Catal. 2005;347:783–791. [Google Scholar]
- 53.a Schmidt B. Synlett. 2004:1541–1544. [Google Scholar]; b Schmidt B, Wildemann H. Eur. J. Org. Chem. 2000:3145–3163. doi: 10.1021/jo005534x. [DOI] [PubMed] [Google Scholar]
- 54.Higashino T, Sakaguchi S, Ishii Y. Org. Lett. 2000;2:4193–4195. doi: 10.1021/ol0067437. [DOI] [PubMed] [Google Scholar]
- 55.Swenton JS, Bradin D, Gates BD. J. Org. Chem. 1991;56:6156–6163. [Google Scholar]
- 56.a Nelson SG, Wang K. J. Am. Chem. Soc. 2006;128:4232–4233. doi: 10.1021/ja058172p. [DOI] [PubMed] [Google Scholar]; b Stevens BD, Nelson SG. J. Org. Chem. 2005;70:4375–4379. doi: 10.1021/jo050225y. [DOI] [PubMed] [Google Scholar]; c Nelson SG, Bungard CJ, Wang K. J. Am. Chem. Soc. 2003;125:13000–13001. doi: 10.1021/ja037655v. [DOI] [PubMed] [Google Scholar]
- 57.Tanaka K, Okazaki E, Shibata Y. J. Am. Chem. Soc. 2009;131:10822–10823. doi: 10.1021/ja9038449. [DOI] [PubMed] [Google Scholar]
- 58.a Eilbracht P, Gersmeier A, Lennartz D, Huber T. Synthesis. 1995:330–334. [Google Scholar]; b Sattelkau T, Eilbracht P. Tetrahedron Lett. 1998;39:1905–1908. [Google Scholar]; c Sattelkau T, Eilbracht P. Tetrahedron Lett. 1998;39:9647–9648. [Google Scholar]; d Sattelkau T, Hollmann C, Eilbracht P. Synlett. 1996:1221–1223. [Google Scholar]; e Dygutsch DP, Eilbracht P. Tetrahedron. 1996;52:5461–5468. [Google Scholar]
- 59.Li X, Keon AE, Sullivan JA, Ovaska TV. Org. Lett. 2008;10:3287–3290. doi: 10.1021/ol8011343. and references therein. [DOI] [PubMed] [Google Scholar]
- 60.Nakamura I, Bajracharya GB, Yamamoto Y. Chem. Lett. 2005;34:174–175. [Google Scholar]
- 61.Lee SI, Fukumoto Y, Chatani N. Chem. Commun. 2010;46:3345–3347. doi: 10.1039/c000747a. [DOI] [PubMed] [Google Scholar]
- 62.a Kim KM, Kim HR, Ryu EK. Heterocycles. 1993;36:497–505. [Google Scholar]; b Kim HJ, Seo G, Kim J, Choic KH. Bull. Korean Chem. Soc. 2004;25:1726–1728. [Google Scholar]; c Reich NW, Yang C, Shi Z, He C. Synthesis. 2006:1278–1280. [Google Scholar]; d Bernard AM, Cocco MT, Onnis V, Piras PP. Synthesis. 1997:41–43. [Google Scholar]; e Grant VH, Liu B. Tetrahedron Lett. 2005;46:1237–1239. [Google Scholar]; f Saidi MR. Heterocycles. 1982;19:1473–1475. [Google Scholar]; g Feoktistov VM, Bunina-Krivorukova LI, Bal'yan KV. Zh. Org. Khim. 1978;14:807–811. [Google Scholar]; h Majumdar KC, Chattopadhyay SK. Can. J. Chem. 2006;84:469–475. [Google Scholar]
- 63.a Adrio LA, Hii KK(M) Chem. Commun. 2008:2325–2327. doi: 10.1039/b719465j. [DOI] [PubMed] [Google Scholar]; b Nguyen R, Yao Z, Li C. Org. Lett. 2006;8:2397–2399. doi: 10.1021/ol0607692. [DOI] [PubMed] [Google Scholar]; c Youn SW, Eom JI. J. Org. Chem. 2006;71:6705–6707. doi: 10.1021/jo061221b. [DOI] [PubMed] [Google Scholar]; d Matsui M, Yamamoto H. Bull. Chem. Soc. Jpn. 1995;68:2657–2661. [Google Scholar]; e Ahluwalia VK, Karora K. J. Chem. Soc. Perkin Trans. 1982;1:335–338. [Google Scholar]; f Bigi F, Carlonia S, Maggi R, Muchetti C, Rastelli M, Sartori G. Synthesis. 1998:301–304. [Google Scholar]
- 64.Binder JT, Kirsch SF. Org. Lett. 2006;8:2151–2153. doi: 10.1021/ol060664z. [DOI] [PubMed] [Google Scholar]
- 65.a Sherry BD, Maus L, Laforteza BN, Toste FD. J. Am. Chem. Soc. 2006;128:8132–8133. doi: 10.1021/ja061344d. [DOI] [PubMed] [Google Scholar]; b Sherry BD, Toste FD. J. Am. Chem. Soc. 2004;126:15978–15979. doi: 10.1021/ja044602k. [DOI] [PubMed] [Google Scholar]
- 66.Saito A, Konishi T, Hanzawa Y. Org. Lett. 2010;12:372–374. doi: 10.1021/ol902716n. [DOI] [PubMed] [Google Scholar]
- 67.Saito A, Oda S, Fukaya H, Hanzawa Y. J. Org. Chem. 2009;74:1517–1524. doi: 10.1021/jo8022523. [DOI] [PubMed] [Google Scholar]
- 68.Liu YK, Qian JQ, Lou SJ, Xu ZY. Synlett. 2009:2971–2976. [Google Scholar]
- 69.Marion N, Nolan SP. Angew. Chem. 2007;119:2806–2809. [Google Scholar]; Angew. Chem. Int. Ed. 2007;46:2750–2752. doi: 10.1002/anie.200604773. [DOI] [PubMed] [Google Scholar]
- 70.Shu X-Z, Shu D, Schienebeck CM, Tang W. Chem. Soc. Rev. 2012;41:7698–7711. doi: 10.1039/c2cs35235d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Maule n P, Krinsky JL, Toste FD. J. Am. Chem. Soc. 2009;131:5413–5420. doi: 10.1021/ja900456m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.a Yeom H-S, Yoon S-J, Shin S. Tetrahedron Lett. 2007;48:4817–4820. [Google Scholar]; b Buzas A, Istrate F, Gagosz F. Org. Lett. 2006;8:1957–1959. doi: 10.1021/ol0606839. [DOI] [PubMed] [Google Scholar]
- 73.a Zhang L, Wang S. J. Am. Chem. Soc. 2006;128:1442–1443. doi: 10.1021/ja057327q. [DOI] [PubMed] [Google Scholar]; b Zhang L. J. Am. Chem. Soc. 2005;127:16804–16805. doi: 10.1021/ja056419c. [DOI] [PubMed] [Google Scholar]
- 74.a Wang S, Zhang L. Org. Lett. 2006;8:4585–4587. doi: 10.1021/ol0618151. [DOI] [PubMed] [Google Scholar]; b Wang S, Zhang L. J. Am. Chem. Soc. 2006;128:8414–8415. doi: 10.1021/ja062777j. [DOI] [PubMed] [Google Scholar]; c Barluenga J, Riesgo L, Vicente R, Lopez L, Tomas MJ. J. Am. Chem. Soc. 2007;129:7772–7773. doi: 10.1021/ja072864r. [DOI] [PubMed] [Google Scholar]
- 75.Buzas A, Gagosz F. J. Am. Chem. Soc. 2006;128:12614–12615. doi: 10.1021/ja064223m. [DOI] [PubMed] [Google Scholar]
- 76.Wang S, Zhang G, Zhang L. Synlett. 2010:692–706. [Google Scholar]
- 77.Marco-Contelles J, Soriano E. Chem. Eur. J. 2007;13:1350–1357. doi: 10.1002/chem.200601522. [DOI] [PubMed] [Google Scholar]
- 78.Schwier T, Sromek AW, Yap DML, Chernyak D, Gevorgyan V. J. Am. Chem. Soc. 2007;129:9868–9878. doi: 10.1021/ja072446m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Itami K, Yamazaki D, Yoshida J. Org. Lett. 2003;5:2161–2164. doi: 10.1021/ol0346234. [DOI] [PubMed] [Google Scholar]
- 80.Watson SP, Knox GR, Heron NM. Tetrahedron Lett. 1994;35:9763–9766. [Google Scholar]
- 81.a Wipf P, Waller DL, Reeves JT. J. Org. Chem. 2005;70:8096–8102. doi: 10.1021/jo051211v. [DOI] [PubMed] [Google Scholar]; b Wipf P, Ribe S. Org. Lett. 2001;3:1503–1505. doi: 10.1021/ol015816z. [DOI] [PubMed] [Google Scholar]
- 82.a Zaed AM, Swift MD, Sutherland A. Org. Biomol. Chem. 2009;7:2678–2680. doi: 10.1039/b907341h. [DOI] [PubMed] [Google Scholar]; b Swift MD, Donaldson A, Sutherland A. Tetrahedron Lett. 2009;50:3241–3244. [Google Scholar]; c Swift MD, Sutherland A. Org. Lett. 2007;9:5239–5242. doi: 10.1021/ol702299c. [DOI] [PubMed] [Google Scholar]
- 83.Anderson CE, Overman LE. J. Am. Chem. Soc. 2003;125:12412–12413. doi: 10.1021/ja037086r. [DOI] [PubMed] [Google Scholar]
- 84.Borrelly S, Paquette LA. J. Org. Chem. 1993;58:2714–2717. [Google Scholar]
- 85.Dauben WG, Michno DM. J. Org. Chem. 1977;42:682–685. [Google Scholar]
- 86.Trost BM, Jonasson C, Wuchrer M. J. Am. Chem. Soc. 2001;123:12736–12737. doi: 10.1021/ja011351w. [DOI] [PubMed] [Google Scholar]
- 87.D'Souza DM, Rominger F, M ller TJJ. Chem. Commun. 2006:4096–4098. doi: 10.1039/b609669g. [DOI] [PubMed] [Google Scholar]
- 88.Gonda J. Angew. Chem. 2004;116:3600–3608. [Google Scholar]; Angew. Chem. Int. Ed. 2004;43:3516–3524. doi: 10.1002/anie.200301718. and references therein. [DOI] [PubMed] [Google Scholar]
- 89.a Heescher C, Schollmeyer D, Nubbemeyer U. Eur. J. Org. Chem. 2013:4399–4404. [Google Scholar]; b Friedemann NM, Härter A, Brandes S, Grob S, Gerlach D, Münch W, Schollmeyer D, Nubbemeyer U. Eur. J. Org. Chem. 2012:2346–2358. and references therein. [Google Scholar]
- 90.a Yoon TP, MacMillan DWC. J. Am. Chem. Soc. 2001;123:2911–2912. doi: 10.1021/ja015612d. [DOI] [PubMed] [Google Scholar]; b Lambert TH, MacMillan DWC. J. Am. Chem. Soc. 2002;124:13646–13647. doi: 10.1021/ja028090q. [DOI] [PubMed] [Google Scholar]; c Dong VM, MacMillan DWC. J. Am. Chem. Soc. 2001;123:2448–2449. doi: 10.1021/ja005925t. [DOI] [PubMed] [Google Scholar]
- 91.Vedejs E, Gingras M. J. Am. Chem. Soc. 1994;116:579–588. [Google Scholar]
- 92.Honda K, Yasui H, Inoue S. Synlett. 2003:2380–2382. [Google Scholar]
- 93.Kuhn C, Skaltsounis L, Monneret C, Florent J-C. Eur. J. Org. Chem. 2003:2585–2595. [Google Scholar]
- 94.a Boyarskikh V, Nyong A, Rainier JD. Angew. Chem. 2008;120:5454–5457. doi: 10.1002/anie.200801336. [DOI] [PubMed] [Google Scholar]; Angew. Chem. Int. Ed. 2008;47:5374–5377. doi: 10.1002/anie.200801336. [DOI] [PubMed] [Google Scholar]; b Nyong AM, Rainier JD. J. Org. Chem. 2005;70:746–748. doi: 10.1021/jo0482413. [DOI] [PubMed] [Google Scholar]; c Novikov AV, Kennedy AR, Rainier JD. J. Org. Chem. 2003;68:993–996. doi: 10.1021/jo026582f. [DOI] [PubMed] [Google Scholar]; d Novikov AV, Sabahi A, Nyong AM, Rainier JD. Tetrahedron: Asymmetry. 2003;14:911–915. [Google Scholar]
- 95.a Paquette LA, Friedrich D, Rogers RD. J. Org. Chem. 1991;56:3841–3849. [Google Scholar]; b Philippo CMG, Paquette LA. J. Am. Chem. Soc. 1991;113:2762–2764. [Google Scholar]
- 96.Bae HJ, Baskar B, An SE, Cheong JY, Thangadurai DT, Hwang I, Rhee YH. Angew. Chem. 2008;120:2295–2298. doi: 10.1002/anie.200705117. [DOI] [PubMed] [Google Scholar]; Angew. Chem. Int. Ed. 2008;47:2263–2266. doi: 10.1002/anie.200705117. [DOI] [PubMed] [Google Scholar]
- 97.Horowitz RM, Geissman TA. J. Am. Chem. Soc. 1950;72:1518–1522. [Google Scholar]
- 98.Rueping M, Antonchick AP. Angew. Chem. 2008;120:10244–10247. [Google Scholar]; Angew. Chem. Int. Ed. 2008;47:10090–10093. doi: 10.1002/anie.200803610. [DOI] [PubMed] [Google Scholar]
- 99.Ito T, Overman LE, Wang J. J. Am. Chem. Soc. 2010;132:3272–3273. doi: 10.1021/ja100607z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Stas S, Tehrani KA, Laus G. Tetrahedron. 2008;64:3457–3463. [Google Scholar]
- 101.a Agami C, Francois C, Puchot-Kadouri C. Synlett. 1998:449–456. [Google Scholar]; b Agami C, Couty F, Poursoulis M. Synlett. 1992:847–848. [Google Scholar]
- 102.McCormack MP, Shalumova T, Tanski JM, Water SP. Org. Lett. 2010;12:3906–3909. doi: 10.1021/ol101606v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.a Overman LE. Acc. Chem. Res. 1992;25:352–359. [Google Scholar]; b Martin CL, Overman LE, Rhode JM. J. Am. Chem. Soc. 2008;130:7568–7569. doi: 10.1021/ja803158y. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Martin CL, Overman LE, Rohde JM. J. Am. Chem. Soc. 2010;132:4894–4906. doi: 10.1021/ja100178u. [DOI] [PMC free article] [PubMed] [Google Scholar]; d Jacobsen EJ, Levin J, Overman LE. J. Am. Chem. Soc. 1988;110:4329–4336. [Google Scholar]; e Overman LE, Robertson GM, Robichaud AJ. J. Am. Chem. Soc. 1991;113:2598–2610. [Google Scholar]
- 104.Overman LE. Tetrahedron. 2009;65:6432–6446. doi: 10.1016/j.tet.2009.05.067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.a Brummond KM, Hong S. J. Org. Chem. 2005;70:907–916. doi: 10.1021/jo0483567. [DOI] [PubMed] [Google Scholar]; b Brummond KM, Lu J. Org. Lett. 2001;3:1347–1349. doi: 10.1021/ol010029n. [DOI] [PubMed] [Google Scholar]
- 106.Cooke A, Bennett J, McDaid E. Tetrahedron Lett. 2002;43:903–905. [Google Scholar]
- 107.Garc a-Cuadrado D, Barluenga S, Winssinger N. Chem. Commun. 2008:4619–4621. doi: 10.1039/b807869f. [DOI] [PubMed] [Google Scholar]
- 108.a Lolkema LDM, Semeyn C, Ashek L, Hiemstra H, Speckamp WN. Tetrahedron. 1994;50:7129–7140. [Google Scholar]; b Lolkema LDM, Hiemstra H, Semeyn C, Speckamp WN. Tetrahedron. 1994;50:7115–7128. [Google Scholar]
- 109.a Jasti R, Rychnovsky SD. J. Am. Chem. Soc. 2006;128:13640–13648. doi: 10.1021/ja064783l. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Jasti R, Rychnovsk SD. Org. Lett. 2006;8:2175–2178. doi: 10.1021/ol0606738. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Jasti R, Anderson CD, Rychnovsky SD. J. Am. Chem. Soc. 2005;127:9939–9945. doi: 10.1021/ja0518326. [DOI] [PubMed] [Google Scholar]; d Rychnovsky SD, Marumoto S, Jaber JJ. Org. Lett. 2001;3:3815–3818. doi: 10.1021/ol0168465. [DOI] [PubMed] [Google Scholar]
- 110.a Crosby SR, Harding JR, King CD, Parker GD, Willis CL. Org. Lett. 2002;4:577–580. doi: 10.1021/ol0102850. [DOI] [PubMed] [Google Scholar]; b Roush WR, Dilley GJ. Synlett. 2001:0955–0959. [Google Scholar]; c Semeyn C, Blaauw RH, Himestra H, Speckamp WN. J. Org. Chem. 1997;62:3426–3427. [Google Scholar]
- 111.a Chan KP, Loh TP. Org. Lett. 2005;7:4491–4494. doi: 10.1021/ol051951q. [DOI] [PubMed] [Google Scholar]; b Marumoto S, Jaber JJ, Vitale JP, Rychnovsky SD. Org. Lett. 2002;4:3919–3922. doi: 10.1021/ol026751i. [DOI] [PubMed] [Google Scholar]
- 112.a Sumida S, Ohga M, Mitani J, Nokami J. J. Am. Chem. Soc. 2000;122:1310–1313. [Google Scholar]; b Nokami J, Yoshizane K, Matsuura H, Sumida S. J. Am. Chem. Soc. 1998;120:6609–6610. [Google Scholar]
- 113.a Loh T-P, Hu Q-Y, Han K-T, Cheng H-S. Org. Lett. 2001;3:2669–2672. doi: 10.1021/ol016228o. [DOI] [PubMed] [Google Scholar]; b Cheng HS, Loh TP. J. Am. Chem. Soc. 2003;125:4990–4991. doi: 10.1021/ja029700p. [DOI] [PubMed] [Google Scholar]; c Loh T-P, Hu Q-Y, Ma L-T. J. Am. Chem. Soc. 2001;123:2450–2451. doi: 10.1021/ja005831j. [DOI] [PubMed] [Google Scholar]
- 114.a Malkov AV, Kabeshov MA, Barlog M, Kocovsky P. Chem. Eur. J. 2009;15:1570–1573. doi: 10.1002/chem.200802224. [DOI] [PubMed] [Google Scholar]; b Loh T-P, Hu Q-Y, Ma L-T. Org. Lett. 2002;4:2389–2391. doi: 10.1021/ol026136e. [DOI] [PubMed] [Google Scholar]; c Kee C-LK, Lee C-HA, Tan K-T, Loh T-P. Org. Lett. 2004;6:1281–1283. doi: 10.1021/ol049633z. [DOI] [PubMed] [Google Scholar]; d Yongin SK. J. Org. Chem. 2006;71:4823–4828. doi: 10.1021/jo060517e. [DOI] [PubMed] [Google Scholar]; e Loh TP, Lee CLK, Tan KT. Org. Lett. 2002;4:2985–2987. doi: 10.1021/ol0263999. [DOI] [PubMed] [Google Scholar]
- 115.a Pereira CL, Chen Y, McDonald FE. J. Am. Chem. Soc. 2009;131:6066–6067. doi: 10.1021/ja9009265. [DOI] [PubMed] [Google Scholar]; b Chen Y-H, McDonald FE. J. Am. Chem. Soc. 2006;128:4568–4569. doi: 10.1021/ja061082f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.a Ramachandran PV, Pratihar D. Org. Lett. 2007;9:2087–2090. doi: 10.1021/ol0705806. [DOI] [PubMed] [Google Scholar]; b Ramachandran PV, Pratihar D, Biswas D. Chem. Commun. 2005:1988–1989. doi: 10.1039/b418996e. [DOI] [PubMed] [Google Scholar]
- 117.Bolla ML, Patterson B, Rychnovsky SD. J. Am. Chem. Soc. 2005;127:16044–16045. doi: 10.1021/ja056483u. [DOI] [PubMed] [Google Scholar]
- 118.a Dalgard JE, Rychnovsky SD. Org. Lett. 2005;7:1589–1591. doi: 10.1021/ol050270s. [DOI] [PubMed] [Google Scholar]; b Dalgard JE, Rychnovsky SD. J. Am. Chem. Soc. 2004;126:15662–15663. doi: 10.1021/ja044736y. [DOI] [PubMed] [Google Scholar]
- 119.Baldwin JE, Urban FJ. J. Chem. Soc. D. 1970:165–166. For a rare example of an all-carbon rearrangement, see. [Google Scholar]
- 120.Hoffmann RW. Angew. Chem. 1979;91:625–634. [Google Scholar]; Angew. Chem. Int. Ed. Engl. 1979;18:563–572. [Google Scholar]
- 121.Hiersemann M. Eur. J. Org. Chem. 2001:483–491. [Google Scholar]
- 122.Nakai T, Mikami K. Chem. Rev. 1986;86:885–902. [Google Scholar]
- 123.Doyle MP. Chem. Rev. 1986;86:919–939. [Google Scholar]
- 124.Zhang Y, Wang J. Coord. Chem. Rev. 2010;254:941–953. [Google Scholar]
- 125.Sweeney JB. Chem. Soc. Rev. 2009;38:1027–1038. doi: 10.1039/b604828p. [DOI] [PubMed] [Google Scholar]
- 126.Li A-H, Dai L-X, Aggarwal VK. Chem. Rev. 1997;97:2341–2372. doi: 10.1021/cr960411r. [DOI] [PubMed] [Google Scholar]
- 127.Doyle MP, Tamblyn WH, Baheri V. J. Org. Chem. 1981;46:5094–5102. [Google Scholar]
- 128.a Greenman KL, Carter DS, Van Vranken DL. Tetrahedron. 2001;57:5219–5225. [Google Scholar]; b Carter DS, Van Vranken DL. Org. Lett. 2000;2:1303–1305. doi: 10.1021/ol005740r. [DOI] [PubMed] [Google Scholar]
- 129.Kitagaki S, Yanamoto Y, Okubo H, Nakajima M, Hashimoto S. Heterocycles. 2000;54:623–628. [Google Scholar]
- 130.Wee AGH, Shi Q, Wang Z, Hatton K. Tetrahedron: Asymmetry. 2003;14:897–909. [Google Scholar]
- 131.a Chappie TA, Weekly RM, McMills MC. Tetrahedron Lett. 1996;37:6523–6526. [Google Scholar]; b Moody CJ, Taylor RJ. Tetrahedron Lett. 1988;29:6005–6008. [Google Scholar]
- 132.a Pirrung MC, Werner JA. J. Am. Chem. Soc. 1986;108:6060–6062. doi: 10.1021/ja00279a076. [DOI] [PubMed] [Google Scholar]; b Roskamp EJ, Johnson CR. J. Am. Chem. Soc. 1986;108:6062–6063. doi: 10.1021/ja00279a077. [DOI] [PubMed] [Google Scholar]
- 133.Clark JS. Tetrahedron Lett. 1992;33:6193–6196. [Google Scholar]
- 134.a Doyle MP, Griffin JH, Chinn MS, van Leusen D. J. Org. Chem. 1984;49:1917–1925. [Google Scholar]; b Doyle MP, Forbes DC, Vasbinder MM, Peterson CS. J. Am. Chem. Soc. 1998;120:7653–7654. and references therein. [Google Scholar]; c Kitagaki S, Yanamoto Y, Tsutsui H, Anada M, Nakajima M, Hashimoto S. Tetrahedron Lett. 2001;42:6361–6364. [Google Scholar]; d Tsutsui H, Matsuura M, Makino K, Nakamura S, Nakajima M, Kitagaki S, Hashimoto S. Isr. J. Chem. 2001;41:283–295. [Google Scholar]; e Shimada N, Nakamura S, Anada M, Shiro M, Hashimoto S. Chem. Lett. 2009;38:488–489. [Google Scholar]
- 135.Li Z, Davies HML. J. Am. Chem. Soc. 2010;132:396–401. doi: 10.1021/ja9075293. [DOI] [PubMed] [Google Scholar]
- 136.a Clark JS, Hodgson PB. Chem. Commun. 1994:2701–2702. [Google Scholar]; b Clark JS, Hodgson PB. Tetrahedron Lett. 1995;36:2519–2522. [Google Scholar]; c Clark JS, Hodgson PB, Goldsmith MD, Street LJ. J. Chem. Soc. Perkin Trans. 2001;1:3312–3324. [Google Scholar]
- 137.Wright DL, Weekly RM, Groff R, McMills MC. Tetrahedron Lett. 1996;37:2165–2168. [Google Scholar]
- 138.Muroni D, Mucedda M, Saba A. Heterocycles. 2009;78:635–644. [Google Scholar]
- 139.a Hodgson DM, Angrish D, Erickson SP, Kloesges J, Lee CH. Org. Lett. 2008;10:5553–5556. doi: 10.1021/ol802334y. also. [DOI] [PubMed] [Google Scholar]; b Hodgson DM, Angrish D, Labande AH. Chem. Commun. 2006:627–628. doi: 10.1039/b515943a. [DOI] [PubMed] [Google Scholar]; c Hodgson DM, Angrish D. Adv. Synth. Catal. 2006;348:2509–2514. [Google Scholar]
- 140.a de March P, Moreno-Mañas M, Ripoll I, Florencio F, García-Blanco S, Martínez-Carrera S. Tetrahedron Lett. 1986;27:3673–3674. [Google Scholar]; b de March P, Moreno-Mañas M, Roca JL. J. Org. Chem. 1988;53:5149–5151. [Google Scholar]
- 141.Hoye TR, Dinsmore CJ. Tetrahedron Lett. 1992;33:169–172. [Google Scholar]
- 142.Murphy GK, West FG. Org. Lett. 2006;8:4359–4361. doi: 10.1021/ol061772o. [DOI] [PubMed] [Google Scholar]
- 143.Kato Y, Miki K, Nishino F, Ohe K, Uemura S. Org. Lett. 2003;5:2619–2621. doi: 10.1021/ol034731q. [DOI] [PubMed] [Google Scholar]
- 144.Davies PW, Albrecht SJ-C. Chem. Commun. 2008:238–240. doi: 10.1039/b714813e. [DOI] [PubMed] [Google Scholar]
- 145.Soheili A, Tambar U. J. Am. Chem. Soc. 2011;133:12956–12959. doi: 10.1021/ja204717b. [DOI] [PubMed] [Google Scholar]
- 146.Grigg R, Markandu J. Tetrahedron Lett. 1991;32:279–282. [Google Scholar]
- 147.a Aburano D, Inagaki F, Tomonaga S, Mukai C. J. Org. Chem. 2009;74:5590–5594. doi: 10.1021/jo901141t. [DOI] [PubMed] [Google Scholar]; b Mukai C, Hirose T, Teramoto S, Kitagaki S. Tetrahedron. 2005;61:10983–10994. [Google Scholar]
- 148.Carter RG, Bourland TC. Chem. Commun. 2000:2031–2032. [Google Scholar]
- 149.Komatsu N, Nishibayashi Y, Uemura S. Tetrahedron Lett. 1993;34:2339–2342. [Google Scholar]
- 150.a Bao H, Qi X, Tambar UK. J. Am. Chem. Soc. 2011;133:1206–1208. doi: 10.1021/ja110500m. [DOI] [PubMed] [Google Scholar]; b Bao H, Qi X, Tambar UK. Synpacts. 2011:1789–1792. [Google Scholar]
- 151.Murakami M, Katsuki T. Tetrahedron Lett. 2002;43:3947–3949. [Google Scholar]
- 152.Takada H, Nishibayashi Y, Ohe K, Uemura S, Baird CP, Sparey TJ, Taylor PC. J. Org. Chem. 1997;62:6512–6518. [Google Scholar]
- 153.Bach T, Körber C. J. Org. Chem. 2000;65:2358–2367. doi: 10.1021/jo991569p. [DOI] [PubMed] [Google Scholar]
- 154.Bao H, Tambar U. J. Am. Chem. Soc. 2012;134:18495–18498. doi: 10.1021/ja307851b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Nokami J, Mandai T, Imakura Y, Nishiuchi K, Kawada M, Wakabayshi S. Tetrahedron Lett. 1981;22:4489–4490. [Google Scholar]
- 156.Nokami J, Taniguchi T, Ogawa Y. Chem. Lett. 1995:43–44. Examples in total synthesis. [Google Scholar]
- 157.Hagiwara H, Isobe K, Numamae A, Hoshi T, Suzuki T. Synlett. 2006:1601–1603. [Google Scholar]
- 158.Nasveschuk CG, Rovis T. Org. Biomol. Chem. 2008;6:240–254. doi: 10.1039/b714881j. [DOI] [PubMed] [Google Scholar]
- 159.a Padwa A, Beall LS, Eidell CK, Worsencroft KJ. J. Org. Chem. 2001;66:2414–2421. doi: 10.1021/jo001684w. [DOI] [PubMed] [Google Scholar]; b Saba A. Tetrahedron Lett. 2003;44:2895–2898. [Google Scholar]; c Vanecko JA, West FG. Org. Lett. 2005;7:2949–2952. doi: 10.1021/ol050915o. [DOI] [PubMed] [Google Scholar]
- 160.Thornton AR, Blakey SB. J. Am. Chem. Soc. 2008;130:5020–5021. doi: 10.1021/ja7111788. [DOI] [PubMed] [Google Scholar]
- 161.Shu X-Z, Ji K-G, Zhao S-C, Zheng Z-J, Chen J, Lu L, Liu X-Y, Liang Y-M. Chem. Eur. J. 2008;14:10556–10559. doi: 10.1002/chem.200801591. [DOI] [PubMed] [Google Scholar]
- 162.Dhayalan V, Clement JA, Jagan R, Mohanakrishnan AK. Eur. J. Org. Chem. 2009:531–546. [Google Scholar]
- 163.a Davies HML, Stafford DG, Hansen T. Org. Lett. 1999;1:233–236. doi: 10.1021/ol9905699. [DOI] [PubMed] [Google Scholar]; b Davies HML, Jin Q. J. Am. Chem. Soc. 2004;126:10862–10863. doi: 10.1021/ja047185k. [DOI] [PubMed] [Google Scholar]; c Davies HML, Dai X, Long MS. J. Am. Chem. Soc. 2006;128:2485–2490. doi: 10.1021/ja056877l. [DOI] [PubMed] [Google Scholar]
- 164.Ajamian A, Gleason JL. Angew. Chem. 2004;116:3842–3848. [Google Scholar]; Angew. Chem. Int. Ed. 2004;43:3754–3760. doi: 10.1002/anie.200301727. [DOI] [PubMed] [Google Scholar]
- 165.Heathcock CH. Proc. Natl. Acad. Sci. USA. 1996;93:14323–14327. doi: 10.1073/pnas.93.25.14323. [DOI] [PMC free article] [PubMed] [Google Scholar]