

Abstract
Nitric oxide (NO) as a cellular signaling molecule and vasodilator regulates a range of physiological and pathological processes. Nitrite (NO2−) is recycled in vivo to generate nitric oxide, particularly in physiologic hypoxia and ischemia. The cytochrome c oxidase (CcO) binuclear hemea3/CuB active site is one entity known to be responsible for cellular nitrite conversion to nitric oxide. We recently reported that a partially reduced heme/Cu assembly reduces nitrite ion, producing NO; the heme serves as the reductant and cupric ion provides a Lewis Acid interaction with nitrite, facilitating nitrite (N−O) bond cleavage (Hematian et al., J Am Chem Soc 134:18912–18915, 2012). To further investigate this nitrite reductase (NIR) chemistry, copper(II)-nitrito complexes with tri-and tetra-dentate ligands were used in this study, where either O,O'-bidentate or O-unidentate modes of nitrite binding to the cupric center are present. To study the role of the reducing ability of the ferrous heme center, two different tetraarylporphyrinate-iron(II) complexes, one with electron donating para-methoxy peripheral substituents, (TMPP)FeII, and the other with electron withdrawing 2,6-difluorophenyl substituents, (F8)FeII, were employed. The results show that differing nitrite coordination modes to copper(II) ion leads to varying kinetic behavior. Here, also, the ferrous heme is in all cases the source of the reducing equivalent required to take nitrite to nitric oxide, but the reduction ability of the heme center does not play a key role in the observed overall reaction rate. Based on our observations, reaction mechanisms are proposed and discussed in terms of heme/Cu heterobinuclear structures.
Introduction
The nitrite anion (NO2−) is biologically important, for example in denitrifying bacteria where it forms from nitrate (NO3−) reduction catalyzed by a molybdoenzyme. Then, either copper or heme containing enzymes reduce the nitrite to nitric oxide (NO, nitrogen monoxide), prior to its being reductively coupled to give nitrous oxide (N2O) and eventually N2 [1–4]. However, in mammalian aerobic organisms, until relatively recently, nitrite was considered as a biologically inert one-electron-oxidative end product of endogenous nitric oxide (NO) metabolism. In fact, this supposedly inert anion can be and is recycled in vivo to generate nitric oxide and constitutes a biochemical circulating reservoir for this important signaling molecule, in particular under conditions of physiologic hypoxia and ischemia [5–10].
The classical biological source of NO is nitric oxide synthase (NOS) oxidation of L-arginine [11, 12]. This nitrite reductase chemistry represents an important alternative biological source of NO [1–4, 13]. Nitrite reduction to nitric oxide possesses both pH and oxygen sensor chemistry [13] because of its requirement for one electron and two protons.
Remarkably, the L-arginine-NOS pathway is oxygen dependent, whereas the nitrite-NO pathway is gradually activated as the oxygen tensions falls. Therefore, nitrite-derived nitric oxide formation can be considered as a back-up system to ensure that there is sufficient NO production in hypoxic conditions where the NO generation (for its signaling functions) in tissues is independent of NOS activity [14].
Besides a non-enzymatic conversion of nitrite to nitric oxide via acidic disproportionation [5, 15], nitrite as the major intravascular and tissue storage form of NO is readily reduced to nitric oxide along a physiological oxygen and pH gradient by a number of proteins/enzymes such as hemoglobin (Hb)[16–18], myoglobin (Mb) [19, 20], neuroglobin [21], xanthine oxidoreductase [8], aldehyde dehydrogenase [8], cytochrome c oxidase (CcO) (see below), cytochrome c [22] and possibly cytochrome P-450 [23–25] (Fig. 1).
Figure 1.

Nitrite is reduced to NO by a variety of metal containing enzymes along the physiological oxygen (O2) gradient. Under normal cellular [O2] tensions (normoxia), the critically important signaling molecule nitric oxide is predominantly produced by nitric oxide synthase (NOS). Under hypoxic (low [O2] concentrations) conditions, those associated with cellular stress, nitrite anion can also serve as an alternative source of NO generation, via enzymatic reduction processes, including that associated with cytochrome c oxidase. See text.
Cytochrome c oxidase, the terminal enzyme of the mitochondrial respiratory chain known to catalyze the four-electron reduction of oxygen to water at a heme/copper heterobinuclear centered active site [26–28], has recently been shown to have a novel enzymatic role catalyzing the reduction of nitrite to nitric oxide [29, 30]. It is noteworthy that unlike the CcO oxidase reaction, which involves four electrons, only one electron is required for the CcO nitrite reductase reaction. This nitrite reductase activity is significantly elevated under hypoxic conditions and increases with lower pH and higher nitrite concentrations that are found in hypoxia [31–34]. These findings are consistent with the following reaction:
However, more recent studies have revealed that the catalytic formation of nitric oxide by CcO also occurs under normoxia and physiological nitrite concentrations and has been detected in a variety of eukaryotes, including yeast, rat liver, human endothelial cells, bovine heart and calf liver, plants, and algae [29, 31, 35].
In this report, we describe several examples of heme/Cu assembly coordination complexes which mediate nitrite reductase chemistry, the one-electron reduction of nitrite to NO. This follows our initial report[36] on this type of chemistry showing a copper(II) complex with coordinated nitrite anion reacts with a ferrous heme, producing nitric oxide. Here, we provide additional examples of this newly discovered chemistry, where the exact nature of the heme and/or the copper chelating ligand is modified. For this reaction chemistry, this approach provides (i) differing modes of binding of nitrite to the cupric center, and (ii) hemes with varying reducing abilities. For the former, we have examples of nitrite bound either to the cupric center with a tripodal tetradentate pyridyl-alkylamine ligand, or a related tridentate alkylamine ligand. For the latter, we compare systems where the heme possesses electron-withdrawing peripheral substituents vs. one that is electron donating. The hope is that via such a systematic approach, we can learn details and fundamental concepts concerning how a heme/Cu assembly may affect nitrite reduction chemistry.
Scheme 1 shows the structures of complexes employed in the present study. The copper complexes are [(tmpa)CuII(NO2)][B(C6F5)4] (tmpa ≡ tris(2-pyridylmethylamine)[36] and a new compound [(AN)CuII(NO2)](CF3SO3) (AN ≡ 3,3’-iminobis (N,N-dimethylpropylamine)); an X-ray crystal structure reveals the binding of nitrite to be different than that found for the tmpa containing complex. The hemes utilized in this study include the iron(II) complex (F8)FeII (F8 ≡ tetrakis(2,6-difluorophenyl)porphyrinate(2–))[37, 38] and a heme compound bearing an electron-rich porphyrinate, (TMPP)FeII (TMPP ≡ tetrakis(4-methoxyphenyl)porphyrinate(2–)). As shown in Fig. 2, the reactivity observed by employing pairwise combinations of copper-nitrite complex and heme is that reaction of a 1:2 Cu/Fe complex mixture leads to highly efficient formation of nitric oxide, as trapped by the second reduced heme to give a stable ferrous heme-nitrosyl; the initially present copper(II) combines with the ferric heme formed from the redox reaction where nitrite is reduced to NO, and a μ-oxo FeIII-O-CuII complex is generated as the other product.
Scheme 1.

Nitrite reduction to nitric oxide mediated by heme/copper assemblies: (Top) Structures of copper and heme complexes employed in the present study. (Bottom) Schematic reaction equation representing the reductase chemistry.
Figure 2.

Displacement ellipsoid plot (50% probability level) of the copper complexes, showing the atom-labeling scheme. Relevant bond lengths (Å) and angles (deg) of a) [(AN)CuII(Cl)]+, Cu-N(1), 2.028 (2); Cu-N(2), 1.998 (2); Cu-N(3), 2.031 (2); Cu-Cl(1), 2.227 (6); N(1)-Cu-N(2), 96.5 (7); N(1)-Cu-N(3), 138.6 (4); N(2)-Cu-N(3), 95.7 (7); N(1)-Cu-C1(1), 102.6 (6); N(2)-Cu-Cl(1), 126.6 (8); N(3)-Cu-C1(1), 101.0 (8) and b) [(AN)CuII(NO2)]+, Cu-N(1), 2.011 (4); Cu-N(2), 1.993 (2); Cu-N(3), 2.043 (2); Cu-O(1), 2.347 (3); Cu-O(2), 2.002 (2); N(1)-Cu-N(2), 97.6 (1); N(1)-Cu-N(3), 136.8 (1); N(2)-Cu-N(3), 94.8 (9); N(1)-Cu-O(1), 109.0 (1); N(1)-Cu-O(2), 92.9 (1); N(2)-Cu-O(1), 95.7 (9); N(2)-Cu-O(2), 152.0 (6); N(3)-Cu-O(1), 110.6 (7); N(3)-Cu-O(2), 95.0 (1); O(1)-Cu-O(2), 56.2 (8).
In fact, the heme/Cu CcO active site also is responsible for the oxidation NO to nitrite [36, 39], when NO is in excess and/or [O2] levels increase (recovery from hypoxia). Our research program in large part concerns itself with the interplay between reductive or oxidative chemistry at a given heme/Cu center, as modulated by metal ion center coordination and redox properties. Thus, as part of these efforts, obtaining an in-depth understanding of the mechanism underlying heme/Cu nitrite reductase activity is critical.
Materials and Methods
All chemicals and solvents were purchased as commercially available analytical grade unless otherwise stated. Diethyl ether (Et2O) and methanol (MeOH) were used after passing them through a 60 cm long column of activated alumina (Innovative Technologies, Inc.) under argon. Acetonitrile (MeCN) and pentane were dried by distillation over calcium hydride and acetone was freshly distilled from Drierite (anhydrous calcium sulfate) under argon prior to use. Tetrahydrofuran (THF) and 2-Methyltetrahydrofuran (MeTHF) (Sigma, 673277, inhibitor free) were purified and dried by distillation from sodium/benzophenone ketyl under argon. Dioxygen was dried by passing through a short column of supported P4O10 (Aquasorb, Mallinkrodt). Nitrogen monoxide (NO) gas was obtained from Matheson Gases and purified following methods previously described in the literature [40–42]. Addition of NO(g) to metal complex solutions was effected by transfer via a three-way long syringe needle. Preparation and handling of air sensitive compounds were performed under an argon atmosphere using standard Schlenk techniques or in an MBraun Labmaster 130 inert atmosphere (<1 ppm O2, <1 ppm H2O) drybox filled with nitrogen. Deoxygenation of solvents was effected by either repeated freeze/pump/thaw cycles or bubbling with argon for 45 – 60 min. ESI-MS spectra were collected by the Mass Spectrometry Facility at The Johns Hopkins University. Elemental analyses were performed by Columbia Analytical Services (Tucson, AZ). Infrared spectra (IR) were obtained using a Thermo Scientific Nicolet Nexus 670 FT-IR spectrophotometer. UV-vis spectra were recorded on a Cary-50 Bio spectrophotometer equipped with a fiber optic coupler (Varian). Spectrophotometer cells used were made by Quark Glass with column and pressure/vacuum side stopcock and 1 cm path length. 1H-NMR spectra were acquired using a Bruker 400 MHz spectrometer. Chemical shifts were reported as δ values relative to an internal standard (Me4Si) and the residual solvent proton peak. Electron paramagnetic resonance (EPR) spectra were recorded on a Bruker EMX spectrometer controlled with a Bruker ER 041 × G microwave bridge operating at X-band (~9.4 GHz).
[(tmpa)CuII(NO2)][B(C6F5)4] [36], [(tmpa)CuI(MeCN)][B(C6F5)4] [36], [(AN)CuI][B(C6F5)4] [43], and (F8)FeII [37, 38] were synthesized and characterized following methods previously described in the literature. (TMPP)FeIII(Cl) were purchased from TriPorTech GmbH-Germany.
[(AN)CuII(Cl)](CF3SO3)
Under an argon atmosphere using a Schlenk flask, a solution of AN (375 mg, 2.00 mmol) in 20 mL of freshly distilled MeCN was added to solid Cu(CF3SO3)2 (362 mg, 1.00 mmol) and CuCl2 (134 mg, 1.00 mmol). The resulting blue solution was allowed to stir for 30 min at room temperature. The solvent was removed in vacuo, and in the drybox the solid residue was dissolved in a minimal volume of THF. The solution was then filtered, and the filtrate was layered with pentane affording turquoise blue needles suitable for X-ray crystal structure analysis. After vacuum-drying the crystals weighed 695 mg (80% yield). Anal. Calcd for C11H25ClCuF3N3O3S: C, 30.34; H, 5.79; N, 9.65. Found: C, 3.54; H, 6.07; N, 9.52. UV-vis spectra (λmax, nm (εmax, M−1cm−1)) in acetone (Fig. S1): 720 (250); 1025 (227) and in MeOH (Fig. S2): 722 (227); 1000 (206). EPR spectra: X-band spectrometer (ν = 9.428 GHz) in acetone at 22 K (Fig. S3): g2 = 2.116, g3 ≈ 2.040 and in THF:MeCN (4:1) at 15 K (Fig. S4): g| = 2.254, A1 = 120 G, g2 = 2.114, g3 = 2.034.
[(AN)CuII(NO2)](CF3SO3)
In the drybox, silver nitrite (77 mg, 0.50 mmol) was added to [(AN)CuII(Cl)](CF3SO3) (218 mg, 0.50 mmol) in 10 mL MeOH resulting a green solution. The reaction mixture was then sonicated for 3 hours to ensure complete precipitation of AgCl, followed by filtration in the drybox. Slow addition of Et2O (30 mL) to the filtrate resulted in crystallization and the green crystals were dried under vacuum to afford 205 mg (92%) of product. Anal. Calcd for C11H25CuF3N4O5S: C, 29.63; H, 5.65; N, 12.56. Found: C, 30.13; H, 5.98; N, 12.26. UV-vis spectra (λmax, nm (εmax, M−1cm−1)) in acetone (Fig. S1): 702 (327) and in MeOH (Fig. S2): 702 (309). EPR spectrum: X-band spectrometer (ν = 9.428 GHz) in acetone at 22 K (Fig. S5): g2 = 2.108, g3 ≈ 2.051. FT-IR spectrum (solid) (Fig. S6): νas(NO2) 1370 cm−1, νs(NO2) 1110 cm−1, and δ(NO2) 835 cm−1. We should note that analogous compounds have been previously described, [(AN)CuII(NO2)](NO3) (with X-ray structure) and [(AN)CuII(NO2)][B(C6F5)4] [44].
(TMPP)FeII • 1.5 H2O
Under an argon atmosphere, to a solution of (TMPP)FeIII(Cl) (413 mg, 0.50 mmol) in deoxygenated CH2Cl2 (150 mL) was added 100 mL of deoxygenated saturated sodium hydrosulfite aqueous solution. The two solutions were vigorously mixed with argon bubbling for about 30 min, during which time the color of the organic later changed from dark brownish-red to bright red. The reaction mixture was allowed to sit for ample time (~20 min) to allow for the separation of the two layers. Then the organic layer was allowed to slowly pass through a Schlenk filter tube (coarse porosity) and dried over a small plug of anhydrous sodium sulfate (~ 6 g). The solvent was removed under vacuum yielding 298 mg (73%) of product. Anal. Calcd for C48H39FeN4O5.5: C, 70.68; H, 4.82; N, 6.87. Found: C, 70.88; H, 4.83; N, 6.73. 1H-NMR (pyridine-d5, 400 MHz): δ 8.83 (s, 8H, pyrrole-H), 8.13 (d, 8H, o-phenyl-H), 7.29 (d, 8H, m-phenyl-H), 4.91 (s, 3H, H2O), 3.88 (s, 12H, methoxy-H). UV-vis spectra (λmax, nm (εmax, M−1cm−1)) (Fig. S7) in acetone: 429 (243 000); 540 (11 400), and in THF (Fig. S8): 430 (256 000); 542 (8 800).
[(TMPP)FeIII-O-CuII(tmpa)][B(C6F5)4] • 3H2O • 2 MeTHF
In a 50 mL Schlenk flask equipped with a stir bar, in the drybox, (TMPP)FeII • 1.5 H2O (40 mg, 0.05 mmol) and [(tmpa)CuI(MeCN)][B(C6F5)4] (53 mg, 0.05 mmol) were dissolved in 10 mL air-free freshly distilled MeTHF. Then dry dioxygen was bubbled through the solution for 2 min accompanied by an immediate color change from dark red to dark green. The mixture was allowed to stir for 20 min at room temperature. The solvent was then removed under vacuum and solid was further dried under vacuum for additional 6 h yielding 86 mg (84%) of [(TMPP)FeIII-O-CuII(tmpa)][B(C6F5)4]. Anal. Calcd for C100H80BCuF20FeN8O10: C, 58.19; H, 3.91; N, 5.43. Found: C, 58.06; H, 3.86; N, 5.26. UV-vis spectra (λmax, nm (εmax, M−1cm−1)) (Fig. S8) in acetone: 443 (252 000); 564 (17 300); 605 (14 700) and in MeCN: 441 (189 000); 561 (14 800); 603 (12 100). ESI-MS in acetone (Fig. S9): 1157.3 (TMPP)Fe-O-Cu(tmpa); 788.2 (TMPP)Fe; 1593.4 [(TMPP)Fe]2O.
In-Situ Generation of (TMPP)FeII(NO)
The ferrous nitrosyl complex, (TMPP)FeII(NO), was generated in-situ by bubbling excess NO(g) through the solution of (TMPP)FeII under a nitrogen atmosphere at room temperature. UV-vis spectra (λmax, nm (εmax, M−1cm−1)) (Fig. 10) in acetone: 410 (142 000); 539 (11 900); 614 (4 800) and in MeCN (Fig. S11): 410 (138 000); 535 (11 500); 610 (4 800). FT-IR spectrum in acetone: ν(NO) 1677 cm−1 (Note; a published value in DMF is 1666 cm−1 [45])
Reaction of two equiv (F8)FeII with [(AN)CuII(NO2)](CF3SO3)
In the drybox, to a solution of (F8)FeII • H2O (20.0 mg, 0.024 mmol) in acetone (100 mL) was added [(AN)CuII(NO2)](CF3SO3) (5.4 mg, 0.012 mmol). After stirring for 3 h, the solvent was removed to yield a brown solid. During the reaction, 305 µL aliquots were withdrawn by syringe every 10 min, diluted with acetone to 5.0 mL, and a UV-vis spectrum was recorded (Fig. 3a). This procedure allowed for quantitative determination of [(F8)FeIII-O-CuII(AN)]+ complex as one of the products and was necessary in order to avoid the breakage (by hydrolysis) of the oxo-bridge occurring at low concentrations. Formation of a one-to-one mixture of the heme-nitrosyl species (F8)FeII(NO) (λmax = 399 nm) and the μ-oxo (λmax = 439 nm) complex was observed. FT-IR spectrum (solid): νNO = 1685 cm−1.
Figure 3.

a) UV-vis spectra of (F8)FeII (8 µM) ( , λmax = 420 (Soret), 541 nm) and reaction solution of (F8)FeII and [(AN)CuII(NO2)](CF3SO3) after stirring for 10 min (15 µM) (
, λmax = 420 (Soret), 541 nm) and reaction solution of (F8)FeII and [(AN)CuII(NO2)](CF3SO3) after stirring for 10 min (15 µM) ( ) and 1 h (15 µM) (
) and 1 h (15 µM) ( , λmax = 399, 439 (Soret), 558 nm) in acetone at RT. b) EPR spectrum of the products of the reaction of (F8)FeII and [(AN)CuII(NO2)](CF3SO3) in acetone at 20 K (2mM).
, λmax = 399, 439 (Soret), 558 nm) in acetone at RT. b) EPR spectrum of the products of the reaction of (F8)FeII and [(AN)CuII(NO2)](CF3SO3) in acetone at 20 K (2mM).
Reaction of two equiv (TMPP)FeII with [(tmpa)CuII(NO2)][B(C6F5)4]
In a drybox, to a solution of (TMPP)FeII • 1.5 H2O (19.7 mg, 0.024 mmol) in acetone (100 mL) was added [(tmpa)CuII(NO2)][B(C6F5)4], (12.9 mg, 0.012 mmol) and after stirring for 3 h, the solvent was removed to yield a brown solid. UV-vis samples preparation during the reaction was carried out in a manner analogous to the procedure described above. UV-vis analysis directly indicates formation of a one-to-one mixture of the heme-nitrosyl species (TMPP)FeII(NO) (λmax = 410 nm) and the μ-oxo (λmax = 443 nm) complex (Fig. 4a). FT-IR spectrum in acetone: νNO = 1677 cm−1.
Figure 4.

a) UV-vis spectra of (TMPP)FeII (10 µM) ( , λmax = 429 (Soret), 540 nm) and reaction solution of (TMPP)FeII and [(tmpa)CuII(NO2)][B(C6F5)4], after stirring for 10 min (10 µM) () and 1 h (10 µM) (, λmax = 410, 443 (Soret), 564 and 605 nm) in acetone at RT. After stirring for 3 h the green spectrum was recorded. b) EPR spectrum of the products of the reaction of (TMPP)FeII and [(tmpa)CuII(NO2)][B(C6F5)4] in acetone at 20 K (2mM).
, λmax = 429 (Soret), 540 nm) and reaction solution of (TMPP)FeII and [(tmpa)CuII(NO2)][B(C6F5)4], after stirring for 10 min (10 µM) () and 1 h (10 µM) (, λmax = 410, 443 (Soret), 564 and 605 nm) in acetone at RT. After stirring for 3 h the green spectrum was recorded. b) EPR spectrum of the products of the reaction of (TMPP)FeII and [(tmpa)CuII(NO2)][B(C6F5)4] in acetone at 20 K (2mM).
Reaction of two equiv (TMPP)FeII with [(AN)CuII(NO2)](CF3SO3)
Addition of [(AN)CuII(NO2)](CF3SO3) (5.4 mg, 0.012 mmol) to a solution of (TMPP)FeII • 1.5 H2O (19.7 mg, 0.024 mmol) in acetone (100 mL) in a drybox was followed by stirring for 3 h. The solvent was then removed to yield a brown solid. The solid product was then dissolved in 10 mL CH2Cl2 and extracted with 25 mL of aqueous NaCl solution (6 mM). The absence of nitrite ion in the aqueous layer was determined by employing QUANTOFIX semiquantitative nitrite test strips [36]. UV-vis samples preparation during the reaction was analogous to the procedure described above. UV-vis displays a Soret band at (λmax= 419 nm) indicating no μ-oxo complex formation (Fig. 5). FT-IR spectrum in acetone: νNO = 1677 cm−1.
Figure 5.

UV-vis spectra of (TMPP)FeII (10 µM) (black, λmax = 429 (Soret), 540 nm) and reaction solution of (TMPP)FeII and [(AN)CuII(NO2)](CF3SO3), after stirring for 10 min (10 µM) ( ) and 1 h (10 µM) (, λmax = 410, 443 (Soret), 564 and 605 nm) in acetone at RT. Also, see text.
) and 1 h (10 µM) (, λmax = 410, 443 (Soret), 564 and 605 nm) in acetone at RT. Also, see text.
Reaction of [(AN)CuI][B(C6F5)4] with Nitrite
In the drybox, to a 25 mL acetone solution of [(AN)CuI][B(C6F5)4] (11.6 mg, 0.012 mmol), (Bu)4N(NO2) (3.6 mg, 0.012 mmol) was added, followed by stirring overnight. UV-vis spectra of the reaction mixture showed no absorbance in the 500–1100 nm range (Figure S12) indicating that no redox reaction occurred.
Reaction of (TMPP)FeII with Nitrite
To a 25 mL acetone solution of (TMPP)FeII • 1.5 H2O (10.2 mg, 0.012 mmol), in the drybox, (Bu)4N(NO2) (3.6 mg, 0.012 mmol) was added. The mixture was stirred for several hours during which no significant color or UV-vis absorbance changes were observed (Figure S13) confirming that nitrite reduction does not occur in the absence of Cu(II) as a Lewis Acid.
X-ray Structure Determination
X-ray structure determination for both Cu complexes [(AN)CuII(Cl)](CF3SO3) and [(AN)CuII(NO2)](CF3SO3) was performed at the X-ray diffraction facility at the Johns Hopkins University. CIF files have been deposited with the Cambridge Crystallographic Data Centre (CCDC). CCDC 961739 and 961740 contain the supplementary crystallographic data for this article. These data can be obtained free of charge from the CCDC via http://www.ccdc.cam.ac.uk/data_request/cif. All reflection intensities were measured at 110(2) K using the Agilent Technologies KM4/Xcalibur (detector: Sapphire3) with enhance graphite-monochromated Mo Kα radiation (λ = 0.71073 Å) under the program CrysAlisPro (Version 1.171.35.11, Agilent Technologies, 2011). The program CrysAlisPro (Version 1.171.35.11, Agilent Technologies, 2011) was used to refine the cell dimensions. Data reduction was done using the program CrysAlisPro (Version 1.171.35.11, Agilent Technologies, 2011). The structure was solved with the program SHELXS-97 [46] and was refined on F2 with SHELXL-97 [46]. Analytical numeric absorption corrections based on a multifaceted crystal model were applied using CrysAlisPro (Version 1.171.35.11, Agilent Technologies, 2011). The temperature of the data collection was controlled using the system Cryojet (manufactured by Oxford Instruments). The H atoms (unless otherwise specified) were placed at calculated positions using the instructions AFIX 23 or AFIX 137 with isotropic displacement parameters having values 1.2 or 1.5 times Ueq of the attached C atoms. For both complexes, the H atom attached to N2 was found from difference Fourier maps, and the N–H bond distance was restrained using the DFIX instruction.
The structure of [(AN)CuII(Cl)](CF3SO3) is ordered. The absolute configuration was established by anomalous-dispersion effects in diffraction measurements on the crystal, and the Flack parameter refined to −0.017(9). The structure of [(AN)CuII(NO2)](CF3SO3) is partly disordered. The triflate counterion is found disordered over 2 orientations [occupancy factor of the major component = 0.586(4)]. The fragment C4–C3–N1–C1–C2 was also found to be disordered over two orientations [occupancy factor of the major component = 0.759(4)].
[(AN)CuII(Cl)](CF3SO3), FW = 435.39, blue-green lath, 0.49 × 0.21 × 0.12 mm3, orthorhombic, P212121 (no. 19), a = 10.3160(2), b = 10.9961(2), c = 15.9762(4) Å, V = 1812.27(7) Å3, Z = 4, Dx = 1.596 g cm−3, μ = 1.510 mm−1, abs. corr. range: 0.752–0.863. 9260 Reflections were measured up to a resolution of (sin θ/λ)max = 0.62 Å−1. 3665 Reflections were unique (Rint = 0.0245), of which 3421 were observed [I > 2σ(I)]. 215 Parameters were refined. R1/wR2 [I > 2σ(I)]: 0.0224/0.0548. R1/wR2 [all refl.]: 0.0258/0.0561. S = 1.056. Residual electron density found between −0.24 and 0.34 e Å−3.
[(AN)CuII(NO2)](CF3SO3), FW = 445.95, blue-green irregular rod, 0.42 × 0.16 × 0.12 mm3, monoclinic, P21/n (no. 14), a = 10.1675(2), b = 15.6928(3), c = 11.7887(2) Å, β = 90.9584(16)°, V = 1880.70(6) Å3, Z = 4, Dx = 1.575 g cm−3, μ = 1.329 mm−1, abs. corr. range: 0.706–0.887. 13996 Reflections were measured up to a resolution of (sin θ/λ)max = 0.62 Å−1. 3802 Reflections were unique (Rint = 0.0396), of which 3154 were observed [I > 2σ(I)]. 342 Parameters were refined using 179 restraints. R1/wR2 [I > 2σ(I)]: 0.0354/0.0910. R1/wR2 [all refl.]: 0.0452/0.0961. S = 1.053. Residual electron density found between −0.42 and 0.53 e Å−3.
Results
Copper complex syntheses and x-ray structures
The nitrite complex, [(AN)CuII(NO2)](CF3SO3), was synthesized by reacting silver nitrite with the chloride precursor [(AN)CuII(Cl)](CF3SO3), and X-ray structures of both compounds were obtained, Fig. 2. The chloride complex of copper(II) is tetra-coordinated, with ligation from the three aliphatic nitrogen atoms of the ligand (N(1), N(2), and N(3)), along with the chloride ligand (Cl1). Using the structural analysis criterion developed by Houser and co-workers [47], the τ4 value is 0.69, making the best description of this complex to be of the "seesaw" type. A three-coordinate copper(I) structure with AN has been previously described [43]. The cupric center in the nitrite complex [(AN)CuII(NO2)](CF3SO3) is, however, penta-coordinated by the N donor atoms of 3,3’-iminobis (N,N-dimethyl-propylamine) ligand, and O(1) and O(2) of an anisobidentate nitrito ligand; the Cu–O(nitrito) bond distances are unequal, with O1 of the nitrito ligand exhibiting a much weaker interaction with the cupric ion center, Cu–O1 = 2.35 and Cu–O2 = 2.00 Å (see Fig. 2).
By contrast, in our previously structurally characterized complex with tetradentate TMPA ligand, [(tmpa)CuII(NO2)][B(C6F5)4] (Scheme 1), the nitrito ligand is essentially unidentate, with highly different Cu–O(nitrito) bond distances, 1.93 and 2.93 Å.[36] Pentacoordination in this complex is completed via the four N-donors of TMPA and the single O-atom of the nitrito ligand, see diagram below.

The infrared spectrum (Supplementary Material) of solid [(AN)CuII(NO2)](CF3SO3) features nitrite ligand vibrations further confirming the anisobidentate ONO− binding (Δν = 260 cm−1) [48, 49]. Although nitrite vibrations can only be assigned with certainty by isotope labeling, they are tentatively attributed here through the spectra differences between the cupric nitrite complex and its chloride precursor. Consistent with the X-ray structures observed, EPR spectra of these complexes in frozen solution indicate either tetragonal or rhombically distorted structures, depending on solvent (Supplementary Material).
Nitrite reductase reactivity; cupric nitrite complexes plus ferrous hemes
In our initial report on this topic [36], we studied the reaction of [(tmpa)CuII(NO2)]+ with two equiv of the ferrous heme (F8)FeII, under anaerobic conditions, in acetone at RT and based on spectroscopic analyses, a one-to-one mixture of the heme-nitrosyl species (F8)FeII(NO) and the μ-oxo complex [(F8)FeIII-O-CuII(tmpa)]+ are produced (Scheme 2, top). Based on the results of control experiments, the reduced heme provides the one-electron required for the reaction, where nitrite (formally NO+) is reduced and one O-atom derived from the nitrite is trapped as an oxo-bridged FeIII-O-CuII product. See below for further discussion on possible mechanisms of reaction.
Scheme 2.

To expand on the initial study, we decided to compare and contrast the reactivity now using a copper(II)-nitrito complex with tri- instead of tetra-dentate ligand. We know from many years of study of dioxygen reduction chemistry employing copper ion complexes, that ligand denticity, along with other factors such as the exact nature of the donor (e.g., alkylamine vs. pyridyl) and the chelate ring sizes (e.g., five- vs. six-membered), govern the structures obtained, the redox properties of derived CuII/CuI complexes and thus overall reactivity [50–55]. When [(AN)CuII(NO2)]+ with tridentate N-chelate is reacted with two equiv (F8)FeII, the same overall reaction occurs, Scheme 2. The corresponding μ-oxo complex [(F8)FeIII-O-CuII(AN)]+ (λmax= 439 nm) forms in good yield (> 80 %), based on the quantitative analyses of UV-Vis and EPR spectra (Fig. 3). [(F8)FeIII-O-CuII(AN)]+ was previously structurally and spectroscopically characterized [56], but this species is somewhat unstable with respect to hydrolysis due to its very basic μ-oxo ligand (especially compared to its analog [(F8)FeIII-O-CuII(tmpa)]+) [57, 58]. Thus, some bridge-protonation and bridge breaking occurs, leading to the mononuclear complex (F8)FeIII(OH) and (AN)CuII-X (X = solvent or hydroxide) species. Here, small amounts of these complexes are detected by a combination of UV-vis and EPR spectroscopies. The other product, (F8)FeII(NO), resulting from capture of NO by one of the two ferrous hemes present in the reaction mixture, is obtained in essentially quantitative yields, based on UV-vis (λmax= 399 nm) [36, 59], EPR (Fig. 3) and IR (νNO = 1685 cm−1) spectroscopies.
When comparing this new system employing the nitrite complex [(AN)CuII(NO2)]+ to that of the reaction using [(tmpa)CuII(NO2)]+, we observe the reaction to be roughly twice as fast for the former, based on monitoring of the disappearance of the UV-vis band for (F8)FeII (λmax= 420 nm). This rate effect may be due to the differing nitrite coordination mode, bidentate vs. unidentate in the AN vs. the TMPA copper complexes, see Discussion. It is worth mentioning that a detailed kinetic analysis was not performed, due to the exceptional instability of the product [(F8)FeIII-O-CuII(AN)]+ when studied at concentrations employed for UV-vis monitoring (see Methods section).
In the second set of experiments, in order to study the role of the reducing ability of the ferrous heme center in nitrite reduction to nitric oxide, we employed a different heme with strong electron donating peripheral substituents, (TMPP)FeII. This ferrous heme was generated by reduction of (TMPP)FeIII(Cl) under anaerobic conditions and characterized by UV-vis, 1H-NMR, and elemental analyses. Also, as one of the expected reaction products, the μ-oxo complex [(TMPP)FeIII-O-CuII(tmpa)]+ (Scheme 3) was synthesized by bubbling dry dioxygen through a one-to-one mixture of the reduced heme and copper mononuclear complexes and it was characterized by UV-vis, ESI-MS, and elemental analyses. [(TMPP)FeIII-O-CuII(tmpa)][B(C6F5)4] has a distinctive red-shifted Soret band (~ 443 nm) [57, 60], that which is quite different from other high-spin ferric hemes such as is observed for the μ-oxo porphyrin-iron(III) dimer, [(TMPP)FeIII]2O [61] (λmax ≈ 412 nm in acetone), and (TMPP)FeIII(OH) (λmax ≈ 434 nm) (Supplementary Material). The authentic sample of [(TMPP)FeII(NO) (νNO = 1677 cm−1), as the other expected product, was also generated in-situ by bubbling excess NO(g) into the solution of [(TMPP)FeII in acetone under a nitrogen atmosphere and it was characterized by UV-vis and IR spectroscopies.
Scheme 3.

Reacting one equiv of the cupric nitrite complex, [(tmpa)CuII(NO2)]+ with two equiv of the more electron rich porphyrin, (TMPP)FeII, results in same overall redox reaction, converting nitrite to nitric oxide, Scheme 3. We observe the reaction proceeds with about the same rate as the combination of (F8)FeII and [(tmpa)CuII(NO2)]+ does, implying that the greater electron donating ability of this ferrous heme does not accelerate the nitrite reduction reaction. The quantitative analyses of UV-Vis and EPR spectra of reaction products and authentic samples confirm generation of a one-to-one mixture of the ferrous heme nitrosyl (TMPP)FeII(NO) (λmax= 410 nm) and the corresponding μ-oxo complex [(TMPP)FeIII-O-CuII(tmpa)]+ (λmax= 443 nm) in high yield, Fig. 4.
In the final system investigated here, we employed the copper(II)-nitrite complex with tridentate (rather than tetradentate) chelate, with the electron-rich ferrous heme. Thus, reaction of [(AN)CuII(NO2)]+ with two equiv (TMPP)FeII, was studied, Scheme 4. IR spectroscopy (νNO = 1677 cm−1) directly indicated nitrosyl complex, (TMPP)FeII(NO), production. However, according to the UV-vis spectra observed, no μ-oxo complex [(TMPP)FeIII-O-CuII(tmpa)]+ formation occurred (Fig. 5). This may be due to its very basic μ-oxo ligand resulting in instability toward protonation and hydrolysis. This was further validated when all attempts toward the synthesis of the authentic μ-oxo complex by oxidation of reduced complexes (by O2 bubbling, as above) were unsuccessful, even when employing Me-THF as a 'dryer' solvent. The species which is instead generated is the ***μ-hydroxo complex [(TMPP)FeIII-(OH)-CuII(tmpa)]+ (λmax = 417 nm), possessing a distinctive Soret band, different from that for the mononuclear complex, (TMPP)FeIII(OH) (λmax = 434 nm). In spite of the difficulties in this system, comparison of UV-vis spectra for the actual reaction to those of authentic compounds indicates the nitrite reduction chemistry outlined in Scheme 4 occurs in very good yield. Nitrite analysis also confirms the conversion of nitrite to nitric oxide is near quantitative, as no trace of nitrite was detected following the reaction.
Scheme 4.

The rate of ferrous heme disappearance for this system (Scheme 4) could be semi-quantitatively described as being a few times faster than for the reaction of the same heme with the [(tmpa)CuII(NO2)]+ analogue, Scheme 3. This observation is the same as that observed for the reaction of the more electron poor heme, (F8)FeII, where nitrito complex with tridentate chelate AN, [(AN)CuII(NO2)]+, is again faster than with [(tmpa)CuII(NO2)]+ (bearing a tetradentate chelate). Thus, in both cases, faster reduction of nitrite to nitric oxide was observed for when the ligation of nitrite is in the bidentate vs. unidentate coordination mode.
Discussion
In the Results section, we have demonstrated the heme/Cu assembly mediated reduction of nitrite to nitric oxide, utilizing two different ferrous hemes reacting with either of two cupric-nitrite complexes, one having tetradentate N4 (TMPA) chelation, while the other copper complex employs a tridentate N3 (AN) ligand. Very clean and high yielding reactions occur giving NO, as trapped by our ferrous heme, and an oxo-bridged heme/Cu product is also produced. The reaction stoichiometry is again indicated in the diagram below. Control experiments previously reported, for (F8)FeII and [(tmpa)CuI(MeCN)]+ [36], showed that neither of these reduced complexes, nor the combination of the two, are capable of nitrite reduction to NO. [(AN)CuI]+ or (TMPP)FeII by themselves are also unreactive to nitrite ion (Supplementary Material) [62].
We do believe that these reactions to a significant extent relate to what happens in cytochrome c oxidase nitrite reductase biochemistry. In our synthetic coordination chemistry system, a second heme equivalent is present in order to trap NO produced, allowing for 100 % conversion. If less than two molar equiv of ferrous heme are added, the reaction only occurs partially, because NO produced reacts very rapidly with any ferrous heme still not reacted with the cupric-nitrite complex. In the enzyme, of course this does not occur, but since only oxidized metal ions are present in the active site following nitrite reduction, i.e., Fe(III) and Cu(II), the NO(g) produced escapes since NO binds at best weakly to these oxidized metal ions.
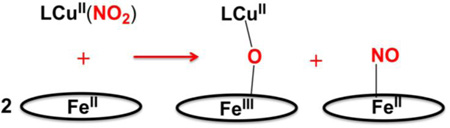
As concerns possible reaction mechanisms, we can start by drawing two significant observations from the results presented. The first is that the nature of the heme, its reducing ability, does not seem to matter in determining the course of the overall reaction, and more importantly it does not affect the observed reaction rate (but of course in the absence of detailed kinetics). Yet, we can deduce that the electron required for nitrite (formally NO+) reduction comes from the heme-Fe(II). We know this because control experiments related to these systems, reported earlier [36], show that a ferric heme with added nitrite, does not react with either LCuI complex, see diagram below.

In fact, from electrochemical measurements [36], we know that for the case of ferrous heme (F8)FeII, the LCuI complexes are chemically stronger reductants. So, the ferrous heme supplies the one-electron required in the reaction, but when using the stronger reductant, (TMPP)FeII, as compared to (F8)FeII, the ferrous heme reduction of nitrite bound to copper(II) does not proceed at an observably greater overall reaction rate. Thus, the electron-transfer reduction apparently does not occur in the rate-determining step.
A second perhaps key observation is that in a relative sense, the reactions of [(AN)CuII(NO2)]+ with ferrous hemes are faster than those with [(tmpa)CuII(NO2)]+. We believe that this is an effect of the differing coordination of nitrite in the two cupric complexes. This quite likely leads to differing approaches of the cupric nitrite complex to form a ferrous heme-(nitrite)-cupric assembly, Scheme 5. As precedent for this supposition, enzymatic nitrite reduction in heme enzymes has in fact been proposed to occur either by nitrite N-coordination or O-ligation to the iron and that these lead to alternative courses of reaction with accompanying varying kinetic behavior [63–66].
Scheme 5.


With [(AN)CuII(NO2)]+, O,O'-bidentate nitrite coordination (vide supra) allows for easier N-atom approach to the ferrous heme, i.e., favoring N-(nitro)-coordination to the iron(II) ion (Scheme 5a), quite likely in the rate determining step. Then, electron transfer from iron(II) to the bridged nitrite moiety could readily lead to a [(AN)CuII(oxo)] (formally oxo = O2−) species and a short-lived ferric nitrosyl complex; the latter would readily lose NO(g), especially in the presence of an exogenous (the second equivalent) ferrous heme and the ferric ion can easily couple to the cupric-oxo species giving the very kinetically stable μ-oxo compounds [(porphyrinate)FeIII-O-CuII(AN)]+ (porphyrinate = F8 or TMPP) (Scheme 5a).
As observed in studies on nitrite reduction by heme protein bacterial nitrite reductases and/or myoglobin [63–66], the approach of the nitrite to the ferrous heme, either via the N- or O-atom, is controlled by factors related to the binding pocket, especially nitrite O-atom H-bonding from nearby protein amino-acid residues. In essence, the cupric ion in the present heme/Cu assemblies plays the analogous role, as a Lewis Acid surrogate of a proton which (i) orients/directs the nitrite anion to favor iron N-binding (for the (AN)CuII complex) or O-binding (in the case of (tmpa)CuII) and (ii) acts as a strong oxo-acceptor (Note: Cu(II) is very oxophilic), which facilitates nitrite (N–O) bond cleavage leading to subsequent formation of nitric oxide along with a μ-oxo heme/Cu complex. We cannot however rule out a mechanism (not shown) in which the exogenous ferrous heme reduces (by electron-transfer) the intermediate ferric-nitrosyl (just mentioned) followed by coupling of the CuII-oxo moiety to the newly produced ferric ion.
For reaction of [(tmpa)CuII(NO2)]+ with either ferrous heme, we envision that the O-unidentate nitrite copper(II) coordination very well could lead to different reaction mechanisms, as outlined in Scheme 5b and 5c. Now, the nitrito O-atom far from and not coordinated to the cupric center approaches the Fe(II) ion, whereupon (i) electron-transfer occurs leading to a short-lived ferric-oxo species, (ii) nitric oxide forms, escapes and is trapped by the second ferrous heme, and (iii) a cupric complex is generated. The FeIII-O− couples to the LCuII complex giving [(porphyrinate)FeIII-O-CuII(tmpa)]+ (Scheme 5b). An alternative but interesting mechanistic proposal would be something like that depicted in Scheme 5c, where the furthest from copper nitrito O-atom approaches the Fe(II) ion and formally two-electron O-atom transfer occurs, giving a high-valent Fe(IV)=O species, NO(g) and a corresponding cuprous complex; NO(g) is again trapped by a second ferrous heme while the Fe(IV)=O and copper(I) moieties couple to give the product [(porphyrinate)FeIII-O-CuII(tmpa)]+. Some precedent for such a mechanism exists. For instance, oxo transfer to an active site iron has been previously suggested as a mechanistic alternative in NO reductase (NOR) biochemistry [67, 68]. Also, we have previously demonstrated that ligand-copper(I) complexes do react with (porphyrinate)Fe(IV)=O species to generate the corresponding μ-oxo heme/Cu complex [56].
In addition to the courses of reaction depicted in Scheme 5b and 5c, we suggest one other possibility, which is that in all cases, either with [(tmpa)CuII(NO2)]+ or [(AN)CuII(NO2)]+, nitrite ion N-atom coordination to the ferrous heme is what leads to a productive reaction, with nitrite (N–O) bond cleavage and NO(g) formation. Such coordination is favored based on known (bio)chemistry and expectations from "hard-soft" acid-base considerations. So, even for the chemistry with [(tmpa)CuII(NO2)]+, a successful reaction may require nitrite dissociation from the cupric ion and/or isomerization-rearrangement so the nitrite N-atom can reach and bind the reduced heme. Perhaps this accounts for the generally slower kinetics observed for the reactions of [(tmpa)CuII(NO2)]+ with either ferrous heme complex. Further studies will be required to address these mechanistic considerations.
Conclusion
Our interest in small molecule activation at heme-copper centers, such as O2-reduction [69], NO reductive coupling [41, 59], NO oxidation by an oxidized heme/Cu center to give nitrite and nitrite reduction by a partially reduced heme/Cu assembly to nitric oxide [36], has led us to expand on our previous initial report for the latter, nitrite reductase chemistry with heme/Cu assemblies. Such chemistry occurs in the mitochondria of cells, providing for generation of additional NO, as needed by cellular demands, such as in hypoxia. Here, with combinations of ferrous or cupric-nitrite complexes, we have shown that NO2− can be readily reduced to nitric oxide, with the electron coming from the ferrous heme. Clean, stoichiometric and high-yielding examples of such reactions were presented as accompanied by detailed chemical and spectroscopic characterization. While detailed kinetic analyses could not be performed, qualitative but clear trends in reaction rates and results as a function of heme or copper ligation tendencies and/or redox properties were observed. On these bases, possible reaction mechanisms were proposed and discussed, highlighting how the details of particular coordination chemistry situations may influence the course of reactions. Of particular note is that the cupric ion binding mode of nitrite is controlled by the denticity of the chelate on copper, leading to how the coordination of nitrite to the ferrous heme takes place, via either N- or O-atom binding. Such discrimination in enzymes with nitrite reductase activity occurs via heme pocket features, especially H-bonding to nitrite anion. In cytochrome c oxidase, the copper ion may serve exactly such a role, (i) orienting the nitrite for subsequent binding to the reduced heme and (ii) acting as an oxo-acceptor, as shown by the findings in the present synthetic chemistry.
Supplementary Material
Acknowledgment
We are grateful to the USA National Institutes of Health, GM60353, for support of this research.
References
- 1.Schopfer MP, Wang J, Karlin KD. Inorg. Chem. 2010;49:6267–6282. doi: 10.1021/ic100033y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Tavares P, Pereira AS, Moura JJG, Moura I. J. Inorg. Biochem. 2006;100:2087–2100. doi: 10.1016/j.jinorgbio.2006.09.003. [DOI] [PubMed] [Google Scholar]
- 3.Zumft WG. Microbiol. Molec. Biol. Revs. 1997;61:533–616. doi: 10.1128/mmbr.61.4.533-616.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Poole RK. Biochem. Soc. Trans. 2005;33:176–180. doi: 10.1042/BST0330176. [DOI] [PubMed] [Google Scholar]
- 5.Samouilov A, Kuppusamy P, Zweier JL. Arch. Biochem. Biophys. 1998;357:1–7. doi: 10.1006/abbi.1998.0785. [DOI] [PubMed] [Google Scholar]
- 6.Dezfulian C, Raat N, Shiva S, Gladwin MT. Cardiovasc. Res. 2007;75:327–338. doi: 10.1016/j.cardiores.2007.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lundberg JO, Gladwin MT, Ahluwalia A, Benjamin N, Bryan NS, Butler A, Cabrales P, Fago A, Feelisch M, Ford PC, Freeman BA, Frenneaux M, Friedman J, Kelm M, Kevil CG, Kim-Shapiro DB, Kozlov AV, Lancaster JR, Lefer DJ, McColl K, McCurry K, Patel RP, Petersson J, Rassaf T, Reutov VP, Richter-Addo GB, Schechter A, Shiva S, Tsuchiya K, van Faassen EE, Webb AJ, Zuckerbraun BS, Zweier JL, Weitzberg E. Nat. Chem. Biol. 2009;5:865–869. doi: 10.1038/nchembio.260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.van Faassen EE, Bahrami S, Feelisch M, Hogg N, Kelm M, Kim-Shapiro DB, Kozlov AV, Li H, Lundberg JO, Mason R, Nohl H, Rassaf T, Samouilov A, Slama-Schwok A, Shiva S, Vanin AF, Weitzberg E, Zweier J, Gladwin MT. Medicinal Research Reviews. 2009;29:683–741. doi: 10.1002/med.20151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Fukuto JM, Carrington SJ, Tantillo DJ, Harrison JG, Ignarro LJ, Freeman BA, Chen A, Wink DA. Chem. Res. Toxic. 2012;25:769–793. doi: 10.1021/tx2005234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kozlov AV, Staniek K, Nohl H. FEBS Letters. 1999;454:127–130. doi: 10.1016/s0014-5793(99)00788-7. [DOI] [PubMed] [Google Scholar]
- 11.Crane BR. Biochem. Soc. Trans. 2008;036:1149–1154. doi: 10.1042/BST0361149. [DOI] [PubMed] [Google Scholar]
- 12.Zhu Y, Silverman RB. Biochemistry. 2008;47:2231–2243. doi: 10.1021/bi7023817. [DOI] [PubMed] [Google Scholar]
- 13.Gladwin MT, Schechter AN, Kim-Shapiro DB, Patel RP, Hogg N, Shiva S, Cannon RO, Kelm M, Wink DA, Espey MG, Oldfield EH, Pluta RM, Freeman BA, Lancaster JR, Feelisch M, Lundberg JO. Nature Chemical Biology. 2005;1:308–314. doi: 10.1038/nchembio1105-308. [DOI] [PubMed] [Google Scholar]
- 14.Gladwin MT, Shiva S. Circulation Research. 2009;104:1136–1138. doi: 10.1161/CIRCRESAHA.109.198911. [DOI] [PubMed] [Google Scholar]
- 15.Zweier JL, Samouilov A, Kuppusamy P. Biochim. Biophys. Acta. 1999;1411:250–262. doi: 10.1016/s0005-2728(99)00018-3. [DOI] [PubMed] [Google Scholar]
- 16.Cosby K, Partovi KS, Crawford JH, Patel RP, Reiter CD, Martyr S, Yang BK, Waclawiw MA, Zalos G, Xu XL, Huang KT, Shields H, Kim-Shapiro DB, Schechter AN, Cannon RO, Gladwin MT. Nat. Med. 2003;9:1498–1505. doi: 10.1038/nm954. [DOI] [PubMed] [Google Scholar]
- 17.Shiva S, Huang Z, Grubina R, Sun JH, Ringwood LA, MacArthur PH, Xu XL, Murphy E, Darley-Usmar VM, Gladwin MT. Circulation Research. 2007;100:654–661. doi: 10.1161/01.RES.0000260171.52224.6b. [DOI] [PubMed] [Google Scholar]
- 18.Shiva S, Rassaf T, Patel RP, Gladwin MT. Cardiovasc. Res. 2011;89:566–573. doi: 10.1093/cvr/cvq327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Rassaf T, Flogel U, Drexhage C, Hendgen-Cotta U, Kelm M, Schrader J. Circulation Research. 2007;100:1749–1754. doi: 10.1161/CIRCRESAHA.107.152488. [DOI] [PubMed] [Google Scholar]
- 20.Totzeck M, Hendgen-Cotta UB, Luedike P, Berenbrink M, Klare JP, Steinhoff HJ, Semmler D, Shiva S, Williams D, Kipar A, Gladwin MT, Schrader J, Kelm M, Cossins AR, Rassaf T. Circulation. 2012;126:325–334. doi: 10.1161/CIRCULATIONAHA.111.087155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Tiso M, Tejero Js, Basu S, Azarov I, Wang X, Simplaceanu V, Frizzell S, Jayaraman T, Geary L, Shapiro C, Ho C, Shiva S, Kim-Shapiro DB, Gladwin MT. J. Biol. Chem. 2011;286:18277–18289. doi: 10.1074/jbc.M110.159541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Basu S, Azarova NA, Font MD, King SB, Hogg N, Gladwin MT, Shiva S, Kim-Shapiro DB. J. Biol. Chem. 2008;283:32590–32597. doi: 10.1074/jbc.M806934200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gladwin MT. Nature Chemical Biology. 2005;1:245–246. doi: 10.1038/nchembio1005-245. [DOI] [PubMed] [Google Scholar]
- 24.Lundberg JO, Weitzberg E, Gladwin MT. Nat. Rev. Drug. Discov. 2008;7:156–167. doi: 10.1038/nrd2466. [DOI] [PubMed] [Google Scholar]
- 25.de Mel A, Murad F, Seifalian AM. Chem. Rev. 2011;111:5742–5767. doi: 10.1021/cr200008n. [DOI] [PubMed] [Google Scholar]
- 26.Babcock GT, Wikström M. Nature. 1992;356:301–309. doi: 10.1038/356301a0. [DOI] [PubMed] [Google Scholar]
- 27.Ferguson-Miller S, Babcock GT. Chem. Rev. 1996;96:2889–2908. doi: 10.1021/cr950051s. [DOI] [PubMed] [Google Scholar]
- 28.Kim E, Chufán EE, Kamaraj K, Karlin KD. Chem. Rev. 2004;104:1077–1133. doi: 10.1021/cr0206162. [DOI] [PubMed] [Google Scholar]
- 29.Poyton RO, Ball KA. Discov Med. 2011;57:154–159. [PubMed] [Google Scholar]
- 30.Shiva S. Redox Biology. 2013;1:40–44. doi: 10.1016/j.redox.2012.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Castello PR, David PS, McClure T, Crook Z, Poyton RO. Cell Metabolism. 2006;3:277–287. doi: 10.1016/j.cmet.2006.02.011. [DOI] [PubMed] [Google Scholar]
- 32.Gupta KJ, Stoimenova M, Kaiser WM. Journal of Experimental Botany. 2005;56:2601–2609. doi: 10.1093/jxb/eri252. [DOI] [PubMed] [Google Scholar]
- 33.Castello PR, Woo DK, Ball K, Wojcik J, Liu L, Poyton RO. Proc. Nat. Acad. Sci. 2008;105:8203–8208. doi: 10.1073/pnas.0709461105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Poyton RO, Castello PR, Ball KA, Woo DK, Pan N. Ann. N. Y. Acad. Sci. 2009;1177:48–56. doi: 10.1111/j.1749-6632.2009.05046.x. [DOI] [PubMed] [Google Scholar]
- 35.Gupta KJ, Igamberdiev AU. Mitochondrion. 2011;11:537–543. doi: 10.1016/j.mito.2011.03.005. [DOI] [PubMed] [Google Scholar]
- 36.Hematian S, Siegler MA, Karlin KD. J. Am. Chem. Soc. 2012;134:18912–18915. doi: 10.1021/ja3083818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kopf M-A, Neuhold Y-M, Zuberbühler AD, Karlin KD. Inorg. Chem. 1999;38:3093–3102. [Google Scholar]
- 38.Ghiladi RA, Kretzer RM, Guzei I, Rheingold AL, Neuhold Y-M, Hatwell KR, Zuberbühler AD, Karlin KD. Inorg. Chem. 2001;40:5754–5767. doi: 10.1021/ic0105866. [DOI] [PubMed] [Google Scholar]
- 39.Torres J, Sharpe MA, Rosquist A, Cooper CE, Wilson MT. FEBS Letters. 2000;475:263–266. doi: 10.1016/s0014-5793(00)01682-3. [DOI] [PubMed] [Google Scholar]
- 40.Ford PC, Lorkovic IM. Chem. Rev. 2002;102:993–1017. doi: 10.1021/cr0000271. [DOI] [PubMed] [Google Scholar]
- 41.Wang J, Schopfer MP, Sarjeant AAN, Karlin KD. J. Am. Chem. Soc. 2009;131:450–451. doi: 10.1021/ja8084324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Schopfer MP, Mondal B, Lee D-H, Sarjeant AAN, Karlin KD. J. Am. Chem. Soc. 2009;131:11304–11305. doi: 10.1021/ja904832j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Liang H-C, Zhang CX, Henson MJ, Sommer RD, Hatwell KR, Kaderli S, Zuberbuehler AD, Rheingold AL, Solomon EI, Karlin KD. J. Am Chem. Soc. 2002;124:4170–4171. doi: 10.1021/ja0125265. [DOI] [PubMed] [Google Scholar]
- 44.Park GY, Deepalatha S, Puiu SC, Lee D-H, Mondal B, Narducci Sarjeant AA, del Rio D, Pau MYM, Solomon EI, Karlin KD. J. Biol. Inorg. Chem. 2009;14:1301–1311. doi: 10.1007/s00775-009-0575-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Vogel KM, Kozlowski PM, Zgierski MZ, Spiro TG. J. Am. Chem. Soc. 1999;121:9915–9921. [Google Scholar]
- 46.Sheldrick GM. Acta Crystallographica Section A. 2008;64:112–122. doi: 10.1107/S0108767307043930. [DOI] [PubMed] [Google Scholar]
- 47.Yang L, Powell DR, Houser RP. Dalton Trans. 2007:955–964. doi: 10.1039/b617136b. [DOI] [PubMed] [Google Scholar]
- 48.Mukhopadhyay U, Bernal I, Massoud SS, Mautner FA. Inorg. Chim. Acta. 2004;357:3673–3682. [Google Scholar]
- 49.Nakamoto K. Infrared and Raman spectra of inorganic and coordination compounds. 5th Ed. New York: Wiley Interscience; 1997. pp. 48–53. [Google Scholar]
- 50.Quant Hatcher L, Karlin KD. J. Biol. Inorg. Chem. 2004;9:669–683. doi: 10.1007/s00775-004-0578-4. [DOI] [PubMed] [Google Scholar]
- 51.Hatcher LQ, Karlin KD. Adv. Inorg. Chem. 2006;58:131–184. [Google Scholar]
- 52.Chufan EE, Mondal B, Gandhi T, Kim E, Rubie ND, Moenne-Loccoz P, Karlin KD. Inorg. Chem. 2007;46:6382–6394. doi: 10.1021/ic700363k. [DOI] [PubMed] [Google Scholar]
- 53.Kakuda S, Peterson RL, Ohkubo K, Karlin KD, Fukuzumi S. J. Am. Chem. Soc. 2013;135:6513–6522. doi: 10.1021/ja3125977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Das D, Lee Y-M, Ohkubo K, Nam W, Karlin KD, Fukuzumi S. J. Am. Chem. Soc. 2013;135:2825–2834. doi: 10.1021/ja312523u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Itoh S. "Chemical Reactivity of Copper Active-Oxygen Complexes". In: Itoh S, Karlin KD, editors. Copper-Oxygen Chemistry. Hoboken: John Wiley & Sons, Inc.; 2011. pp. 225–282. [Google Scholar]
- 56.Halime Z, Kieber-Emmons MT, Qayyum MF, Mondal B, Gandhi T, Puiu SC, Chufán EE, Sarjeant AAN, Hodgson KO, Hedman B, Solomon EI, Karlin KD. Inorg. Chem. 2010;49:3629–3645. doi: 10.1021/ic9020993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Fox S, Nanthakumar A, Wikström M, Karlin KD, Blackburn NJ. J. Am. Chem. Soc. 1996;118:24–34. [Google Scholar]
- 58.Obias HV, van Strijdonck GPF, Lee D-H, Ralle M, Blackburn NJ, Karlin KD. J. Am. Chem. Soc. 1998;120:9696–9697. [Google Scholar]
- 59.Wang J, Schopfer MP, Puiu SC, Sarjeant AAN, Karlin KD. Inorg. Chem. 2010;49:1404–1419. doi: 10.1021/ic901431r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Karlin KD, Nanthakumar A, Fox S, Murthy NN, Ravi N, Huynh BH, Orosz RD, Day EP. J. Am. Chem. Soc. 1994;116:4753–4763. [Google Scholar]
- 61.Helms JH, Terhaar LW, Hatfield WE, Harris DL, Jayaraj K, Toney GE, Gold A, Mewborn TD, Pemberton JR. Inorg. Chem. 1986;25:2334–2337. [Google Scholar]
- 62.In further control experiments, combinations of reduced complexes, (F8)FeII/[(AN)CuI]+, (TMPP)FeII/[(tmpa)CuI(MeCN)]+ and (TMPP)FeII/[(AN)CuI]+, were studied. There, we observed slow reactivity toward nitrite ion, hours compared to minutes for the FeII/CuII-nitrite 'parent' reactions, and the final solutions appeared to be a complex mixtures.
- 63.Yi J, Heinecke J, Tan H, Ford PC, Richter-Addo GB. J. Am. Chem. Soc. 2009;131:18119–18128. doi: 10.1021/ja904726q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Silaghi-Dumitrescu R. Inorg. Chem. 2004;43:3715–3718. doi: 10.1021/ic035403p. [DOI] [PubMed] [Google Scholar]
- 65.Williams PA, Fulop V, Garman EF, Saunders NFW, Ferguson SJ, Hajdu J. Nature. 1997;389:406–412. doi: 10.1038/38775. [DOI] [PubMed] [Google Scholar]
- 66.Perissinotti LL, Marti MA, Doctorovich F, Luque FJ, Estrin DA. Biochemistry. 2008;47:9793–9802. doi: 10.1021/bi801104c. [DOI] [PubMed] [Google Scholar]
- 67.Blomberg LM, Blomberg MRA, Siegbahn PEM. Biochim. Biophys. Acta. 2006;1757:240–252. doi: 10.1016/j.bbabio.2006.04.008. [DOI] [PubMed] [Google Scholar]
- 68.Blomberg LM, Blomberg MRA, Siegbahn PEM. J. Biol. Inorg. Chem. 2007;12:79–89. doi: 10.1007/s00775-006-0166-x. [DOI] [PubMed] [Google Scholar]
- 69.Halime Z, Kotani H, Li Y, Fukuzumi S, Karlin KD. Proc. Nat. Acad. Sci. 2011;108:13990–13994. doi: 10.1073/pnas.1104698108. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.