

Background: Smoke components can generate 1) oxidative stress; 2) complement activation; 3) endoplasmic reticulum stress; and 4) lipid dysregulation.
Results: In smoke-exposed RPE cells all four measures were activated, and reversed by antioxidants and blocking alternative complement pathway signaling.
Conclusion: Oxidative stress and complement act synergistically in age-related macular degeneration (AMD) pathogenesis.
Significance: Identifying mechanisms of lipid deposition will aid to develop new therapeutic approaches for AMD.
Keywords: Complement System, ER Stress, Lipodystrophy, Mitochondria, Oxidative Stress, Unfolded Protein Response, Age-related Macular Degeneration, Smoking
Abstract
Age-related macular degeneration (AMD) is a complex disease caused by genetic and environmental factors, including genetic variants in complement components and smoking. Smoke exposure leads to oxidative stress, complement activation, endoplasmic reticulum (ER) stress, and lipid dysregulation, which have all been proposed to be associated with AMD pathogenesis. Here we examine the effects of smoke exposure on the retinal pigment epithelium (RPE). Mice were exposed to cigarette smoke or filtered air for 6 months. RPE cells grown as stable monolayers were exposed to 5% cigarette smoke extract (CSE). Effects of smoke were determined by biochemical, molecular, and histological measures. Effects of the alternative pathway (AP) of complement and complement C3a anaphylatoxin receptor signaling were analyzed using knock-out mice or specific inhibitors. ER stress markers were elevated after smoke exposure in RPE of intact mice, which was eliminated in AP-deficient mice. To examine this relationship further, RPE monolayers were exposed to CSE. Short term smoke exposure resulted in production and release of complement C3, the generation of C3a, oxidative stress, complement activation on the cell membrane, and ER stress. Long term exposure to CSE resulted in lipid accumulation, and secretion. All measures were reversed by blocking C3a complement receptor (C3aR), alternative complement pathway signaling, and antioxidant therapy. Taken together, our results provide clear evidence that smoke exposure results in oxidative stress and complement activation via the AP, resulting in ER stress-mediated lipid accumulation, and further suggesting that oxidative stress and complement act synergistically in the pathogenesis of AMD.
Introduction
Age-related macular degeneration (AMD)2 is the leading cause of blindness in people over the age of 60 in the Western world. AMD is a late-onset maculopathy that ultimately results in the loss of central vision. The early phase of AMD is characterized by the presence of drusen, which are deposits rich in lipoproteins located between the retinal pigment epithelium (RPE) and Bruch's membrane (BrM). The late phase of AMD can present itself as either the atrophic, dry; or the neovascular, wet form (1, 2). The atrophic form of the disease or geographic atrophy results in loss of RPE cells, followed by the loss of underlying photoreceptors and overlying choriocapillaris. The neovascular form or choroidal neovascularization is characterized by growth of new choroidal blood vessels through the BrM and RPE into the subretinal space. Because new vessels tend to be more leaky than established ones, the resulting fluid leakage into either the subretinal or sub-RPE space can lead to impairment of retinal function and increased photoreceptor cell death (3). Hence one of the main target cells for pathology in both early and late AMD is the RPE.
Based on the presence of oxidized lipids and complement components in drusen and BrM, the hypothesis of oxidative/inflammatory stress-mediated AMD was postulated (4). This hypothesis gained support when some of the main genetic risk factors were identified as polymorphisms occurring in complement genes, including the alternative pathway (AP) inhibitor, CFH (complement factor H (5–8)), and others. Hence based on the current literature, the following sequence is proposed: the trigger for pathophysiology is provided by chronic oxidative stress resulting in alterations in the target tissues (photoreceptors, RPE, BrM, and choriocapillaris); tissue damage leads to chronic inflammation, involving the innate immune system (e.g. the complement system); chronic inflammation subsequently leads to cellular damage in the target tissues, extracellular matrix modifications, and the accumulation of extracellular debris including lipids; finally leading to the development of drusen and pigmentary changes (early AMD) followed by the progression to geographic atrophy and/or choroidal neovascularization (late AMD) (9, 10). According to this hypothesis, in the presence of complement abnormalities that lead to an overactive alternative pathway, the tissue effects of chronic inflammation are exacerbated.
The complement system is an evolutionary ancient part of the innate and adaptive immune system, and developed to participate in clearing pathogens and non-self cells from the organism. To execute this function, there are three initiation pathways (classical, lectin, and alternative) that all lead to a common terminal pathway via the generation of homologous complement C3 convertases (11). Activation of the complement system results in the generation of three classes of effector molecules: the soluble anaphylatoxins C3a and C5a, which signal through their respective G-protein coupled receptors (C3aR and C5aR) and are involved in chemotaxis and mediating inflammatory responses; the cell-bound opsonins C3b, C3d, and iC3b, which are derived from C3 upon cleavage, and are involved in removal of pathogens and cells; and the terminal membrane attack complex, which forms a nonspecific pore in the cell membrane, and is involved in cell lysis. Self-cells are protected from complement activation by the expression of membrane-bound complement inhibitory molecules and soluble serum inhibitors that prevent self-cell complement-mediated injury. However, because the levels, as well as the cellular localization, of these inhibitors can be influenced by environmental factors such as oxidative stress (12) or cigarette smoke (13, 14), self-cell surfaces can become targets of complement activation. For example, in AMD, changes in levels and localization of CFH and CD55 (15), CD46 (16), as well as CD59 (17) have been reported in RPE, BrM, and choroid, and have been associated with increased cellular deposition of complement C3 and membrane attack complex in tissue samples (4, 18) of patients.
Smoking has been recognized as the only modifiable risk factor for AMD. Smoking increases the risk for developing AMD (19), and promotes the progression of AMD from the atrophic to the neovascular form (20, 21); smoking cessation on the other hand can reduce both disease risk and rate of progression (22). Smoking is thought to contribute to disease mainly by generating oxidative stress in the target tissues by producing free radicals (23) and depleting the antioxidant system (reviewed in Ref. 24). However, important for this study, cigarette smoke has been shown to be able to directly activate C3 by modifying C3 in a way that reduces its ability to bind to CFH (25), and serum levels of complement components are elevated in smokers (26). Neufeld and colleagues (27) have shown complement activation in the RPE/choroid in mice exposed to long-term smoke inhalation, including the presence of the anaphylatoxin C3a and the deposition membrane attack complex. Our follow-up study, using the same long term smoke model and comparing effects in wild type mice and mice without a functional AP (complement factor B knock-out mice, CFB−/−), showed clearly that ocular pathology generated by smoke exposure in mouse is dependent upon AP activation (28).
The endoplasmic reticulum (ER) functions include the synthesis of secretory and membrane proteins, the production of lipids and sterols, and calcium homeostasis. Many different environmental and genetic factors can cause ER stress; relevant for this study, smoking, as well as complement activation, has been suggested as triggers. Acute and chronic smoke exposure can adversely affect protein synthesis (29), lipid metabolism (30), and calcium homeostasis (78), resulting in ER stress. Specifically, smoking results in misfolding of nascent polypeptides in the ER as well as accumulation of protein aggregates; both of which trigger specific compensatory mechanisms to reduce or reverse the accumulation of unfolded proteins (unfolded protein response, UPR) (31), or removal of protein aggregates (autophagy) (32), respectively. Interestingly, in passive Heymann nephritis, a model of membranous nephropathy, membrane attack complex-induced podocyte injury has been found to be associated with and might be mediated by ER stress (summarized by Ref. 33). The UPR is a highly conserved response designed to relieve stress and restore ER homeostasis (34). As a first line of defense, the UPR reduces overall protein synthesis while selectively increasing synthesis of proteins involved in folding. To reduce the amount of unfolded proteins or those that did not get modified properly, damaged proteins are routed into the proteasome or to the autophagosomes for degradation. Finally, the UPR can lead to the expansion of the ER size as well as its capacity. The UPR is monitored and mediated by three receptor systems localized in the ER membrane, PERK (eukaryotic translation initiation factor 2-α kinase 3), IRE1 (inositol requiring enzyme 1), and ATF6 (activating transcription factor 6) (for reviews see Refs. 29 and 35)). All three sensors share the same entry molecule GRP78 (glucose-regulated protein of 78 kDa) on the ER luminal surface (36), but trigger unique downstream events associated with unique marker genes. The relative contribution and the sequence of activation within the three pathways is cell-type and stress-dependent; which is analyzed typically by examining up-regulation of mRNA, splice variants, or protein expression of factors downstream of PERK, ATF6, and IRE1. Here we use GRP78, CHOP (to represent IRE1 activation), sXBP1 (to represent ATF6 activation), and eIF2α (to represent PERK involvement) as specific markers.
Lipid accumulation is one of the hallmarks of age-related macular degeneration, building up as extracellular lesions in BrM. The analysis of lipid composition in BrM has suggested that the lipoprotein particles are different from those found in plasma. They contain free and esterified cholesterol (37), phosphatidylcholine, and apolipoprotein B100 (38), and differ in composition such as their enrichment for esterified cholesterol and the percent phospholipid being comprised of phosphatidylcholine (39). It is suggested that the lipoproteins in BrM may be derived in part from the photoreceptor outer segments, but are processed significantly by the RPE (40). Relevant for this study, lipid accumulation has been associated with ER stress and the UPR. ER stress has been shown to activate transcription factors know as sterol regulatory element-binding proteins (SREBP), which increase the expression of proteins required for the biosynthesis and uptake of cholesterol and triglycerides. Although the mechanism of ER stress-mediated SREBP activation is unclear, players from the UPR including eIF2α and GRP78 have been implicated (reviewed in Ref. 41) and interestingly, sterol regulatory element-binding transcription factor 1 (SREBF-1) can be translated by an internal ribosome entry site (42).
Given the aforementioned lines of evidence, we tested whether lipid accumulation in the RPE can be triggered by smoke exposure. More specifically, we asked whether lipid accumulation is dependent upon complement activation and ER stress. Furthermore, the presence of a feedback loop between complement and oxidative stress is explored. Finally, we tested the hypothesis that complement alternative pathway inhibition provides protection in cigarette smoke-induced oxidative and ER stress, and lipid accumulation.
EXPERIMENTAL PROCEDURES
Animals
CFB−/− mice on a C57BL/6J background were generously provided by V. Michael Holers (University of Colorado Health Science Center, Denver, CO) (43); C57BL/6J mice were purchased (Jackson Laboratory, Bar Harbor, ME). Animals were housed under a 12:12 h, light:dark cycle with access to food and water ad libitum. At 8 weeks of age male mice were divided into two groups (n = 12 per group and genotype); with the control group remaining in a filtered air environment, the experimental groups being subjected to cigarette smoke. Cigarette smoke exposure was carried out (5 h per day, 5 days per week) by burning 3R4F reference cigarettes (University of Kentucky, Louisville, KY) using a smoking machine (model TE-10; Teague Enterprises) for 6 months. The average concentration of total suspended particulates was 130 mg/m3 and was monitored twice daily. For more details, see Ref. 28.
Reagents
The reagents used in these studies included pooled normal human serum (Quidel) as a source of complement proteins. To block the alternative pathway of complement activation, a targeted inhibitory protein (CR2-fH) was produced as previously described (44). This agent targets the inhibitory domain of factor H to sites of C3d deposition and effectively blocks alternative pathway activation. To block anaphylatoxin C3a-receptor signaling, the experiments were performed in the presence and absence of a selective, high affinity, competitive C3a-receptor antagonist (Calbiochem; N2-[(2,2-diphenylethoxy)acetyl]-l-arginine, TFA) following the manufacturer's recommended dose; to prevent oxidative stress, the antioxidant NAC (N-acetylcysteine; Sigma) was used. Antibodies specific for CHOP, GRP78, eIF2α, and GAPDH were purchased from Cell Signaling Technology (Danvers, MA), XBP1 was from Santa Cruz (Dallas, TX); the antibody to determine C3d deposition was generated by us and used as described (45).
Cell Culture System
Experiments were performed on ARPE-19 cells, a human RPE cell line that displays the differentiated phenotype of RPE cells, and forms a polarized monolayer on Transwell filters (Costar) (46). To form monolayers, cells were expanded in DMEM:F-12 (Invitrogen) with 10% fetal bovine serum (FBS) and 1× penicillin:streptomycin. After the cells reached confluence, they were grown in serum-reduced media until they reached stable barrier facilities, based on transepithelial resistance (TER) measurements. TER values reach a stable plateau of 40–45 Ω cm2 within 2–3 weeks after confluence (46). FBS was removed completely for the final days prior to measurements, which does not alter survival or monolayer formation (47), such that complement-sufficient normal human serum can be used as a source of complement. TER was determined by measuring the resistance across the monolayer with an EVOM volt-ohmmeter (World Precision Instruments) as described previously, subtracting the TER for filters without cells and then multiplying by the surface area of the filters.
Cigarette Smoke Extract (CSE)
Cigarette smoke extract was generated as described (48); by bubbling smoke from four 3R4F cigarettes (University of Kentucky) through 50 ml of media (DMEM:F-12 plus 1× penicillin:streptomycin) using a Shapiro cigarette smoke machine (Washington University, St. Louis, MO). Media was sterile-filtered (0.22 μm) and used immediately. CSE was diluted with fresh media to create 5, 10, and 20% final concentrations. Stable ARPE-19 cell monolayers were treated with CSE applied to the apical compartment as described under “Results.” Treatment was performed apically, because we have shown previously that the apical side of ARPE-19 monolayers is more susceptible to oxidative stress-mediated complement activation than the basal side (46).
Immunocytochemistry
Intracellular CHOP, GRP78, and XBP1 in ARPE-19 cells was examined by immunofluorescence microscopy. Cells were grown on Transwell plates as described above. After specific treatments, cells were fixed in PBS containing 4% paraformaldehyde and nonspecific binding sites were blocked with 1% normal goat serum and 3% BSA in PBS (as a preabsorption buffer) for 2 h. The cells were incubated overnight at 4 °C with primary antibody followed by incubation for 1 h at room temperature with FITC-conjugated secondary antibody (1:200; Zymed Laboratories Inc., Invitrogen). As a negative control, primary antibodies were omitted. Surface staining of C3d was examined as described (45). The membranes were cut out of the wells, placed on a microscope slide, coverslipped with antifade, and imaged using confocal microscopy (Leica SP2).
Lipid Staining
Cells grown on Transwell plates or enucleated eyes from control and smoke-exposed mice were fixed in PBS containing 4% paraformaldehyde. Eyecups were cryoprotected in 30% sucrose in PBS, frozen in TissueTek O.C.T. (Fisher Scientific, Waltham, MA), and cut into 14-μm cryostat sections. For general lipid staining, RPE monolayers and mouse ocular sections were stained with Nile Red (Sigma). Nile Red (stock solution: 0.5 mg/ml in acetone) was applied to sections in phosphate-buffered saline at 1.75 μg/ml (49). Neutral lipids were identified using HCS LipidTox (Invitrogen), according to the manufacturer's instructions. For the identification of unesterified and esterified cholesterol, monolayers were incubated with 250 μg/ml of Filipin (stock solution: 2.5 mg/ml in N,N-dimethylformamide; Sigma); to identify the latter, monolayers were incubated with cholesterol esterase (30 μg/ml; for 5 h at 37 °C) to hydrolyze the esterified 3β-hydroxy group of cholesterol to a fatty acid. Staining of monolayers were examined after membranes were cut out of the wells, placed on a microscope slide, coverslipped with antifade, and imaged using confocal miscroscopy (Nile Red) or fluorescence microscopy (Filipin). To quantify the lipid signal in Bruch's membrane, images were thresholded, binarized, and measured in ImageJ software.
ELISA for C3 and C3a Measurements
To measure release of C3 and production of C3a by the cells, cell culture supernatants were centrifuged at 20,000 × g for 5 min. Microplates were coated with the respective C3 and C3a capture antibody (BD Bioscience) and 100 μl of the supernatant was added. The captured proteins were detected with C3- or C3a-specific antibodies conjugated to horseradish peroxidase, followed by development with chromogenic substrate OPD (Sigma). Product development was assayed by measuring absorbance at 492 nm. Aliquots were assayed in duplicate, and values were compared with a C3 and anaphylatoxin dose-response curve.
Western Blot Analyses
Cell culture supernatants were separated by electrophoresis on a 10% BisTris polyacrylamide gel (Invitrogen), and proteins were transferred to a nitrocellulose membrane. The membrane was probed with polyclonal antibodies to GRP78, CHOP, XBP1, and GAPDH (loading control), and antibody binding was visualized using a chemiluminescence detection kit (Amersham Biosciences Life Science). Density of the bands was analyzed and normalized against GAPDH control using the Alpha Innotech Fluorchem 9900 imaging system while running Alpha Ease FC Software 3.3 (Alpha Innotech, San Leandro, CA).
Measurements of Oxidative Stress
Cells were analyzed for reactive oxygen species (ROS) production, using 2,7-dichlorofluorescein diacetate dye (DCFDA; Molecular Probes/Invitrogen) according to the manufacturer's protocol. In short, after treatment of monolayers on Transwell filters, cells were collected, stained with 20 mm DCFDA dye, washed, and analyzed using a spectrophotometer (excitation wavelength, 485 nm; emission wavelength, 535 nm). Lipid peroxidation was measured determining the production of malondialdehyde (MDA), a natural biproduct of lipid peroxidation. Cells were collected after the respective treatments and homogenized in MDA lysis buffer (Abcam). MDA present in the cells was reacted with thiobarbituric acid to generate the MDA-thiobarbituric acid adduct and quantified colorimetrically using a spectrophotometer (λ = 532 nm). Standard curves for both ROS and MDA measurements were prepared as recommended.
Reverse Transcription PCR
RPE/choroid/sclera fractions (henceforth referred to as RPE/choroid) were isolated from control and smoke-exposed animals and stored at −80 °C until they were used. Cells were collected after treatment and stored at −80 °C.
Quantitative RT-PCR analyses were performed as previously described in detail (50). In short, real-time PCR analyses were performed in triplicate in a GeneAmp® 5700 Sequence Detection System (Applied Biosystems) using standard cycling conditions. Quantitative values were obtained by the cycle number, normalizing genes of interest to β-actin, and determining the fold-difference between experimental and control samples. Fold difference values were compared using Z-test analyses, accepting a significance of p < 0.05. See Table 1 for a list of primers used.
TABLE 1.
RT-PCR primer sequences
| Gene name | Symbol | Forward primer | Reverse primer |
|---|---|---|---|
| Actin β (human) | ACTB | 5′-AAATCTGGCACCACACCTTC-3′ | 5′-GGGGTGTTGAAGGTCTCAAA-3′ |
| Sterol regulatory element-binding transcription factor 1 (human) | SREBF1 | 5′-CTGCTGTCCACAAAAGCAAA-3′ | 5′-GGTCAGTGTGTCCTCCACCT-3′ |
| Tight junction protein ZO-1 (human) | TJP1 | 5′-CGCAGCCACAACCAATTCAT-3′ | 5′-TCAGGCGAAAGGTAAGGGAC-3′ |
| DNA damage-inducible transcript 3 protein (human) | CHOP | 5′-CACCACACCTGAAAGCAGACT-3′ | 5′-CCTCTTGCAGGTCCTCATACC-3′ |
| Binding immunoglobulin protein (human) | BIP | 5′-GAACGTCTGATTGGCGATGC-3′ | 5′-TCAACCACCTTGAACGGCAA-3′ |
| Complement component 3 (human) | C3 | 5′-ACTCTGTGCCCCCTGTAGTG-3′ | 5′-TGGTCAGGGACATCTGTTTG-3′ |
| X-Box binding protein 1 (human) | XBP | 5′-TTACGAGAGAAAACTCATGGCC-3′ | 5′-GGGTCCAAGTTGTCCAGAATGC-3′ |
| Actin β (mouse) | Actb | 5′-GCTACAGCTTCACCACCACA-3′ | 5′-TCTCCAGGGAGGAAGAGGAT-3′ |
| Sterol regulatory element-binding transcription factor 1 (mouse) | Srebf1 | 5′-GTGAGCCTGACAAGCAATCA-3′ | 5′-GGTGCCTACAGAGCAAGAGG-3′ |
| Binding immunoglobulin protein (mouse) | Bip | 5′-TGCAGCAGGACATCAAGTTC-3′ | 5′-TTTCTTCTGGGGCAAATGTC-3′ |
| DNA damage-inducible transcript 3 protein (mouse) | Chop | 5′-CTGCACCAAGCATGAACAGT-3′ | 5′-CTACCCTCAGTCCCCTCCTC-3′ |
Semi-quantitative RT-PCR was used to amplify the spliced and unspliced XBP1 mRNA. XBP1 primers were used as described by Samali et al. (51) and are listed in Table 1. PCR products were electrophoresed on a 2.5% agarose gel. β-Actin was used as a loading control. The size difference between the spliced and unspliced XBP1 is 26 nucleotides.
Statistics
Data are expressed as mean ± S.E. of the mean. Statview software was used for statistical analysis. The Fisher PLSD was used to compare trends over time. Comparison of two conditions was performed using the Student's t test.
RESULTS
Effects of Chronic Cigarette Smoke Exposure on ER Stress and Lipid Homeostasis in Vivo
In a previous publication (28) we have shown that 6 months of smoke exposure impairs retinal function as determined by electroretinography and contrast sensitivity measurements in C57BL/6J mice. In these published studies, gene expression analyses on the RPE/choroid fraction indicated increased complement activation and oxidative stress, concomitant with reduced mitochondrial function and autophagy. RPE/BrM alterations were analyzed by electron microscopy, and demonstrated increased mitochondrial size, mislocalization within the RPE, and a thickening of BrM. These features are indicative of altered mitochondrial function and stress, as well as altered degradation of cellular components. Importantly, mice deficient in AP signaling (lacking CFB) were protected from these cigarette smoke inhalation-mediated alterations (28).
Here we investigated whether these same animals exhibited ER stress and lipid dysregulation. ER stress markers, Grp78 and Chop, were elevated ∼2.5- and ∼8.5-fold, respectively, in the RPE/choroid fraction of smoke-exposed C57BL/6J wild type mice, whereas in CFB−/− mice, expression of these markers was not significantly altered as compared with normal (Fig. 1). Srebf-1, which can be up-regulated by UPR transcription factors and is indicative of altered cholesterol and fatty acid homeostasis, was also increased ∼4.5-fold in the RPE/choroid fraction of smoke-exposed wild type mice, but not in CFB−/− mice (Fig. 1).
FIGURE 1.

Gene expression changes in ocular tissues between WT and CFB−/− mice following CSE. Analysis of marker gene expression in WT and CFB−/− mice, using quantitative RT-PCR on cDNA generated from a RPE/choroid/sclera fraction. Quantitative values were obtained by cycle number (Ct value), determining the difference between the mean experimental and control (β-actin) ΔCt values for smoke versus room air-exposed mice within each genotype (fold-difference). Candidates were examined from two categories, ER stress (Grp78 and Chop) and lipid metabolism (Srebf-1). Significant changes were identified in all three genes for C57BL/6J mice, whereas gene expression was minimally affected in CFB−/− animals. Data are expressed as mean ± S.E. (n = 3 per condition, *, p < 0.05).
A separate cohort of smoke-exposed and control C57BL/6J mice was analyzed for lipid deposition. Cross-sections were stained for lipids in RPE/choroid using Nile Red, Filipin staining (49), as well as LipidTox. Nile Red is a general lipid dye that equally binds to free fatty acids, triglycerides, cholesterol, and to some degree phospholipids; LipidTox binds with high specificity to neutral lipids; whereas Filipin recognizes cholesterol. Esterified and unesterified cholesterols can be distinguished with Filipin after incubation with cholesterol esterase, which removes the ester moiety from cholesterol. Based on the outer segment composition, strong lipid staining could be observed in the outer segment layer for both Nile Red (data not shown) and LipidTox in both the control air- and smoke-exposed animals (Fig. 2, A and B). Lipid droplets were found to be present distal to the RPE, a layer most likely representing the thickened Bruch's membrane in smoke-exposed, but not in control sections. Quantification of the lipid signal in Bruch's membrane (see Fig. 2, a and b, for examples of thresholded and binarized images) revealed that ∼5-fold higher levels were present in retinal sections from smoke-exposed when compared with control mice (487 ± 58 versus 100 ± 11; n = 3 per condition; p < 0.005). However, no staining could be obtained using the Filipin dye to identify either esterified or unesterified cholesterol (data not shown). This observation is consistent with data presented by Curcio and co-workers (52), who have shown convincingly that whereas mice have the ability to generate lipids in the RPE, they fail to retain it in the ocular structures.
FIGURE 2.

Lipid deposition in eyes exposed to smoke. Localization of neutral lipids was identified using LipidTox staining, comparing C57BL/6J mice exposed to room air (A) or smoke (B). Lipid particles were seen in Bruch's membrane (BrM) and the basal side of the RPE in smoke-exposed mice when compared with controls. Insets highlight RPE/BrM and corresponding binarized images from control and smoke-exposed eyes (a and b). Abbreviations: RPE, retinal pigment epithelium; OS, outer segments; IS, inner segments; ONL, outer nuclear layer; OPL, outer plexiform layer; INL, inner nuclear layer; IPL, inner plexiform layer; RGC, retinal ganglion cell layer.
Cigarette Smoke-induced Lipid Deposition in Cultured RPE Cells
To further examine the mechanism of smoke-induced ER stress and lipid homeostasis, ARPE-19 cell monolayers were exposed to FBS-free media bubbled with 5% cigarette smoke (CSE). Daily treatment with CSE for 5 days resulted in significant lipid deposition as visualized with Nile Red staining. Nile Red-positive staining was found inside the cells (Fig. 3B), as well as on the Transwell plate itself (Fig. 3, b), indicative of basolateral secretion of the lipids by the RPE cells. Similar staining was obtained using the neutral lipid stain LipidTox (data not shown). Because one of the main neutral lipids present in BrM is cholesterol, staining was repeated using Filipin. Filipin-positive material could be documented on smoke-exposed ARPE-19 cells both before and after esterase treatment (Fig. 3, D and E). This level of lipid deposition had no effect on barrier function of the monolayer, as measured by TER (data not shown), although mRNA for the tight-junction protein ZO-1 was slightly, but significantly increased (Fig. 7A).
FIGURE 3.

Lipid deposition in ARPE-19 cell monolayers exposed to smoke. Localization of lipids was identified using Nile Red (A, B, and F–I), and unesterified (C and D) and esterified cholesterol (E) was characterized, comparing cells exposed to control media (A and C) or smoke (B and D–G). Nile Red-positive lipid particles were seen in RPE cells after smoke exposure (B) when compared with controls (A). Lipid deposits could also be detected on the filters of the Transwell plates after cells were completely detached, confirming basolateral release of lipids from these cells (b, inset). Lipid particles contained both unesterified (D) and esterified cholesterol (E). Nile Red-positive lipid particles were eliminated in cells pretreated with the alternative pathway inhibitor CR2-fH (F) or specific C3a-receptor antagonist (G) for 1 h prior to smoke exposure. Monolayers treated daily with tunicamycin to induce ER stress were found to stain strongly for Nile Red-positive lipids (H); levels that did not change after pretreatment with CR2-fH (I).
FIGURE 7.

Gene expression changes related to ER stress and lipid metabolism in ARPE-19 cells exposed to smoke. Analysis of marker gene expression using quantitative RT-PCR (A and C) or regular PCR (B) on cDNA generated from ARPE-19. Some wells had been pretreated with CR2-fH or C3a-receptor antagonist for 1 h prior to smoke exposure. A, quantitative RT-PCR for ER stress (GRP78 and CHOP) and lipid metabolism (SREBF-1) markers were performed and quantified as described in the legend for Fig. 1. Significant changes were identified in all three genes for cells exposed to smoke; mRNA levels were reduced after complement inhibition. B, XBP1 gets spliced in response to ER stress, reducing the mRNA product tested here from 289 to 263 base pairs (bp). Smoke exposure increased XBP1 splicing; tunicamycin was used as a positive control. C, quantitative RT-PCR for ER stress markers (GRP78 and CHOP) was performed on cells exposed to smoke, or CSE and pretreatment with the antioxidant NAC. Significant changes were identified in all three genes for cells exposed to smoke; mRNA levels were reduced after NAC treatment. Data are expressed as mean ± S.E. (n = 3 per condition, *, p < 0.05).
Smoke Exposure Induces Oxidative Stress in RPE Cells
One of the main effects of smoke exposure is considered to be the induction of oxidative stress, leading to free-radical production (23) and a depletion of the antioxidant system (reviewed in Ref. 24). Here we confirmed that 2 h of CSE exposure resulted in a rapid increase in the amount of ROS measurable using 2,7-dichlorofluorescein diacetate (Fig. 4A); levels were still elevated by 24 h after CSE exposure. ROS have been shown to degrade polyunsaturated lipids, forming malondialdehyde. Reactive oxygen species can initiate lipid peroxidation or the oxidative degradation of lipids. Here we documented lipid peroxidation by measuring the formation of MDA 2 h after CSE, a natural side product of this process. CSE resulted in an ∼8-fold increase in MDA when compared with control media (Fig. 4B).
FIGURE 4.

Oxidative stress in ARPE-19 cells exposed to smoke. Cells were grown into mature monolayers, exposed to CSE, and collected for analysis after 2 or 24 h. Some wells were pretreated with CR2-fH or C3a-receptor antagonist for 1 h. A, ROS were determined using DCFDA. Levels were elevated by CSE after 2 or 24 h and reduced after complement inhibition. B, oxidative degradation of lipids was determined by examining lipid peroxidation by measuring the formation of MDA, a natural side product of this process. After 2 h, levels were elevated by CSE and reduced after complement inhibition. Data are expressed as mean ± S.E. (n = 3 per condition, *, p < 0.05).
Cigarette Smoke-induced Complement Activation in RPE Cells
Cigarette smoke has been shown previously to directly activate C3, leading to activation of the AP, by modifying C3 in a way that reduces its ability to bind to CFH (25). In support of this finding, in lung epithelial cells, exposure of 10% serum and CSE resulted in complement activation as evidenced by the generation of C4a, C3a, and C5a (53). If the AP were to be activated in this process, the cleavage of C3 into C3a and C3b exposes a highly reactive thiol ester bond on C3b, which allows it to react with and deposit onto membranes. The membrane association of C3b can trigger the AP pathway; yet upon inactivation of C3b by factor I, cofactor and CR1 activity will produce C3d. The same authors demonstrated that exposure of cells to CSE and serum resulted in C3d deposition as demonstrated by immunostaining (53).
Because the RPE in vivo exhibits complement activation products after CSE (27), and has been documented to express many complement components (18), we asked whether monolayers stimulated with CSE secrete their own complement C3. Measurement of supernatants from ARPE-19 cells stimulated with CSE for 4 and 24 h demonstrated a significant increase in C3 secretion into both the basal and apical supernatant (Fig. 5A). Furthermore, quantitative RT-PCR analysis showed increased C3 mRNA levels at 24 h post-CSE exposure (Fig. 5C), suggesting that CSE induces C3 transcription. In addition, measurements of C3a revealed that short term CSE resulted in the generation of biologically active anaphylatoxin C3a (Fig. 5B). However, whereas C3 was secreted equally toward both sides, generation of anaphylatoxin was limited to the apical compartment, an expected finding, because CSE was only applied to the apical side and therefore components within the smoke extract presumably directly modified and activated C3. Hence all additional experiments measuring complement activation were limited to the analysis of apical supernatants. Overall, CSE for 4 h resulted in the release of ∼13 ng/ml of C3, of which ∼5% became cleaved to generate C3a (∼0.7 ng/ml). Finally, CSE resulted in significant complement C3d deposition on the apical surface of ARPE-19 cell monolayer as shown by immunocytochemistry as early as 2 h after CSE exposure (data not shown), lasting for at least 24 h (Fig. 6).
FIGURE 5.

Complement activation in ARPE-19 cells exposed to smoke. Cells were grown into mature monolayers, exchanged to serum-free media 24 h prior to the experiment, exposed to CSE in serum-free media, and collected for ELISA analysis after 4 h, or quantitative RT-PCR after 24 h. A and B, ELISA was performed, using plates coated with C3- or C3a-specific capture antibodies. Some wells were pretreated with CR2-fH, C3a-receptor antagonist, or antioxidant (NAC) for 1 h. To determine polarity of C3 (A) and C3a production (B), apical and basal supernatants were collected after CSE. C3 secretion was increased toward both sides; C3a production was limited to the side of CSE. Levels of apical C3 and C3a were elevated by CSE and reduced after application of inhibitors. C, C3a mRNA levels were elevated by CSE and reduced after application of inhibitors. Data are expressed as mean ± S.E. (n = 3 per condition, *, p < 0.05); n.d., not determined.
FIGURE 6.

Complement deposition in ARPE-19 cells exposed to smoke. Cells were grown into mature monolayers, exchanged to serum-free media 24 h prior to the experiment, exposed to CSE in serum-free media, and fixed in 4% paraformaldehyde after 2 or 24 h. Immunocytochemistry was performed using a well characterized mouse monoclonal antibody that recognizes human C3d on control (B) and CSE cells (D) at 24 h. A mouse monoclonal antibody against nitrophenol was used as control (A and C).
Effects of Cigarette Smoke Exposure on ER Stress and Lipid Homeostasis in RPE Cells
To examine whether CSE triggers ER stress in RPE monolayers, cells were examined for ER stress and lipid homeostasis by quantitative RT-PCR 24 h after the exposure to CSE. ER stress markers, GRP78 and CHOP, were elevated ∼3- and ∼13-fold, respectively; SREBF-1 was also increased ∼3-fold (Fig. 7A). Interestingly, these fold-differences were similar to those identified in vivo within the mouse RPE (Fig. 1). Analysis of XBP1 activation requires monitoring of the production of the spliced version by regular PCR. Exposure to CSE resulted in the formation of the spliced XBP1 (sXBP1), similar to that generated by tunicamycin, a known ER stress inducer (Fig. 7B). GRP78, CHOP, and XBP1 are not only elevated at the mRNA level, but protein expression is significantly increased (Fig. 8, A and B), and can be visualized in the ER by confocal microscopy, using antibodies specific for GRP78, CHOP, and XBP1 (Fig. 9). Finally, eIF2α activation, which requires phosphorylation, was increased after CSE (Fig. 8, A and B).
FIGURE 8.

Protein expression changes related to ER stress in ARPE-19 cells exposed to smoke. Monolayers were treated as for Fig. 7, but examined by Western blotting (A), and protein levels were quantified (B). Protein levels for GRP78, CHOP, and XBP1 were elevated after smoke exposure or tunicamycin (TM) treatment (positive control). Phosphorylation of the β-subunit of the transcription factor eIF2, which is part of the cellular ER stress response, was significantly increased after smoke exposure. Pretreatment with CR2-fH or C3a-receptor antagonist prevented the increase in GRP78, CHOP, and XBP1 expression and returned the levels of phosphorylated eIF2α to baseline levels. Tunicamycin (TM) was used as a positive control. Data are expressed as mean ± S.E. (n = 3 per condition, *, p < 0.05).
FIGURE 9.

Immunocytochemical analysis of ER stress in ARPE-19 cells exposed to smoke. Cells were grown into mature monolayers, exchanged to serum-free media 24 h prior to the experiment, exposed to CSE in serum-free media, and fixed in 4% paraformaldehyde after 24 h. Immunocytochemistry was performed using ER stress markers on control (A) and CSE cells (B–D) and imaged by confocal microscopy (Leica SP2). CHOP (B)-, GRP78 (C)-, and XBP1 (D)-positive staining could be identified in CSE cells; control cells were found to be negative (only staining for CHOP is shown for control cells).
Effects of Complement Inhibition on CSE-induced Pathology
Because cigarette smoke has been shown to be able to directly activate C3 by modifying C3 in a way that reduces its ability to bind to CFH (25), activating the alternative pathway, the contribution of the AP to anaphylatoxin production, oxidative stress, ER stress, and lipid deposition was investigated. Pre-treatment with specific antagonist for the C3a-receptor or an inhibitor for the AP pathway (CR2-fH) would be expected to ameliorate pathology. Prior to the exposure with 5% CSE, cells were pretreated for 1 h with either C3aR antagonist, or the AP pathway inhibitor CR2-fH.
Treatment of monolayers with CR2-fH or C3a-receptor blockers completely prevented the CSE-stimulated release of C3 into the apical media, prevented the accumulation of the anaphylatoxin C3a as well as the increase in C3 mRNA (Fig. 5, A–C). Likewise, ROS and lipid peroxidation were found to be completely inhibited in the presence of complement inhibition at 2 h or significantly reduced at 24 h post-CSE (Fig. 4, A and B). CR2-fH and anaphylatoxin-receptor antagonist prevented the increase in markers for ER stress and lipid metabolism at both the mRNA and protein level (Figs. 7A and 8, A and B). Finally, cells pre-treated with CR2-fH had negligible lipid deposition as documented by Nile Red staining; likewise C3a-receptor antagonism effectively blocked lipid deposition (Fig. 3, F and G). Treatment with the inhibitors alone in the absence of CSE had no effect (data not shown).
Relationship between Complement Activation, Oxidative Stress, ER Stress, and Lipid Deposition
The main findings thus far are as follows: short term CSE resulted in the production and release of complement C3, the generation of the anaphylatoxin C3a, and AP activation; short term CSE triggered oxidative as well as ER stress; long term exposure resulted in lipid accumulation; and finally, all these readouts could be reduced by blocking anaphylatoxin receptor and AP signaling. Next, we aimed to establish the relationship between complement activation, oxidative, and ER stress as well as lipid deposition.
To determine whether CSE results in complement-mediated ER stress, experiments were repeated in the presence of C3a-receptor inhibition or by blocking the AP pathway using CR2-fH. ER stress and lipid homeostasis markers GRP78, CHOP, and SREBF-1 mRNA levels were found to be significantly reduced by pretreatment with either complement inhibitor (Fig. 7A). Likewise, protein levels for GRP78, CHOP, and XBP1, as well as, for phosphorylated eIF2α were significantly decreased (Fig. 8, A and B).
To determine whether CSE results in long term oxidative stress, or whether CSE-mediated complement activation provides a feedback loop as suggested in the literature (9), ROS and lipid peroxidation measurements were repeated in the presence of complement inhibition (Fig. 4, A and B). ROS and lipid peroxidation were found to be completely inhibited in the presence of complement inhibition at 2 h and significantly reduced at 24 h post-CSE exposure, supporting the notion that persistent inflammation causes oxidative stress (9). Pretreatment of cells with of NAC, an antioxidant amino acid derivative, for 1 h prior to CSE prevented the increase in ER-stress marker gene expression (GRP78 and CHOP) at 24 h (Fig. 7C). Likewise, it prevented the increase of C3 protein secretion and gene transcription as marked by mRNA expression (Fig. 5, A and C).
To determine whether long term ER stress can trigger complement activation, cells were treated daily with tunicamycin, which resulted in lipid deposition similar to that generated by CSE (Fig. 3H). However, pretreatment with CR2-fH, which completely eliminated CSE-mediated lipid deposition, had no effect on tunicamycin-mediated lipid deposition (Fig. 3I). Hence a feedback from ER stress to complement activation does not appear to exist in these cells.
DISCUSSION
The main results of the current study were as follows: 1) C57BL/6J mice exposed to long term smoke inhalation exhibited signs of ER stress (UPR) and lipid dysregulation based on gene expression profiles; 2) the same genes were altered after CSE treatment of RPE monolayers in culture; 3) CSE triggered the production and release of C3 from RPE cells, which led to the activation of the AP and subsequent production of C3a involving an anaphylatoxin-receptor-dependent autocrine feedback loop; 4) CSE-mediated complement activation resulted in oxidative and ER stress; 5) oxidative stress and complement activation are linked through a feedback loop; 6) long term CSE resulted in lipid accumulation both in vivo and in vitro; 7) lipid accumulation and secretion in vitro was found to be complement-dependent; 8) which together supports the following pathway: smoke-mediated activation of AP signaling, resulting in oxidative stress, followed by ER stress, and leading to lipid deposition (Fig. 10).
FIGURE 10.
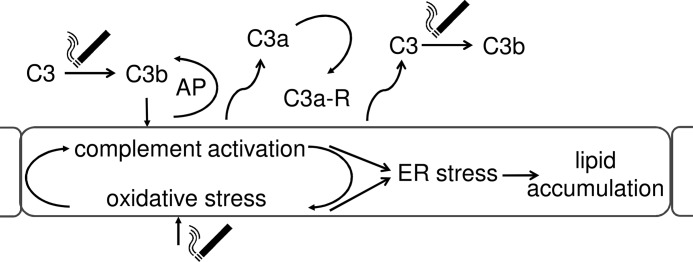
Complement activation in RPE cells increases lipid deposition. Smoke exposure activates C3, triggering the activation of the AP. AP of complement activation results in the generation of anaphylatoxins C3a, which trigger anaphylatoxin-receptor-dependent ER stress. Complement activation, however, also generates oxidative stress and increases C3 release, thereby amplifying the response to smoke exposure. ER stress finally leads to lipid deposition, one of the known hallmarks of AMD. Thus, it is expected that complement inhibition in early AMD results in a reduction in lipid production and deposition in RPE and Bruch's membrane.
Risk factors for AMD include age, smoking, as well as an overactive complement system due to SNPs in complement regulatory proteins or complement activators. Aging may contribute to disease pathogenesis in part by affecting mitochondrial metabolism. Mitochondrial DNA mutations and respiratory chain dysfunction have been shown to accompany normal aging, impairing cellular homeostasis. Smoking is thought to generate oxidative stress, and although the RPE has available effective defenses against oxidative damage, such as high amounts of antioxidants (54), the aged RPE loses that capability (55, 56). Finally, an overactive complement system, possibly due to the loss of membrane-bound and soluble inhibitors available to the RPE (15–17) can lead to formation of immune deposits on the RPE (57) and drusen as well as damage of or alterations in RPE cells (46, 58), which together can lead to a breakdown of the blood retina barrier, choroidal neovascularization, and vision loss (reviewed in Ref. 59). Here we provide evidence that smoking triggers complement activation on the surface of RPE cells, leading to ER stress and lipid dysregulation.
Smoke exposure in a mouse model has been shown to lead to pathological changes in BrM (60), complement activation at the level of the RPE-choroid (27), with pathology being dependent upon the activation of the AP (28). In primary human RPE cells, previous work has shown that exposure to CSE in serum-free media resulted in lipid peroxidation, senescent changes, and alterations in extracellular matrix after 72 h (61). Although the authors did not examine the underlying mechanism that triggered these changes, the focus of the discussion was primarily on oxidative stress. In subconfluent ARPE-19 cells, benzo(a)pyrene, a toxic element present in cigarette smoke, was shown to result in mitochondrial DNA damage, and increased lysosomal and exocytotic activity. Interestingly, mRNA levels for CFH, C3, CFB, and CD59 were increased under those conditions (27). Finally, exposure of subconfluent RPE cells to CSE or hydroquinone, another toxic component of cigarette smoke, among other effects, induced expression of VEGF, as well as indicators of redox imbalance in serum-free media after 24 h (62). Dysregulation of the complement system in mouse on the other hand has been associated with changes in retinal structure and function (63, 64), alterations in retinal perfusion (65); but no data are available on UPR or lipid dysregulation in that context. Complement activation on RPE cells has been shown to lead to VEGF secretion followed by loss of barrier function (46, 66) as well as secretion and activation of membrane metalloproteinases (58). Here we extended the analysis on complement-mediated effects in RPE cells by demonstrating that CSE resulted in the activation of the complement system on the surface of RPE cells (C3d deposition), generation and release of complement C3 concomitant with the generation of C3a, followed by activation of the AP amplification loop. Complement activation, presumably via C3a-receptor signaling, resulted in ER stress and the UPR, leading to lipid accumulation and secretion. All UPR measures, as well as lipid accumulation and oxidative stress, were significantly reduced by blocking anaphylatoxin receptor signaling, or the AP amplification loop.
ER stress and the UPR have been examined in the context of retinal degeneration, in particular with regards to misfolded proteins in photoreceptors and the RPE (reviewed by Ref. 67); and hence therapeutic approaches have included the search for small molecular chaperones to promote protein folding (reviewed by Ref. 35). A recent review article focused on the role of ER stress in AMD and its potential for triggering choroidal neovascularization (68), as AMD risk factors, such as oxidative, proteotoxic, and metabolic stress as well as the presence of cytokines can trigger ER stress and the UPR; and VEGF production in human RPR cells has been shown to be activated by ATF4 (69), one of the transcription factors produced by the UPR. However, whereas a link has been presented between the UPR and complement activation in kidney disease (33), similar evidence is missing in AMD. Our data provide a clear and novel link between CSE-mediated complement activation and the UPR, leading to lipid accumulation.
Regions of the aging or pathological eye that exhibit lipid accumulation include BrM (49) and the RPE (70); yet the origin of the lipids in drusen, oil droplets, and BrM deposits is not entirely clear. As the two main lipid components in these structures are esterified cholesterol and phosphatidylcholine (38, 49), the lipid composition of BrM appears to be more similar to that of plasma than of photoreceptors (71). Hence, because photoreceptor outer segments, the main cargo to be recycled or removed by the RPE, are rich in docosahexaenoic acid, there would be a need for additional processing steps within the RPE to convert the fatty acids into neutral lipids (40). Alternatively, cholesterol from other sources might contribute to the lipids contained in the lipoprotein particles secreted by the RPE (40). Here we provide data that suggests that cholesterol uptake regulated via the transcription factors SREBF-1, and triggered by complement activation, might be involved in lipid dysregulation in the RPE. Interestingly, similar to the work reported here, Johnson and co-workers (72) showed that primary RPE cells grown in long term cultures accumulate sub-RPE deposits rich in apolipoprotein E. Taken together, whereas the data in the literature as well as our data do not provide clear evidence that the lipid deposits are derived from reprocessed serum lipids rather than from RPE plasma membrane, these cumulative data clearly support the hypothesis that photoreceptor phagocytosis is not required. We hope to determine the source of lipids in future studies.
Here we demonstrated complement-mediated effects that require signaling via the anaphylatoxin C3a-receptor. C3a is generated by the C3 convertases, and is inactivated by the removal of the C-terminal arginine amino acid, producing C3a desArg. In both mouse and human there is a single receptor for C3a (C3a-receptor), which can also serve to mediate signaling for C3a desArg. Ligand binding and signaling by the C3a-receptor has been well characterized, a process that involves pertussis toxin-sensitive Gαi signaling (73). Receptor signaling through the anaphylatoxin receptors can result in changes in intracellular signaling calcium levels, trigger prolonged activation of protein phosphorylation such as ERK1/2 and Akt, and may also involve arrestin binding to the phosphorylated receptor. Importantly, C3a-receptors are expressed by the RPE (18). Here we observed both the generation of C3a by CSE, as well as C3a-receptor engagement. The commercially available C3a-receptor antagonist was found to reduce oxidative and ER stress as well as lipid accumulation. In addition, we observed that either blocking the AP, or C3a-receptor inhibition prevented the CSE-induced C3 production and release and concomitant anaphylatoxin production. Similar results were reported in human macrophages, in which C3 secretion was found to be triggered by application of C3a, an effect that was enhanced by co-administering oxidized LDL (74). Other pathways for control of C3 expression and secretion have been reported, including cytokine signaling (75–77). It will be of interest to examine the potential synergistic effects of CSE and cytokine stimulation.
As indicated above, the hypothesis of oxidative/inflammatory stress-mediated AMD included a feedback loop between chronic oxidative stress and chronic inflammation or complement activation (4). However, in RPE tissue, this feedback loop has not yet been investigated. Here we have shown that CSE results in both oxidative stress and complement activation. Blocking complement activation was found to eliminate oxidative stress and, vice versa, blocking oxidative stress prevented C3 production and release, the prerequisite for complement activation on the cell surface and anaphylatoxin production and signaling. Finally, a similar loop has not yet been characterized in any other tissue or cell type.
Taken together, there is growing evidence linking oxidative stress, smoking, ER stress, lipid dysfunction, as well as complement activation to the development and progression of AMD. The data presented here show that cigarette smoke exposure in RPE cells leads to ER stress and lipid accumulation, and provide the first direct evidence that the AP of complement is required for these alterations to occur. In addition, we provide the first experimental evidence of the postulated feedback loop between oxidative stress and complement. Although we have shown that oxidative stress renders cells susceptible to complement activation (46), here we have provided evidence that blocking oxidative stress reduces local complement production and activation. Finally, our data suggest that lipid dysregulation in AMD may be amenable to anti-complement-based therapies.
Acknowledgments
Animal studies were conducted in a facility constructed with support from the National Institutes of Health Grant C06RR015455.
This work was supported, in whole or in part, by National Institutes of Health Grant R01EY019320, Department for Veterans Affairs merit Award RX000444, the Beckman Initiative for Macular Research, an unrestricted grant to the Medical University of South Carolina from Research to Prevent Blindness (RPB), New York, NY, and Foundation Fighting Blindness, Columbia, MD (to B. R.); and National Institutes of Health Grant R01 091944 from the NHLBI and Flight Attendant Medical Research Institute (FAMRI) CIA Grant 092079 (to C. A.).
- AMD
- age-related macular degeneration
- RPE
- retinal pigment epithelium
- AP
- alternative pathway
- CFB
- complement factor B
- CFH
- complement factor H
- UPR
- unfolded protein response
- GRP78
- glucose-regulated protein of 78 kDa
- NAC
- N-acetylcysteine
- TER
- transepithelial resistance
- CSE
- cigarette smoke extract
- ROS
- reactive oxygen species
- BisTris
- 2-[bis(2-hydroxyethyl)amino]-2-(hydroxymethyl)propane-1,3-diol
- ER
- endoplasmic reticulum
- DCFDA
- 2,7-dichlorofluorescein diacetate
- MDA
- malondialdehyde
- SREBP
- sterol regulatory element-binding protein.
REFERENCES
- 1. Ferris F. L., 3rd, Wilkinson C. P., Bird A., Chakravarthy U., Chew E., Csaky K., Sadda S. R., and Beckman Initiative for Macular Research Classification C. (2013) Clinical classification of age-related macular degeneration. Ophthalmology 120, 844–851 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Brown M. M., Brown G. C., Stein J. D., Roth Z., Campanella J., Beauchamp G. R. (2005) Age-related macular degeneration: economic burden and value-based medicine analysis. Can. J. Ophthalmol. 40, 277–287 [DOI] [PubMed] [Google Scholar]
- 3. Freund K. B., Zweifel S. A., Engelbert M. (2010) Do we need a new classification for choroidal neovascularization in age-related macular degeneration? Retina 30, 1333–1349 [DOI] [PubMed] [Google Scholar]
- 4. Hageman G. S., Luthert P. J., Victor Chong N. H., Johnson L. V., Anderson D. H., Mullins R. F. (2001) An integrated hypothesis that considers drusen as biomarkers of immune-mediated processes at the RPE-Bruch's membrane interface in aging and age-related macular degeneration. Prog. Retin. Eye Res. 20, 705–732 [DOI] [PubMed] [Google Scholar]
- 5. Edwards A. O., Ritter R., 3rd, Abel K. J., Manning A., Panhuysen C., Farrer L. A. (2005) Complement factor H polymorphism and age-related macular degeneration. Science 308, 421–424 [DOI] [PubMed] [Google Scholar]
- 6. Hageman G. S., Anderson D. H., Johnson L. V., Hancox L. S., Taiber A. J., Hardisty L. I., Hageman J. L., Stockman H. A., Borchardt J. D., Gehrs K. M., Smith R. J., Silvestri G., Russell S. R., Klaver C. C., Barbazetto I., Chang S., Yannuzzi L. A., Barile G. R., Merriam J. C., Smith R. T., Olsh A. K., Bergeron J., Zernant J., Merriam J. E., Gold B., Dean M., Allikmets R. (2005) A common haplotype in the complement regulatory gene factor H (HF1/CFH) predisposes individuals to age-related macular degeneration. Proc. Natl. Acad. Sci. U.S.A. 102, 7227–7232 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Haines J. L., Hauser M. A., Schmidt S., Scott W. K., Olson L. M., Gallins P., Spencer K. L., Kwan S. Y., Noureddine M., Gilbert J. R., Schnetz-Boutaud N., Agarwal A., Postel E. A., Pericak-Vance M. A. (2005) Complement factor H variant increases the risk of age-related macular degeneration. Science 308, 419–421 [DOI] [PubMed] [Google Scholar]
- 8. Klein R. J., Zeiss C., Chew E. Y., Tsai J. Y., Sackler R. S., Haynes C., Henning A. K., SanGiovanni J. P., Mane S. M., Mayne S. T., Bracken M. B., Ferris F. L., Ott J., Barnstable C., Hoh J. (2005) Complement factor H polymorphism in age-related macular degeneration. Science 308, 385–389 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Zarbin M. A., Rosenfeld P. J. (2010) Pathway-based therapies for age-related macular degeneration: an integrated survey of emerging treatment alternatives. Retina 30, 1350–1367 [DOI] [PubMed] [Google Scholar]
- 10. Whitcup S. M., Sodhi A., Atkinson J. P., Holers V. M., Sinha D., Rohrer B., Dick A. D. (2013) The role of the immune response in age-related macular degeneration. Int. J. Inflam. 2013, 348092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Müller-Eberhard H. J. (1988) Molecular organization and function of the complement system. Annu. Rev. Biochem. 57, 321–347 [DOI] [PubMed] [Google Scholar]
- 12. Thurman J. M., Ljubanovi D., Royer P. A., Kraus D. M., Molina H., Barry N. P., Proctor G., Levi M., Holers V. M. (2006) Altered renal tubular expression of the complement inhibitor Crry permits complement activation after ischemia/reperfusion. J. Clin. Invest. 116, 357–368 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Yang P., Tyrrell J., Han I., Jaffe G. J. (2009) Expression and modulation of RPE cell membrane complement regulatory proteins. Invest. Ophthalmol. Vis. Sci. 50, 3473–3481 [DOI] [PubMed] [Google Scholar]
- 14. Yin W., Ghebrehiwet B., Weksler B., Peerschke E. I. (2008) Regulated complement deposition on the surface of human endothelial cells: effect of tobacco smoke and shear stress. Thromb. Res. 122, 221–228 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Fett A. L., Hermann M. M., Muether P. S., Kirchhof B., Fauser S. (2012) Immunohistochemical localization of complement regulatory proteins in the human retina. Histol. Histopathol. 27, 357–364 [DOI] [PubMed] [Google Scholar]
- 16. Vogt S. D., Curcio C. A., Wang L., Li C. M., McGwin G., Jr., Medeiros N. E., Philp N. J., Kimble J. A., Read R. W. (2011) Retinal pigment epithelial expression of complement regulator CD46 is altered early in the course of geographic atrophy. Exp. Eye Res. 93, 413–423 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Ebrahimi K. B., Fijalkowski N., Cano M., Handa J. T. (2013) Decreased membrane complement regulators in the retinal pigmented epithelium contributes to age-related macular degeneration. J. Pathol. 229, 729–742 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Anderson D. H., Radeke M. J., Gallo N. B., Chapin E. A., Johnson P. T., Curletti C. R., Hancox L. S., Hu J., Ebright J. N., Malek G., Hauser M. A., Rickman C. B., Bok D., Hageman G. S., Johnson L. V. (2010) The pivotal role of the complement system in aging and age-related macular degeneration: hypothesis re-visited. Prog. Retin. Eye Res. 29, 95–112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Lois N., Abdelkader E., Reglitz K., Garden C., Ayres J. G. (2008) Environmental tobacco smoke exposure and eye disease. Br. J. Ophthalmol. 92, 1304–1310 [DOI] [PubMed] [Google Scholar]
- 20. Chakravarthy U., Augood C., Bentham G. C., de Jong P. T., Rahu M., Seland J., Soubrane G., Tomazzoli L., Topouzis F., Vingerling J. R., Vioque J., Young I. S., Fletcher A. E. (2007) Cigarette smoking and age-related macular degeneration in the EUREYE study. Ophthalmology 114, 1157–1163 [DOI] [PubMed] [Google Scholar]
- 21. Mitchell P., Wang J. J., Smith W., Leeder S. R. (2002) Smoking and the 5-year incidence of age-related maculopathy: the Blue Mountains Eye Study. Arch. Ophthalmol. 120, 1357–1363 [DOI] [PubMed] [Google Scholar]
- 22. Khan J. C., Thurlby D. A., Shahid H., Clayton D. G., Yates J. R., Bradley M., Moore A. T., Bird A. C., and Genetic Factors in AMD Study. (2006) Smoking and age related macular degeneration: the number of pack years of cigarette smoking is a major determinant of risk for both geographic atrophy and choroidal neovascularisation. Br. J. Ophthalmol. 90, 75–80 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Church D. F., Pryor W. A. (1985) Free-radical chemistry of cigarette smoke and its toxicological implications. Environ. Health Perspect. 64, 111–126 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. van der Vaart H., Postma D. S., Timens W., ten Hacken N. H. (2004) Acute effects of cigarette smoke on inflammation and oxidative stress: a review. Thorax 59, 713–721 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Kew R. R., Ghebrehiwet B., Janoff A. (1985) Cigarette smoke can activate the alternative pathway of complement in vitro by modifying the third component of complement. J. Clin. Invest. 75, 1000–1007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Sanai M., Hussain N. (2011) Levels of inflammatory markers (complement C3, complement C4 and C-reactive protein) in smokers. Afr. J. Biotechnol. 10, 19211–19217 [Google Scholar]
- 27. Wang A. L., Lukas T. J., Yuan M., Du N., Handa J. T., Neufeld A. H. (2009) Changes in retinal pigment epithelium related to cigarette smoke: possible relevance to smoking as a risk factor for age-related macular degeneration. PLoS ONE 4, e5304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Woodell A., Coughlin B., Kunchithapautham K., Casey S., Williamson T., Ferrell W. D., Atkinson C., Jones B. W., Rohrer B. (2013) Alternative complement pathway deficiency ameliorates chronic smoke-induced functional and morphological ocular injury. PLoS ONE 8, e67894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Kelsen S. G. (2012) Respiratory epithelial cell responses to cigarette smoke: the unfolded protein response. Pulm. Pharmacol. Ther 25, 447–452 [DOI] [PubMed] [Google Scholar]
- 30. Tweed J. O., Hsia S. H., Lutfy K., Friedman T. C. (2012) The endocrine effects of nicotine and cigarette smoke. Trends Endocrinol. Metab. 23, 334–342 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Walter P., Ron D. (2011) The unfolded protein response: from stress pathway to homeostatic regulation. Science 334, 1081–1086 [DOI] [PubMed] [Google Scholar]
- 32. Ishida Y., Yamamoto A., Kitamura A., Lamandé S. R., Yoshimori T., Bateman J. F., Kubota H., Nagata K. (2009) Autophagic elimination of misfolded procollagen aggregates in the endoplasmic reticulum as a means of cell protection. Mol. Biol. Cell 20, 2744–2754 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Kitamura M. (2008) Endoplasmic reticulum stress and unfolded protein response in renal pathophysiology: Janus faces. Am. J. Physiol. Renal Physiol. 295, F323–334 [DOI] [PubMed] [Google Scholar]
- 34. Meusser B., Hirsch C., Jarosch E., Sommer T. (2005) ERAD: the long road to destruction. Nat. Cell Biol. 7, 766–772 [DOI] [PubMed] [Google Scholar]
- 35. Haeri M., Knox B. E. (2012) Endoplasmic reticulum stress and unfolded protein response pathways: potential for treating age-related retinal degeneration. J. Ophthalmic. Vis. Res. 7, 45–59 [PMC free article] [PubMed] [Google Scholar]
- 36. Ni M., Zhang Y., Lee A. S. (2011) Beyond the endoplasmic reticulum: atypical GRP78 in cell viability, signalling and therapeutic targeting. Biochem. J. 434, 181–188 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Haimovici R., Gantz D. L., Rumelt S., Freddo T. F., Small D. M. (2001) The lipid composition of drusen, Bruch's membrane, and sclera by hot stage polarizing light microscopy. Invest. Ophthalmol. Vis. Sci. 42, 1592–1599 [PubMed] [Google Scholar]
- 38. Wang L., Clark M. E., Crossman D. K., Kojima K., Messinger J. D., Mobley J. A., Curcio C. A. (2010) Abundant lipid and protein components of drusen. PLoS ONE 5, e10329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Curcio C. A., Millican C. L., Bailey T., Kruth H. S. (2001) Accumulation of cholesterol with age in human Bruch's membrane. Invest. Ophthalmol. Vis. Sci. 42, 265–274 [PubMed] [Google Scholar]
- 40. Ebrahimi K. B., Handa J. T. (2011) Lipids, lipoproteins, and age-related macular degeneration. J. Lipids 2011, 802059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Colgan S. M., Hashimi A. A., Austin R. C. (2011) Endoplasmic reticulum stress and lipid dysregulation. Expert Rev. Mol. Med. 13, e4. [DOI] [PubMed] [Google Scholar]
- 42. Damiano F., Alemanno S., Gnoni G. V., Siculella L. (2010) Translational control of the sterol-regulatory transcription factor SREBP-1 mRNA in response to serum starvation or ER stress is mediated by an internal ribosome entry site. Biochem. J. 429, 603–612 [DOI] [PubMed] [Google Scholar]
- 43. Matsumoto M., Fukuda W., Circolo A., Goellner J., Strauss-Schoenberger J., Wang X., Fujita S., Hidvegi T., Chaplin D. D., Colten H. R. (1997) Abrogation of the alternative complement pathway by targeted deletion of murine factor B. Proc. Natl. Acad. Sci. U.S.A. 94, 8720–8725 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Huang Y., Qiao F., Atkinson C., Holers V. M., Tomlinson S. (2008) A novel targeted inhibitor of the alternative pathway of complement and its therapeutic application in ischemia/reperfusion injury. J. Immunol. 181, 8068–8076 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Thurman J. M., Kulik L., Orth H., Wong M., Renner B., Sargsyan S. A., Mitchell L. M., Hourcade D. E., Hannan J. P., Kovacs J. M., Coughlin B., Woodell A. S., Pickering M. C., Rohrer B., Holers V. M. (2013) Detection of complement activation using monoclonal antibodies against C3d. J. Clin. Invest. 123, 2218–2230 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Thurman J. M., Renner B., Kunchithapautham K., Ferreira V. P., Pangburn M. K., Ablonczy Z., Tomlinson S., Holers V. M., Rohrer B. (2009) Oxidative stress renders retinal pigment epithelial cells susceptible to complement-mediated injury. J. Biol. Chem. 284, 16939–16947 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Ablonczy Z., Crosson C. E. (2007) VEGF modulation of retinal pigment epithelium resistance. Exp. Eye Res. 85, 762–771 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Mulligan R. M., Atkinson C., Vertegel A. A., Reukov V., Schlosser R. J. (2009) Cigarette smoke extract stimulates interleukin-8 production in human airway epithelium and is attenuated by superoxide dismutase in vitro. Am. J. Rhinol. Allergy 23, e1–4 [DOI] [PubMed] [Google Scholar]
- 49. Rudolf M., Curcio C. A. (2009) Esterified cholesterol is highly localized to Bruch's membrane, as revealed by lipid histochemistry in wholemounts of human choroid. J. Histochem. Cytochem. 57, 731–739 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Lohr H. R., Kuntchithapautham K., Sharma A. K., Rohrer B. (2006) Multiple, parallel cellular suicide mechanisms participate in photoreceptor cell death. Exp. Eye Res. 83, 380–389 [DOI] [PubMed] [Google Scholar]
- 51. Samali A., Fitzgerald U., Deegan S., Gupta S. (2010) Methods for monitoring endoplasmic reticulum stress and the unfolded protein response. Int. J. Cell Biol. 2010, 830307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Dithmar S., Curcio C. A., Le N. A., Brown S., Grossniklaus H. E. (2000) Ultrastructural changes in Bruch's membrane of apolipoprotein E-deficient mice. Invest. Ophthalmol. Vis. Sci. 41, 2035–2042 [PubMed] [Google Scholar]
- 53. Davis K. S., Casey S. E., Mulligan J. K., Mulligan R. M., Schlosser R. J., Atkinson C. (2010) Murine complement deficiency ameliorates acute cigarette smoke-induced nasal damage. Otolaryngol. Head Neck Surg. 143, 152–158 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Rózanowska M., Jarvis-Evans J., Korytowski W., Boulton M. E., Burke J. M., Sarna T. (1995) Blue light-induced reactivity of retinal age pigment: in vitro generation of oxygen-reactive species. J. Biol. Chem. 270, 18825–18830 [DOI] [PubMed] [Google Scholar]
- 55. Liles M. R., Newsome D. A., Oliver P. D. (1991) Antioxidant enzymes in the aging human retinal pigment epithelium. Arch. Ophthalmol. 109, 1285–1288 [DOI] [PubMed] [Google Scholar]
- 56. Tate D. J., Jr., Oliver P. D., Miceli M. V., Stern R., Shuster S., Newsome D. A. (1993) Age-dependent change in the hyaluronic acid content of the human chorioretinal complex. Arch. Ophthalmol. 111, 963–967 [DOI] [PubMed] [Google Scholar]
- 57. Joseph K., Kulik L., Coughlin B., Kunchithapautham K., Bandyopadhyay M., Thiel S., Thielens N. M., Holers V. M., Rohrer B. (2013) Oxidative stress sensitizes RPE cells to complement-mediated injury in a natural antibody-, lectin pathway-, and phospholipid epitope-dependent manner. J. Biol. Chem. 288, 12753–12765 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Bandyopadhyay M., Rohrer B. (2012) Matrix metalloproteinase activity creates pro-angiogenic environment in primary human retinal pigment epithelial cells exposed to complement. Invest. Ophthalmol. Vis. Sci. 53, 1953–1961 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Zipfel P. F., Lauer N., Skerka C. (2010) The role of complement in AMD. Adv. Exp. Med. Biol. 703, 9–24 [DOI] [PubMed] [Google Scholar]
- 60. Espinosa-Heidmann D. G., Suner I. J., Catanuto P., Hernandez E. P., Marin-Castano M. E., Cousins S. W. (2006) Cigarette smoke-related oxidants and the development of sub-RPE deposits in an experimental animal model of dry AMD. Invest. Ophthalmol. Vis. Sci. 47, 729–737 [DOI] [PubMed] [Google Scholar]
- 61. Yu A. L., Birke K., Burger J., Welge-Lussen U. (2012) Biological effects of cigarette smoke in cultured human retinal pigment epithelial cells. PLoS ONE 7, e48501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Bertram K. M., Baglole C. J., Phipps R. P., Libby R. T. (2009) Molecular regulation of cigarette smoke induced-oxidative stress in human retinal pigment epithelial cells: implications for age-related macular degeneration. Am. J. Physiol. Cell Physiol. 297, C1200–1210 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Coffey P. J., Gias C., McDermott C. J., Lundh P., Pickering M. C., Sethi C., Bird A., Fitzke F. W., Maass A., Chen L. L., Holder G. E., Luthert P. J., Salt T. E., Moss S. E., Greenwood J. (2007) Complement factor H deficiency in aged mice causes retinal abnormalities and visual dysfunction. Proc. Natl. Acad. Sci. U.S.A. 104, 16651–16656 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Yu M., Zou W., Peachey N. S., McIntyre T. M., Liu J. (2012) A novel role of complement in retinal degeneration. Invest. Ophthalmol. Vis. Sci. 53, 7684–7692 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Lundh von Leithner P., Kam J. H., Bainbridge J., Catchpole I., Gough G., Coffey P., Jeffery G. (2009) Complement factor H is critical in the maintenance of retinal perfusion. Am. J. Pathol. 175, 412–421 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Kunchithapautham K., Bandyopadhyay M., Dahrouj M., Thurman J. M., Rohrer B. (2012) Sublytic membrane-attack complex activation and VEGF secretion in retinal pigment epithelial cells. Adv. Exp. Med. Biol. 723, 23–30 [DOI] [PubMed] [Google Scholar]
- 67. Lin J. H., Lavail M. M. (2010) Misfolded proteins and retinal dystrophies. Adv. Exp. Med. Biol. 664, 115–121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Salminen A., Kauppinen A., Hyttinen J. M., Toropainen E., Kaarniranta K. (2010) Endoplasmic reticulum stress in age-related macular degeneration: trigger for neovascularization. Mol. Med. 16, 535–542 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Roybal C. N., Yang S., Sun C. W., Hurtado D., Vander Jagt D. L., Townes T. M., Abcouwer S. F. (2004) Homocysteine increases the expression of vascular endothelial growth factor by a mechanism involving endoplasmic reticulum stress and transcription factor ATF4. J. Biol. Chem. 279, 14844–14852 [DOI] [PubMed] [Google Scholar]
- 70. Ishida B. Y., Bailey K. R., Duncan K. G., Chalkley R. J., Burlingame A. L., Kane J. P., Schwartz D. M. (2004) Regulated expression of apolipoprotein E by human retinal pigment epithelial cells. J. Lipid Res. 45, 263–271 [DOI] [PubMed] [Google Scholar]
- 71. Wang L., Li C. M., Rudolf M., Belyaeva O. V., Chung B. H., Messinger J. D., Kedishvili N. Y., Curcio C. A. (2009) Lipoprotein particles of intraocular origin in human Bruch membrane: an unusual lipid profile. Invest. Ophthalmol. Vis. Sci. 50, 870–877 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Johnson L. V., Forest D. L., Banna C. D., Radeke C. M., Maloney M. A., Hu J., Spencer C. N., Walker A. M., Tsie M. S., Bok D., Radeke M. J., Anderson D. H. (2011) Cell culture model that mimics drusen formation and triggers complement activation associated with age-related macular degeneration. Proc. Natl. Acad. Sci. U.S.A. 108, 18277–18282 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Klos A., Wende E., Wareham K. J., Monk P. N. (2013) International Union of Pharmacology. LXXXVII: complement peptide C5a, C4a, and C3a receptors. Pharmacol. Rev. 65, 500–543 [DOI] [PubMed] [Google Scholar]
- 74. Mogilenko D. A., Kudriavtsev I. V., Trulioff A. S., Shavva V. S., Dizhe E. B., Missyul B. V., Zhakhov A. V., Ischenko A. M., Perevozchikov A. P., Orlov S. V. (2012) Modified low density lipoprotein stimulates complement C3 expression and secretion via liver X receptor and Toll-like receptor 4 activation in human macrophages. J. Biol. Chem. 287, 5954–5968 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Damman J., Nijboer W. N., Schuurs T. A., Leuvenink H. G., Morariu A. M., Tullius S. G., van Goor H., Ploeg R. J., Seelen M. A. (2011) Local renal complement C3 induction by donor brain death is associated with reduced renal allograft function after transplantation. Nephrol. Dial. Transplant. 26, 2345–2354 [DOI] [PubMed] [Google Scholar]
- 76. Platel D., Guiguet M., Briere F., Bernard A., Mack G. (1994) Human interleukin-6 acts as a co-factor for the up-regulation of C3 production by rat liver epithelial cells. Eur. Cytokine Netw. 5, 405–410 [PubMed] [Google Scholar]
- 77. Luo C., Chen M., Xu H. (2011) Complement gene expression and regulation in mouse retina and retinal pigment epithelium/choroid. Mol. Vis. 17, 1588–1597 [PMC free article] [PubMed] [Google Scholar]
- 78. Yun L., Nicholas H., Mark A., Madesh M., Steven K. (2012) Cigarette smoke-induced reactive oxygen species (ROS) production in human airway epithelial cells is calcium and NADPH-oxidase (NOX) dependent. in C53.COPD pathogenesis: in vitro and in vivo studies, pp. A4561–A4561, American Thoracic Society, New York [Google Scholar]