

Background: Pharmacological chaperone (PC) therapy has been proposed for lysosomal storage diseases.
Results: Wild type and mutant β-galactosidases exhibit similar enzymological properties. The recognition mechanism of glycomimetic PC candidates involves both sugar-like and substituent moieties.
Conclusion: Crystal structures reveal the molecular basis for high binding potency of PC compounds.
Significance: Enzymological properties, binding affinities, and recognition modes are biophysically and structurally characterized.
Keywords: Crystal Structure, Galactose, Glycosidases, Lysosomal Storage Disease, X-ray Crystallography
Abstract
GM1 gangliosidosis and Morquio B disease are autosomal recessive diseases caused by the defect in the lysosomal β-galactosidase (β-Gal), frequently related to misfolding and subsequent endoplasmic reticulum-associated degradation. Pharmacological chaperone (PC) therapy is a newly developed molecular therapeutic approach by using small molecule ligands of the mutant enzyme that are able to promote the correct folding and prevent endoplasmic reticulum-associated degradation and promote trafficking to the lysosome. In this report, we describe the enzymological properties of purified recombinant human β-GalWT and two representative mutations in GM1 gangliosidosis Japanese patients, β-GalR201C and β-GalI51T. We have also evaluated the PC effect of two competitive inhibitors of β-Gal. Moreover, we provide a detailed atomic view of the recognition mechanism of these compounds in comparison with two structurally related analogues. All compounds bind to the active site of β-Gal with the sugar-mimicking moiety making hydrogen bonds to active site residues. Moreover, the binding affinity, the enzyme selectivity, and the PC potential are strongly affected by the mono- or bicyclic structure of the core as well as the orientation, nature, and length of the exocyclic substituent. These results provide understanding on the mechanism of action of β-Gal selective chaperoning by newly developed PC compounds.
Introduction
Human β-d-galactosidase (EC 3.2.1.23, β-Gal)3 is a lysosomal hydrolase that catalyzes removal of terminal β-linked galactose in GM1 ganglioside and keratan sulfate (1–3). In humans, deficiency of β-Gal enzyme causes GM1 gangliosidosis and Morquio B disease, two lysosomal storage diseases (LSDs) characterized by the progressive accumulation of metabolites in the cell (4–6). GM1 gangliosidosis is a severe neurodegenerative disease that is classified into three types, infantile, juvenile, and adult, depending on the onset and severity (7). Morquio B disease is a rare bone disease without central nervous system involvement (7). Currently, more than 160 mutations in the human β-Gal gene have been identified as factors causative of its deficiency (8, 9).
Two principal treatment strategies are currently approved or in clinical trials for LSDs. The first is enzyme replacement therapy, where the deficient enzyme is supplied by regular injection of purified recombinant human enzyme (10–12). However, little or no improvement has been observed in the central nervous system affectations in LSD patients because the enzyme cannot cross the blood-brain barrier. The second is substrate reduction therapy, which uses an orally available small molecule that inhibits the biosynthesis of the glycosphingolipid (13). However, this treatment is nonspecific, leading to serious side effects. An alternative treatment, pharmacological chaperone (PC) therapy, has been proposed for GM1 gangliosidosis, Morquio B disease, and other LSDs (14). This therapy uses a small molecule ligand that can bind to the mutant protein and stabilize the correct conformation of the protein at neutral pH in the endoplasmic reticulum, allowing it to be transported to the lysosome where the ligand dissociates at acidic pH and in the presence of excess substrate.
Galactose, the catalytic product of α- or β-Gal, the iminosugar 1-deoxygalactonojirimycin (DGJ; Fig. 1), a mimic of galactose, and some derivatives have been well studied as PCs for these LSDs (15, 16). However, DGJ has promiscuity for a number of galactopyranoside-processing isoenzymes, which may hamper clinical development.
FIGURE 1.
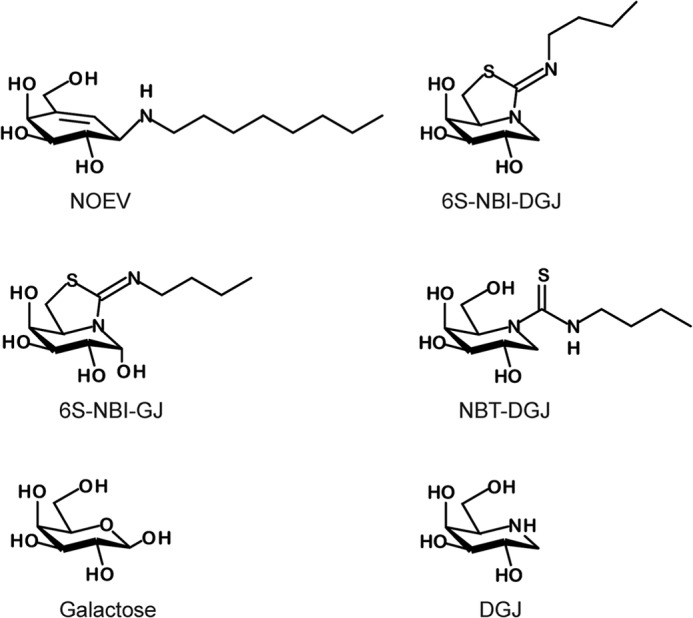
Structures of DGJ derivatives and PC compounds discussed in this study.
We had previously reported the valienamine derivative N-octyl-4-epi-β-valienamine (NOEV) and the sp2-iminosugar derivative 5N,6S-(N′-butyliminomethylidene)-6-thio-1-deoxygalactonojirimycin (6S-NBI-DGJ; Fig. 1), as effective PC compounds that improve human mutant β-Gal activity in cultured cells (8, 17–20). The effect of NOEV and 6S-NBI-DGJ on reducing the accumulation of GM1 ganglioside was mutation specific. In fact, NOEV reduced the accumulation in β-GalR201C but not in β-GalI51T cells, whereas 6S-NBI-DGJ was effective in both cell lines (8, 20). On the other hand, the inhibitory activity of NOEV overcame the chaperone activity at micromolar concentrations, whereas the chaperone effect of 6S-NBI-DGJ steadily increased in a dose-dependent manner up to milimolar concentrations (20). The presence of an extra pseudoanomeric hydroxyl in 5N,6S-(N′-butyliminomethylidene)-6-thiogalactonojirimycin (6S-NBI-GJ; Fig. 1) results in a selectivity shift toward β-glucosidase (β-glucocerebrosidase), which has been exploited for the development of efficient pharmacological chaperones for Gaucher disease (21–23). The monocyclic N-(N′-butylthiocarbamoyl)-1-deoxygalactonojirimycin (NBT-DGJ; Fig. 1) derivative turned out to be instead a selective inhibitor of lysosomal α-Gal with potential as a pharmacological chaperone for Fabry disease (17).
We reported the crystal structure of human β-Gal (24). Getting more structural information on how this set of compounds interact with human β-Gal and establishing PC·enzyme complex structure-activity relationships provide useful information about the features governing chaperone-enzyme interactions at the molecular level and how they could be implemented in the design of new generations of PC drug candidates for GM1 gangliosidosis.
In this study, we examined the enzymological properties of recombinant human β-GalWT and two representative mutations in Japanese patients, β-GalR201C and β-GalI51T. To date, the chaperone ability for candidate compounds was evaluated against lysates from cultured human fibroblasts or transiently transfected cells. In this report, all assays were performed using purified recombinant human β-Gal, so that the chaperone effect can be estimated without any interference. We also determined crystal structures of the complexes of the strong β-Gal inhibitors, NOEV and 6S-NBI-DGJ, and of the much weaker ligands 6S-NBI-GJ and NBT-DGJ bound to human β-GalWT. β-GalI51T mutant structures complexed with galactose or 6S-NBI-DGJ were also determined.
EXPERIMENTAL PROCEDURES
β-Gal Inhibitors/Chaperones
The valienamine derivative NOEV, and the three sp2-iminosugar derivatives 6S-NBI-DGJ, 6S-NBI-GJ, and NBT-DGJ, were synthesized following previously reported procedures (Fig. 1) (17, 20, 25, 26). All compounds are stable at room temperature and soluble in water up to 10 mm (NOEV and 6S-NBI-DGJ) or 20 mm (6S-NBI-GJ and NBT-DGJ). The molecular weights are 287.4, 260.4, 278.4, and 276.4, respectively. Stock solution was prepared in sterile H2O and stored at −20 °C. NOEV and 6S-NBI-DGJ inhibit human β-Gal in vivo with high selectivity, whereas 6S-NBI-GJ and NBT-DGJ are very weak inhibitors of β-Gal but low micromolar inhibitors of β-glucocerebrosidase or α-Gal, respectively.
Cloning, Expression, and Purification of Human β-Gal
The details of cloning, expression, and purification of β-Gal were reported previously (27). Mutations were introduced by PrimeSTAR Max DNA polymerase (Takara) using oligonucleotide primers following the manufacturer's protocol. Briefly, β-GalWT and two mutant proteins, β-GalR201C and β-GalI51T (residues 24–677), fused to the N-terminal hexahistidine and FLAG tag, were expressed in yeast Pichia pastoris KM71 and purified to homogeneity in a nickel-Sepharose column. In the course of purification, polysaccharide moieties attached to the protein were trimmed off by endoglycosidase Hf treatment. For crystallization, β-Gals were subjected to limited proteolysis with bovine trypsin and further purified by cation-exchange column chromatography, and Superdex 200 size exclusion column (GE Healthcare) equilibrated with buffer A (0.01 m MES, pH 6.0, 0.1 m NaCl). Finally, purified β-Gals were concentrated to 10 mg/ml in buffer A with or without 10 mm galactose.
Kinetics of Purified β-Gals and Evaluation of Inhibitors
β-Gal activity was measured by using 4-methylumbelliferyll-β-d-galactopyranoside in buffer B (0.15 m sodium citrate, pH 4.5, and 0.2 m NaCl) as a substrate, and incubating at 37 °C for 6 min. The reaction was terminated by adding 0.2 m glycine-NaOH buffer (pH 10.7). The liberated 4-methylumbelliferyll was measured with a fluorescence plate reader (excitation 355 nm; emission 460 nm; fluoroskan ascent, Thermo electron Corp.).
A kinetic experiment on purified β-Gal was performed using various concentrations (75–375 mm) of 4-methylumbelliferyll-β-Gal as a substrate to determine the maximum velocity (Vmax) and Michaelis constant (Km) values. For inhibition assays, the ligand compounds (galactose, 25 mm; DGJ, 250 μm; NOEV, 2.5 μm; 6S-NBI-DGJ, 250 μm; 6S-NBI-GJ, 2.5 mm; and NBT-DGJ, 2.5 mm) were added to the purified β-Gal, and then the activity of β-Gal was measured as described above. The inhibition constant (Ki) values (Table 1) were calculated from the plots of slope versus inhibitor concentration.
TABLE 1.
Enzymological parameters of the wild-type and mutant β-Gals, and inhibitory constant values of PC compounds
The enzyme activity of β-Gal was fluorometrically determined with 4-methylumbelliferyll-β-d-galactopyranoside as a substrate.
| Specific activity | Vmax | Ki |
Ki |
NOEV | 6S-NBI-DGJ | 6S-NBI-GJ | NBT-DGJ | ||
|---|---|---|---|---|---|---|---|---|---|
| Galactose | DGJ | ||||||||
| μmol/min/mg | μm/min | mm | mm | μm | |||||
| WT | 15.6 | 1.78 | 0.49 | 4.14 | 61.8 | 1.1 | 68.3 | 253.1 | 372.7 |
| R201C | 13.1 | 1.74 | 0.44 | 5.61 | 49.0 | 0.94 | 53.3 | 320.4 | 388.3 |
| I51T | 16.3 | 1.82 | 0.55 | 4.91 | 44.7 | 1.2 | 68.8 | 319.6 | 329.0 |
Effects of Pharmacological Chaperones on the Stability of the β-Gals
To compare the stability of the β-GalWT and that of mutant proteins under various pH conditions in a buffer, 10 μg/ml of β-Gal was incubated in either 0.1 m sodium citrate buffer (pH 3.5–7.0) or 0.1 m Hepes buffer, pH 7.5–8.0, with 0.1 m NaCl at 48 °C for 40 min. After incubation, β-Gal activity was assayed immediately under pH 4.5. To assess the effects of PC compounds on the stability of WT and mutant β-Gal, various concentrations (galactose, 0.2, 2, and 20 mm; DGJ, 2, 20, and 200 μm; NOEV, 0.02, 0.2, and 2 μm; 6S-NBI-DGJ 2, 20, and 200 μm) of PC compounds were added to 10 μg/ml of β-Gal in buffer C (0.1 m sodium citrate, pH 7.0, and 0.1 m NaCl), followed by incubation at 48 °C for various times (0, 10, 20, and 40 min). As a control, the same experiment was carried out with or without PCs (galactose, 20 mm; DGJ, 200 μm; NOEV, 2 μm; 6S-NBI-DGJ, 200 μm) under the acidic condition (0.1 m sodium citrate, pH 4.8, and 0.1 m NaCl). Values are averages of three individual experiments.
CD Spectrum
Circular dichroism (CD) spectra were recorded at 20 °C on a Jasco J-720W spectropolarimeter equipped with a Julabo F25-ED temperature controller. The wild type and two mutant proteins of β-Gal were diluted to 0.1 mg/ml in 20 mm sodium acetate, pH 5.0, with or without galactose (50 mm). CD spectra were collected over the 200–280 nm wavelength range and with a resolution of 0.1 nm, a bandwidth of 1 nm, and a response time of 1 s. Final spectra were the sum of 16 scans accumulated at a speed of 50 nm/min.
Isothermal Titration Calorimetry
Isothermal titration calorimeter MicroCal iTC200 was employed to determine the affinities between β-Gal and the two PCs, NOEV or 6S-NBI-DGJ, at 25 °C. The calorimetric cell was filled with 50 (for NOEV) or 100 μm (for 6S-NBI-DGJ) β-Gal, and PCs (0.5 mm NOEV and 1 mm 6S-NBI-DGJ) were injected into the cell with a 60-μl syringe. The released heat was measured by integrating the calorimetric output curves. Data were fit with the model for one set of binding sites in Origin (OriginLab).
Crystallization and Data Collection
Crystallization experiments were performed by sitting-drop vapor diffusion methods at 4 °C. Crystals of β-GalWT·6S-NBI-DGJ complex were grown from the equivolume of 10 mg/ml of β-GalWT in buffer A containing 1 mm 6S-NBI-DGJ and the reservoir solution containing 20–26% PEG3350, 0.2 m ammonium sulfate, and 0.1 m Hepes, pH 7.5. The others were first prepared as β-GalWT or β-GalI51T·galactose complex crystals, and then the crystals were soaked into the solution containing each PC compound. Crystals of β-GalWT or β-GalI51T·galactose complex were grown from the equivolume of 10 mg/ml of β-GalWT or β-GalI51T in buffer A containing 10 mm galactose and the reservoir solution containing 20–26% PEG3350, 0.2 m ammonium sulfate, and 0.1 m Hepes, pH 7.5. Then, β-GalWT or β-GalI51T-galactose crystals were transferred into mother liquor (25% PEG3350, 0.2 m ammonium sulfate, and 0.1 m Hepes, pH 7.5) supplemented with PC compounds (NOEV or 6S-NBI-DGJ, 1 mm; 6S-NBI-GJ or NBT-DGJ, 10 mm) and incubated for 3–55 min (NOEV, 55 min; 6S-NBI-DGJ, 40 min; 6S-NBI-GJ, 15 min; NBT-DGJ, 3 min) at 4 °C.
Diffraction datasets were collected at beamline BL-17A and AR-NE3A at the Photon Factory (Tsukuba, Japan). Prior to data collection, the crystals of the each PC compound·β-Gal complex were soaked for a few seconds in the reservoir solution supplemented with the corresponding PC compound (galactose 10 mm; NOEV and 6S-NBI-DGJ, 1 mm; 6S-NBI-GJ and NBT-DGJ, 2 mm) and 15% ethylene glycol, and flash-cooled to 95 K. The datasets were processed with the HKL2000 package (Table 2) (28).
TABLE 2.
Data collection and crystallographic refinement statistics
| β-Gal |
||||||
|---|---|---|---|---|---|---|
| WT | WT | WT | WT | I51T | I51T | |
| PC compound | NOEV | 6S-NBI-DGJ | 6S-NBI-GJ | NBT-DGJ | Galactose | 6S-NBI-DGJ |
| Data collection | ||||||
| X-ray source | PF-BL17A | PF-BL17A | PF-AR NE3A | PF-AR NE3A | PF-AR NE3A | PF-AR NE3A |
| Wavelength (Å) | 0.98000 | 0.98000 | 1.00000 | 1.00000 | 1.00000 | 1.00000 |
| Resolution range (Å) | 50–2.1 (2.14–2.10) | 50–2.2 (2.24–2.20) | 50–2.0 (2.03–2.00) | 50–2.3 (2.34–2.30) | 50–2.2 (2.19–2.15) | 50–2.3 (2.34–2.30) |
| Space group | P21 | P21 | P21 | P21 | P21 | P21 |
| Unit cell parameters | ||||||
| a (Å) | 94.8 | 95.0 | 95.0 | 95.1 | 95.0 | 95.0 |
| b (Å) | 115.6 | 116.5 | 116.0 | 116.3 | 115.9 | 116.2 |
| c (Å) | 140.3 | 140.4 | 140.6 | 141.8 | 140.3 | 140.7 |
| β (°) | 92.3 | 92.2 | 92.3 | 92.4 | 92.2 | 92.3 |
| No. of observed reflections | 709,280 | 367,282 | 743,452 | 597,215 | 515,585 | 505,251 |
| No. of unique reflections | 172,353 | 139,532 | 199,457 | 133,461 | 160,267 | 134,623 |
| Redundancy | 4.1 (3.8) | 2.6 (2.5) | 3.7 (3.6) | 4.5 (4.5) | 3.2 (3.3) | 3.8 (3.7) |
| Completeness (%) | 98.6 (96.4) | 90.4 (93.7) | 97.5 (96.4) | 98.3 (98.1) | 96.4 (98.6) | 99.1 (100.0) |
| Average I/σ(I) | 16.7 (3.2) | 9.0 (2.0) | 15.8 (2.2) | 13.5 (2.5) | 15.5 (3.5) | 11.5 (2.3) |
| Rmergea | 0.136 (0.602) | 0.148 (0.571) | 0.128 (0.605) | 0.171 (0.763) | 0.154 (0.478) | 0.173 (0.677) |
| Refinement | ||||||
| Resolution range (Å) | 47.4–2.1 | 43.4–2.2 | 25.9–2.0 | 26.0–2.3 | 27.3–2.2 | 25.3–2.3 |
| Protein residues | 2,415 | 2,416 | 2,416 | 2,416 | 2,416 | 2,416 |
| No. of atoms | 19,291 | 19,302 | 19,305 | 19,243 | 19,280 | 19,263 |
| Ligand | 80 | 68 | 72 | 72 | 48 | 68 |
| N-Acetylglucosamine | 224 | 224 | 224 | 224 | 224 | 224 |
| Water molecules | 1,513 | 1,248 | 1,596 | 1,093 | 1,429 | 1,241 |
| SO42−/Cl−/EGb | 40/4/32 | 40/4/32 | 40/4/32 | 40/4/32 | 40/4/32 | 40/4/32 |
| Average B factor (Å2) | 22.9 | 21.9 | 28.8 | 32.0 | 28.9 | 29.0 |
| Rworkc /Rfreed (%) | 17.9/22.4 | 17.7/23.0 | 18.6/23.3 | 17.4/24.0 | 19.6/24.2 | 17.9/24.0 |
| Root mean square deviation | ||||||
| Bond length (Å) | 0.012 | 0.013 | 0.014 | 0.016 | 0.014 | 0.016 |
| Bond angles (°) | 1.54 | 1.59 | 1.69 | 1.84 | 1.70 | 1.86 |
| Ramachandran plot (%) | ||||||
| Favored | 96.6 | 96.6 | 97.3 | 96.0 | 96.5 | 96.1 |
| Allowed | 3.4 | 3.3 | 2.6 | 3.8 | 3.4 | 3.8 |
| Disallowed | 0.0 | 0.1 | 0.1 | 0.2 | 0.1 | 0.1 |
Values in parentheses are for the shell with the highest resolution.
a Rmerge(I) = Σ|I − 〈I〉|/ΣI,where I is the diffraction intensity.
b EG indicates ethylene glycole.
c R = Σ|Fo − Fc|/ΣFo, where Fo and Fc are the observed and calculated structure amplitudes, respectively.
d Rfree is an R value for a 5% subset of all reflections, but was not used in the refinement.
Structure Determination and Crystallographic Refinement
Structures of β-GalWT complexed with PC compounds (NOEV, 6S-NBI-DGJ, 6S-NBI-GJ, or NBT-DGJ) and β-GalI51T complexed with galactose or 6S-NBI-DGJ were determined by the molecular replacement method using the β-GalWT-galactose complex structure (PDB code 3THC) as a search model and the program Molrep implemented in the CCP4 suite (29). Model building and adjustment were carried out using program COOT (30). Crystallographic refinement was performed using the program REFMAC (31) implemented in the CCP4 package until the R factor was converged (Table 2). The atomic coordinates and structure factors have been deposited in the Protein Data Bank (3WEZ (β-GalWT-NOEV), 3WF0 (β-GalWT-6S-NBI-DGJ), 3WF1 (β-GalWT-6S-NBI-GJ), 3WF2 (β-GalWT-NBT-DGJ), 3WF3, (β-GalI51T-galactose) and 3WF4 (β-GalI51T-6S-NBI-DGJ)) (supplemental data).
RESULTS
Enzymological Properties of β-Gal
The Michaelis-Menten parameters of the purified recombinant β-Gal were determined (Table 1). The Vmax values of β-GalWT, β-GalR201C, and β-GalI51T were 1.8, 1.7, and 1.8 μm/min, and the Km values were 0.5, 0.4, and 0.6 mm, respectively. The results suggest that the wild type and mutant proteins show similar enzyme activity and substrate affinity. These observations further support the promise of PCs capable of rescuing the mutant protein from endoplasmic reticulum-associated degradation and promoting trafficking to the lysosome for the treatment of GM1 gangliosidosis. The corresponding inhibition constant (Ki) values are presented in Table 1. Data for d-galactose and DGJ have been collected for comparative purposes. Galactose is a very weak inhibitor, although DGJ shows 100-fold stronger inhibition than galactose. NOEV exhibited the most potent inhibition of β-Gal among the series, with Ki close to 1 μm. 6S-NBI-DGJ inhibits β-Gal to the same degree as DGJ, whereas the structurally related derivatives 6S-NBI-GJ and NBT-DGJ showed much weaker inhibitory properties. Consistent with the degree of the inhibition, a isothermal titration calorimetry experiment demonstrated that NOEV exhibited a strong binding affinity with the dissociation constant of submicromolar, whereas 6S-NBI-DGJ exhibited a ∼100-fold reduced affinity (supplemental Fig. S1). It should be noted that all the compounds inhibited WT and mutant β-Gal with virtually identical potencies.
The Stability of WT and Mutant β-Gal under Various pH Conditions
The activity of WT and mutant β-Gal after heat treatment (48 °C, 40 min) was examined under various pH conditions (Fig. 2). As previously reported (32), each β-Gal was more active at acidic pH (4.5–5.5), whereas the catalytic activity was greatly reduced under strong acidic (pH < 3.5) and basic (pH > 8.0) conditions. When comparing wild type and mutant enzymes, it was found that β-GalI51T was only slightly more sensitive to heat-induced inactivation in the whole range of pH values from 3.5 to 8.0, whereas the activity of β-GalR201C was significantly reduced under various pH conditions compared with β-GalWT. As a conclusion from these results, β-Gal mutations associated with GM1 gangliosidosis lead to the reduction of enzyme stabilities but do not lose the catalytic activity at all, suggesting that both β-GalR201C and β-GalI51T could process GM1 ganglioside and prevent/reduce its accumulation provided they are properly transferred to the lysosome with the help of PCs.
FIGURE 2.
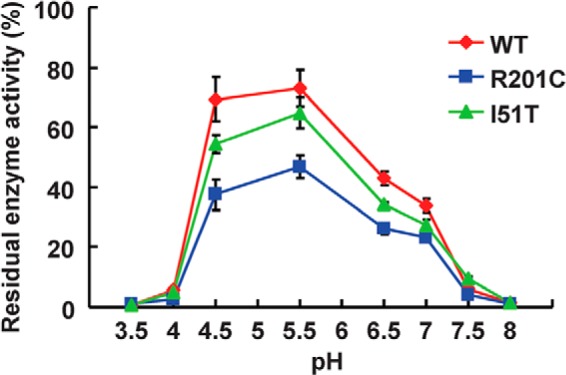
Enzymatic activity of the WT and two mutant proteins of β-Gal under various pH conditions. The purified WT and two mutant proteins of β-Gal were incubated under the indicated pH conditions in a buffer at 48 °C for 40 min. Then, enzyme assay was performed under pH 4.5 conditions. The residual enzyme activity is shown as a percentage of the values at the time 0. Values are averages of three individual experiments and expressed as the percent ratio of the activity at a particular pH condition to the value at the time 0.
Effects of the Pharmacological Chaperones on the Stability of the β-Gal
The ability of a ligand to prevent heat-induced inactivation of a given glycosidase has been previously used as an indication of its pharmacological chaperone potential (33). In our case, galactose, DGJ, NOEV, and 6S-NBI-DGJ were added to β-Gal at neutral pH in buffers, and then the remaining enzyme activities were examined after heat treatment (Fig. 3). All compounds were able to increase the residual activities of the β-Gal in a dose-dependent manner. β-GalWT activity was decreased to ∼20% after 40 min at 48 °C incubation, but galactose-treated β-Gal retained ∼70% activity. In accordance with the inhibitory activity, NOEV-treated β-Gal, and DGJ and 6S-NBI-DGJ-treated β-Gal reached a similar activity improvement at ∼10,000- and ∼100-fold lower concentrations, respectively, compared with galactose, suggesting that these compounds bear potential as PCs. Little chaperone effects were observed under the acidic condition (supplemental Fig. S2).
FIGURE 3.

Effects of PC compounds on the stability of the WT and two mutant proteins of β-Gals at pH 7.0. The purified WT and two mutant proteins of β-Gal were incubated in 0.1 m sodium citrate, pH 7.0, with or without galactose, DGJ, NOEV, and 6S-NBI-DGJ at 48 °C for the incubated times. Then, the enzyme assay was performed at pH 4.5. The residual enzyme activity is shown as a percentage of the values at the time 0. Values are averages of three individual experiments and are expressed as the percent ratio of the activity at a particular pH condition to the value at the time 0.
Structural Basis of Pharmacological Chaperone Interaction with Human β-Gal
To gain atomic insight into the PC compounds binding to β-Gal, we determined crystal structures of β-GalWT complexed with NOEV, 6S-NBI-DGJ, 6S-NBI-GJ, and NBT-DGJ. The overall structures of β-Gal remained largely unchanged among these four complexes and all structures showed a strong overall agreement with galactose-bound β-GalWT (PDB code 3THC) (24) with root mean square deviations ranging from 0.17 to 0.26 Å.
β-Gal was folded into three domains: the TIM barrel domain, β-domain 1, and β-domain 2 (supplemental Fig. S3). The galactose mimetics were embedded in the same ligand binding pocket of the TIM barrel domain in β-Gal, with the alkyl chain at the exocyclic nitrogen atom oriented toward the entrance of the active site pocket (Fig. 4, A–D). In the case of NOEV, all hydroxyl groups made direct hydrogen bonds with β-Gal in a similar manner to galactose. In addition, a hydrogen bond was formed between the exocyclic N atom of NOEV and Glu-188 of β-Gal, stabilizing the orientation of the octyl chain. The hydrophobic tail extended along the protein surface consisting of Tyr-485, Trp-273, Leu-274, His-276, and Asn-321 (Fig. 4A). The terminal methyl group (C16 carbon atom) was in contact with the CE1 atom of His-276, and CB and CG atoms of Asn-321 (Fig. 5A).
FIGURE 4.

Binding of PC compounds to the active site of human β-Gal. A–D, left, PC compounds bound to β-Gal viewed from the entrance of active site pocket: A, NOEV; B, 6S-NBI-DGJ; C, 6S-NBI-GJ; D, NBT-DGJ. Hydrogen bonds defined by ccp4 mg (37) are shown by dashed lines. In B, a unique side chain arrangement of Tyr-333 in the 6S-NBI-DGJ complex is shown in crimson. The Fo − Fc electron density maps of PC compounds are shown in gray. A and B are contoured at 2.0 σ, C at 1.0 σ, and D at 1.5 σ. Right, schematic representation of the interaction between β-Gal and each PC compound: A, NOEV; B, 6S-NBI-DGJ; C, 6S-NBI-GJ; and D, NBT-DGJ. Hydrogen bonds are shown as dotted lines, and van der Waals interactions are shown by arcs.
FIGURE 5.
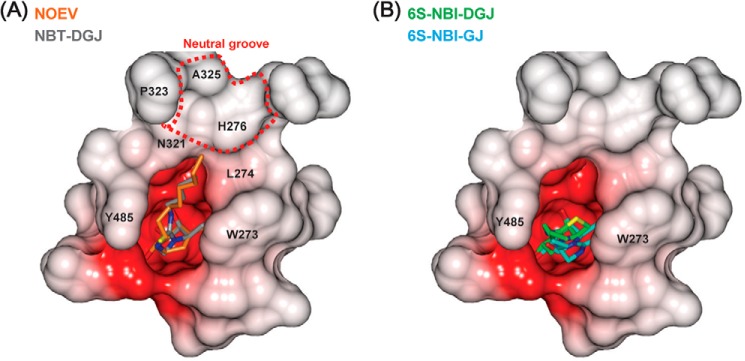
Comparison of the binding of the PC compounds. Electrostatic potential surface of the surrounding of the active site pocket. A, NOEV (orange) complex structure is superimposed on NBT-DGJ (gray) complex. B, 6S-NBI-DGJ (green) and 6S-NBI-GJ (cyan) are drawn in a similar manner as in A.
Similar to NOEV, hydroxyls OH2, OH3, and OH4 of the sp2-iminosugar 6S-NBI-DGJ made direct hydrogen bonds with β-Gal (Fig. 4B). The exocyclic N atom of 6S-NBI-DGJ also made a hydrogen bond with Glu-188 of β-Gal, which orients the butyl chain toward the hydrophobic pocket flanked by Tyr-485 and Trp-273 (Fig. 5B). Contrary to NOEV, for which the endocyclic double-bond imposes a half-chair conformation at the valienamine ring, the 6-membered ring of 6S-NBI-DGJ exhibits an almost ideal chair conformation close to that encountered for galactose in the galactose·β-Gal complex. The 5-membered ring is fused to the 6-membered ring at an angle of 110° (supplemental Fig. S4). Because 6S-NBI-DGJ lacks the primary hydroxyl equivalent to the OH6 group in galactose, the side chain orientation of Tyr-333 is shifted so as to fill the corresponding space. This feature is unique to the 6S-NBI-DGJ·β-Gal complex structure.
The presence of a pseudoanomeric hydroxyl group, OH1, in the galactonojirimycin analogue 6S-NBI-GJ led to a very significant decrease in the binding affinity toward β-Gal as compared with the DGJ congener 6S-NBI-DGJ. Actually this compound is a rather selective inhibitor of β-glucosidase (21). Whereas 6S-NBI-GJ has been shown to exist exclusively in the 4C1 chair conformation in water solution, with OH1 axially oriented in the α-configuration (34, 35), in the corresponding complex with β-Gal the opposite β-configuration was encountered, with the 6-membered ring and the 5-membered ring in the same plane (supplemental Fig. S4). The β-oriented OH1 group is now involved in hydrogen bonding with Glu-188 (Fig. 4C), with the butyl chain extended to Tyr-485 and Trp-273 (Fig. 5B).
The monocyclic derivative NBT-DGJ recovered the hydrogen bonding interaction between the primary hydroxyl OH6 and Tyr-333 (Fig. 4D). Actually, all hydroxyl groups of NBT-DGJ made direct hydrogen bonds with β-Gal as in the NOEV·β-Gal complex. However, the exocyclic N and S atoms had no interactions with β-Gal, which is in agreement with the weak binding affinity observed in the kinetic inhibition studies.
We also determined crystal structures of β-GalI51T complexed with galactose or 6S-NBI-DGJ (Fig. 6). The quality of the electron density maps is sufficiently high to allow modeling of the SNP residue (Fig. 6B). These overall structures are essentially identical to those of β-GalWT bound to the corresponding ligand. In fact, the main chain atoms in each complex could be superimposed on those of the galactose or 6S-NBI-DGJ complex with root mean square deviations of 0.16 and 0.17 Å, respectively. CD spectrum demonstrated that structures of the wild type and mutant proteins are essentially the same regardless of the presence or absence of the PC compounds (supplemental Fig. S5).
FIGURE 6.
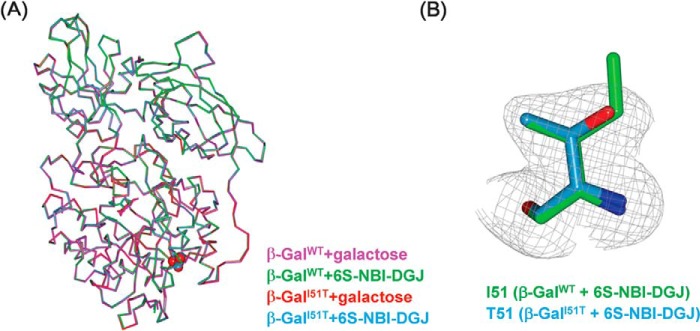
Structures of β-GalI51T complexed with galactose or 6S-NBI-DGJ. A, superimposition of the overall structures of the β-GalWT-galactose (magenta), β-GalWT-6S-NBI-DGJ (green), β-GalI51T-galactose (red), and β-GalI51T-6S-NBI-DGJ (cyan). Mutated residue I51T is shown in space-filling representation. B, the Fo − Fc electron density map (gray) corresponding to mutated residue Thr-51 of β-GalI51T-6S-NBI-DGJ is contoured at 3.0 σ. Shown is the Ile-51 residue of β-GalWT-6S-NBI-DGJ (green) and the Thr-51 residue of β-GalI51T-6S-NBI-DGJ (cyan).
DISCUSSION
In this study, we determined the enzymological properties of purified recombinant human β-Gal (β-GalWT) and two representative mutant proteins, β-GalR201C and β-GalI51T. Moreover, we determined the crystal structures of β-GalWT complexed with four ligand compounds, two of which, NOEV and 6S-NBI-DGJ, have shown pharmacological chaperone activity for several GM1 gangliosidosis-associated mutations, whereas the other two, 6S-NBI-GJ and NBT-DGJ, showed only weak affinity for the enzyme despite having a structure closely related to that of 6S-NBI-DGJ. Crystal structures of β-GalI51T complexed with galactose or 6S-NBI-DGJ have also been determined. Although NOEV and 6S-NBI-DGJ exhibit micromolar and tens of micromolar Ki values, these compounds were shown to significantly enhance the β-Gal activities (up to 6-fold) in GM1 fibroblasts, demonstrating their efficacy in vitro. It was also demonstrated that the PC compounds ameliorated accumulation of GM1 ganglioside in a mouse model (human β-GalR201C), including the brain, after oral administration (8, 18, 20).
The Ile-51 mutation is located in the inner region of β-Gal, whereas the Arg-201 mutation is located instead on the lateral face of the TIM barrel domain, being exposed to the solvent. In both the Arg-201 and Ile-51 mutant proteins, the mutated amino acid is far from the active site, indicating that the mutations are unlikely to affect the active site directly. Accordingly, β-GalI51T and β-GalR201C showed similar enzymological parameters as β-GalWT (Table 1). This coincides with the fact that no significant conformational change in β-GalI51T was observed in the crystal structure of the mutant protein as compared with the wild type enzyme (Fig. 6A). By contrast, β-GalR201C was more unstable than β-GalWT and β-GalI51T under various pH conditions (Fig. 2). Arg-201 forms a salt bridge to Asp-198 (24) and the loss of this salt bridge probably affects the stability of the protein, increasing denaturation propensity. These enzymological properties, obtained using recombinant β-Gal, were consistent with previously reported data (32).
The structural study provides an atomic basis for the binding mechanism of active site-directed pharmacological chaperones to β-Gal, by highlighting the importance of the sugar-like aglycon moiety, the nature of the exocyclic substituent and the conformational properties of the ligand. NOEV and 6S-NBI-DGJ were recognized in a similar manner; however, NOEV was a tight binding inhibitor, whereas 6S-NBI-DGJ binds >60-fold weaker (Table 1). This high affinity of NOEV results from the exocyclic N atom recognition, the length and the orientation of the extended hydrophobic tail and half-chair conformation imposed by the double bond. The sp2-iminosugar type inhibitor 6S-NBI-DGJ is characterized by a rigid bicyclic core derived from DGJ. 6S-NBI-DGJ lacks OH6, exhibits a different length and orientation of the hydrophobic tail, and adopts the chair conformation, resulting in the weaker affinity. As a consequence, the corresponding hydrogen bond interaction with Tyr-333 is missing, which is expected to have a detrimental impact in complex stability. Noteworthy, this scenario leads to a shift of Tyr-333 in the 6S-NBI-DGJ·β-Gal complex, occupying the space where OH6 is located in the corresponding complex with NOEV, revealing a certain degree of flexibility at this region of β-Gal. The shortened alkyl substituent in 6S-NBI-DGJ as compared with NOEV, butyl instead of octyl, is also expected to affect the binding affinity. In fact, N-alkyl-4-epi-β-valienamine derivatives with longer alkyl chains than NOEV have been shown to exhibit higher affinity to bovine β-Gal (36), whereas N-hexyl-4-epi-β-valienamine had 3-fold weaker affinity than NOEV (14). Consistently, the N′-octyl analogue of 6S-NBI-DGJ was also found to be a 4-fold stronger inhibitor of bovine β-Gal than 6S-NBI-DGJ, but it was discarded for PC chaperone studies with the human lysosomal enzyme due to toxicity issues (17). Probably the longer alkyl chain interacts with the neutral groove, formed by His-276, Asn-321, Pro-323, and Ala-325, in a wider surface area than the butyl chain in 6S-NBI-DGJ, thereby increasing the binding affinity (Fig. 5). Unlike the octyl chain in NOEV, the butyl chain does not extend over the neutral groove at the entrance of the active site in β-Gal (Fig. 5B), which in part is responsible for its moderate binding affinity to β-Gal.
The present study revealed that the stability of the mutant proteins (R201C, I51T) coincides with the severity of the disease and the PC effect toward β-GalWT and β-GalR201C are well consistent with the previous data (8, 20). However, NOEV has been shown to exhibit differences in their PC effects toward some β-Gal mutant proteins in vitro (20). This behavior does not correlate, however, with the present data on the stabilization effect toward heat-induced inactivation of purified recombinant β-Gal, where NOEV proved more efficient than 6S-NBI-DGJ irrespective of the mutation. Mutant enzyme rescued by PCs is a complex process that also involves stabilization of the already folded enzyme to promote trafficking and maturation. How close are the folded states is probably strongly mutation-dependent, which may explain the above discrepancy between in vitro and cellulo results. Further investigation will be required.
The hydrogen bond interactions involving OH2, OH3, and OH4 of the sugar-like moiety and two glutamic acid residues (Glu-129 and Glu-268) at the active site of β-Gal are critical for binding in both the NOEV and 6S-NBI-DGJ complexes and likely define the d-galacto configurational selectivity of the enzyme. A third glutamic acid, Glu-188, interacts with the exocyclic basic nitrogen atom. This hydrogen bond contributes substantially to β-Gal binding affinity and its absence is likely to be at the origin of the much lower inhibitory potencies of the structurally related sp2-iminosugars 6S-NBI-GJ and NBT-DGJ as well as of the monosaccharide galactose. The weakening of the β-Gal binding affinity for NOEV and 6S-NBI-DGJ observed at acidic pH (8, 17) can be rationalized in terms of the expected decrease in the strength of the key hydrogen bonds after protonation of the glutamic acid residues in the protein and the basic nitrogen functionality in the chaperone.
The sharp decrease in β-Gal binding affinity for 6S-NBI-GJ and NBT-DGJ as compared with 6S-NBI-DGJ further illustrates the strong dependence of the inhibitory/chaperone activity of sp2-iminosugar glycomimetics on subtle chemical modifications. The findings presented here may be particularly useful for the rational design of second generation PCs for the treatment of the mutant β-Gal-associated LSDs GM1 gangliosidosis and Morquio B disease. Thus, according to the x-ray data, structural changes at the exocyclic substituent in the 6S-NBI-GJ scaffold are expected to be tolerated by the enzyme, offering potential for drug optimization. In addition, the incorporation of an appropriate substituent at the five-membered ring methylene carbon might lead to favorable interactions with Tyr-333, which could be exploited in the design of higher affinity ligands. Research in that direction is currently sought in our laboratories.
Supplementary Material
Acknowledgment
We thank the beamline staffs at the Photon Factory for assistance with data collection.
This work was supported by Grants-in-Aid from the Ministry of Education, Culture, Science, Sports, and Technology of Japan (to U. O. and T. S.) and Grants 13680918 and 14207106 (to Y. S.), Ministry of Health, Labor and Welfare of Japan H20-Kokoro-022 and H22-Nanji-Ippan-002, and Japan Science and Technology Agency Grant AS232Z00009G (to Y. S.), grants from the Takeda Science Foundation and Mochida Memorial Foundation for Medical and Pharmaceutical Research (to U. O. and T. S.), and Spanish Ministerio de Economía y Competitividad contracts SAF2010-15670 and CTQ2010-15848, the Fundación Ramón Areces, the Junta de Andalucía, the European Regional Development Funds (FEDER), the Eurpean Social Funds (FSE), and the Center for Research, Technology and Innovation of the University of Seville (CITIUS).

This article contains supplemental data and Figs. S1–S5.
The atomic coordinates and structure factors (codes 3WEZ, 3WF0, 3WEZ, 3WF0, 3WF1, 3WF2, 3WF3, and 3WF4) have been deposited in the Protein Data Bank (http://wwpdb.org/).
- β-Gal
- β-galactosidase
- GM1
- monosialotetrahexosylganglioside
- LSD
- lysosomal storage disease
- DGJ
- 1-deoxygalactonojirimycin
- PC
- pharmacological chaperone
- NOEV
- N-octyl-4-epi-β-valienamine
- 6S-NBI-DGJ
- 5N,6S-(N′-butyliminomethylidene)-6-thio-1-deoxygalactonojirimycin
- 6S-NBI-GJ
- 5N,6S-(N′-butyliminomethylidene)-6-thiogalactonojirimycin
- NBT-DGJ
- N-(N′-butylthiocarbamoyl)-1-deoxygalactonojirimycin.
REFERENCES
- 1. Alpers D. H. (1969) On β-galactosidase activity. Gastroenterology 56, 985–986 [PubMed] [Google Scholar]
- 2. Asp N. G., Dahlqvist A. (1972) Human small intestine-galactosidases: specific assay of three different enzymes. Anal. Biochem. 47, 527–538 [DOI] [PubMed] [Google Scholar]
- 3. Distler J. J., Jourdian G. W. (1973) The purification and properties of β-galactosidase from bovine testes. J. Biol. Chem. 248, 6772–6780 [PubMed] [Google Scholar]
- 4. Callahan J. W. (1999) Molecular basis of GM1 gangliosidosis and Morquio disease, type B: structure-function studies of lysosomal β-galactosidase and the non-lysosomal β-galactosidase-like protein. Biochim. Biophys. Acta 1455, 85–103 [DOI] [PubMed] [Google Scholar]
- 5. Oshima A., Yoshida K., Shimmoto M., Fukuhara Y., Sakuraba H., Suzuki Y. (1991) Human β-galactosidase gene mutations in Morquio B disease. Am. J. Hum. Genet. 49, 1091–1093 [PMC free article] [PubMed] [Google Scholar]
- 6. Yoshida K., Oshima A., Shimmoto M., Fukuhara Y., Sakuraba H., Yanagisawa N., Suzuki Y. (1991) Human β-galactosidase gene mutations in GM1-gangliosidosis: a common mutation among Japanese adult/chronic cases. Am. J. Hum. Genet. 49, 435–442 [PMC free article] [PubMed] [Google Scholar]
- 7. Suzuki Y., Nanba E., Matsuda J., Higaki K., Oshima A. (2013) β-Galactosidase deficiency (β-galactosidosis): GM1-gangliosidosis and Morquio B disease in The Online Metabolic and Molecular Bases of Inherited Disease (Valle D., Beaudet A. L., Vogelstein B., Kinzler K. W., Antonarakis S. F., Ballabio A., eds) Chapter 151, McGraw-Hill, New York [Google Scholar]
- 8. Higaki K., Li L., Bahrudin U., Okuzawa S., Takamuram A., Yamamoto K., Adachi K., Paraguison R. C., Takai T., Ikehata H., Tominaga L., Hisatome I., Iida M., Ogawa S., Matsuda J., Ninomiya H., Sakakibara Y., Ohno K., Suzuki Y., Nanba E. (2011) Chemical chaperone therapy: chaperone effect on mutant enzyme and cellular pathophysiology in β-galactosidase deficiency. Hum. Mutat. 32, 843–852 [DOI] [PubMed] [Google Scholar]
- 9. Hofer D., Paul K., Fantur K., Beck M., Bürger F., Caillaud C., Fumic K., Ledvinova J., Lugowska A., Michelakakis H., Radeva B., Ramaswami U., Plecko B., Paschke E. (2009) GM1 gangliosidosis and Morquio B disease: expression analysis of missense mutations affecting the catalytic site of acid β-galactosidase. Hum. Mutat. 30, 1214–1221 [DOI] [PubMed] [Google Scholar]
- 10. Brady R. O. (2006) Enzyme replacement for lysosomal diseases. Annu. Rev. Med. 57, 283–296 [DOI] [PubMed] [Google Scholar]
- 11. Desnick R. J., Schuchman E. H. (2012) Enzyme replacement therapy for lysosomal diseases: lessons from 20 years of experience and remaining challenges. Annu. Rev. Genomics Hum. Genet. 13, 307–335 [DOI] [PubMed] [Google Scholar]
- 12. Parenti G. (2009) Treating lysosomal storage diseases with pharmacological chaperones: from concept to clinics. EMBO Mol. Med. 1, 268–279 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Platt F. M., Jeyakumar M. (2008) Substrate reduction therapy. Acta Paediatr. 97, 88–93 [DOI] [PubMed] [Google Scholar]
- 14. Fan J. Q. (2008) A counterintuitive approach to treat enzyme deficiencies: use of enzyme inhibitors for restoring mutant enzyme activity. Biol. Chem. 389, 1–11 [DOI] [PubMed] [Google Scholar]
- 15. Guce A. I., Clark N. E., Rogich J. J., Garman S. C. (2011) The molecular basis of pharmacological chaperoning in human α-galactosidase. Chem. Biol. 18, 1521–1526 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Suzuki Y. (2013) Chaperone therapy update: Fabry disease, GM1-gangliosidosis and Gaucher disease. Brain Dev. 35, 515–523 [DOI] [PubMed] [Google Scholar]
- 17. Aguilar-Moncayo M., Takai T., Higaki K., Mena-Barragán T., Hirano Y., Yura K., Li L., Yu Y., Ninomiya H., García-Moreno M. I., Ishii S., Sakakibara Y., Ohno K., Nanba E., Ortiz Mellet C., García Fernandez J. M., Suzuki Y. (2012) Tuning glycosidase inhibition through aglycone interactions: pharmacological chaperones for Fabry disease and GM1 gangliosidosis. Chem. Commun. 48, 6514–6516 [DOI] [PubMed] [Google Scholar]
- 18. Matsuda J., Suzuki O., Oshima A., Yamamoto Y., Noguchi A., Takimoto K., Itoh M., Matsuzaki Y., Yasuda Y., Ogawa S., Sakata Y., Nanba E., Higaki K., Ogawa Y., Tominaga L., Ohno K., Iwasaki H., Watanabe H., Brady R. O., Suzuki Y. (2003) Chemical chaperone therapy for brain pathology in GM1-gangliosidosis. Proc. Natl. Acad. Sci. U.S.A. 100, 15912–15917 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Ogawa S., Matsunaga Y. K., Suzuki Y. (2002) Chemical modification of the β-glucocerebrosidase inhibitor N-octyl-β-valienamine: synthesis and biological evaluation of 4-epimeric and 4-O-(β-d-galactopyranosyl) derivatives. Bioorg. Med. Chem. 10, 1967–1972 [DOI] [PubMed] [Google Scholar]
- 20. Takai T., Higaki K., Aguilar-Moncayo M., Mena-Barragán T., Hirano Y., Yura K., Yu L., Ninomiya H., García-Moreno M. I., Sakakibara Y., Ohno K., Nanba E., Ortiz Mellet C., García Fernández J. M., Suzuki Y. (2013) A bicyclic 1-deoxygalactonojirimycin derivative as a novel pharmacological chaperone for GM1 gangliosidosis. Mol. Ther. 21, 526–532 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Aguilar-Moncayo M., García-Moreno M. I., Trapero A., Egido-Gabás M., Llebaria A., García Fernández J. M., Ortiz Mellet C. (2011) Bicyclic (galacto)nojirimycin analogues as glycosidase inhibitors: effect of structural modifications in their pharmacological chaperone potential towards β-glucocerebrosidase. Org. Biomol. Chem. 9, 3698–3713 [DOI] [PubMed] [Google Scholar]
- 22. Benito J. M., García Fernández J. M., Ortiz Mellet C. (2011) Pharmacological chaperone therapy for Gaucher disease: a patent review. Expert Opin. Ther. Pat. 21, 885–903 [DOI] [PubMed] [Google Scholar]
- 23. Luan Z., Higaki K., Aguilar-Moncayo M., Ninomiya H., Ohno K., García-Moreno M. I., Ortiz Mellet C., García Fernández J. M., Suzuki Y. (2009) Chaperone activity of bicyclic nojirimycin analogues for Gaucher mutations in comparison with N-(n-nonyl)deoxynojirimycin. ChemBioChem 10, 2780–2792 [DOI] [PubMed] [Google Scholar]
- 24. Ohto U., Usui K., Ochi T., Yuki K., Satow Y., Shimizu T. (2012) Crystal structure of human β-galactosidase: structural basis of GM1 gangliosidosis and morquio B diseases. J. Biol. Chem. 287, 1801–1812 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Aguilar M., Gloster T. M., García-Moreno M. I., Ortiz Mellet C., Davies G. J., Llebaria A., Casas J., Egido-Gabás M., García Fernandez J. M. (2008) Molecular basis for β-glucosidase inhibition by ring-modified calystegine analoges. ChemBioChem 9, 2612–2618 [DOI] [PubMed] [Google Scholar]
- 26. Kuno S., Takahashi A., Ogawa S. (2013) Concise syntheses of potent chaperone drug candidates, N-octyl-4-epi-β-valinenamine (NOEV) and its 6-deoxy derivative, from (+)-proto-quercitol. Carbohydr. Res. 368, 8–15 [DOI] [PubMed] [Google Scholar]
- 27. Usui K., Ohto U., Ochi T., Shimizu T., Satow Y. (2012) Expression, purification, crystallization and preliminary X-ray crystallographic analysis of human β-galactosidase. Acta Crystallogr. Sect. F Struct. Biol. Cryst. Commun. 68, 73–77 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Otwinowski Z., Minor W. (1997) Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol. 276, 307–326 [DOI] [PubMed] [Google Scholar]
- 29. Collaborative Computational Project, Number 4. (1994) The CCP4 suite: programs for protein crystallography. Acta Crystallogr. D Biol. Crystallogr. 50, 760–763 [DOI] [PubMed] [Google Scholar]
- 30. Emsley P., Cowtan K. (2004) Coot: model-building tools for molecular graphics. Acta Crystallogr. D Biol. Crystallogr. 60, 2126–2132 [DOI] [PubMed] [Google Scholar]
- 31. Murshudov G. N., Vagin A. A., Dodson E. J. (1997) Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr. D Biol. Crystallogr. 53, 240–255 [DOI] [PubMed] [Google Scholar]
- 32. Coelho C. J., Spopelsa A. M., Tobo P. R., Severini M. H., Silva C. D., Giugliani R. (1999) Biochemical studies on leukocyte and fibroblast human β-galactosidase. Clin. Biochem. 32, 1801–1812 [DOI] [PubMed] [Google Scholar]
- 33. Sawkar A. R., Cheng W. C., Beutler E., Wong C. H., Balch W. E., Kelly J. W. (2002) Chemical chaperones increase the cellular activity of N370S β-glucosidase: a therapeutic strategy for Gaucher disease. Proc. Natl. Acad. Sci. U.S.A. 99, 15428–15433 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Díaz Pérez V. M., García Moreno M. I., Ortiz Mellet C., Fuentes J., Díaz Arribas J. C., Cañada F. J., García Fernández J. M. (2000) Generalized anomeric effect in action: synthesis and evaluation of stable reducing indolizidine glycomimetics as glycosidase inhibitors. J. Org. Chem. 65, 136–143 [DOI] [PubMed] [Google Scholar]
- 35. Sánchez-Fernández E. M., Rísquez-Cuadro R., Ortiz Mellet C., García Fernández J. M., Nieto P. M., Angulo J. (2012) sp2-Iminosugar O-, S-, and N-glycosides as conformational mimics of α-linked disaccharides: implications for glycosidase inhibition. Chem. Eur. J. 18, 8527–8539 [DOI] [PubMed] [Google Scholar]
- 36. Suzuki Y., Ogawa S., Sakakibara Y. (2009) Chaperone therapy for neuronopathic lysosomal diseases: competitive inhibitors as chemical chaperones for enhancement of mutant enzyme activities. Perspect. Med. Chem. 3, 7–19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. McNicholas S., Potterton E., Wilson K. S., Noble M. E. (2011) Presenting your structures: the CCP4mg molecular-graphics software. Acta Crystallogr. D Biol. Crystallogr. 67, 386–394 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.