

Abstract
The progesterone receptor (PR) plays roles in normal mammary development and breast cancer formation, where it may exert both stimulatory and inhibitory actions. Previously, the breast cancer susceptibility gene product BRCA1 was found to interact with and inhibit the transcriptional activity of estrogen receptor-α. In this study, we found that exogenous wild-type BRCA1 inhibited the activity of the PR in transient transfection assays utilizing a mouse mammary tumor virus-Luc reporter. Wild-type BRCA1 inhibited the activity of endogenous PR in human breast cancer cells (T47D and MCF-7) and inhibited the activity of exogenous PR-A, PR-B, and [PR-A plus PR-B] isoforms. On the other hand, knockdown of endogenous BRCA1 using small interfering RNA enhanced the progesterone-stimulated activity of the PR by about 4-fold. We documented an in vivo association of the endogenous BRCA1 with PR isoforms A and B and a direct in vitro interaction between BRCA1 and PR, which was partially mapped. Whereas down-regulation of the coactivator p300 contributes to the BRCA1-mediated repression of estrogen receptor-α, this mechanism does not contribute to inhibition of PR activity, because exogenous p300 did not rescue the BRCA1 repression of PR activity. The BRCA1-PR interaction has functional consequences. Thus, we showed that BRCA1 inhibits the expression of various endogenous progesterone-responsive genes and inhibits progesterone-stimulated proliferation of T47D cells. Finally, exogenous progesterone caused an exaggerated proliferative response in the mammary glands of mice harboring a mammary-targeted conditional deletion of the full-length isoform of Brca1. These findings suggest that BRCA1 regulates the activity of progesterone, a major hormone of pregnancy that may also participate in mammary carcinogenesis.
The Breast Cancer and ovarian susceptibility gene 1 (BRCA1) on chromosome 17q21 was identified based on its genetic linkage to familial early-onset breast and ovarian cancer syndromes (1). In addition to breast and ovarian cancer, BRCA1 mutation carriers were found to exhibit significantly increased risk for other hormone-responsive cancer types, including endometrial and cervical cancers in women and prostate cancer in men under 65 yr of age (2–4). These findings suggest a possible linkage between BRCA1 and steroid hormone-responsive cancer types (reviewed in Ref. 5). The molecular pathways through which BRCA1 mediates its tumor suppressor function are mostly unknown. Much research has focused on the potential role of BRCA1 as a caretaker gene in maintaining genomic stability. This caretaker function may be related, in part, to established BRCA1 functions in several DNA damage signaling, repair, and DNA damage-responsive cell cycle checkpoints (reviewed in Ref. 6).
Whereas many tumor suppressor genes function in DNA damage response, cell cycle, and apoptosis pathways, these types of functions, by themselves, do not explain the strong predilection of BRCA1 mutation carriers to develop specific tumor types, particularly hormone-dependent cancers, as opposed to a more generalized spectrum of cancers. Thus, we and others have tried to identify additional functions for BRCA1 to explain its association with specific cancer types. In this regard, we found that BRCA1 inhibits estrogen receptor (ER-α) signaling by inhibiting the conserved carboxyl-terminal transcriptional activation function (AF-2) of ER-α, which is linked to the ligand-binding domain (7). The BRCA1 repression of ER-α activity is manifested by the inhibition of estrogen-stimulated expression of pS2, cathepsin D, and a variety of other estrogen-responsive genes (8, 9). The inhibition of ER-α activity is due, in part, to a direct interaction of the BRCA1 and ER-α proteins and, in part, to BRCA1-mediated down-regulation of expression of p300, a transcriptional coactivator of ER-α (8, 10, 11). It has also been reported that BRCA1 mediates ligand-independent repression of ER-α activity (i.e. that the absence of BRCA1 permits activation of ER-α in the absence of estrogen) (12). In addition, BRCA1 was found to interact directly with ER-α and inhibit estrogen-stimulated production of vascular endothelial growth factor (13). In a similar vein, BRCA1 was found to interact directly with and stimulate androgen receptor activity in prostate cancer cells, leading to increased androgen-responsive gene expression (14, 15).
During the course of studies on the functional interaction between BRCA1 and ER-α, we observed a functional interaction between BRCA1 and another steroid hormone receptor, the progesterone receptor (PR). The PR is a transcriptional target of ER-α and is thought to play a key role in mammary differentiation, the normal menstrual cycle, and in the mammary and uterine changes associated with pregnancy (16, 17). Its role in breast cancer is not clearly defined. However, the observation that hormone replacement therapy (HRT) regimens containing a combination of estrogen and a progestin confers an increase in breast cancer risk relative to HRT regimens containing estrogen alone (18, 19) suggests that, in some contexts, the PR may stimulate the development of human breast cancer. The linkage between PR signaling and breast cancer is considered further in Discussion. In this manuscript, we present findings suggesting that the tumor suppressor BRCA1 can interact with the PR and regulate PR action in cultured mammary epithelial cells and in an animal model.
Results
BRCA1 Inhibits Endogenous PR Activity
We used a mouse mammary tumor virus (MMTV)-Luc reporter to examine the effect of an exogenous wild-type BRCA1 gene (wtBRCA1) on PR activity. This reporter contains a degenerate glucocorticoid response element that is activated by several different types of steroid hormones (progesterone, androgens, and glucocorticoids), but is not activated to any significant extent by estrogen (20). Briefly, cells were cotransfected overnight with MMTV-Luc reporter and with either wtBRCA1, empty pcDNA3 vector, or no vector, treated ± progesterone (P, 100 nM) for T = 24 h, and assayed for luciferase activity. After subtracting the blank, the luciferase values were expressed relative to negative control conditions (MMTV-Luc reporter only, no progesterone).
Figure 1A shows the response of T47D human breast cancer cells, which are PR positive and progesterone responsive (21). Progesterone stimulated reporter activity by about 20- to 150-fold in different experiments, and the empty pcDNA3 vector had little or no effect on progesterone-stimulated reporter activity. In contrast to the pcDNA3 vector, wtBRCA1 strongly inhibited progesterone-stimulated PR activity. We could not detect any effect of wtBRCA1 on basal PR activity, but the basal luciferase activity was very close to the blank reading in the absence of progesterone. Similar results were obtained using MCF-7, an estrogen-responsive human breast cancer cell type that also contains PR, but in smaller quantities than T47D (Fig. 1B). In different experiments, the fold-stimulation of PR activity by progesterone in MCF-7 cells was about 5- to 10-fold, in all cases significantly less than in T47D cells.
Fig. 1. Overexpression of BRCA1 Inhibits PR Activity.
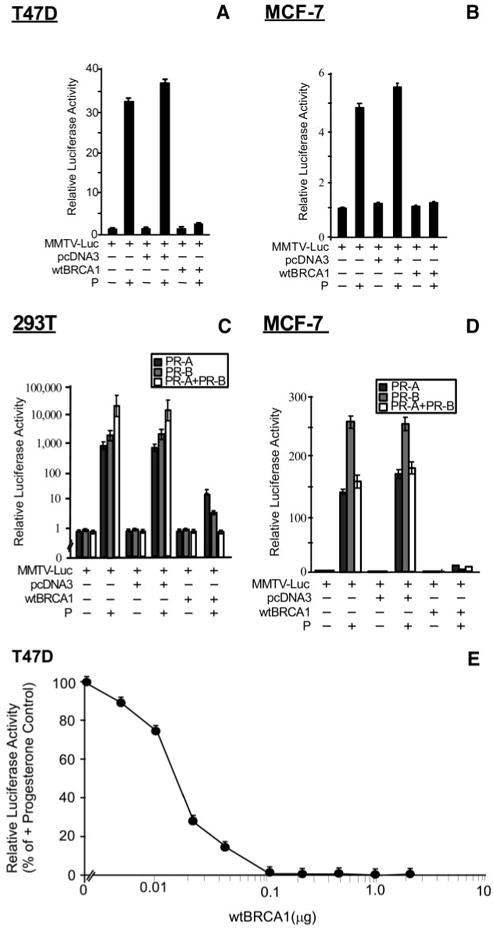
Subconfluent proliferating cells in 24-well dishes were transiently transfected overnight with the MMTV-Luc reporter plus the indicated expression vectors (0.25 μg per well for each transfected vector; see Materials and Methods), washed, postincubated with progesterone (100 nM for 24 h) in medium containing 5% CSS, and harvested for luciferase assays. Panels A and B show assays of T47D (A) and MCF-7 (B) human breast cancer cells. Panels C and D show assays of 293T human embryonal kidney (C) and MCF-7 (D) cells transiently transfected with expression vectors for PR-A, PR-B, or both. Luciferase values are expressed relative to control cells transfected with the MMTV-Luc reporter only and are means ± SEMs of quadruplicate wells. In each case, wtBRCA1 significantly inhibited progesterone-stimulated PR activity relative to the empty pcDNA3 vector or no vector controls (P < 0.001; two-tailed t test). Each experiment was performed at least twice to assure reproducibility of the findings. Panel E shows the dose dependence of wtBRCA1-mediated inhibition of PR activity in T47D cells. Assays were performed as described above, except using different quantities of the wtBRCA1 expression plasmid. The total transfected DNA content was equalized in all assays by the addition of the empty pcDNA3 plasmid. The luciferase values plotted are expressed relative to the + progesterone positive control (i.e. 0 wtBRCA1 plasmid, 100 nM progesterone). P, Progesterone.
BRCA1 Inhibits Exogenous PR-A and PR-B Isoforms
T47D and MCF-7 cells express both isoforms of PR, PR-A and PR-B, so it was not possible to determine from Fig. 1, A and B, whether wtBRCA1 has differential effects on PR-A vs. PR-B activity. To independently evaluate the effects of BRCA1 on PR-A vs. PR-B activity, we performed a third experiment using 293T embryonal kidney cells (which are PR negative) cotransfected with MMTV-Luc and either PR-A and/or PR-B expression vectors. In this experiment, progesterone greatly stimulated MMTV-Luc reporter activity in cells transfected with PR-A and/or PR-B, indicating that each PR isoform could activate the MMTV-Luc reporter (Fig. 1C). Consistently, we found that wtBRCA1 blocked the progesterone-stimulated activity of PR-A alone, PR-B alone, or a combination of PR-A plus PR-B. Here, the combination of PR-A plus PR-B yielded a higher total level of progesterone-stimulated MMTV-Luc activity than did either isoform by itself, but net activity of the combination of the two isoforms was abrogated by wtBRCA1.
We performed a similar experiment using MCF-7 cells, which express low levels of endogenous PR. Here, exogenous PR-A or PR-B caused a marked increase in progesterone-stimulated reporter activity (compare the fold-stimulation in Fig. 1D vs. Fig. 1B). Interestingly, in MCF-7 cells, the combination of PR-A and PR-B gave a net progesterone-stimulated MMTV-Luc activity similar to that of PR-A alone and less than that of PR-B alone. Again, wtBRCA1 strongly inhibited the progesterone-stimulated activity of the exogenous PR-A and/or PR-B (Fig. 1D). These findings indicate that wtBRCA1 effectively inhibits the ability of either or both isoforms of PR to activate a progesterone-responsive reporter.
Dose-Dependent Inhibition of PR Activity by BRCA1
Next, we performed wtBRCA1 plasmid dose-response studies to evaluate the dose dependence of the wtBRCA1-mediated inhibition of endogenous PR activity in T47D cells. These studies revealed that a wtBRCA1 plasmid dose of 5 ng was sufficient to give a detectable reduction (∼10%) of progesterone-stimulated PR activity, whereas 50% inhibition of PR activity required about 15 ng of wtBRCA1 plasmid (Fig. 1E). A wtBRCA1 plasmid dose of 100 ng per well or more gave close to 100% inhibition of progesterone-stimulated PR activity. The dose of wtBRCA1 plasmid used to repress PR activity in most experiments was 0.25 μg (= 250 ng) per well, which should be sufficient to give maximal inhibition.
Cancer-Associated Mutant BRCA1 Does Not Inhibit PR Activity
T300G is a breast cancer-associated mutation of BRCA1 that encodes a full-length protein with a single amino acid substitution (61Cys → Gly) that disrupts the N-terminal RING domain. Previous studies revealed that the T300G mutation abrogates the ability of BRCA1 to repress ER-α activity (8). Here, we found that in contrast to wtBRCA1, an expression vector encoding BRCA1-T300G had little or no effect on progesterone-stimulated PR activity (Fig. 2A). Western blotting of T47D cells transfected with wtBRCA1, BRCA1-T300G, or the empty pcDNA3 vector revealed that the BRCA1-T300G protein was expressed to a similar degree as the wtBRCA1 protein (Fig. 2B). Neither the empty pcDNA3 vector nor treatment with progesterone had any obvious effect on BRCA1 protein levels. These findings indicate that a functionally defective BRCA1 protein that is well expressed fails to block PR signaling.
Fig. 2. Cancer-Associated Point Mutant BRCA1 Fails to Inhibit PR Activity.
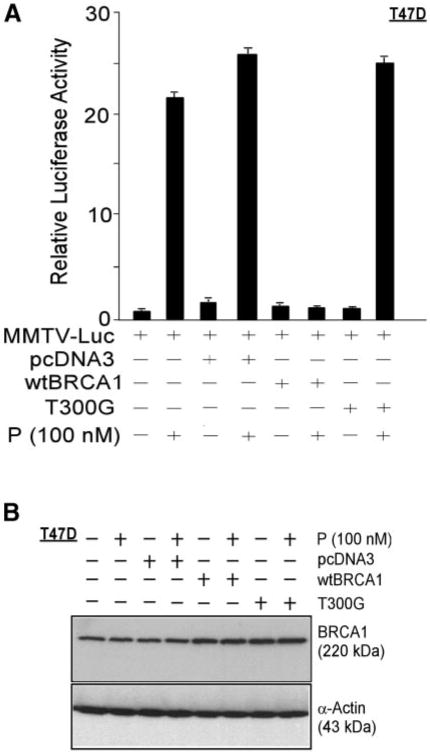
Luciferase assays were performed as described in the legend to Fig. 1, using either empty pcDNA3 vector, wtBRCA1, or a cancer-associated full-length point mutant BRCA1 [T300G (= 61Cys → Gly)] with a mutation that disrupts the N-terminal RING domain. The mutant BRCA1 failed to inhibit progesterone-stimulated PR activity, and the measured progesterone-stimulated PR activity values were significantly higher for the T300G mutant than for wtBRCA1 (P < 0.001; two-tailed t test) (panel A). Panel B shows the expression of BRCA1 in T47D cells transfected overnight (as above) with pcDNA3 or wtBRCA1 (5 μg of DNA per well in six-well dishes), postincubated for 24 h in medium containing 5% CSS without or with progesterone (100 nM), and harvested for Western blotting to detect BRCA1 or α-actin (control for loading and transfer). P, Progesterone.
Knockdown of Endogenous BRCA1 Stimulates PR Activity
We used a previously validated BRCA1 small interfering RNA (siRNA) (22, 23) to determine whether the endogenous BRCA1 protein can also regulate PR activity. Treatment with BRCA1-siRNA reduces BRCA1 protein levels to less than 25% of control by 72 h. The knockdown of BRCA1 protein levels by BRCA1-siRNA in T47D cells is illustrated in Fig. 3A. A scrambled-sequence control siRNA had no effect on BRCA1 protein levels (data not shown). T47D cells were pre-treated with BRCA1 vs. control (scrambled sequence) siRNA (100 nM × 72 h) and then assayed for PR activity, as in the previous experiments. Whereas the control siRNA had little or no effect on progesterone-stimulated PR activity, the BRCA1-siRNA caused a significant (>4-fold) increase in progesterone-stimulated PR activity (P < 0.001, two-tailed t test) (see Fig. 3B). The BRCA1-siRNA also partially reversed the wtBRCA1-mediated inhibition of PR activity (P < 0.001). However, the BRCA1-siRNA did not cause ligand-independent activation of PR.
Fig. 3. Knockdown of BRCA1 Using siRNA Stimulates PR Activity.
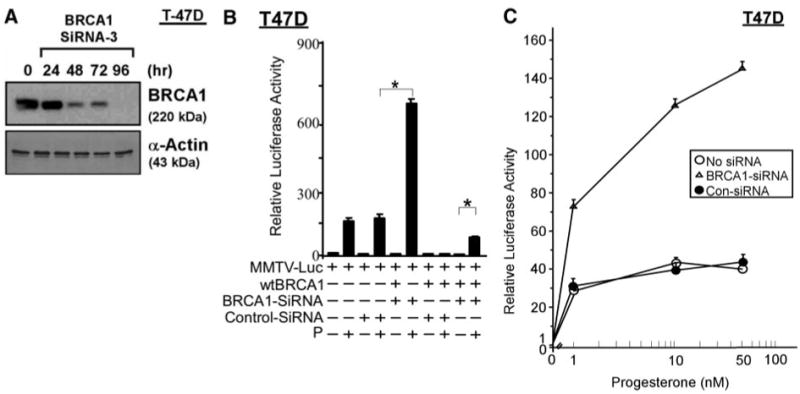
A, Western blot showing loss of BRCA1 protein in BRCA1-siRNA-treated T47D cells. Subconfluent proliferating T47D cells were treated with BRCA1-siRNA (100 nM) for the indicated time intervals (as described in Materials and Methods), harvested, and subjected to Western blotting to detect BRCA1 and α-actin (control for loading and transfer). B and C, BRCA1-siRNA confers enhanced progesterone-stimulated PR activity. Subconfluent proliferating T47D cells in 24-well dishes were pretreated with BRCA1-siRNA or control-siRNA (100 nM for 72 h), and then transfected overnight with the MMTV-Luc reporter ± wtBRCA1 and assayed for PR activity using 100 nM progesterone, as described in the legend to Fig. 1. Luciferase values are expressed relative to the no progesterone negative control and are means ± SEMs of quadruplicate wells. Asterisk above brackets indicates statistically significant differences in progesterone-stimulated PR activity (P < 0.001; two-tailed t test). A second experiment gave similar results. Panel C shows data that were similarly obtained, except using progesterone doses of 1, 10, or 50 nM. At each of these doses, PR activity was significantly higher in the presence of BRCA1-siRNA than in the presence of control-siRNA or no siRNA (vehicle only) (P < 0.001). P, Progesterone; Con, control.
Although the data in Fig. 3B were obtained utilizing a concentration of 100 nM of progesterone, we also observed a significantly increased PR activity in the presence of BRCA1-siRNA at lower concentrations of progesterone from 1–50 nM (P < 0.001) (Fig. 3C). These findings suggest that the endogenous BRCA1 protein can inhibit progesterone-stimulated PR activity over a wide range of progesterone concentrations. In conjunction with the data shown in Fig. 1E, they also suggest that BRCA1 regulates PR activity over a wide range of BRCA1 protein levels.
Physical Interaction between BRCA1 and PR
Because BRCA1 interacts directly with two other steroid hormone receptors [estrogen receptor-α (ER-α) and androgen receptor], we sought to determine whether BRCA1 can also interact with the progesterone receptor, and whether the interaction is isoform specific. We used IP-Western blotting, as described previously (8, 10), to determine whether the endogenous BRCA1 associates with the endogenous PR in vivo, in T47D cells, which express both PR-A and PR-B. These studies used subconfluent proliferating cells in standard growth medium to determine whether the endogenous BRCA1 and PR isoforms can be coprecipitated. The BRCA1 IP was performed using a combination of three BRCA1 monoclonal antibodies (see Materials and Methods). The BRCA1 IP coprecipitated BRCA1 with both PR-A (85 kDa) and PR-B (130 kDa), whereas the control (normal IgG) IP precipitated neither BRCA1 nor PR-A, nor PR-B (see Fig. 4A). As an additional control, an unprecipitated cell lysate is shown on the same Western blot. Conversely, an anti-PR antibody (but not the control antibody) precipitated PR-A, PR-B, and BRCA1 (Fig. 4B). These findings suggest that BRCA1 associates with both PR-A and PR-B under standard cell growth conditions in medium containing 5% fetal calf serum (which may contain small quantities of various steroid hormones, including progesterone).
Fig. 4. Association of Endogenous BRCA1 and PR Isoforms in Vivo in T47D Cells.
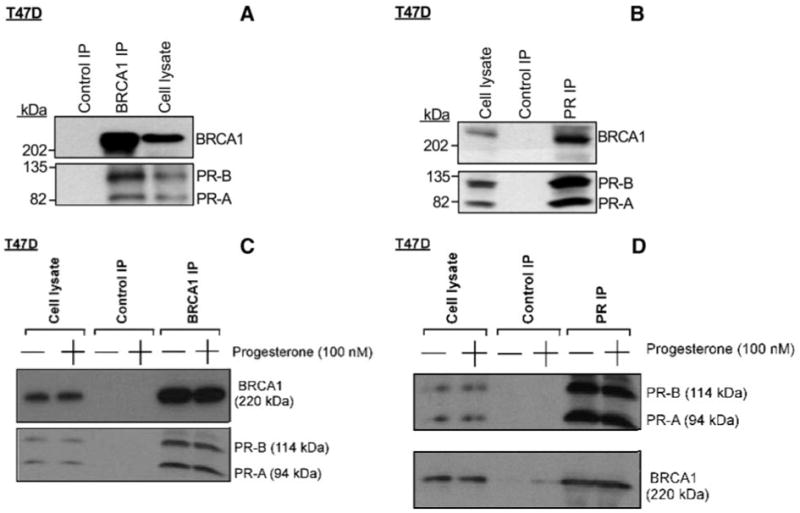
mcSubconfluent proliferating T47D cells in standard growth medium (DMEM plus 5% fetal calf serum) were harvested, subjected to anti-BRCA1 IP (A) or anti-PR IP (B), and Western blotted to detect BRCA1 and PR using an antibody that detects both PR-A and PR-B. As negative controls, IPs carried out using normal (nonimmune) IgG and nonprecipitated T47D cell lysates were Western blotted. Several repeat experiments gave similar results. To test the effect of progesterone on the BRCA1-PR interaction, T47D cells were placed in DMEM containing 5% CSS for several days and then incubated in fresh DMEM containing 5% CSS without (vehicle only) or with 100 nM progesterone for 24 h. The cells were then subjected to anti-BRCA1 IP (C) or anti-PR IP (D), as described above. For each experiment (A–D), the nonprecipitated cell lysate lanes contained 50 μg cell protein, whereas the IPs were performed using 1000 μg of cell protein.
To examine the effect of progesterone on the in vivo association between BRCA1 and PR, we performed a similar experiment, except that T47D cells were cultured in medium containing 5% charcoal-stripped serum (CSS) for several days and then incubated in fresh medium containing 5% CSS ± progesterone (100 nM) for 24 h, before harvesting for IP-Western blotting. As shown in Fig. 4C (BRCA1 IP) and Fig. 4D (PR IP), treatment with progesterone had little or no effect on the BRCA1-PR in vivo association. These findings suggest that the BRCA1-PR interaction is ligand independent.
Next we used glutathione-S-transferase (GST) capture assays to determine whether the BRCA1 and PR proteins directly interact in vitro. We used a set of GST-PR fusion proteins to pull down in vitro translated (IVT) full-length wtBRCA1. The domain structure of the different GST-PR proteins tested is depicted in Fig. 5A. The expression of the GST-PR proteins was determined by anti-GST Western blotting (Fig. 5B). Note that GST-PR 1–165 (which contains the AF-3 domain of PR-B) contains the portion of PR-B that is missing in the PR-A isoform. In this study, IVT wtBRCA1 was captured by GST-PR 166–456 and GST-PR 457–687, but not by the other two GST-PR proteins (Fig. 5C). As a negative control, GST alone failed to capture wtBRCA1. Consistent with the finding that BRCA1 inhibits the activity of both PR isoforms and that BRCA1 associates with either isoform in vivo, the in vitro interaction of BRCA1 and PR involved amino acids 166– 687, which are present in PR-A and PR-B. Several repeat experiments gave identical results. Each of these assays was performed in the absence of progesterone, suggesting that the BRCA1-PR physical interaction does not require presence of the ligand.
Fig. 5. Direct Interaction of BRCA1 and PR, Demonstrated by GST Capture Assays.
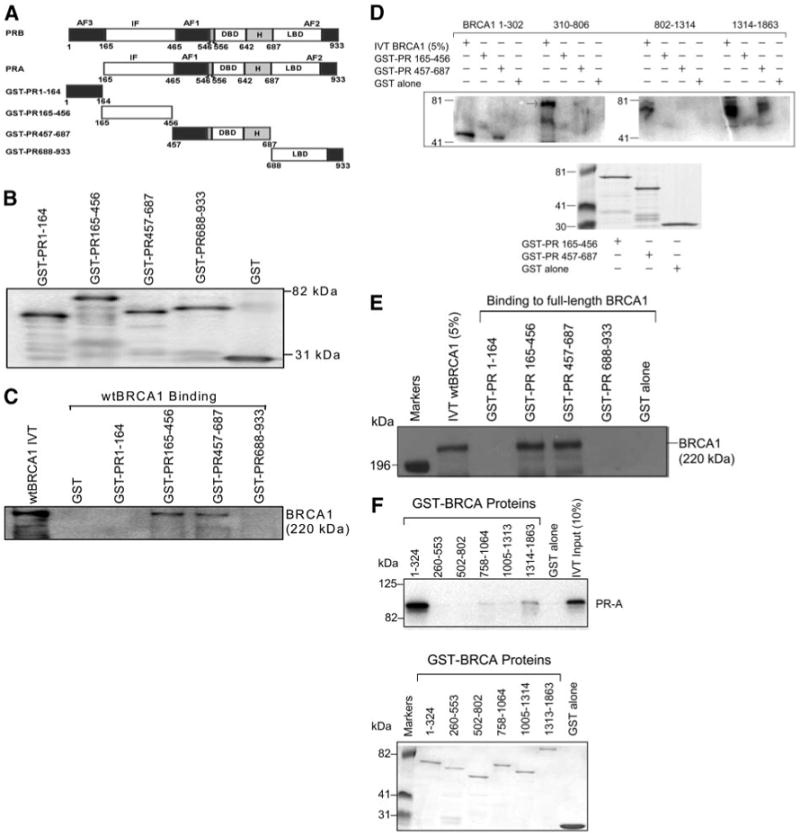
A, Schematic diagram of GST-PR proteins. The domain structure of the two PR isoforms, PR-A and PR-B, and that of the four GST-PR proteins used in this study are shown. DBD, DNA-binding domain; H, hinge region; IF, inhibitory function domain; LBD, ligand-binding domain. Diagrams were derived from Ref. 24. B, Expression of GST-PR proteins. The GST-PR proteins were expressed from E. coli transformed with the p-GEX vectors containing the different PR cDNAs. The chimeric GST-PR proteins were detected by Western blotting using an anti-GST antibody. C, Capture of wtBRCA1 by different GST-PR proteins. IVT [35S]methionine-labeled full-length wtBRCA1 was incubated with beads coated with different GST-PR proteins or GST alone (negative control). The captured proteins were eluted in Laemmli sample buffer and subjected to SDS-PAGE autoradiography. The input lane contains 10% of the quantity of IVT wtBRCA1 used for each GST capture assay. Several independent repeat experiments gave similar results. D, Capture of different IVT BRCA1 protein fragments by GST-PR proteins. The ability of two GST-PR proteins (GST-PR 165–456 and GST-PR 457–687) or GST alone (negative control) to capture four different IVT [35S]methionine-labeled BRCA1 protein fragments was tested (top panel). The bottom panel is a Coomassie blue-stained gel showing the expression of each GST-PR protein. Several independent repeat experiments gave similar results. E, Further demonstration of capture of IVT full-length wtBRCA1 by two distinct GST-PR proteins. GST capture experiments were performed as described in panel C except that nonradioactive IVT full-length BRCA1 was used and the captured BRCA1 was detected by Western blotting, using a specific anti-BRCA1 antibody (C-20). F, Capture of IVT full-length PR-A by different GST-BRCA1 proteins. A series of GST-BRCA1 protein fragments or GST alone (negative control) was used to capture IVT [35S]methionine-labeled PR-A (top). A Coomassie blue-stained gel showing the expression of each GST-BRCA1 protein is provided (bottom).
We next tested the ability of GST-PR 166–456 and GST-PR 457–687 to capture a series of different IVT BRCA1 protein fragments. Here, we found that GST-PR 457–687 reproducibly captured two BRCA1 fragments, one from the N terminus (amino acids 1–302) and one from the C terminus (amino acids 1314–1863) (Fig. 5D). We were unable to document reproducible capture of IVT BRCA1 proteins fragments by GST-PR 166–456. However, as noted above, the ability of GST-BRCA1 166–456 to capture full-length wtBRCA1 was noted in multiple experiments, including those performed using radiolabeled IVT wtBRCA1 or unlabeled wtBRCA1 detected using anti-BRCA1 antibody C-20 (Fig. 5, C and E, respectively). These suggest that this interaction may be confirmation sensitive. Finally, we tested the ability of a set of different GST-BRCA1 proteins to capture IVT full-length PR-A (because the in vivo and in vitro experiments indicate that amino acids 1–164, which are present only in PR-B, are not required for interaction with BRCA1). Here, we found that the N terminus (amino acids 1–324) and C terminus (amino acids 1314–1863) of BRCA1 capture PR-A (Fig. 5F). These findings are consistent with the finding that similar IVT BRCA1 protein fragments are captured by GST-PR 457–687. Differences in the binding of ER-α vs. PR to BRCA1 are considered in Discussion.
Collaboration of ER- α and PR in Stimulating PR Signaling
Recently, an interaction between the PR and ER-α that further stimulates PR activity was described (24). Here, we tested the ability of ER-α to stimulate PR activity and the ability of BRCA1 to repress the stimulation in T47D cells (Fig. 6A) and MCF-7 cells (Fig. 6B). Both T47D and MCF-7 cells are ER-α positive and estrogen responsive. Whereas estrogen [17β-estradiol (E2), 1 μM] by itself had little or no effect on MMTV-Luc reporter activity, E2 modestly but significantly enhanced the progesterone-stimulated PR activity, by about 30– 50% (P < 0.01, two-tailed t tests). Exogenous wtBRCA1 inhibited the (progesterone + E2)-stimulated PR activity (Fig. 6, A and B) as efficiently as it inhibited the progesterone-stimulated PR activity (Fig. 1, A and B). These findings suggest that in the presence of both E2 and progesterone, the E2-stimulated component of PR activity is as susceptible to inhibition by BRCA1 as the progesterone-stimulated component.
Fig. 6. Collaboration of ERs and PRs in Stimulation of PR Activity.
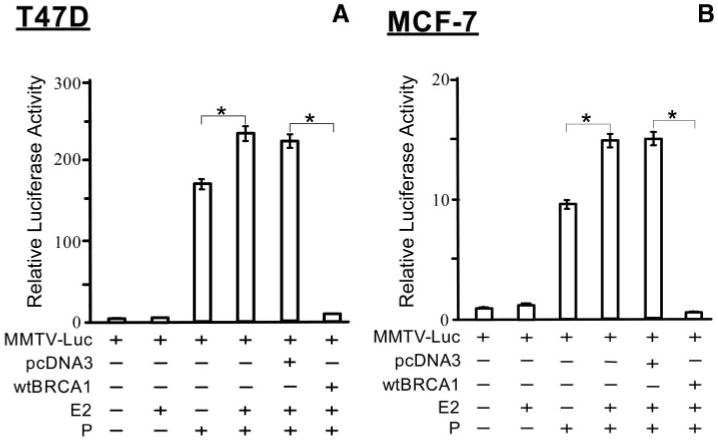
Subconfluent proliferating T47D cells (A) or MCF-7 cells (B) were transfected overnight with the MMTV-Luc reporter plasmid and with or without the wtBRCA1 or empty pcDNA3 plasmids. Assays were performed as described in the legend to Fig. 1, except that after transfection, the cells were treated with E2 (1 μM) and/or progesterone (100 nM) for 24 h, before harvesting for luciferase assays. Luciferase values are expressed relative to the negative control (0 E2, 0 progesterone) and are means ± SEMs of four replicate wells. Asterisks above brackets represent statistically significant differences (P < 0.01 to 0.001; two-tailed t test). P, Progesterone.
p300 Rescues BRCA1-Mediated Repression of ER-α But Not PR Activity
Previously, we showed that BRCA1 down-regulates the expression of the nuclear receptor coactivator p300 and that exogenous p300 rescues the BRCA1 repression of ER-α activity, through a mechanism that is distinct from the p300 coactivator function (10, 25). Here, we tested whether p300, which interacts with PR as well as BRCA1 and ER-α, could also rescue the BRCA1 repression of PR activity. As shown in Fig. 7A, p300 failed to rescue the wtBRCA1-mediated repression of progesterone (100 nM)-stimulated PR activity in T47D cells, as determined using the MMTV-Luc reporter to obtain a readout of PR activity. As a positive control, p300 did rescue the wtBRCA1 repression of estrogen (1 μM)-stimulated ER-α activity in T47D cells, as determined using the estrogen-response element-thymidine kinase luciferase (ERE-TK-Luc) reporter in assays performed at the same time. In this experiment, p300 enhanced the E2-stimulated activity in the absence of wtBRCA1, consistent with its activity as an ER-α coactivator. However, p300 did not stimulate the progesterone-inducible activity of the PR. A second experiment yielded similar results.
Fig. 7. The Nuclear Receptor Coactivator p300 Rescues BRCA1 Inhibition of ER-α But Not PR Activity.
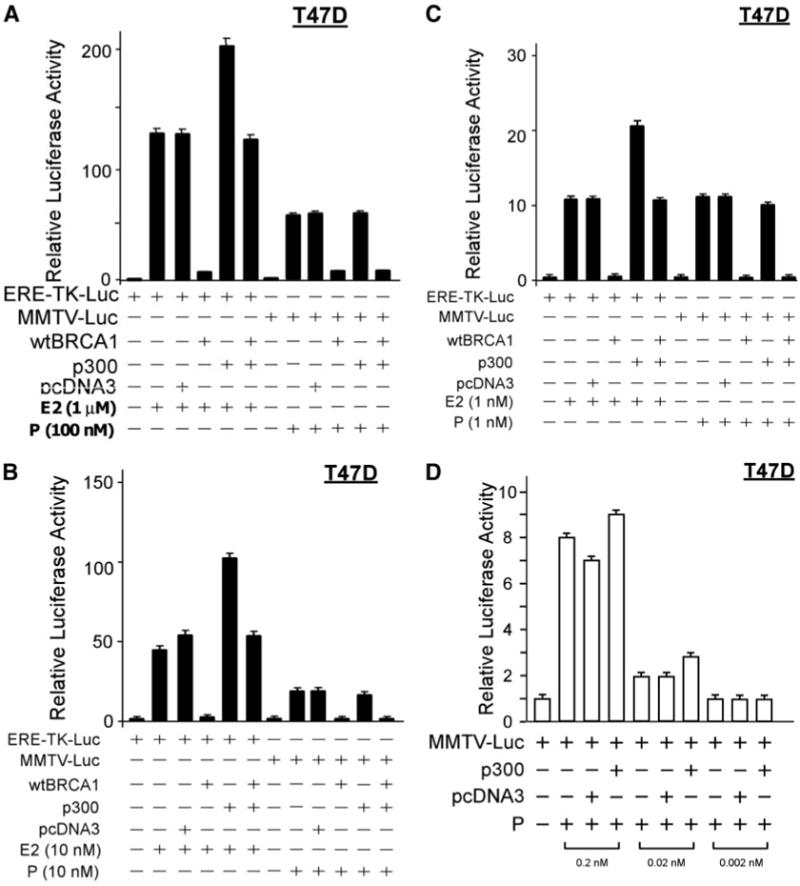
Subconfluent proliferating T47D cells were transfected overnight with either an estrogen (ERE-TK-Luc)- or progesterone (MMTV-Luc)-responsive reporter and cotransfected ± wtBRCA1 and ± wild-type p300 expression vectors, using the empty pcDNA3 vector as the control vector. The cells were then stimulated with the indicated doses of E2 or progesterone for 24 h, harvested, and assayed for luciferase activity. The luciferase values are expressed relative to the negative control (0 E2 or 0 progesterone) and are means ± SEMs of four replicate wells. Panels A–C show data for cells treated with 1 μM E2 or 100 nM progesterone (A), 10 nM E2 or 10 nM progesterone (B), and 1 nM E2 or 1 nM progesterone (C). Panel D shows data for cells treated with 0.2, 0.02, or 0.002 nM progesterone. P, Progesterone.
Because the effects of nuclear receptor cofactors may be dependent upon the concentration of the ligand, we performed additional experiments using lower concentrations of estrogen and progesterone. In studies using 10 nM or E2 and 10 nM of progesterone (Fig. 7B) or using 1 nM of E2 and 1 nM of progesterone (Fig. 7C), p300 still failed to rescue the wtBRCA1 repression of PR but continued to rescue the wtBRCA1-mediated repression of ER-α activity. In the absence of wtBRCA1, p300 stimulated ER-α but not PR activity at 10 and 1 nM of the respective ligands. However, at lower doses of progesterone (0.2 and 0.02 nM), we did observe a modest degree of coactivator activity of p300 (Fig. 7D). At the lowest dose of progesterone tested (0.002 nM), there was no ligand-inducible PR activity (Fig. 7D). These findings suggest that p300 lacks the ability to rescue BRCA1 repression of PR activity as it does ER-α activity.
BRCA1 Inhibits Progesterone-Inducible Gene Expression
Previous studies have identified a number of progesterone-regulated endogenous cellular genes, including progesterone-responsive genes regulated similarly by both PR isoforms, and genes differentially regulated by PR-A vs. PR-B (26). The studies described above indicate that BRCA1 regulates PR signaling through a heterologous and degenerate progesterone response element in the MMTV-Luc reporter. Here, we evaluated the ability of BRCA1 to regulate the expression of various endogenous progesterone-inducible genes. In this experiment, the expression of six known progesterone-responsive genes in T47D cells was measured using a validated method of rigorously controlled semiquantitative RT-PCR assays (22, 23, 27) to determine mRNA levels. All PCR reactions were individually optimized so that each reaction occurred within the linear range of product amplification. Glyceraldehyde-3-phosphate dehydrogenase (GAPDH), the expression of which was not affected by BRCA1 or progesterone, was used as a control gene.
T47D cells were transfected overnight with wtBRCA1 vs. empty pcDNA3 vector, treated ± progesterone (100 nM × 6 h), and tested for expression of the following progesterone-regulated genes (24): Bcl-XL (BCL2-like 1, long isoform), FKBP54 (54-kDa PR-associated immunophilin), STAT5A (signal transducer and activator of transcription 5A), HSD11β2 (hydroxysteroid 11-β dehydrogenase 2), HEF1 (human enhancer of filamentation 1), and VIL2 (villin 2, also called ezrin). For each of these genes, untransfected cells and cells transfected with pcDNA3 showed a significant progesterone-stimulated increase in mRNA levels, whereas cells transfected with wtBRCA1 showed little or no progesterone stimulation of these genes (Fig. 8A). Two control genes (GAPDH and β2-macroglobulin) were unaffected by progesterone or wtBRCA1. Two independent experiments showed very similar results. In addition, we have also shown that several low-abundance genes not known to be regulated by PR were not induced by progesterone or down-regulated by BRCA1 (Fig. 8B). These include: G1P2 (interferon-induced protein IFI-15K), OAS3 (oligoadenylate synthase 3), and stanniocalcin 1 (STC1). These findings confirm that BRCA1 regulation of PR activity is not limited to an artificial heterologous reporter (MMTV-Luc) but also applies to a series of endogenous progesterone-responsive genes.
Fig. 8. BRCA1 Inhibits Progesterone-Induced Gene Expression.
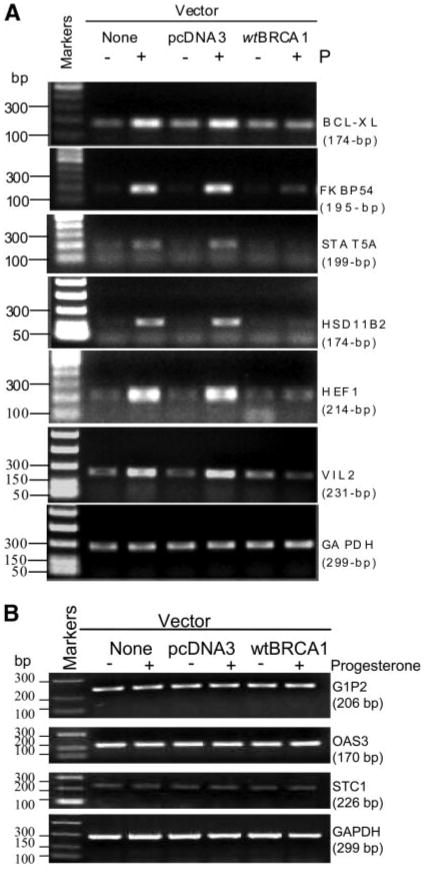
A, BRCA1 inhibits progesterone-stimulated expression of known PR-regulated genes. The experiment shown is representative of three independent experiments, all of which gave similar results. B, BRCA1 does not inhibit expression of several genes not known to be PR regulated. Methodology: subconfluent proliferating T47D cells were placed in phenol red-free DMEM containing 2% CSS for 48 h. The cells were then transfected overnight with wtBRCA1, empty vector (pcDNA3), or no vector (vehicle only), using Lipofectamine 2000 (Invitrogen, Carlsbad, CA), washed, and postincubated for 24 h at 37 C to allow gene expression. The cells were then treated ± progesterone (100 ng/ml for 6 h) in phenol-free DMEM containing 2% CSS. The cells were harvested, and rigorously controlled semiquantitative RT-PCR was carried out, as described in Materials and Methods. Each reaction was optimized so that all reactions fell within the linear range of product amplification. GAPDH was used as a control gene. BCL-XL, BCL2-like 1, long isoform; FKBP54, 54-kDa progesterone receptor-associated immunophilin; G1P2, interferon-inducible protein IFI-15K; GAPDH, glyceraldehyde-3-phosphate dehydrogenase; HEF1, human enhancer of filamentation 1; HSD11B2, hydroxysteroid (11-β) dehydrogenase 2; OAS3, oligoadenylate synthetase 3; STAT5A, signal transducer and activator of transcription 5A; STC1, stanniocalcin 1; VIL2, villin 2 (also called ezrin); P, progesterone.
BRCA1 Inhibits Progesterone-Stimulated Cell Proliferation in T47D Cells
To assess the effect of BRCA1 on progesterone-stimulated cell proliferation, T47D cells were transfected overnight with wtBRCA1 or empty pcDNA3 vector (control), washed, allowed to recover from transfection for several hours, seeded into six-well dishes at 104 cells per well in 5% CSS on d 0, and allowed to attach to the culture dish and grow for 24 h. On d 1, the medium was replaced with 2% CSS ± progesterone (100 nM), and triplicate wells were counted each day on d 1 to d 4. In the absence of progesterone, there was some residual growth in 2% CSS, which was observed in both wtBRCA1 and pcDNA3-transfected cells (Fig. 9A). In the presence of progesterone, the control (pcDNA3)-transfected cells showed significantly higher cell counts than the wtBRCA1-transfected cells on d 4 (P < 0.01, two-tailed t test). The results shown in Fig. 9A are the averages of three independent experiments, each of which showed significantly higher cell counts in pcDNA3-transfected/progesterone-treated cells than in wtBRCA1-transfected/progesterone-treated cells on d 4.
Fig. 9. BRCA1 Inhibits Progesterone-Stimulated Proliferation of T47D Cells.
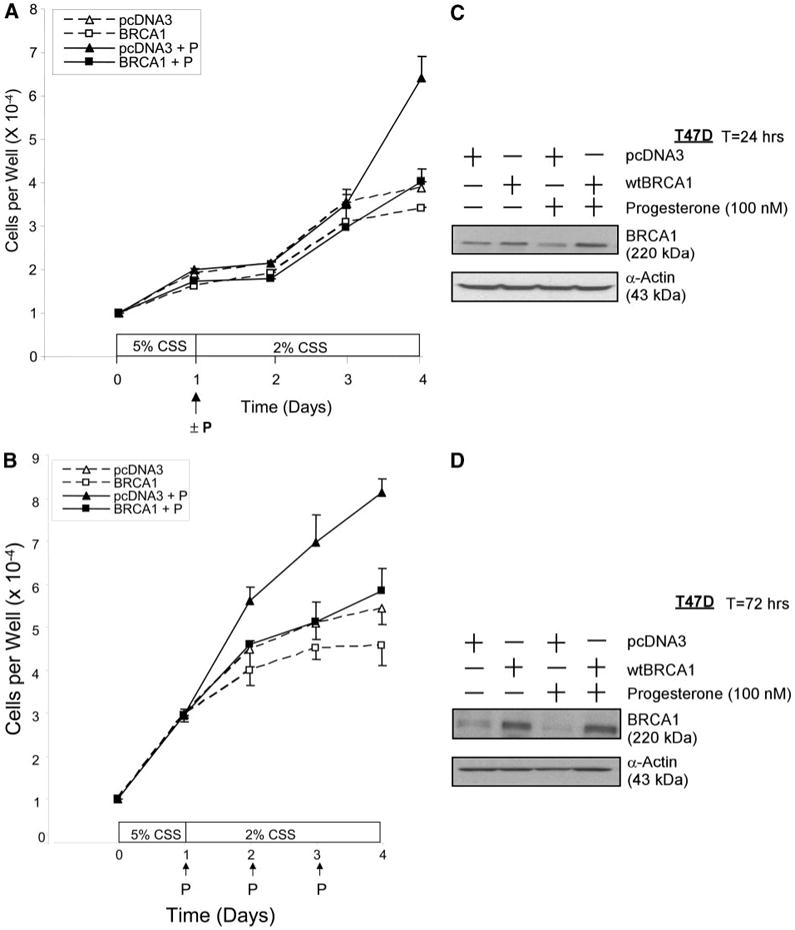
In panel A, T47D cells were transfected overnight with wtBRCA1 or empty pcDNA3 vector (control), washed, allowed to recover from transfection for several hours, seeded into six-well dishes (104 cells per well) in 5% CSS (d 0), and allowed to attach and grow for 24 h. At this time (d 1), the medium was replaced with 2% CSS ± progesterone (100 nM). Triplicate wells were counted daily on d 1–d 4. Values plotted are means ± SEMs of the mean cell counts from three independent experiments. For each individual experiment, cell counts were determined based on n = 3 replicate wells per experimental condition and time point. Cell counts on d 4 were significantly higher in (pcDNA3 + progesterone)-treated cells than in (wtBRCA1 + progesterone)-treated cells (P < 0.01; two-tailed t test). In panel B, the experiments were performed the same way as in panel A, except that the medium was replaced daily with fresh medium ± fresh progesterone (100 nM) starting on d 1. Again, the values plotted are means ± SEMs of three independent experiments. Panels C and D show the expression of BRCA1 in T47D cells transfected with pcDNA3 or wtBRCA1 (5 μg per well in six-well dishes), postincubated for 24 h (C) or 72 h (D) in medium containing CSS without or with progesterone (100 nM), and harvested for Western blotting. P, Progesterone; T, time.
Because progesterone may be metabolized rapidly, we performed a similar series of growth experiments in which the progesterone was replenished every day. Figure 9B shows the average of three independent experiments, each of which yielded similar results. Here, the pcDNA3-transfected/progesterone-treated cells showed significantly higher cell counts than the wtBRCA1-transfected/progesterone-treated cells on d4(P < 0.01) as well asd3(P < 0.04). These findings suggest that BRCA1 inhibits progesterone-stimulated proliferation of T47D cells.
Progesterone Stimulates Mammary Growth in Brca1-Deficient Mice
To study the interaction of progesterone and Brca1 in vivo, we used a well-characterized mouse model featuring a conditional (Co) mammary-targeted deletion of Brca1 exon 11 (Refs. 28–30; reviewed in Refs. 31 and 32). This model employs Cre-mediated recombination of two floxed Brca1 exon 11 alleles, targeted to the mammary epithelium via the MMTV-long terminal repeat promoter. Exon 11 codes for about 60% of the Brca1 protein, and the 90-kDa Brca1 Δ exon 11 protein resulting from the deletion is functionally defective. Brca1Co/CoMMTV-Cre mice exhibit a low incidence of mammary tumorigenesis (0 to <25% by age 12–13 months) that is significantly increased (37–75% by age 1 yr) in the setting of a heterozygous p53 deletion (p53+/− genotype) (28, 32). The mammary adenocarcinomas that develop in the Brca1Co/CoMMTV-Cre mice exhibit genetic changes similar to those found in human BRCA1 mutation-related breast cancers (29, 30, 32). In our hands, whole mounts of Brca1-deficient mice [age 6 wk (midpuberty)] consistently showed altered mammary morphology, characterized by increased numbers of terminal end buds and increased mammary growth, as compared with wild-type (Brca1+/+) nontransgenic mice (data not shown). This finding suggests that Brca1-deficient mammary glands are more susceptible to the proliferative action of endogenous hormones during development than are Brca1-competent glands.
To test the effect of Brca1 on the response of the mammary gland to progesterone, two studies were performed. In the first study, adult virgin female Brca1Co/CoMMTV-Cre/p53+/+ mice or wild-type nontransgenic mice with intact ovaries at about 1 yr of age were implanted with a progesterone pellet (10 mg/60-d slow release) or a placebo pellet. In the second study, female Brca1Co/CoMMTV-Cre/p53+/+ mice and wild-type nontransgenic mice about 1 yr of age were ovariectomized, allowed to recover, and then treated with either a placebo pellet, an estrogen pellet (0.72 mg/60-d slow release), or a combination of progesterone (10 mg/60-d slow release) and estrogen (0.72 mg/60-d slow release) pellets. In each study, at 4 wk after pellet administration, the mice were weighed, euthanized, and necropsied, and the mammary glands were removed. We studied the mammary gland responses in Brca1Co/CoMMTV-Cre mice with wild-type p53 (p53+/+), to avoid having to deal with the variable of p53 gene status.
In the first study (mice with intact ovaries), there was a significant increase in mammary gland volume in mice with a conditional deletion of Brca1 exon 11 targeted to mammary epithelial cells (n = 5) as compared with wild-type control mice (n = 3) exposed to progesterone (P < 0.02; t test) (Fig. 10). There was no significant difference in mammary gland volume between placebo-exposed Brca1Co/CoMMTV-Cre and wild-type mice. Progesterone exposure was associated with a modest increase in mammary gland volume in the wild-type progesterone-exposed group as compared with the wild-type placebo-exposed group, but the difference was not statistically significant (P > 0.2). There were no statistically significant differences in the mean weights of the mice among the four different groups. A significant increase in the quantified tertiary branching density was found in the intact Brca1Co/CoMMTV-Cre mice as compared with wild-type control mice exposed to progesterone (P < 0.04; t test) (see Fig. 11 legend). There was no significant increase in the density of secondary branching structures (data not shown). In addition, there was no significant difference in the density of tertiary branching of placebo-exposed Brca1Co/CoMMTV-Cre vs. wild-type intact mice. All of the treatment groups demonstrated similar expression of PR (illustrated in Fig. 11, E–H, insets).
Fig. 10. Progesterone Exposure Stimulates Mammary Growth in Brca1-Deficient Mice with Intact Ovaries.
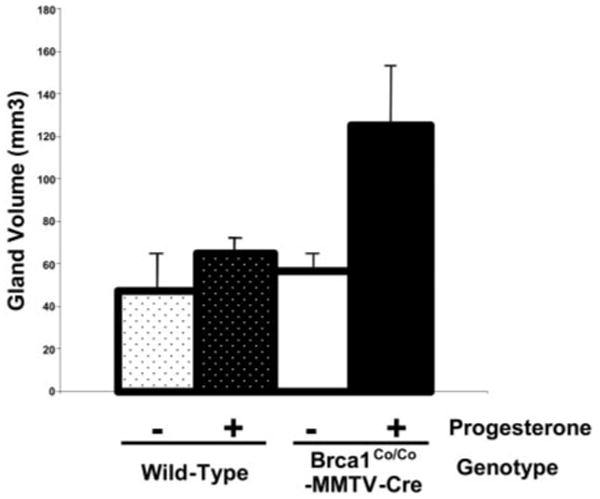
mcGroups of postpubertal female Brca1Co/CoMMTV-Cre with normal levels of p53 (p53+/+) and wild-type nontransgenic mice were anesthetized and implanted sc with a 10-mg 60-d constant release progesterone pellet or a placebo pellet. Four weeks after pellet administration, the mice were euthanized and mammary glands were removed. Total mammary gland volume were measured and compared in the Brca1Co/CoMMTV-Cre and wild-type mice given either progesterone or placebo. Progesterone was associated with a statistically significant increase in mammary gland volume in the Brca1Co/CoMMTV-Cre (P < 0.02; t test) but not in the wild-type mice. Mammary gland volume was calculated after measuring length, width, and depth of dissected fourth mammary gland. See Materials and Methods for details.
Fig. 11. Effect of Progesterone on Mammary Gland Morphology in Brca1-Deficient Mice with Intact Ovaries.
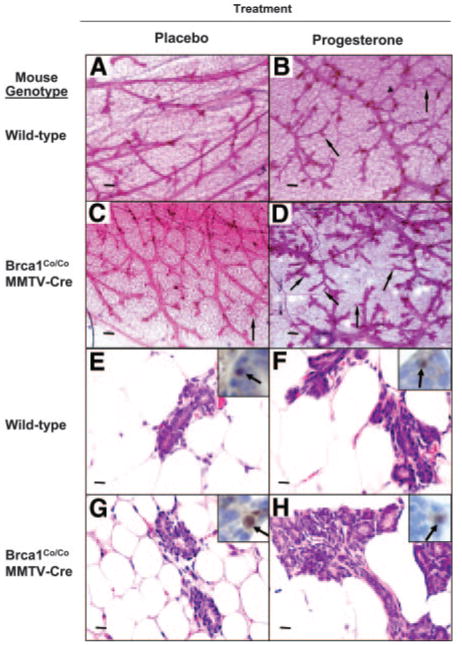
Whole mounts of the inguinal (no. 4) mammary glands from wild-type or Brca1Co/CoMMTV-Cre mice were prepared 4 wk after administration of a placebo pellet (A and C) or a progesterone pellet (10 mg/60-d release) (B and D). The density of tertiary branching was increased in the mammary glands of Brca1Co/CoMMTV-Cre mice treated with exogenous progesterone (thin arrows; see D compared with A–C). Magnification, ×4; scale bars, 200 μm. The quantitative values of density of tertiary branching (units of tertiary branches per mm2; see Materials and Methods section) were as follows: wild-type, placebo 44 ± 14; wild-type, progesterone 40 ± 8; Brca1Co/CoMMTV-Cre, placebo 98 ± 73; Brca1Co/CoMMTVCre, progesterone 172 ± 49. H & E-stained histological sections (magnification, ×40; scale bars = 20 μm) and immunohistochemical detection of nuclear localized PR (insets) are illustrated in panels E–H.
In ovariectomized mice, the quantified mammary epithelial cell density was significantly increased in the Brca1Co/CoMMTV-Cre mice in both the estrogen (P < 0.005; t test) and estrogen plus progesterone (P < 0.004; t test) treatment groups, as compared with the respective ovariectomized wild-type mouse treatment groups (see Fig. 12 legend and illustrations). In ovariectomized Brca1Co/CoMMTV-Cre mice, mammary epithelial cell density was significantly higher in mice that received the combination of estrogen and progesterone as compared with those treated with estrogen alone (P < 0.002; t test). More extensive dense alveolar-like growth was found in the estrogen plus progesterone treated Brca1Co/CoMMTV-Cre female mice (Fig. 12, F and L, thick arrows), whereas wild-type mice treated with estrogen plus progesterone developed more normal appearing alveolar-like structures (Fig. 12, C and I, thin arrows). Estrogen treatment alone resulted in a statistically significant increase in mammary epithelial cell density as compared with placebo treatment (P < 0.02; t test) in Brca1Co/CoMMTV-Cre female mice. In contrast, ovariectomized wild-type mice demonstrated no significant difference in mammary epithelial cell density after treatment with either estrogen or the combination of estrogen and progesterone. These findings indicate that a deficiency of Brca1 confers an exaggerated progesterone-induced growth response in the mammary glands of both intact and ovariectomized adult female mice.
Fig. 12. Effect of Hormonal Treatments on the Mammary Glands of Ovariectomized Brca1-Deficient Mice.
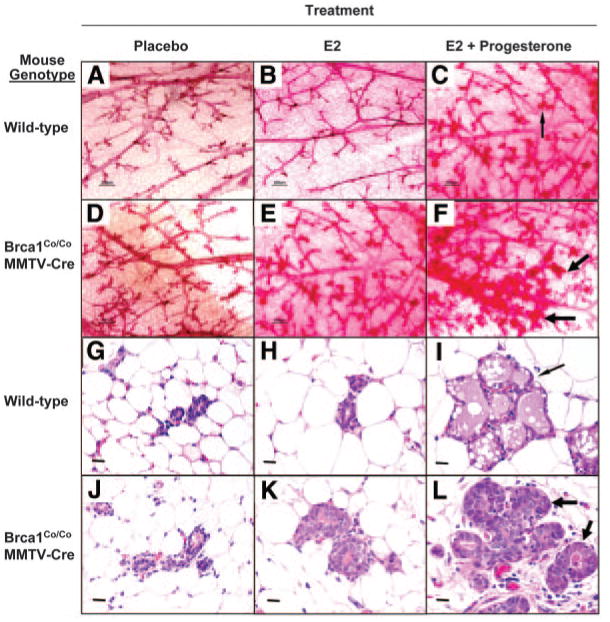
Whole mounts were prepared from the inguinal (no. 4) mammary glands of ovariectomized placebo control (A and D), E2-treated (B and E), and E2 plus progesterone-treated (C and F) wild-type and Brca1Co/CoMMTV-Cre female mice. Dense alveolar-like growth was observed in the Brca1Co/CoMMTV-Cre female mice that were exposed to estrogen plus progesterone (thick arrows, F). Magnification, ×4; scale bars, 200 μm. Representative H & E-stained sections of inguinal (no. 4) mammary glands of ovariectomized mice are shown for placebo control (G and J), E2-treated (H and K), and E2 plus progesterone-treated (I and L) wild-type and Brca1Co/CoMMTV-Cre female mice. These sections illustrate the increased density of mammary epithelial cells (MECs) in the alveolar-like structures in the estrogen plus progesterone-treated Brca1Co/CoMMTV-Cre female mice (thick arrows, L). Magnification, ×40; scale bars = 20 μm. Quantitation of MEC density (number of MECs per ×40 field, see Materials and Methods section) yielded the following values: wild-type ovariectomized (OVX) placebo control, 81 ± 9; wild-type OVX E2 treated, 66 ± 13; wild-type OVX E2 plus progesterone-treated, 136 ± 21; Brca1Co/CoMMTV-Cre OVX placebo control, 91 ± 5; Brca1Co/CoMMTV-Cre OVX E2 treated, 149 ± 13; Brca1Co/CoMMTV-Cre OVX E2 plus progesterone treated, 273 ± 17.
Discussion
We showed that wtBRCA1 inhibits the activity of the endogenous PR in PR-positive cells and of the exogenous PR-A and PR-B isoforms in PR-negative cells. Knockdown of endogenous BRCA1 by siRNA significantly enhanced progesterone-stimulated PR activity, suggesting that the endogenous BRCA1 protein inhibits PR activity. In contrast, a cancer-associated point mutant BRCA1 (T300G) failed to inhibit PR activity. wtBRCA1 blocked the progesterone-stimulated expression of various endogenous progesterone-responsive genes (eg., Bcl-XL, STAT5A, and others) but had no effect on the expression of several non-PR-regulated low-abundance genes. The BRCA1 inhibition of PR activity may be due, in part, to a direct interaction with PR. Thus, BRCA1 was found to associate with both PR-A and PR-B in vivo in T47D cells, and GST capture assays revealed that BRCA1 binds to at least two sites within the interior of the PR protein (amino acids 166–687), a region that is shared by PR-A and PR-B. At least two sites on BRCA1 were found to interact with PR, one within the N terminus (amino acids 1–302) and the other within the C terminus (amino acids 1314–1863). These interactions were demonstrated using bidirectional GST capture assays.
The BRCA1 binding site(s) on PR was located outside of the AF-2/ligand-binding domain region (amino acids 687–933). In contrast, BRCA1 binding to ER-α is mediated by the AF-2/ligand-binding domain region of ER-α (8). As for ER-α, BRCA1 association with and binding to PR did not require the presence of ligand. Our findings suggest that BRCA1 may function as a corepressor for both PR and ER-α. This hypothesis is supported by finding that BRCA1 fully represses PR activity in the presence of (E2 + progesterone), indicating that the E2-stimulated/ER-α mediated enhancement of progesterone-stimulated PR activity, which was described previously (24), is blocked by wtBRCA1. These studies do not establish the stoichiometry of the BRCA1-PR interaction, but the finding that BRCA1 can interact with two different fragments of PR (amino acids 166–456 and 457–687) suggests that it may interact with and inactivate the dimerized form of the PR, which is the active form of the PR that binds to the progesterone response element of target genes (33, 34). Consistent with this hypothesis, it was shown that Brca1 expression is increased in proliferating mammary epithelial cells undergoing differentiation during puberty and pregnancy (35–37).
In the case of ER-α, in MCF-7 cells, endogenous BRCA1 is present on the promoter of an ER-α target gene (e.g. pS2 or cathepsin D), and the addition of ligand (E2) causes an increase in the quantity of ER-α and a loss of BRCA1 from the promoter (12). Here, overexpression of BRCA1 may abrogate the loss of BRCA1 from E2-responsive promoters. The same might be true of the PR, although this remains to be documented. In any case, the pools of BRCA1 and PR at the promoters of progesterone-responsive genes are probably more important than the bulk BRCA1-PR complexes detected by IP. Because the vast majority of BRCA1 is localized in the nucleus, it is likely that BRCA1 does not prevent the initial activation of ER-α or PR but blocks their transcriptional activity within the nucleus.
Although BRCA1 repression of ER-α was dependent upon the transcriptional coactivator p300 (10), BRCA1 repression of PR occurred independently of p300, because unlike ER-α, repression of PR activity was not rescued by overexpression of p300. In the absence of exogenous wtBRCA1, p300 stimulated ER-α activity by up to 2-fold at different concentrations of E2. However, p300 had no effect on PR activity at higher doses of progesterone and only a modest effect at the lowest doses. We note here that the coactivator action of p300 is dependent upon nuclear receptor type, cell type, and the specific experimental context. In several prior studies documenting the ability of p300 or CREB-binding protein (CBP) to function as a coactivator for PR, the ability of p300/CBP to enhance PR transcriptional activity was reported in HeLa cells but not in T47D cells (38–40). Thus, it is likely that in our studies of T47D cells, the total levels of p300 and its functional homolog CBP were sufficient so that p300 was not limiting for PR activity. Taken together, our findings suggest that the mechanism of BRCA1-mediated repression of PR activity is not identical to that of BRCA1-mediated repression of ER-α activity.
Studies using a mouse model with a mammary-targeted Brca1 deficiency revealed that in comparison with wild-type (Brca1+/+) mice, virgin female adult mice with a mammary-targeted deletion of the full-length Brca1 isoform showed an exaggerated growth response to exogenous progesterone supplied through a slow-release pellet. The progesterone-induced changes in mammary gland morphology in Brca1-deficient mice with intact ovaries were somewhat similar to the physiological effects of endogenous progesterone stimulation during early pregnancy, which include growth of both the mammary gland fat pad and epithelial cell ductal tree as well as an increase in tertiary side branching (41, 42). Our findings suggest that a Brca1 deficiency blocks the normal restraint on mammary growth in virgin mice exposed to exogenous progesterone.
We further observed an exaggerated mammary epithelial cell growth response to the combination of estrogen and progesterone in the ovariectomized mice that was different from that found in mice with intact ovaries. In these mice, there was no significant growth of the mammary gland fat pad. However, mammary epithelial cell density was significantly increased, with the formation of lobular-alveolar structures similar to those formed during late pregnancy. The response of the ovariectomized wild-type mice to continuous estrogen treatment was predictable from previous studies. Thus, ovariectomized wild-type mice that were continuously treated with estrogen demonstrate a blunted growth response 4 wk after the initiation of treatment, even though transient increases in mammary epithelial cell proliferation were found at 1–3 d (43). In contrast, Brca1-deficient mice demonstrated a measurable and persistent growth response to estrogen alone with an even more profound growth effect due to the combination of estrogen and progesterone. In both experimental models, the findings suggest that Brca1 may function to limit the physiological growth response to progesterone.
A physiological role for Brca1 in mammary development has been suggested based on studies of the pattern of Brca1 expression during murine development. Thus, as noted above, Brca1 expression is increased in proliferating mammary epithelial cells undergoing differentiation during puberty and pregnancy (35–37). A role for BRCA1 in mammary differentiation is further suggested by the findings that BRCA1 expression is increased when cultured mammary epithelial cells are induced to differentiate in vitro by a hormonal cocktail and is further stimulated by exogenous wtBRCA1 (44, 45). Taken together, these findings suggest that the tumor suppressor activity of BRCA1 may be exerted at key windows of time (e.g. puberty and pregnancy), during which BRCA1 regulates mammary epithelial cell proliferation and promotes mammary differentiation. In this model of tumor suppression, BRCA1 plays a key role in regulating the responses to E2 and progesterone through their receptors.
A role for the PR in breast cancer etiology is suggested by several findings: 1) early first pregnancy confers a reduced risk for breast cancer in the general population; 2) epidemiological studies suggest that the addition of a progestational agent to estrogen in HRT adds a small but significant increment of risk for breast cancer (18, 19); and 3) animal model studies suggest a growth-stimulatory and mammary cancer-promoting role for the PR (reviewed in Refs. 41 and 42). Thus, studies using general and isoform-specific PR knockout mouse models indicate that: 1) the PR is required for pregnancy-associated mammary ductal side branching and lobuloalveolar development; and 2) knockout of the PR conferred resistance to carcinogen-induced mammary cancers (42). Moreover, mammary epithelial cell proliferation during the mouse estrus and human menstrual cycle requires progesterone stimulation (41), further implicating progesterone in stimulation of mammary epithelial cell proliferation.
Although pregnancy is associated with increased circulating progesterone, the greatly altered hormonal milieu makes it impossible to impute the reduced breast cancer risk associated with early first pregnancy in humans to progesterone alone. However, in rodents, a 3-wk exposure to a combination of (E2+progesterone), but not to either agent alone, promotes resistance to chemical carcinogen-induced mammary cancer (46). The mechanism of the resistance to carcinogenesis is unclear, but (E2+progesterone) appears to induce molecular alterations similar to pregnancy that affect the ability of mammary epithelial cells to undergo proliferation and differentiation in response to subsequent stimuli (46, 47). One of these protective alterations may be up- regulation of Brca1 expression in the mammary epithelial cells, although the cause and effect are not proven.
In this regard, epidemiological studies suggest that in women with BRCA1 mutations, pregnancy does not lower breast cancer risk (reviewed in Ref. 48). In fact, in this population of women, early first pregnancy may accelerate the development of breast cancer (46). This finding suggests that the restriction of ER-α and PR activity due to the endogenous wtBRCA1 alleles may contribute to the risk reduction caused by pregnancy. Here, we note that the hypothesis that BRCA1 mediates the breast cancer-suppressive effect of early pregnancy in humans or (E2+progesterone) treatment in rodents remains to be proven.
The hypothesis of a hormone (E2 and progesterone)/BRCA1 interaction is not supported by the findings of various studies that most BRCA1 mutant breast cancers are hormone receptor (ER-α and PR) negative (49, 50). Moreover, in the NSABP-P1 Tamoxifen Breast Cancer Prevention Trial, which admittedly had a relatively small number of BRCA mutation carriers, tamoxifen did not reduce the incidence of breast cancer in BRCA1 carriers. In contrast, tamoxifen significantly reduced the risk of BRCA2 mutant cancers, and it reduced the overall breast cancer risk by about 50% in women judged to be at high risk (51). However, there is clear evidence to support a hormone-BRCA1 interaction in human breast cancer [reviewed by Narod (48)]: 1) prophylactic oophorectomy results in a 50% risk reduction for breast cancer in BRCA1 mutation carriers (higher if performed before age 40) (52); 2) pregnancy increases the risk of very-early-onset breast cancer and of breast cancer in BRCA1 mutation carriers; and 3) smoking reduces the risk of BRCA1-associated breast cancer, possibly through an antiestrogenic action.
These apparently conflicting observations may be explained if the loss of ER-α and PR in BRCA1 mutant breast cancers were a late event, with hyperstimulation of mammary epithelial cell proliferation by E2 and progesterone occurring much earlier, due to the inactivation of BRCA1. The loss of ER-α and PR could be a later consequence of genomic instability in cells lacking functional BRCA1. Alternatively, the loss of BRCA1 could promote paracrine stimulation of growth of hormone receptor-negative epithelial cells by hormone receptor-positive cells. Such paracrine type mechanisms may occur during normal mammary development (42).
The hypothesis that loss of PR expression is a late event in BRCA1-dependent carcinogenesis is supported by the observation that PR expression is increased in nontumor mammary epithelial cells adjacent to BRCA1-mutant breast cancers, as compared with sporadic breast cancers (53). Finally, our finding that BRCA1-siRNA stimulates PR activity in breast cancer cells suggests the potential relevancy of a BRCA1-PR interaction to sporadic breast cancer development, because recent studies suggest that BRCA1 mRNA and protein expression is absent or significantly decreased in about 30–40% of sporadic breast cancers (54, 55). Here, the loss of BRCA1 expression may be due to epigenetic mechanisms (e.g. hypermethylation of the BRCA1 promoter) and/or loss of one of the two BRCA1 alleles in the tumor cells (56, 57).
In summary, we showed that BRCA1 interacts physically with the PR and inhibits its transcriptional activity. The interaction does not require progesterone and is not isoform specific. These findings may have implications for understanding the functional interaction between the BRCA1 gene and hormonal control of mammary development and tumorigenesis.
Materials and Methods
Cell Lines and Culture
Human breast cancer cells (T47D and MCF-7), which are PR positive and progesterone responsive, and human embryonal kidney epithelial cells (293T), which are PR negative, were originally obtained from the American Type Culture Collection (Manassas, VA) and cultured as described previously (22, 25). For routine subculture, the cells were grown in DMEM supplemented with fetal calf serum (5% vol/vol), L-glutamine (5 mM), nonessential amino acids (5 mM), penicillin (100 U/ml), and streptomycin (100 μg/ml). All cell culture media were obtained from BioWhittaker (Walkersville, MD).
Expression Vectors and Reporters
The wtBRCA1 expression vector was created by cloning a full-length BRCA1 cDNA into the pcDNA3 vector (Invitrogen, San Diego, CA) using artificially engineered 5′-HindIII and 3′-NotI sites (25). Expression vectors encoding the full-length PR isoform (PR-B) and its amino-terminally truncated major isoform (PR-A) were obtained from Dr. Anna Riegel (Department of Oncology, Georgetown University, Washington, DC). The MMTV-Luc reporter was obtained from Dr. Richard Pes-tell (Department of Oncology, Georgetown University). This reporter contains a degenerate glucocorticoid response element/progesterone response element that is responsive to progesterone, androgens, and glucocorticoids (through the PR, androgen receptor, and glucocorticoid receptor, respectively) but is not responsive to estrogen (20). The ERE-TK-Luc is an estrogen-responsive reporter plasmid consisting of the vitellogenin A2 ERE upstream of a minimal TK promoter and the luciferase gene (7, 8, 10).
siRNAs
Double-stranded siRNA to knock down endogenous BRCA1 protein levels (BRCA1-siRNA) and a scrambled-sequence (control) siRNA were chemically synthesized by Dharmacon, Inc. (Lafayette, CO). The BRCA1-siRNA has previously been experimentally validated (22, 23). The DNA sequences upon which these RNAs based are as follows: BRCA1-siRNA 5′-AATGCCAAAGTAGCTAATGTA-3′; and control-siRNA 5′-GTCACGATAAGACAATGATAT-3′. For BRCA1 knockdown experiments, subconfluent proliferating T47D cells cultured in 24-well dishes were treated with BRCA1-siRNA or control-siRNA (100 nM) using siPORT Amine transfection agent (Ambion, Inc., Austin, TX) as per the manufacturer's instructions. Previous studies have established that a 3-d exposure to BRCA1-siRNA causes reduction of BRCA1 protein levels to less than 25% of the control value, whereas the control siRNA has little or no effect on BRCA1 protein levels (22). Neither siRNA is toxic to cells under experimental conditions, as determined using MTT cell viability assays.
Assay of PR Activity
Subconfluent proliferating cells in 24-well dishes were incubated overnight with 0.25 μg of each indicated vector in serum-free DMEM containing Lipofectamine (Life Technologies, Gaithersburg, MD). The total transfected DNA was kept constant by addition of appropriate control vectors. The cells were washed, incubated in serum-free phenolphthalein-free DMEM containing 5% CSS (obtained from the Lombardi Comprehensive Cancer Center Tissue Culture Shared Resource) (0.2 ml per well) ± progesterone (100 nM) and/or E2 (1 μM) for 24 h, and harvested for luciferase assays. To control for transfection efficiency, plasmid pRSV-β-gal was cotransfected to allow normalization of luciferase values to β-galactosidase activity in the same sample. Luciferase values were usually expressed relative to the unstimulated control (0 progesterone) and represent means ± SEMs of four replicate wells. The results shown are representative of two or more independent experiments. Progesterone and E2 were purchased from Sigma Chemical Co. (St. Louis, MO).
IP
Subconfluent proliferating cells in 100-mm plastic Petri dishes were harvested, and nuclear extracts were prepared according to the method of Dignam (58). IPs were performed as described previously (8, 10). Each IP was carried out using 6 μg of antibody or antibody combination and 1000 μg of extracted protein. The precipitated proteins were collected using protein G beads, washed, collected in boiling Laemmli sample buffer, and subjected to Western blotting. The IP antibodies were as follows: antihuman BRCA1 mouse monoclonal antibody combination (Ab-1 + Ab-2 + Ab-3, Oncogene Research Products, Cambridge, MA); antihuman PR (C-19, catalog no. sc-538, Santa Cruz Biotechnology, Inc., Santa Cruz, CA); or an equal quantity of normal mouse IgG (control IP).
Western Blotting
Equal aliquots of total protein (50 μg per lane) were electro-phoresed on a 4–13% SDS-polyacrylamide gradient gel, transferred to nitrocellulose membranes (Millipore, Billerica, MA), and blotted using primary antibodies directed against the following: BRCA1 (C-20, rabbit polyclonal, Santa Cruz Biotechnology, 1:200 dilution), the human PR (AB-52, catalog no. sc-810, mouse monoclonal IgG1, Santa Cruz, 1:200), or α-actin (I-19, goat polyclonal, Santa Cruz, 1:500). Methodological details are provided elsewhere (8, 10). Proteins were visualized using the enhanced chemiluminescence system [Amersham Biosciences, Buckinghamshire, UK], with colored markers (Bio-Rad Laboratories, Hercules, CA) as molecular size standards.
GST Capture Assays
GST bead assays were performed essentially as described earlier (8, 10). [35S]Methionine-labeled BRCA1 proteins or PR-A were prepared by in vitro transcription (using the T7 promoter of the pcDNA3 vector) and translation. In vitro transcription and translation (IVT) and translation were carried out using the TNT-coupled rabbit reticulocyte lysate system (Promega Corp., Madison, WI), according to the manufacturer's instructions. A set of GST-PR fusion proteins were generated from cDNAs cloned into the p-GEX vector. [The GST-PR expression vectors were provided by Dr. Anna Riegel (Georgetown University).] GST-PR fusion proteins were expressed in Escherichia coli and purified by affinity chromatography. These proteins were visualized by Western blotting, using anti-GST mouse monoclonal antibody 27–4577-01 (Amersham Pharmacia Biotech, Arlington Heights, IL; 1:5000). The BRCA1 1–302, 310–806, 802–1314, and 1314–1863 were generated by PCR cloning, followed by BamH1 and EcoR1 double digestion and insertion into the BamH1 and EcoR1 site of pcDNA3 vector. Expression vectors for GST-BRCA1 protein fragments corresponding to BRCA1 amino acids 1–324, 260–553, 502–802, 758-1064, 1005–1313, and 1314–1863 were generously provided by Dr. Toru Ouchi (Ruttenberg Cancer Center, Mount Sinai School of Medicine, New York, NY). These constructs have been described earlier (59). [35S]Methionine-labeled BRCA1 proteins or PR-A were incubated with GST (negative control) or GST fusion proteins for 4 h at 4 C, recovered using glutathione agarose beads, eluted in boiling sample buffer, and analyzed by SDS-PAGE autoradiography. The GST bead assays were repeated in several independent experiments to assure reproducibility of the findings.
Semiquantitative RT-PCR Analysis
Progesterone-responsive mRNA expression was determined by rigorously controlled semiquantitative RT-PCR assays (8– 10, 22, 23). Briefly, RNA was extracted using the Tripure Isolation Reagent (Roche, Inc., Boulder, CO) followed by the DNase treatment using RQ1 RNase-Free DNase (Promega Corp.). cDNA synthesis carried out using an iScript cDNA Synthesis Kit (Bio-Rad, Hercules, CA), using 2 μg of total cellular RNA. One microliter of cDNA was used for each PCR reaction. All PCR reactions were carried out in a 25-μl reaction volume, containing 10 mM Tris·HCl (pH 8.0), 50 mM KCl, 1.5 mM MgCl2, 2.5 mM of each deoxynucleotide triphosphate, 400 nM of primers, and 0.625 U Taq DNA polymerase (Mirus Takara Biotechnology, Madison, WI). The thermal cycling conditions were as follows: denaturation at 94 C for 30 sec and annealing at 55 C for 30 sec, followed by extension at 72 C for 1 min for a total of 25–29 cycles. The PCR products (15 μl per lane) were resolved on ethidium bromide-stained 2% agarose gel, and the gels were photographed under UV illumination.
For each amplified product, the cycle numbers and PCR conditions were individually adjusted so that all reactions occurred within the linear range of product amplification. The forward and reverse PCR primer pairs (5′ to 3′ direction) and the expected product sizes are listed in Table 1. GAPDH was used as a control gene.
Table 1. Primers Used for Semiquantitative RT-PCR.
| Gene Name | Symbol | Primer Sequences Forward (F) and Reverse (R) | Product Size (bp) | Position in cDNA | PCR Cycles |
|---|---|---|---|---|---|
| BCL2-like 1, long isoform (Bcl-XL) | BCLX | F: 5′-ggctgggatacttttgtgga-3′ R: 5′-gggagggtagagtggatggt-3′ |
174 | 692–866 | 25 |
| 54-kDa Progesterone receptor-associated immunophilin | FKBP54 | F: 5′-tccctcgaatgcaactctct-3′ R: 5′-gccacatctctgcagtcaaa-3′ |
195 | 342–537 | 26 |
| Signal transducer and activator of transcription 5A | STAT5A | F: 5′-acatttgaggagctgcgact-3′ R: 5′-cctccagagacacctgcttc-3′ |
199 | 1078–1277 | 27 |
| Hydroxysteroid 11 β-dehydrogenase 2 | HSD11B2 | F: 5′-gacctgaccaaaccaggaga-3′ R: 5′-gccaaagaaattcacctcca-3′ |
174 | 544–718 | 27 |
| Human enhancer of filamentation 1 | HEF1 | F: 5′-tgggaatctgggaagcatag-3′ R: 5′-tctggtctctcttccctcca-3′ |
214 | 3116–3330 | 29 |
| Villin 2 (ezrin) | VIL2 | F: 5′-ggctgcaggactatgaggag-3′ R: 5′-tggctgtgtattctgcaagc-3′ |
231 | 1165–1396 | 26 |
| Breast cancer susceptibility gene 1 | BRCA1 | F: 5′-gagtaacaagccaaatgaacag-3′ R: 5′-tggatacttaaagccttctgtg-3′ |
457 | 2159–2616 | 29 |
| Interferon-induced protein IFI-15K | G1P2 | F: 5′-tgtcggtgtcagagctgaag-3′ R: 5′-gcccttgttattcctcacca-3′ |
206 | 142–348 | 32 |
| 2′,5′-Oligoadenylate synthetase 3 | OAS3 | F: 5′-gtcaaacccaagccacaagt-3′ R: 5′-tgtaggcacacctggtggta-3′ |
170 | 564–734 | 32 |
| Stanniocalcin 1 | STC1 | F: 5′-ttactcgtcccctttcatcg-3′ R: 5′-tccagctgccaggactactt-3′ |
226 | 2225–2451 | 35 |
| Glyceraldehyde-3-phosphate dehydrogenase | GAPDH | F: 5′-ggctctccagaacatcatccctgc-3′ R: 5′-gggtgtcgctgttgaagtcagagg-3′ |
299 | 633–932 | 27 |
Cell Proliferation Assays
To test the effect of BRCA1 on progesterone-stimulated cell proliferation, subconfluent T47D cells were transfected overnight with wtBRCA1 or empty pcDNA3 vector (15 μg plasmid DNA per 100-mm plastic Petri dish; see above), washed, allowed to recover from the transfection for several hours, harvested, counted, and inoculated into six-well dishes at 10,000 cells per well in DMEM containing 5% CSS. After allowing the cells to attach to the dish and grow for 24 h, the medium was switched to DMEM containing 2% CSS, and the cells were treated without or with progesterone (100 nM). For each experimental condition, three replicate wells were counted daily for a total of 4 d, using a Coulter counter. A total of four independent experiments was performed.
Mouse Model of Mammary-Targeted Brca1 Deficiency
Mice and Genotyping
Brca1 conditional (Co) knockout mice carrying the MMTV-Cre recombinase gene in a p53 wild-type (p53+/+) background (28) were maintained on a C57Bl/6 genetic background. This model utilizes Cre-mediated recombination of two floxed Brca1 exon 11 alleles, targeted to the mammary epithelium via the MMTV long terminal repeat (60). Nontransgenic C57Bl/6 mice were used as controls. All mice were maintained in accordance with institutional guidelines approved by the Georgetown University Animal Care and Use Committee. The presence of the floxed Brca1 alleles, the absence of wtBrca1 alleles, and the presence of the MMTV-Cre transgene were identified using PCR on tail DNA as described previously (28, 32, 60).
Progesterone Treatment Studies
Virgin postpubertal female Brca1Co/CoMMTV-Cre (n = 8) and wild-type controls (n = 5) at about 1 yr of age were anesthetized and implanted sc with a 10 mg/60-d constant-release progesterone pellet or a placebo pellet (Innovative Research of America, Sarasota, FL). Postpubertal female Brca1Co/CoMMTV-Cre (n = 9) and wild-type controls (n = 9) at about 1 yr were ovariectomized. Twelve days after ovariectomy (61), mice were anesthetized and implanted sc with either a 0.72 mg 60-d constant release estrogen pellet, or a 0.72 mg 60-d constant release estrogen pellet and a 10 mg 60-d constant release progesterone pellet or a placebo pellet (Innovative Research of America). Four weeks after pellet placement, the mice were weighed, euthanized, and necropsied, and the mammary glands were removed to assess the mammary gland response to progesterone. One inguinal (no. 4) mammary gland from each animal was dissected and spread on a glass slide at the time of necropsy for whole-mount analyses. Whole-mount fixation was carried out as described previously (32). The mammary gland volume was calculated after measuring length, width, and depth of dissected fourth mammary gland. Mammary gland whole mounts were analyzed to assess the effects of progesterone on the mammary morphology. For quantitative branching analysis, the density of side structures was determined by dividing each gland in half and counting the number of side structures, including secondary and tertiary branches, within six randomly selected ×4 fields distal to the lymph node. The number of tertiary branches in three fields from each side of the gland (six fields total) was counted. The total area counted for each sample was 3.14 mm2. The density of tertiary branching was expressed as the mean number of tertiary branches ± SEM per mm2. The contralateral inguinal mammary gland from each mouse was fixed in 10% buffered formalin overnight at 4 C and embedded in paraffin using standard techniques. Sections (5-μm) were cut for hematoxylin and eosin staining and immunohistochemistry. For mammary epithelial cell density measurements, the number of mammary epithelial cells within 10 randomly selected ×40 fields on hematoxylin and eosin-stained sections were counted, and means and SEMs for each treatment group were determined. Mammary epithelial cell density was expressed as the number of mammary epithelial cells per ×40 field. For PR immunohistochemistry, antigens were exposed to pre-heated BORG solution (BD1000G1, Biocare, Walnut Creek, CA) in a decloaking chamber (Biocare). The slides were allowed to cool for an additional 20 min in the BORG solution, quenched with 3% H2O2, blocked with mouse IgG-blocking reagent, incubated with Mouse On Mouse diluent, and incubated for 1 h with a 1:100 dilution of the PR antibody (NCL-L-PG-AB; Novacastra, Newcastle, UK). The tissues were exposed to biotinylated antirabbit IgG for 30 min at 25 C, elite stain for 30 min, stained with diaminobenzidine peroxidase substrate kit (SK-4100, Vector Laboratories, Inc., Burlingame, CA) for 5 min, counterstained with hematoxylin, and mounted (61). Fields (×4 and ×40) were defined using the Nikon Eclipse E800M microscope (Nikon Instruments, Inc., Melville, NY). Digital photographs were taken using Nikon DMX1200 software (Nikon Instruments, Inc.).
Statistical Analysis
Means and SEMs were calculated. Means were compared using Student's t test and ANOVA using GraphPad Prism version 4.00 for Windows (GraphPad Software, Inc., San Diego, CA). P < 0.05 was considered statistically significant.
Acknowledgments
This work was supported by United States Public Health Service Grants R01-CA82599, R01-CA80000, and RO1-ES09169 and by Grants BASIC99-003255 and BCTR0201295 from the Susan G. Komen Breast Cancer Foundation (to E.M.R.).
Abbreviations
- AF
Activation function
- CBP
cAMP response element-binding protein (CREB)-binding protein
- CSS
charcoal-stripped serum
- E2
17βestradiol
- ER
estrogen receptor
- ERE-TK-luc
estrogen response element-thymidine kinase-luciferase
- GAPDH
glyceraldehyde-3-phosphate dehydrogenase
- GST
glutathione-S-transferase
- HRT
hormone replacement therapy
- IP
immunoprecipitation
- IVT
in vitro translated
- MMTV
mouse mammary tumor virus
- PR
progesterone receptor
- siRNA
small interfering RNA
- wtBRCA1
wild-type BRCA1
References
- 1.Miki Y, Swensen J, Shattuck-Eidens D, Futreal PA, Harshman K, Tavtigian S, Liu Q, Cichran C, Bennett LM, Ding W, Bell R, Rosenthal J, Hussey C, Tran T, McClure M, Frye C, Hattier T, Phelps R, Haugen-Strano A, Katcher Y, Yakumo K, Gholami Z, Shaffer D, Stone S, Bayer S, et al. A strong candidate for the breast and ovarian cancer susceptibility gene BRCA1. Science. 1994;266:66–71. doi: 10.1126/science.7545954. [DOI] [PubMed] [Google Scholar]
- 2.Ford D, Easton DF, Bishop DT, Narod SA, Goldgar DE. Risks of cancer in BRCA1-mutation carriers. Breast Cancer Linkage Consortium. Lancet. 1994;343:692–695. doi: 10.1016/s0140-6736(94)91578-4. [DOI] [PubMed] [Google Scholar]
- 3.Streuwing JP, Hartge P, Wacholder S, Baker SM, Berlin M, McAdams M, Timmerman MM, Brody LC, Tucker MA. The risk of cancer associated with specific mutations of BRCA1 and BRCA2 among Ashkenazi Jews. N Engl J Med. 1997;336:1401–1408. doi: 10.1056/NEJM199705153362001. [DOI] [PubMed] [Google Scholar]
- 4.Thompson D, Easton DF the Breast Cancer Linkage Consortium. Cancer incidence in BRCA1 mutation carriers. J Natl Cancer Inst. 2002;94:1358–1365. doi: 10.1093/jnci/94.18.1358. [DOI] [PubMed] [Google Scholar]
- 5.Rosen EM, Fan S, Pestell RG, Goldberg ID. BRCA1 in hormone responsive cancers. Trends Endocrinol Metab. 2003;14:378–385. doi: 10.1016/j.tem.2003.08.001. Review. [DOI] [PubMed] [Google Scholar]
- 6.Rosen EM, Fan S, Pestell RG, Goldberg ID. The BRCA1 gene in breast cancer. J Cell Physiol. 2003;196:19–41. doi: 10.1002/jcp.10257. Review. [DOI] [PubMed] [Google Scholar]
- 7.Fan S, Wang JA, Meng Q, Yuan RQ, Ma YX, Erdos MR, Pestell RG, Yuan F, Auborn KJ, Goldberg ID, Rosen EM. BRCA1 inhibits estrogen receptor signaling in transfected cells. Science. 1999;284:1354–1356. doi: 10.1126/science.284.5418.1354. [DOI] [PubMed] [Google Scholar]
- 8.Fan S, Wang JA, Ma YX, Yuan RQ, Meng Q, Erdos MR, Pestell RG, Goldberg ID, Rosen EM. 2001 Role of direct interaction in BRCA1 inhibition of estrogen receptor activity. Oncogene. 2001;20:77–87. doi: 10.1038/sj.onc.1204073. [DOI] [PubMed] [Google Scholar]
- 9.Xu J, Fan S, Rosen EM. Regulation of the estrogeninducible gene expression profile by the breast cancer susceptibility gene BRCA1. Endocrinology. 2005;146:2031–2047. doi: 10.1210/en.2004-0409. [DOI] [PubMed] [Google Scholar]
- 10.Fan S, Wang JA, Ma YX, Yuan RQ, Meng Q, Erdos MR, Pestell RG, Goldberg ID, Rosen EM. p300 modulates BRCA1 inhibition of estrogen receptor activity. Cancer Res. 2002;62:141–151. [PubMed] [Google Scholar]
- 11.Ma YX, Tomita Y, Fan S, Wu K, Tong Y, Zhao Z, Song LN, Goldberg ID, Rosen EM. Structural determinants of the BRCA1: estrogen receptor interaction. Oncogene. 2005;24:1831–1846. doi: 10.1038/sj.onc.1208190. [DOI] [PubMed] [Google Scholar]
- 12.Zheng L, Annab LA, Afshari CA, Lee WH, Boyer TG. BRCA1 mediates ligand-independent transcriptional repression of the estrogen receptor. Proc Natl Acad Sci USA. 2001;98:9587–9592. doi: 10.1073/pnas.171174298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kawai H, Li H, Chun P, Avraham S, Avraham HK. Direct interaction between BRCA1 and the estrogen receptor regulates vascular endothelial growth factor (VEGF) transcription and secretion in breast cancer cells. Oncogene. 2002;21:7730–7739. doi: 10.1038/sj.onc.1205971. [DOI] [PubMed] [Google Scholar]
- 14.Yeh S, Hu YC, Rahman M, Lin HK, Hsu CL, Ting HJ, Kang HY, Chang C. Increase of androgen-induced cell death and androgen receptor transactivation by BRCA1 in prostate cancer cells. Proc Natl Acad Sci USA. 2000;97:11256–11261. doi: 10.1073/pnas.190353897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Park JJ, Irvine RA, Buchanan G, Koh SS, Park JM, Tilley WD, Stallcup MR, Press MF, Coetzee GA. Breast cancer susceptibility gene 1 (BRCAI) is a coactivator of the androgen receptor. Cancer Res. 2000;60:5946–5949. [PubMed] [Google Scholar]
- 16.Lessey BA. Two pathways of progesterone action in the human endometrium: implications for implantation and contraception. Steroids. 2003;68:809–815. doi: 10.1016/j.steroids.2003.09.004. Review. [DOI] [PubMed] [Google Scholar]
- 17.Visvader JE, Lindeman GJ. Transcriptional regulators in mammary gland development and cancer. Int J Biochem Cell Biol. 2003;35:1034–1051. doi: 10.1016/s1357-2725(03)00030-x. Review. [DOI] [PubMed] [Google Scholar]
- 18.Fentiman IS. 20 Oral contraceptives, hormone replacement therapy and breast cancer. Int J Clin Pract. 2002;56:755–759. Review. [PubMed] [Google Scholar]
- 19.Sasco AJ. Epidemiology of breast cancer: an environmental disease? APMIS. 2001;109:321–332. doi: 10.1034/j.1600-0463.2001.090501.x. Review. [DOI] [PubMed] [Google Scholar]
- 20.Scheidereit C, von der Ahe D, Cato AC, Wenz M, Suske G, Carlson C, Bosshard H, Westphal HM, Beato M. Protein-DNA interactions at steroid hormone regulated genes. Endocr Res. 1989;15:417–440. doi: 10.3109/07435808909036347. [DOI] [PubMed] [Google Scholar]
- 21.Kalkhoven E, Kwakkenbos-Isbrucker L, de Laat SW, van der Saag PT, van der Burg B. Synthetic progestins induce proliferation of breast tumor cell lines via the progesterone or estrogen receptor. Mol Cell Endocrinol. 1994;102:45–52. doi: 10.1016/0303-7207(94)90096-5. [DOI] [PubMed] [Google Scholar]
- 22.Xiong J, Fan S, Meng Q, Schramm L, Wang C, Bouzahza B, Zhou J, Zafonte B, Goldberg ID, Haddad BR, Pestell RG, Rosen EM. BRCA1 inhibition of telomerase activity in cultured cells. Mol Cell Biol. 2003;23:8668–8690. doi: 10.1128/MCB.23.23.8668-8690.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bae I, Fan S, Meng Q, Rih JK, Kim HJ, Kang HJ, Xu J, Goldberg ID, Jaiswal AK, Rosen EM. BRCA1 induces antioxidant gene expression and resistance to oxidative stress. Cancer Res. 2004;64:7893–7909. doi: 10.1158/0008-5472.CAN-04-1119. [DOI] [PubMed] [Google Scholar]
- 24.Ballare C, Uhrig M, Bechtold T, Sancho E, Di Domenico M, Migliaccio A, Auricchio F, Beato M. Two domains of the progesterone receptor interact with the estrogen receptor and are required for progesterone activation of the c-Src/Erk pathway in mammalian cells. Mol Cell Biol. 2003;23:1994–2008. doi: 10.1128/MCB.23.6.1994-2008.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Fan S, Wang JA, Yuan RQ, Ma YX, Meng Q, Goldberg ID, Rosen EM. BRCA1 as a human prostate tumor suppressor: modulation of proliferation, damage responses, and expression of regulatory proteins. Oncogene. 1998;16:3069–3083. doi: 10.1038/sj.onc.1202116. [DOI] [PubMed] [Google Scholar]
- 26.Richer JK, Jacobsen BM, Manning NG, Abel MG, Wolf DM, Horwitz KB. Differential gene regulation by the two progesterone receptor isoforms in human breast cancer cells. J Biol Chem. 2002;277:5209–5218. doi: 10.1074/jbc.M110090200. [DOI] [PubMed] [Google Scholar]
- 27.Yuan R, Fan S, Achary M, Stewart DM, Goldberg ID, Rosen EM. Altered gene expression pattern in cultured human breast cancer cells treated with hepatocyte growth factor/scatter factor in the setting of DNA damage. Cancer Res. 2001;61:8022–8031. [PubMed] [Google Scholar]
- 28.Xu X, Wagner KU, Larson D, Weaver Z, Li C, Ried T, Hennighausen L, Wynshaw-Boris A, Deng CX. Conditional mutation of Brca1 in mammary epithelial cells results in blunted ductal morphogenesis and tumour formation. Nat Genet. 1999;22:37–43. doi: 10.1038/8743. [DOI] [PubMed] [Google Scholar]
- 29.Brodie SG, Xu X, Qiao W, Li WM, Cao L, Deng CX. Multiple genetic changes are associated with mammary tumorigenesis in Brca1 conditional knockout mice. Oncogene. 2001;20:7514–7523. doi: 10.1038/sj.onc.1204929. [DOI] [PubMed] [Google Scholar]
- 30.Weaver Z, Montagna C, Xu X, Howard T, Gadina M, Brodie SG, Deng CX, Ried T. Mammary tumors in mice conditionally mutant for Brca1 exhibit gross genomic instability and centrosome amplification yet display a recurring distribution of genomic imbalances that is similar to human breast cancer. Oncogene. 2002;21:5097–5107. doi: 10.1038/sj.onc.1205636. [DOI] [PubMed] [Google Scholar]
- 31.Deng CX. Tumor formation in Brca1 conditional mutant mice. Environ Mol Mutagen. 2002;39:171–177. doi: 10.1002/em.10069. Review. [DOI] [PubMed] [Google Scholar]
- 32.Jones LP, Li M, Halama ED, Ma Y, Lubet R, Grubbs CJ, Deng CX, Rosen EM, Furth PA. Promotion of mammary cancer development by tamoxifen in a mouse model of Brca1-mutation-related breast cancer. Oncogene. 2005;24:3554–3556. doi: 10.1038/sj.onc.1208426. [DOI] [PubMed] [Google Scholar]
- 33.Leonhardt SA, Altmann M, Edwards DP. Agonist and antagonists induce homodimerization and mixed ligand heterodimerization of human progesterone receptors in vivo by a mammalian two-hybrid assay. Mol Endocrinol. 1998;12:1914–1930. doi: 10.1210/mend.12.12.0210. [DOI] [PubMed] [Google Scholar]
- 34.Tetel MJ, Jung S, Carbajo P, Ladtkow T, Skafar DF, Edwards DP. Hinge and amino-terminal sequences contribute to solution dimerization of human progesterone receptor. Mol Endocrinol. 1997;11:1114–1128. doi: 10.1210/mend.11.8.9963. [DOI] [PubMed] [Google Scholar]
- 35.Marquis ST, Rajan JV, Wynshaw-Boris A, Xu J, Yin GY, Abel KJ, Weber BL, Chodosh LA. The developmental pattern of Brca1 expression implies a role in differentiation of the breast and other tissues. Nat Genet. 1995;11:17–26. doi: 10.1038/ng0995-17. [DOI] [PubMed] [Google Scholar]
- 36.Lane TF, Deng C, Elson A, Lyu MS, Kozak CA, Leder P. Expression of Brca1 is associated with terminal differentiation of ectodermally and mesodermally derived tissues in mice. Genes Dev. 1995;9:2712–2722. doi: 10.1101/gad.9.21.2712. [DOI] [PubMed] [Google Scholar]
- 37.Rajan JV, Marquis ST, Gardner HP, Chodosh LA. Developmental expression of Brca2 colocalizes with Brca1 and is associated with proliferation and differentiation in multiple tissues. Dev Biol. 1997;184:385–401. doi: 10.1006/dbio.1997.8526. [DOI] [PubMed] [Google Scholar]
- 38.McKenna NJ, Nawaz Z, Tsai SY, Tsai MJ, O'Malley BW. 1998 Distinct steady-state nuclear receptor co-regulator complexes exist in vivo. Proc Natl Acad Sci USA. 1998;95:11697–11702. doi: 10.1073/pnas.95.20.11697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Liu Z, Wong J, Tsai SY, Tsai MJ, O'Malley BW. Sequential recruitment of steroid receptor coactivator-1 (SRC-1) and p300 enhances progesterone receptor-dependent initiation and reinitiation of transcription from chromatin. Proc Natl Acad Sci USA. 2001;98:12426–12431. doi: 10.1073/pnas.231474798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Smith CL, Onate SA, Tsai MJ, O'Malley BW. CREB binding protein acts synergistically with steroid receptor coactivator-1 to enhance steroid receptor-dependent transcription. Proc Natl Acad Sci USA. 1996;93:8884–8888. doi: 10.1073/pnas.93.17.8884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Schairer C. Progesterone receptors—animal models and cell signalling in breast cancer. Implications for breast cancer of inclusion of progestins in hormone replacement therapies. Breast Cancer Res. 2002;4:244–248. doi: 10.1186/bcr540. Review. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Conneely OM, Jericevic BM, Lydon JP. Progesterone receptors in mammary gland development and tumorigenesis. J Mammary Gland Biol Neoplasia. 2003;8:205–214. doi: 10.1023/a:1025952924864. Review. [DOI] [PubMed] [Google Scholar]
- 43.Raafat AM, Hofseth LJ, Li S, Bennett JM, Haslam SZ. A mouse model to study the effects of hormone replacement therapy on normal mammary gland during menopause: enhanced proliferative response to estrogen in late postmenopausal mice. Endocrinology. 1999;140:2570–2580. doi: 10.1210/endo.140.6.6634. [DOI] [PubMed] [Google Scholar]
- 44.Rajan JV, Wang M, Marquis ST, Chodosh LA. Brca2 is coordinately regulated with Brca1 during proliferation and differentiation in mammary epithelial cells. Proc Natl Acad Sci USA. 1996;93:13078–13083. doi: 10.1073/pnas.93.23.13078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kubista M, Rosner M, Kubista E, Bernaschek G, Hengstschlager M. Brca1 regulates in vitro differentiation of mammary epithelial cells. Oncogene. 2002;21:4747–4756. doi: 10.1038/sj.onc.1205580. [DOI] [PubMed] [Google Scholar]
- 46.Medina D, Peterson LE, Moraes R, Gay J. Short-term exposure to estrogen and progesterone induces partial protection against N-nitroso-N-methylurea-induced mammary tumorigenesis in Wistar-Furth rats. Cancer Lett. 2001;169:1–6. doi: 10.1016/s0304-3835(01)00507-9. [DOI] [PubMed] [Google Scholar]
- 47.Yang J, Yoshizawa K, Nandi S, Tsubura A. Protective effects of pregnancy and lactation against N-methyl-N-nitrosourea-induced mammary carcinomas in female Lewis rats. Carcinogenesis. 1999;20:623–628. doi: 10.1093/carcin/20.4.623. [DOI] [PubMed] [Google Scholar]
- 48.Narod SA. Hormonal prevention of hereditary breast cancer. Ann NY Acad Sci. 2001;952:36–43. doi: 10.1111/j.1749-6632.2001.tb02726.x. Review. [DOI] [PubMed] [Google Scholar]
- 49.Armes JE, Trute L, White D, Southey MC, Hammet F, Tesoriero A, Hutchins AM, Dite GS, McCredie MR, Giles GG, Hopper JL, Venter DJ. Distinct molecular pathogeneses of early-onset breast cancers in BRCA1 and BRCA2 mutation carriers: a population-based study. Cancer Res. 1999;59:2011–2017. [PubMed] [Google Scholar]
- 50.Lakhani SR, Van De Vijver MJ, Jacquemier J, Anderson TJ, Osin PP, McGuffog L, Easton DF. The pathology of familial breast cancer: predictive value of immunohistochemical markers estrogen receptor, progesterone receptor, HER-2, and p53 in patients with mutations in BRCA1 and BRCA2. J Clin Oncol. 2002;20:2310–2318. doi: 10.1200/JCO.2002.09.023. [DOI] [PubMed] [Google Scholar]
- 51.King MC, Wieand S, Hale K, Lee M, Walsh T, Owens K, Tait J, Ford L, Dunn BK, Costantino J, Wickerham L, Wolmark N, Fisher B National Surgical Adjuvant Breast and Bowel Project. Tamoxifen and breast cancer incidence among women with inherited mutations in BRCA1 and BRCA2: National Surgical Adjuvant Breast and Bowel Project (NSABP-P1) Breast Cancer Prevention Trial. JAMA. 2001;286:2251–2256. doi: 10.1001/jama.286.18.2251. [DOI] [PubMed] [Google Scholar]
- 52.Rebbeck TR, Lynch HT, Neuhausen SL, Narod SA, Van't Veer L, Garber JE, Evans G, Isaacs C, Daly MB, Matloff E, Olopade OI, Weber BL Prevention and Observation of Surgical End Points Study Group. Prophylactic oophorectomy in carriers of BRCA1 or BRCA2 mutations. N Engl J Med. 2002;346:1616–1622. doi: 10.1056/NEJMoa012158. [DOI] [PubMed] [Google Scholar]
- 53.King TA, Gemignani ML, Li W, Giri DD, Panageas KS, Bogomolniy F, Arroyo C, Olvera N, Robson ME, Offit K, Borgen PI, Boyd J. Increased progesterone receptor expression in benign epithelium of BRCA1-related breast cancers. Cancer Res. 2004;64:5051–5053. doi: 10.1158/0008-5472.CAN-04-1283. [DOI] [PubMed] [Google Scholar]
- 54.Taylor J, Lymboura M, Pace PE, A'hern RP, Desai AJ, Shousha S, Coombes RC, Ali S. An important role for BRCA1 in breast cancer progression is indicated by its loss in a large proportion of non-familial breast cancers. Int J Cancer. 1998;79:334–342. doi: 10.1002/(sici)1097-0215(19980821)79:4<334::aid-ijc5>3.0.co;2-w. [DOI] [PubMed] [Google Scholar]
- 55.Wilson CA, Ramos L, Villasenor MR, Anders KH, Press MF, Clarke K, Karlan B, Chen JJ, Scully R, Livingston D, Zuch RH, Kanter MH, Cohen S, Calzone FJ, Slamon DJ. Localization of human BRCA1 and its loss in high-grade, non-inherited breast carcinomas. Nat Genet. 1999;21:236–240. doi: 10.1038/6029. [DOI] [PubMed] [Google Scholar]
- 56.Esteller M, Silva JM, Dominguez G, Bonilla F, MatiasGuiu X, Lerma E, Bussaglia E, Prat J, Harkes IC, Repasky EA, Gabrielson E, Schutte M, Baylin SB, Herman JG. Promoter hypermethylation and BRCA1 inactivation in sporadic breast and ovarian tumors. J Natl Cancer Inst. 2000;92:564–569. doi: 10.1093/jnci/92.7.564. [DOI] [PubMed] [Google Scholar]
- 57.Staff S, Isola J, Tanner M. Haplo-insufficiency of BRCA1 in sporadic breast cancer. Cancer Res. 2003;63:4978–4983. [PubMed] [Google Scholar]
- 58.Dignam JD, Lebovitz RM, Roeder RG. Accurate transcription initiation by RNA polymerase II in a soluble extract from isolated mammalian nuclei. Nucleic Acids Res. 1983;5:1475–1489. doi: 10.1093/nar/11.5.1475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Ouchi T, Lee SW, Ouchi M, Aaronson SA, Horvath CM. Collaboration of signal transducer and activator of transcription 1 (STAT1) and BRCA1 in differential regulation of IFN- γ target genes. Proc Natl Acad Sci USA. 2000;97:5208–5213. doi: 10.1073/pnas.080469697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Wagner KU, Wall RJ, St-Onge L, Gruss P, Wynshaw-Boris A, Garrett L, Li M, Furth PA, Hennighausen L. Cre-mediated gene deletion in the mammary gland. Nucleic Acids Res. 1997;25:4323–4330. doi: 10.1093/nar/25.21.4323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Frech MS, Halama ED, Tilli MT, Singh B, Gunther EJ, Chodosh LA, Flaws JA, Furth PA. Deregulated estrogen receptor α expression in mammary epithelial cells of transgenic mice results in the development of ductal carcinoma in situ. Cancer Res. 2005;65:681–685. [PMC free article] [PubMed] [Google Scholar]