

Abstract
Humans are composed of hundreds of cell types. As the genomic DNA of each somatic cell is identical, cell type is determined by what is expressed and when. Until recently, little has been reported about the determinants of human cell identity, particularly from the joint perspective of gene evolution and expression. Here, we chart the evolutionary past of all documented human cell types via the collective histories of proteins, the principal product of gene expression. FANTOM5 data provide cell-type–specific digital expression of human protein-coding genes and the SUPERFAMILY resource is used to provide protein domain annotation. The evolutionary epoch in which each protein was created is inferred by comparison with domain annotation of all other completely sequenced genomes. Studying the distribution across epochs of genes expressed in each cell type reveals insights into human cellular evolution in terms of protein innovation. For each cell type, its history of protein innovation is charted based on the genes it expresses. Combining the histories of all cell types enables us to create a timeline of cell evolution. This timeline identifies the possibility that our common ancestor Coelomata (cavity-forming animals) provided the innovation required for the innate immune system, whereas cells which now form the brain of human have followed a trajectory of continually accumulating novel proteins since Opisthokonta (boundary of animals and fungi). We conclude that exaptation of existing domain architectures into new contexts is the dominant source of cell-type–specific domain architectures.
Keywords: CAGE, transcriptome, protein domains, evolution
Introduction
Multicellular life relies on the interplay of different specialized cell types within a single organism. As such, the understanding of these components of multicellularity is at the center of much of modern biology. However, little is known about the origin of these cell types and how they emerged and diversified from a single-celled ancestor (Arendt 2008). As cell types from the same organism are constrained to using an identical genome sequence, the difference between two cell types emerges through differential expression of genes and the proteins that they encode (Barbosa-Morais et al. 2012). Understanding the evolutionary story behind these differences in expression can help identify the order in which cell types evolved (Ponting 2008; Mukhopadhyay et al. 2012); grouping cell types identifies those that have a shared evolutionary past as well as highlighting genes that are critical for modern cell phenotypes.
A typical approach for studying cell-type evolution in an organism is to take a set of features from cells of interest in one organism and then look for similarities between these features and those from other organisms across the tree of life (Arendt 2003). This reveals the branching points where an ancestral form of a cell type might have existed. This however requires a large number of outgroup organisms for a high resolution of branching. Classical studies of this form used features based on cell morphology (Arendt 2003, 2008). The advent of sequencing technology has now enabled the study of biomolecular features. Genetic sequences can be compared using BLAST searches across other genomes which extract evolutionary relation via sequence homology (Mukhopadhyay et al. 2012). Such an approach is called phylostratigraphy (Domazet-Loso et al. 2007). These studies are however affected by the inability of BLAST to resolve distant evolutionary relations between homologs. It is well described that protein structure is better conserved than sequence over evolution (Dayhoff et al. 1975; Russell et al. 1994; Illergård et al. 2009); hence, in this analysis, our features of evolution are protein structures, which are in turn comprised of modular units—protein structure domains (listed as an architecture; fig. 1). A structurally oriented approach that, unlike BLAST, incorporates protein domain architectures will allow for more sensitive detection of homologous proteins in other organisms (Geer et al. 2002; Madera and Gough 2002). In turn, this will increase the quality of resolving the epoch when more ancient proteins were created, not visible through pure sequence homology alone.
Fig. 1.
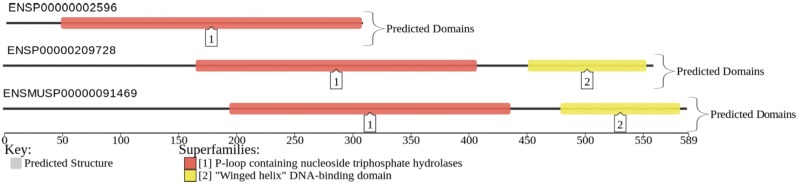
An example of SUPERFAMILY domains (De Lima Morais et al. 2011) (colored) assigned to human protein sequences (black horizontal lines: N′, left, to C′, right) ENSP00000002596 and ENSP00000209728, alongside mouse sequence ENSMUSP00000091469. This sequence of domains constitutes an architecture. Domain architectures can be assigned in this fashion to every protein sequence for every gene in every full-sequenced cellular genome using a library of several thousand domain sequence models that represent over 2,000 SCOP structural superfamilies (Murzin et al. 1995). Two genes in the same genome can readily possess identical architectures. Possession of the same domain architecture in two or more proteins is a sensitive and reliable indicator of evolutionary relation. In this example, the architecture of ENSP00000002596 is considered an evolutionarily distinct object to that of ENSP00000209728 and ENSMUSP00000091469, which share the same architecture. Addition/loss of a domain or a long stretch of unannotated sequence within a protein is considered as the creation of a new architecture. Convergent evolution of these architectures has been shown to be rare (Gough 2005). Figures produced using D2P2 (Oates et al. 2013).
We study cell-type, cell-line, and tissue-specific expression data from the human FANTOM5 (Forrest et al. 2014) set and investigate how different cell types utilize domain architectures created at different epochs in evolutionary history. This analysis comes in two parts: first, looking at domain architectures that are unique to particular cell types, to set upper and lower bounds for when these cell types could have arisen in evolutionary time; second, looking at how the usage of shared domain architectures differs between all cell types, tissues, and samples. In doing this, we create protein innovation usage (PIU) profiles for each cell type over the human evolutionary lineage, and a novel measure of the distance between these histories of protein innovation is presented. Each profile identifies key taxonomic points that were critical in the development of a cell type. By combining all cell-type profiles, we are able to group those that are evolutionarily similar, highlighting cell types that might have evolved in concert. Profiles are then used to create an evolutionary timeline of human cells, enabling a discussion about the relationship between the emergence of cellular phenotypes and the evolution of the underlying components (proteins).
This work is part of the FANTOM5 project (Forrest et al. 2014). Data downloads, genomic tools and copublished manuscripts are summarized here http://fantom.gsc.riken.jp/5/. (last accessed March 17, 2014)
Results and Discussion
Characterization of FANTOM Data
The human libraries from the FANTOM5 (Forrest et al. 2014) data set were collapsed to 492 unique tissues, primary cell types, and cell lines by combining replicas. In the analyses presented later, we specify if only primary cell types were used (156 in total) or if all 492 samples are used. The primary cell-type samples each contain only a single cell type rather than a mixture of cell types, as is the case with tissue samples. Human primary cells are taken from anonymous donors and as a result are not modified in any way, as is the case with the cell-line samples. In the second part of the analysis, we use the whole set of FANTOM5 samples together but find that tissue and cell-line samples naturally cluster apart from primary cells.
Using pooled expression for each gene in each sample, we chose a binary threshold of ln (Tags Per Million) more than 2 to determine whether a gene was expressed. For each sample, we then identified the set of structural domain architectures that are annotated to the longest transcript of each expressed gene. The total number of distinct domain architectures expressed in the union of all cell types is 4,204, annotated to 16,259 distinct genes. This is out of a possible 4,608 distinct domain architectures in all the longest transcripts in ENSEMBL Homo sapiens (build 37). As the genes that are expressed in each cell type are different, the protein (and hence domain architecture) usage also varies. For instance, profiling the “clonetech universal reference RNA,” which is a sample made up of RNA from a mixture of sources, detected the greatest number of distinct domain architectures (3,609) whilst the “tongue epidermis” sample had the fewest (578). The average number of distinct domain architectures for a given sample is 2,652 (see supplementary figure S4, Supplementary Material online, for more detail).
It has been shown in other studies that the effect of alternative splicing is important for both the protein structure and regulatory network (Yura et al. 2006; Barbosa-Morais et al. 2012; Buljan et al. 2012). As this study uses CAGE data and not RNAseq, we have chosen to abstract each transcript from a given gene to a single longest transcript (see Materials and Methods). The most recent common ancestor (MRCA) of this longest transcript represents the lower bound (i.e., most recent) in terms of the introduction of any possible splice variants of a gene. As we are interested in studying evolution in terms of genes and not the evolution of splice variation, we consider this a suitable level of abstraction.
The MRCA of a domain architecture represents the point in evolution at which it is thought to have come into existence. In this study, a domain architecture MRCA can be in 1 of 13 epochs, spanning from H. sapiens back to the last universal common ancestor (LUCA). The distribution of expressed domain architecture MRCAs is not homogeneous; older epochs contain more domain architecture MRCAs than newer ones (supplementary fig. S1, Supplementary Material online).
This work suggests that exaptation of existing domain architectures into new contexts is the dominant source of cell-type–specific domain architectures. There is a trend from LUCA to H. sapiens (supplementary fig. S2, Supplementary Material online) of domain addition to existing architectures being the predominant creation event driving domain architecture innovation. However, few of these domain addition events are specific to one functional role in the cell which is evident as that there are few domain architectures solely unique to one primary cell type (supplementary fig. S4, Supplementary Material online). One explanation is that the increase in functional specialization has occurred as a result of more complex networks of regulation within the cell, as previously suggested (Buljan et al. 2012; Habib et al. 2012), and facilitated by the reuse of functional modules (domains) in different molecular contexts (Moore et al. 2008; Wang and Caetano-Anollés 2009; Moore et al. 2013).
Cell-Type–Specific Domain Architectures
An intuitive question to ask of cellular evolution is at what point in time did each human cell evolve. One way to attempt to answer this question for a given cell type is to identify the earliest point in evolution when all of the proteins which it expresses exist. The result of doing this however points to almost all cell types having appeared, in their current observed form, very recently, that is, since Primates or even Great apes. This is because once a new cell type appears, it continues to evolve indefinitely, and so most cell types express some proteins that have evolved very recently. Accepting that ancestral cells will be of a slightly different form to modern human cells, the issue arises as to how different two cells must be before they can no longer be considered as the same cell type. As measured by overlap in domain architectures, this is approximately 95% (supplementary fig. S8, Supplementary Material online) for the matrix of existing primary cells. Asking again the original question, but looking for the earliest point in time when only 95% of expressed proteins existed, merely points to a slightly earlier point in time when the form of the ancestral cells would be recognizably similar to the modern cell. The fact that most cell types appear at a similar point in time according to this criterion (data not shown) suggests that the rates of evolution (as measured by protein innovation) are not wildly different for most cell types. Thus, to successfully examine cellular evolution on a historical timeline, cell types must be grouped to represent a common ancestral type of origin.
To group related cell types, we make use of the Cell Ontology (Forrest et al. 2014) to group together the 156 primary cell-type samples at two levels: first, we group fibroblast, embryonic, epithelial, mesenchymal, and hematopoietic cells; then break them down into 6, 3, and 12 subgroups, respectively. Even when grouped, extracting an evolutionary history of an ancestral cell is a challenge. Unlike organisms, which speciate and follow independent paths of molecular innovation, cell types are not independent. By example, a very recently evolved protein may be expressed in many related cell types. We overcome this by locating the epoch in evolution when a protein first appears by comparing its domain architecture to those in all other completely sequenced genomes (see Materials and Methods). The groupings of cell types and epochs of protein creation are combined to produce figure 2. This kind of approach is the best possible in the absence of cell-specific expression data for all other genomes as well (which would allow the reconstruction of a full evolutionary tree of cell types).
Fig. 2.

The distribution of cell-specific domain architectures. Two levels of the cell ontology are used to segregate the primary cell samples. This shows how the cell-type–specific domain architectures are distributed amongst the constituent members of the coarse-grained ontologies.
Major Features of Cell-Specific Evolution
We observe that fibroblast cells do not contain many unique protein domain architectures as compared with other cell type groupings. From the 16 fibroblast samples taken from various parts of the body, only three unique architectures are found, and all were already present in the LUCA. These three architectures are comprised of a single repeated domain (Fibronectin type 3, Galactose oxidase central domain, and the PKD domain). Each of these domains appears in other domain architectures adding to the evidence that structural innovation since the last universal ancestor has not been important for fibroblast cells.
Each of the four remaining groupings possesses domain architectures exclusive to just them, with epochs spread across epoch ranges. It is surprising however, that the majority of cell-type–specific domain architectures in the remaining groupings are also already present at the LUCA, before the rise of multicellularity. This means that these domain architectures have undergone specialization over the course of evolution, now only being required in a single cell-specific phenotype.
“Embryonic stem cell” samples contain a unique domain architecture derived from the IRS4 (insulin response substrate 4) gene that came about in H. sapiens. It is a gene whose expression has not been detectable in mice (Qu et al. 1999) and is also linked with tumor growth and proliferation (Cuevas et al. 2009; Mardilovich et al. 2009). However, little is known about the function of this gene, with previous attempts failing to identify a tissue type containing detectable levels of expression (Schreyer 2003). In the FANTOM5 data, this domain architecture (annotated to the IRS4 gene) is reliably detected in each of the “H9 Embryonic stem cell” samples. The human-specific innovation of this protein results from the addition of a Formin homology 2 domain. This domain has previously been identified as a promiscuous protein domain playing a role in lineage-specific structural and signaling interactions in a number of proteins (Cvrcková et al. 2004). dcGO (Fang and Gough 2013) assigns GO terms such as cellular component organization of biogenesis and organelle organization to this domain, meaning that this gene would be a strong target for investigations into H. sapiens-specific embryonic development.
Epithelial cells contain the highest number of domain architectures unique to that grouping in the cell ontology (149). As there is a bias toward epithelial cells in the FANTOM5 data set, this result may not on first inspection seem surprising. Looking more deeply, these epithelial-specific domain architectures are not shared by all of the epithelial cells; in fact, they seem disparate in their usage of domain architecture innovation. This suggests that whilst they come from the same class of cell-type, the location in the body in which they are located also plays a large role in the domain content of their expressed genes. For instance, the nephron epithelial cells (e.g., renal mesangial, renal proximal, and renal glomerular endothelial cells) only contain one nephron-specific domain architecture whilst the columnar epithelial cells (e.g., melanocytes, lens, and ciliary epithelial cells) contain seven unique domain architectures.
Within the hematopoietic cells, there is a large bias toward cell-specific domain architectures occurring early in evolutionary time. The only recent innovation specific to hematopoietic cells has occurred specifically in T-cells, mast cells, and reticulocytes (in the adaptive immune system and blood). There are six domain architectures that arise in T-cells only (especially CD8+, CD4+CD25+CD45RA+, and CD4+ T-cells). These domain architectures arise from TNFRSF4, SPEF2, ZMYND12, UMODL1, and IL12RB2 genes (domain architecture annotation is shown in supplementary fig. S9, Supplementary Material online). From these six, UMODL1 and NKRF arose at Euteleostomi and Euarchontoglires, respectively. NKRF or NF-kappa-B-repressing factor is a transcription factor that mediates transcriptional repression of certain NF-kappa-B-responsive genes. UMODL1 or Uromodulin-like-1 gene has a domain architecture comprising of four protein domains (Fibronectin type 3, Elafin-like, Growth factor receptor domain, and EGF/Laminin). Uromodulin-like 1 protein has been shown to demonstrate a prompt and robust response to CD3/CD28 antibodies in proliferating CD4+ T cells, implicating it in immune response to pathogens (Wang et al. 2012). As this innovation took place in the ancestral Euarchontoglate, T-cells found in organisms more distant from human than Euarchontoglires (Rodents, Rabbits, and Primates) cannot posses the same structural content in their T-cells as we do.
Conversely, monocytes appear to be older, with the last monocyte-specific protein innovations taking place at Coelomata (species containing fluid-filled body cavities). This is also echoed by Langerhan cells, another innate component of the immune system. The last point of innovation specific to this group is at the ancestral Euteleostome (ancestor of all bony vertebrates). For instance, a monocyte-specific domain architecture can be found in NOD2, which is an intracellular sensor for bacteria (Ogura et al. 2001). When compared with other cellular types the cells involved in the innate immune system appear to be the oldest.
Patterns in Usage of Protein Innovation
We have seen above that it is not uncommon for groups of phenotypically similar cell type to posses several domain architectures that are mutually unique to just them. The number of these exclusive architectures across samples, however, is insufficient to study function solely in terms of uniqueness. Instead, we look now toward studying patterns in cell-type usage of all expressed domain architectures, created at different evolutionary epochs.
Over evolutionary time, an evolving cell type has available to it all protein domain architectures within the genome of that era. Some of these might only be expressed in one modern cell type (as investigated earlier), but many more will be expressed in multiple other samples (as seen in sample pairwise domain content overlaps in supplementary fig. S8, Supplementary Material online). These architectures will be accumulated over the evolutionary history of that cell. A single cell-type history is defined by the points in evolutionary time where new protein content used by that cell appeared in the genome. We anticipate modern versions of that cell type to express more architectures from an age crucial to the development of its core internal components. To numerically capture such historic information about the protein innovation in a cell type, we construct a novel measure: the PIU, see Materials and Methods. This relative measure describes how far above or below the average protein innovation at a given epoch those proteins expressed in a given cell type are. Combining the PIU for each of 13 key phyletic divisions in the NCBI taxonomy (Federhen 2012), we create an evolutionary profile, giving an insight into the trend of protein innovation over time. An example profile can be found in figure 3 for the cell type sample CD8+ T-cells.
Fig. 3.
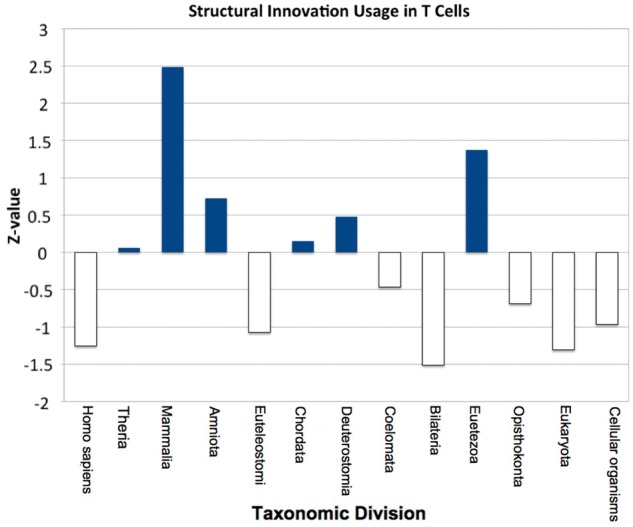
The evolutionary profile for T-cells. Blue bars represent greater than average usage of domain architectures appearing at that evolutionary time, with the height of the bar illustrating magnitude of PIU score. The clear bars show below average usage of architectures of such age. For these T-cells: in Eumetezoa, Amniota, and Mammalia, there is a much greater usage of protein innovation. Conversely, they make less than average use of structural innovation between cellular organisms and Opisthokonta as well as at Bilateria, Euteleostomi, and H. sapiens.
The evolutionary profiles we present identify periods of evolutionary time where prolonged above or below average use of protein innovation has occurred. To group samples with similar evolutionary histories, we use a self-organizing map (SOM) clustering algorithm (presented in supplementary fig. S3, Supplementary Material online). Samples in the same cluster have similar patterns in PIU over human ancestry and hence have similar evolutionary histories. This does not necessarily mean that they are expressing identical proteins created at those epochs simply that they express a similar proportion of their total number of distinct architectures to those dated to that period.
Figure 4 details the results of the SOM clustering over all evolutionary profiles for all 492 unique tissues, cell types, and cell lines. Displayed are stylized profiles of above average, high, and very high (increasing line thickness) usage of protein innovation across epochs, alongside annotation of distinct and enriched architectures. We identified ten clusters, each containing between 3 and 20 subclusters (units). The profiles shown are those for representative samples of the cluster in which they sit. SOM cluster and unit membership, alongside sample evolutionary profiles, can also be explored through an associated webpage located at http://supfam.cs.bris.ac.uk/SUPERFAMILY/trap/, last accessed March 17, 2014).
Fig. 4.

The molecular evolution timeline of all 493 cell types, tissues, and cell lines, grouped into ten major clusters. Displayed are stylized profiles of above average, high, and very high (increasing black line thickness) usage of protein innovation across epochs, alongside annotation of distinct and enriched architectures.
The clusters qualitatively compare well with groupings determined by gene coexpression in the core FANTOM5 article (Forrest et al. 2014); smooth muscle cells are close to epithelial samples and immune system components are all grouped nearby. However, the data here has only 13 dimensions (the ancestral phylogenetic epochs), as opposed to the many thousands of promoters in the genome. Cluster divisions also broadly group samples into biologically meaningful categories. Clusters 1 and 2 group similar evolutionary profiles for many white blood cell types (leukocytes), with a significant division between the adaptive and innate immune system (Fischer’s exact P value < 0.05; supplementary fig. S5, Supplementary Material online). It is interesting to note that in general, cancer cell lines and tissues each separately cluster with their counterparts and not with their primary cell components (clusters 8 and 10). This suggests that phylostratigraphic evolutionary analysis between different classes of biological sample (tissue, cell type, and cell line) is not possible as the ancestral trends in expressed genes are not similar.
There are new domain architectures created at every major division point in the NCBI taxonomy (supplementary figs. S1 and S2, Supplementary Material online); however, tissues and cell types have not made uniform usage of this protein innovation. Evolutionary profiles reveal punctuated evolution of samples; “chondrocytes” (cluster 6) makes use of architectures from Metazoa, Chordata, Euteleostomi, and H. sapiens whilst “whole blood” (cluster 2) draws heavily from proteins created before Bileteria and then again at Mammalia. This reiterates the earlier point that it is not possible to unambiguously date the emergence of a particular cell or tissue type to a single taxonomic era by the majority age of the components that they express. To do so would require equally high quality expression data from many organisms across the full breadth of the tree of life. Using our evolutionary profile information, we can still gain insights into the progression of cell type and tissue evolution, as discussed later.
Evolution of the Human Immune System: The Components Are Old
As discussed earlier, the innate immune system shows strong evidence for being an ancient cell type, with all hematopoietic innovation taking place within adaptive immune cell types. An example profile for T-cells from cluster 2 can be seen in figure 3. To summarize, these cell types contain four periods of above average use of domain-architecture innovation at Metazoa, Chordata, Amniota, and Mammalia (fig. 4). During the expansion of the metazoans, it is known that the first immune system cells evolved to cope with the large number of single-celled pathogens that attempted to infect the newly formed multicellular life forms. The enriched domain architectures that are associated with cluster 1 include Toll-like receptors and leucine-rich repeats, both of which are thought to have played an important role in these early immune cells (Hoffmann 1999; Janeway and Medzhitov 2002)—see supplementary materials (Supplementary Material online) for a full list of superfamilies, many of which have creation points before Chordata. Furthermore, the use of structural innovation in Chordata, Amniota, and Mammalia concurs with what is known about the evolution of the adaptive immune system at these times. Enriched domain architectures for this cluster include the SH2 domain, which is known to regulate the signaling events in the adaptive immune system of Eukarya (Liu et al. 2011) as well as the ADP-ribosylation domain that is known to modulate immune response (Corda and Di Girolamo 2002). This pattern of use of protein innovation (high at Chordata and again at Mammalia) lends evidence to a hypothesis discussed by Cooper and Herrin (2010) that an ancestral immune system existed around the time of the ancestral chordate, with newer cell types developed around the time of the rise of mammals.
Evolution of the Spleen and Thymus: Concerted Evolution with the Brain
Cluster 5 groups tissue samples of the brain together. It is homogenous in this respect, with the exception of “spleen” and “thymus” tissue samples being placed in a subcluster alongside “spinal cord,” “pineal gland,” and “adult globus pallidus” tissue samples. The spleen and thymus are both lymphoid organs that modulate immunity and their presence within a cluster of brain-specific samples suggests concerted evolution in their structural innovation over time. Specifically, whenever the brain has undergone innovation due to selective pressure the same has been true for the spleen and thymus. Both organs are heavily innervated and reflex control of immunity by the central nervous system has been recently demonstrated (Rosas-Ballina et al. 2008; Rosas-Ballina and Tracey 2009). Here, we add new molecular-expression evidence to support such a hypothesis. This is in contrast to the idea that the spleen is solely part of the circulatory system, as was previously hypothesized by anatomic descriptions (Tischendorf 1985).
Conclusions
We have used phylogenetic stratification of the structural domain architectures of proteins expressed in FANTOM5 human cell samples to chart their histories in terms of protein innovation. The results show that human cell types, in the form that they currently exist in, are the product of continuous and ongoing evolution. This means that we do not see a clear point in time when a human cell came into being, because the ancestral cell type would have been of a different form from that which they now exhibit. By considering groupings of related cells, however, we can see the points in evolution when proteins uniquely expressed in the cells of that type were created. Without equivalent data for a large number of genomes, it is not possible to reconstruct an evolutionary tree of cell types; the best possible picture we can achieve is a timeline of cellular evolution. This is however sufficient to allow corroboration, at a molecular level, of some current theories on the evolution of specific cell types and tissues.
It has been suggested previously that species-specific adaptive processes are enabled by de novo creation of genes (Toll-Riera et al. 2009; Bornberg-Bauer et al. 2010) or through domain shuffling/gene fusion (Moore et al. 2008; Buljan et al. 2010; Bornberg-Bauer and Albà 2013). We propose that adaptive processes are the result of reuse of existing domain architectures, homologs of which might not be detectable through use of BLAST (Mistry et al. 2013), rather than de novo creation of the entire gene sequence. This is supported by a search using sensitive indicators of homology across all completely sequenced genomes and demonstration that many different niches are filled by homologs of the same domain architecture. This said we should remain mindful of the fact that of these many structural homologs, some might fall into more or less evolutionarily related groupings; a resolution that we miss here and should be considered for further study.
The findings we present have been made possible for the first time by the cell-type–specific digital gene expression from the FANTOM5 project. A future extension of this work to all cell types in many species, to the same high quality as in FANTOM5, is required to complete the picture of cellular evolution in nature and reconstruct the full evolutionary tree of cell types.
Materials and Methods
There are four core components to our methodology (fig. 5): 1) identification of expressed genes, 2) assignment of domain architectures, 3) creation of evolutionary profiles, and 4) clustering of evolutionary profiles. An enrichment analysis of the clustered domain architectures and associated dcGO ontological terms was also conducted. The details of these processes are explained in the following sections.
Fig. 5.

The pipeline used in presented work: (A) Clustered CAGE tags are aligned to the human genome and mapped to the closest known gene (where possible). (B) When this gene is protein coding, SUPERFAMILY is used to annotate it with its protein domain architecture. (C) Each expressed domain architecture then has its MRCA calculated, which allows for a profile of structural innovation usage for each cell type to be built up (fig. 3). (D) The profiles can then be clustered to find cell types which share a common evolutionary past. These clusters can then be analyzed to discover domain architectures key to bifurcations in cell phenotype.
Identification of Expressed Genes
The FANTOM5 CAGE human data set is a collection of tissue and cell samples (Forrest et al. 2014), providing the genomic transcription start site (TSS), a mapping to the closest Entrez GeneID and expression level of the gene products transcribed. A binary value was set for each gene expressed in each distinct tissue sample above a tags per million (TPM) intensity threshold of 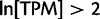 (
(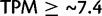 ). This cutoff was in line with previous characterization of the CAGE methodology (Balwierz et al. 2009), which showed a roughly linear relationship, after a tag count of just under ten, between
). This cutoff was in line with previous characterization of the CAGE methodology (Balwierz et al. 2009), which showed a roughly linear relationship, after a tag count of just under ten, between  and the number of TSSs expressing at that magnitude. In cases, where replicates were present, a further stipulation was made that the average binary expression value of a gene amongst replicate samples must exceed 0.75.
and the number of TSSs expressing at that magnitude. In cases, where replicates were present, a further stipulation was made that the average binary expression value of a gene amongst replicate samples must exceed 0.75.
Assignment of Domain Architectures and Epochs
We used a mapping from Entrez GeneID to gene-product protein sequences, provided by UniProt (UniProt Consortium 2010), for which the SUPERFAMILY database (v.1.75) provides domain architecture annotation (De Lima Morais et al. 2011). It is worth highlighting that as an EntrezID is a gene id, as opposed to a gene product id, the mapping from EntrezID to UniprotID is one-to-many. At the expense of not studying at the resolution of per-sample splice-isoform products, we selected the longest transcript from the mapped UniprotIDs. As we are interested in studying evolution in terms of when a gene first appeared in its current form, and not the evolution of splicing, we consider this level of abstraction appropriate.
SUPERFAMILY provides domain architecture assignments to all proteins within 1,559 fully sequenced cellular genomes; 373 eukaryotes at varying taxonomic divisions alongside 1,175 archaea & bacteria (at an outgroup from human at cellular organisms). It also details taxonomic placement of these genomes in accordance with the NCBI taxonomy (Federhen 2012). Dollo parsimony was used to determine the MRCA of the related gene transcript homologs, which was set as the creation point or “epoch” of the domain architecture. The epochs for each of the domain architectures expressed in human fall into 1 of 13 evolutionary eras, matching key taxonomic division points, stretching from H. sapiens to cellular organisms. These were chosen so as to ensure that there was sufficient variability in the number of domain architectures expressed from that age, necessary for the evolutionary profiles (discussed later). A domain architecture that is ubiquitous and found in all kingdoms of life would have an epoch at cellular organisms, whereas a domain architecture found only in four-limbed vertebrates would have an epoch at Tetrapoda.
Further studies that might be more focused on functional characterization of cell transcriptomes, and less on evolutionary analysis, might be interested in considering the effect of splice isoforms. Previous studies have shown that alternative splicing can affect protein structure (Yura et al. 2006); however, this effect is not as strong at the domain architecture level. The point at which an existing gene undergoes domain shuffling, acquiring a new architecture, is a reasonable point at which to declare that the protein no longer exists in its previous form.
Creation of Evolutionary Profiles
Each cell type expresses a collection of domain architectures, created at various evolutionary epochs and with differing abundance. So, as to compare different phyletic profiles of domain architecture usage between samples, we created a per-sample Z-score of the proportion of distinct expressed domain architectures of each epoch, as compared against all other samples (supplementary figs. S6 and S7, Supplementary Material online). This value is also called the PIU in the Discussion section. This assignment across all epochs provides an evolutionary profile, detailing eras of high and low usage of structural domains created at points in time over our evolutionary past. These relate to evolutionary eras of high usage of structural innovation and low usage of structural innovation, respectively.
For a set of samples 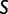 and set of evolutionary epochs
and set of evolutionary epochs 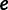 , the number of unique domain architecture expressed in a given sample
, the number of unique domain architecture expressed in a given sample 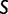 at epoch
at epoch 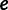 is
is 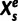 .
.
This means that the total number of expressed architectures for a given sample 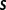 is
is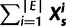 or
or 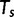 .
.
To calculate the z-value for a sample S at an epoch e, we used the following equation: 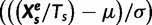 , where
, where 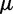 is mean and
is mean and 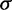 is the standard deviation at each evolutionary epoch over all samples.
is the standard deviation at each evolutionary epoch over all samples.
Clustering of Evolutionary Profiles
A coarse-grained overview of the data was achieved by clustering all experimental samples from the FANTOM data set (i.e., primary cells and cell lines of different tissue origins) by their evolutionary profiles. The clustering was performed using SOM methods (Vesanto 1999). The SOM and its derivatives have been used extensively to cluster and visualize high-dimensional biological data (evolutionary profiles in this setting). We chose distance matrix-based clustering of the SOM to obtain a total of 110 subclusters (units) that were further grouped into ten major clusters in a topology-preserving manner. A crucial feature of this clustering is that they optimize the number of clusters and division criteria to fully respect the inherent structure of the input data (without a priori assumption of the data structure). The clusters and units were visualized using a component plane presentation integrated SOM (Xiao et al. 2003), displaying evolutionary epoch-specific changes of clustered cell types (presented in supplementary fig. S3, Supplementary Material online). Unit and cluster membership can be fully explored using the publically accessible website (see Public Accessibility of Data).
Assignment of Ontological Terms
Domain architecture and gene enrichment was performed by identifying unique items to groupings of samples, whether by a cell ontology (Forrest et al. 2014) or through use of the evolutionary profile SOM clusters.
The dcGO resource (Fang and Gough 2013) was used to provide domain-centric Gene Ontology (GO) for these enriched architectures. The most specific (highest information) terms in dcGO were used.
Public Accessibility of Data
All of the data described in this manuscript, including protein domain architecture assignments to each and every gene transcript of the FANTOM5 human data set and details of cell type placement in the SOM clustering are available as part of a mySQL compatible dump available at http://supfam.cs.bris.ac.uk/SUPERFAMILY/trap/ (last accessed March 17, 2014). Also provided are evolutionary epoch profiles, such as that presented in figure 3 for T-cells, for every distinct cell type. Finally, scripts used to generate these data are available from GitHub (https://github.com/Scriven/TraP, last accessed March 17, 2014) for reuse or inspection under an open source license.
Supplementary Material
Supplementary figures S1–S9 are available at Molecular Biology and Evolution online (http://www.mbe.oxfordjournals.org/).
Acknowledgments
H.K. was responsible for data management. A.R.R.F. was involved in the FANTOM5 concepts and management. O.J.L.R., A.J.S., J.G., and M.E.O. were the major drivers of the novel research directions discussed in this manuscript. A.J.S. and O.J.L.R. performed the data analysis and interpretation, with significant input by M.E.O. in the early stages of the work. O.L.J.R. and A.J.S. prepared the manuscript with J.G., M.E.O., and H.F. making contributions to its structure and refinement. The authors thank all members of the FANTOM5 consortium for contributing to generation of samples and analysis of the data set and thank GeNAS for data production. They also thank David de Lima Morais for useful discussion at the preliminary stages of this work. FANTOM5 was made possible by a research grant for RIKEN Omics Science Center from MEXT to Yoshihide Hayashizaki and a grant of the Innovative Cell Biology by Innovative Technology (Cell Innovation Program) from the MEXT, Japan to Yoshihide Hayashizaki. This work was supported by the Bristol centre for Complexity Sciences (BCCS) studentships from the Engineering and Physical Sciences Research Council (EPSRC) grant EP/E501214 to A.J.S. and M.E.O.; the Biotechnology and Biological Sciences research Council (BBSRC) [BB/ G022771/1 to J.G., O.J.L.R., and H.F.; and MEXT to RIKEN CLST and RIKEN PMI. RIKEN Omics Science Center ceased to exist as of April 1, 2013, due to RIKEN reorganization. This manuscript is part of a series of papers from the FANTOM5 consortium (http://fantom.gsc.riken.jp/).
References
- Arendt D. Evolution of eyes and photoreceptor cell types. Int J Dev Biol. 2003;47:563–571. [PubMed] [Google Scholar]
- Arendt D. The evolution of cell types in animals: emerging principles from molecular studies. Nat Rev Genet. 2008;9:868–882. doi: 10.1038/nrg2416. [DOI] [PubMed] [Google Scholar]
- Balwierz PJ, Carninci P, Daub CO, Kawai J, Hayashizaki Y, Van Belle W, Beisel C, Van Nimwegen E. Methods for analyzing deep sequencing expression data: constructing the human and mouse promoterome with deepCAGE data. Genome Biol. 2009;10:R79. doi: 10.1186/gb-2009-10-7-r79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barbosa-Morais NL, Irimia M, Pan Q, Xiong HY, Gueroussov S, Lee LJ, Slobodeniuc V, Kutter C, Watt S, Colak R, et al. The evolutionary landscape of alternative splicing in vertebrate species. Science. 2012;338:1587–1593. doi: 10.1126/science.1230612. [DOI] [PubMed] [Google Scholar]
- Bornberg-Bauer E, Albà MM. Dynamics and adaptive benefits of modular protein evolution. Curr Opin Struct Biol. 2013;23:466–459. doi: 10.1016/j.sbi.2013.02.012. [DOI] [PubMed] [Google Scholar]
- Bornberg-Bauer E, Huylmans A-K, Sikosek T. How do new proteins arise? Curr Opin Struct Biol. 2010;20:390–396. doi: 10.1016/j.sbi.2010.02.005. [DOI] [PubMed] [Google Scholar]
- Buljan M, Chalancon G, Eustermann S, Wagner GP, Fuxreiter M, Bateman A, Babu MM. Tissue-specific splicing of disordered segments that Embed binding motifs rewires protein interaction networks. Mol Cell. 2012;46:871–883. doi: 10.1016/j.molcel.2012.05.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buljan M, Frankish A, Bateman A. Quantifying the mechanisms of domain gain in animal proteins. Genome Biol. 2010;11:R74. doi: 10.1186/gb-2010-11-7-r74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooper MD, Herrin BR. How did our complex immune system evolve? Nat Rev Immunol. 2010;10:2–3. doi: 10.1038/nri2686. [DOI] [PubMed] [Google Scholar]
- Corda D, Di Girolamo M. Mono-ADP-ribosylation: a tool for modulating immune response and cell signaling. Sci STKE. 2002 doi: 10.1126/stke.2002.163.pe53. 2002:pe53. [DOI] [PubMed] [Google Scholar]
- Cuevas EP, Escribano O, Monserrat J, Martínez-Botas J, Sánchez MG, Chiloeches A, Hernández-Breijo B, Sánchez-Alonso V, Román ID, Fernández-Moreno MD, et al. RNAi-mediated silencing of insulin receptor substrate-4 enhances actinomycin d- and tumor necrosis factor-alpha-induced cell death in hepatocarcinoma cancer cell lines. J Cell Biochem. 2009;108:1292–1301. doi: 10.1002/jcb.22359. [DOI] [PubMed] [Google Scholar]
- Cvrcková F, Novotný M, Pícková D, Zárský V. Formin homology 2 domains occur in multiple contexts in angiosperms. BMC Genomics. 2004;5:44. doi: 10.1186/1471-2164-5-44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dayhoff MO, McLaughlin PJ, Barker WC, Hunt LT. Evolution of sequences within protein superfamilies. Naturwissenschaften. 1975;62:154–161. [Google Scholar]
- De Lima Morais DA, Fang H, Rackham OJL, Wilson D, Pethica R, Chothia C, Gough J. SUPERFAMILY 1.75 including a domain-centric gene ontology method. Nucleic Acids Res. 2011;39:D427–D434. doi: 10.1093/nar/gkq1130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Domazet-Loso T, Brajković J, Tautz D. A phylostratigraphy approach to uncover the genomic history of major adaptations in metazoan lineages. Trends Genet. 2007;23:533–539. doi: 10.1016/j.tig.2007.08.014. [DOI] [PubMed] [Google Scholar]
- Fang H, Gough J. dcGO: database of domain-centric ontologies on functions, phenotypes, diseases and more. Nucleic Acids Res. 2013;41:D536–D544. doi: 10.1093/nar/gks1080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Federhen S. The NCBI taxonomy database. Nucleic Acids Res. 2012;40:D136–D143. doi: 10.1093/nar/gkr1178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forrest ARR, Kawaji H, Rehli M, Baillie JK, de Hoon MJL, Haberle V, Lassmann T, Kulakovskiy IV, Lizio M, Itoh M, et al. A promoter level mammalian expression atlas. Nature. 2014;507:462–470. doi: 10.1038/nature13182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geer LY, Domrachev M, Lipman DJ, Bryant SH. CDART: protein homology by domain architecture. Genome Res. 2002;12:1619–1623. doi: 10.1101/gr.278202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gough J. Convergent evolution of domain architectures (is rare) Bioinformatics. 2005;21:1464–1471. doi: 10.1093/bioinformatics/bti204. [DOI] [PubMed] [Google Scholar]
- Habib N, Wapinski I, Margalit H, Regev A, Friedman N. A functional selection model explains evolutionary robustness despite plasticity in regulatory networks. Mol Syst Biol. 2012;8:619. doi: 10.1038/msb.2012.50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffmann JA. Phylogenetic Perspectives in Innate Immunity. Science. 1999;284:1313–1318. doi: 10.1126/science.284.5418.1313. [DOI] [PubMed] [Google Scholar]
- Illergård K, Ardell DH, Elofsson A. Structure is three to ten times more conserved than sequence—a study of structural response in protein cores. Proteins. 2009;77:499–508. doi: 10.1002/prot.22458. [DOI] [PubMed] [Google Scholar]
- Janeway CA, Medzhitov R. Innate immune recognition. Annu Rev Immunol. 2002;20:197–216. doi: 10.1146/annurev.immunol.20.083001.084359. [DOI] [PubMed] [Google Scholar]
- Liu BA, Shah E, Jablonowski K, Stergachis A, Engelmann B, Nash PD. The SH2 domain-containing proteins in 21 species establish the provenance and scope of phosphotyrosine signaling in eukaryotes. Sci Signal. 2011;4 doi: 10.1126/scisignal.2002105. ra83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madera M, Gough J. A comparison of profile hidden Markov model procedures for remote homology detection. Nucleic Acids Res. 2002;30:4321–4328. doi: 10.1093/nar/gkf544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mardilovich K, Pankratz SL, Shaw LM. Expression and function of the insulin receptor substrate proteins in cancer. Cell Commun Signal. 2009;7:14. doi: 10.1186/1478-811X-7-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mistry J, Finn RD, Eddy SR, Bateman A, Punta M. Challenges in homology search: HMMER3 and convergent evolution of coiled-coil regions. Nucleic Acids Res. 2013;41:e121. doi: 10.1093/nar/gkt263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moore AD, Björklund AK, Ekman D, Bornberg-Bauer E, Elofsson A. Arrangements in the modular evolution of proteins. Trends Biochem Sci. 2008;33:444–451. doi: 10.1016/j.tibs.2008.05.008. [DOI] [PubMed] [Google Scholar]
- Moore AD, Grath S, Schüler A, Huylmans AK, Bornberg-Bauer E. Quantification and functional analysis of modular protein evolution in a dense phylogenetic tree. Biochim Biophys Acta. 2013;1834:898–907. doi: 10.1016/j.bbapap.2013.01.007. [DOI] [PubMed] [Google Scholar]
- Mukhopadhyay S, Grange P, Sengupta AM, Mitra PP. What does the allen gene expression atlas tell us about mouse brain evolution? 2012 arXiv:1206.0324. [Google Scholar]
- Murzin A, Brenner S, Hubbard T, Chothia C. SCOP: A structural classification of proteins database for the investigation of sequences and structures. J Mol Biol. 1995;247:536–540. doi: 10.1006/jmbi.1995.0159. [DOI] [PubMed] [Google Scholar]
- Oates ME, Romero P, Ishida T, Ghalwash M, Mizianty MJ, Xue B, Dosztányi Z, Uversky VN, Obradovic Z, Kurgan L, et al. D2P2: database of disordered protein predictions. Nucleic Acids Res. 2013;41(Database issue):D508–D516. doi: 10.1093/nar/gks1226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ogura Y, Inohara N, Benito A, Chen FF, Yamaoka S, Nunez G. Nod2, a Nod1/Apaf-1 family member that is restricted to monocytes and activates NF-kappaB. J Biol Chem. 2001;276:4812–4818. doi: 10.1074/jbc.M008072200. [DOI] [PubMed] [Google Scholar]
- Ponting CP. The functional repertoires of metazoan genomes. Nat Rev Genet. 2008;9:689–698. doi: 10.1038/nrg2413. [DOI] [PubMed] [Google Scholar]
- Qu BH, Karas M, Koval A, LeRoith D. Insulin receptor substrate-4 enhances insulin-like growth factor-I-induced cell proliferation. J Biol Chem. 1999;274:31179–31184. doi: 10.1074/jbc.274.44.31179. [DOI] [PubMed] [Google Scholar]
- Rosas-Ballina M, Ochani M, Parrish WR, Ochani K, Harris YT, Huston JM, Chavan S, Tracey KJ. Splenic nerve is required for cholinergic antiinflammatory pathway control of TNF in endotoxemia. Proc Natl Acad Sci U S A. 2008;105:11008–11013. doi: 10.1073/pnas.0803237105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosas-Ballina M, Tracey KJ. The neurology of the immune system: neural reflexes regulate immunity. Neuron. 2009;64:28–32. doi: 10.1016/j.neuron.2009.09.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Russell RJ, Hough DW, Danson MJ, Taylor GL. The crystal structure of citrate synthase from the thermophilic Archaeon, Thermoplasma acidophilum. Structure. 1994;2:1157–1167. doi: 10.1016/s0969-2126(94)00118-9. [DOI] [PubMed] [Google Scholar]
- Schreyer S. Insulin receptor substrate-4 is expressed in muscle tissue without acting as a substrate for the insulin receptor. Endocrinology. 2003;144:1211–1218. doi: 10.1210/en.2002-220723. [DOI] [PubMed] [Google Scholar]
- Tischendorf F. On the evolution of the spleen. Experientia. 1985;41:145–152. doi: 10.1007/BF02002606. [DOI] [PubMed] [Google Scholar]
- Toll-Riera M, Bosch N, Bellora N, Castelo R, Armengol L, Estivill X, Albà MM. Origin of primate orphan genes: a comparative genomics approach. Mol Biol Evol. 2009;26:603–612. doi: 10.1093/molbev/msn281. [DOI] [PubMed] [Google Scholar]
- UniProt Consortium. The universal protein resource (UniProt) in 2010. Nucleic Acids Res. 2010;38:D142–D148. doi: 10.1093/nar/gkp846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vesanto J. SOM-based data visualization methods. Intelligent Data Anal. 1999;3:111–126. [Google Scholar]
- Wang M, Caetano-Anollés G. The evolutionary mechanics of domain organization in proteomes and the rise of modularity in the protein world. Structure. 2009 doi: 10.1016/j.str.2008.11.008. (London, England: 1993) 17:66–78. [DOI] [PubMed] [Google Scholar]
- Wang W, Tang Y, Ni L, Kim E, Jongwutiwes T, Hourvitz A, Zhang R, Xiong H, Liu HC, Rosenwaks Z. Overexpression of Uromodulin-like1 accelerates follicle depletion and subsequent ovarian degeneration. Cell Death Dis. 2012;3:e433. doi: 10.1038/cddis.2012.169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao L, Wang K, Teng Y, Zhang J. Component plane presentation integrated self-organizing map for microarray data analysis. FEBS Lett. 2003;538:117–124. doi: 10.1016/s0014-5793(03)00156-x. [DOI] [PubMed] [Google Scholar]
- Yura K, Shionyu M, Hagino K, Hijikata A, Hirashima Y, Nakahara T, Eguchi T, Shinoda K, Yamaguchi A, Takahashi K, et al. Alternative splicing in human transcriptome: functional and structural influence on proteins. Gene. 2006;380:63–71. doi: 10.1016/j.gene.2006.05.015. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.