

Abstract
Objective:
A significant cause of spontaneous hemorrhages in the elderly is cerebral amyloid angiopathy (CAA), which causes degeneration of cerebral vessels, but the mechanisms are unclear.
Methods:
We isolated leptomeningeal vessels from rapidly autopsied brains (the average of postmortem intervals was 3.28 hours) from 9 patients with CAA and 10 age-matched controls, and used molecular, cell biology, and immunohistochemical approaches to examine β-site APP-cleaving enzyme 1 (BACE1) protein expression and enzymatic activities as well as tight junction molecular components in small- and medium-sized arteries of the cerebral cortex and leptomeninges.
Results:
We not only identified that the cerebral vessels, including leptomeningeal and cortical vessels, synthesize and express BACE1, but also found a significant elevation of both BACE1 protein levels and enzymatic activities in leptomeningeal vessels from patients with CAA. Moreover, overexpression of BACE1 in endothelial cells resulted in a significant reduction of occludin, a tight junction protein in blood vessels.
Conclusion:
These findings suggest that in addition to neurons, cerebral vascular cells express functional BACE1. Moreover, elevated vascular BACE1 may contribute to deficiency of occludin in cerebral vessels, which ultimately has a critical role in pathogenesis of CAA and its related hemorrhage.
Cerebral amyloid angiopathy (CAA) is an age-associated condition pathologically characterized by the deposition of β-amyloid (Aβ) protein in the medial layer of primarily small- and medium-sized arteries of the cerebral cortex and leptomeninges. One of its most common complications is CAA-related hemorrhage.1–3 β-site APP-cleaving enzyme 1 (BACE1) is a key enzyme in the generation of Aβ,4–6 and we and others have discovered elevated BACE1 activity in brains with Alzheimer disease (AD)7–9 and have reported a direct involvement of BACE1 in stroke and cell death.10 The degeneration of endothelial cells, smooth muscle cells, and pericytes, and the breakdown of the blood-brain barrier (BBB), can be observed in patients with CAA-related hemorrhage.11,12 In the present study, we found that, in clean isolated cerebral vessels with CAA, (1) BACE1 protein levels and enzyme activity were elevated; (2) tight junction protein ZO-1 and occludin levels were decreased; and (3) overexpressing BACE1 in vascular cells is correlated with the level reduction of occludin proteins. Our pathologic and cellular evidence suggests that the increased BACE1 level enhances cerebral hemorrhage in CAA brains.
METHODS
Standard protocol approvals, registrations, and patient consents.
Human tissue used in this study was collected with written informed consent of all subjects or next of kin and with approval from the ethical standards committee on human tissue samples and the institutional review boards of Sun Health Research Institute.
Collection of human cortical and leptomeningeal tissue.
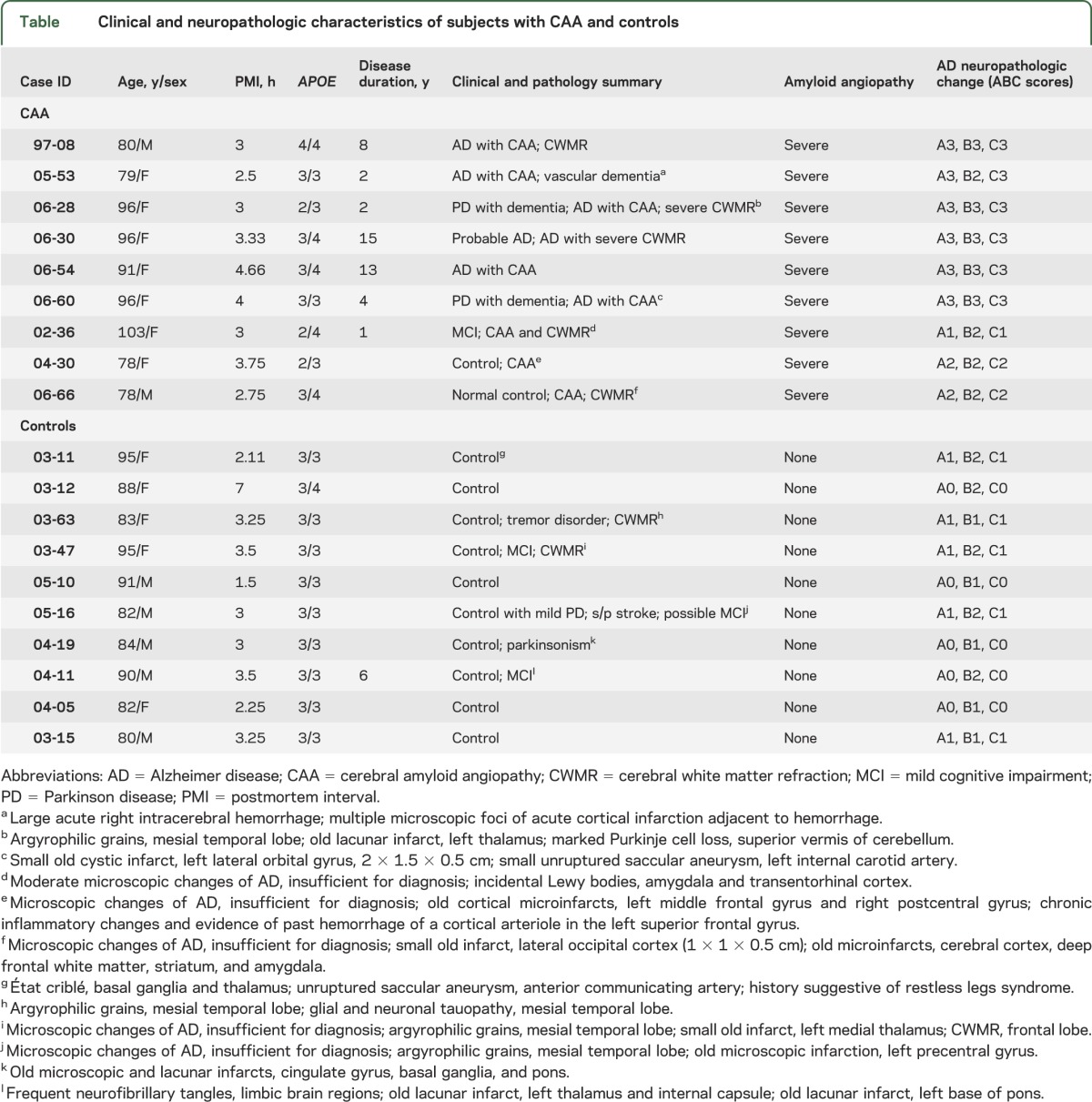
Human cortical and leptomeningeal tissues of subjects with CAA and controls were obtained from the Brain and Body Donation Program.13 CAA was diagnosed by postmortem pathologic examination, and the severity of CAA was graded according to the criteria reported by Vonsattel et al.14 CAA pathology was demonstrated in all autopsied CAA brains in our study (figure 1, E and F). As shown in the table, no CAA pathology was found in our control subjects, while all CAA brains exhibited severe CAA pathology. The average of postmortem intervals was 3.28 hours. We randomly selected 19 cases, consisting of 9 CAA cases and 10 controls. These 2 groups were well matched for age, sex, postmortem intervals, and sample preparations (table).
Figure 1. BACE1 expression in human cortical and leptomeningeal vessels.
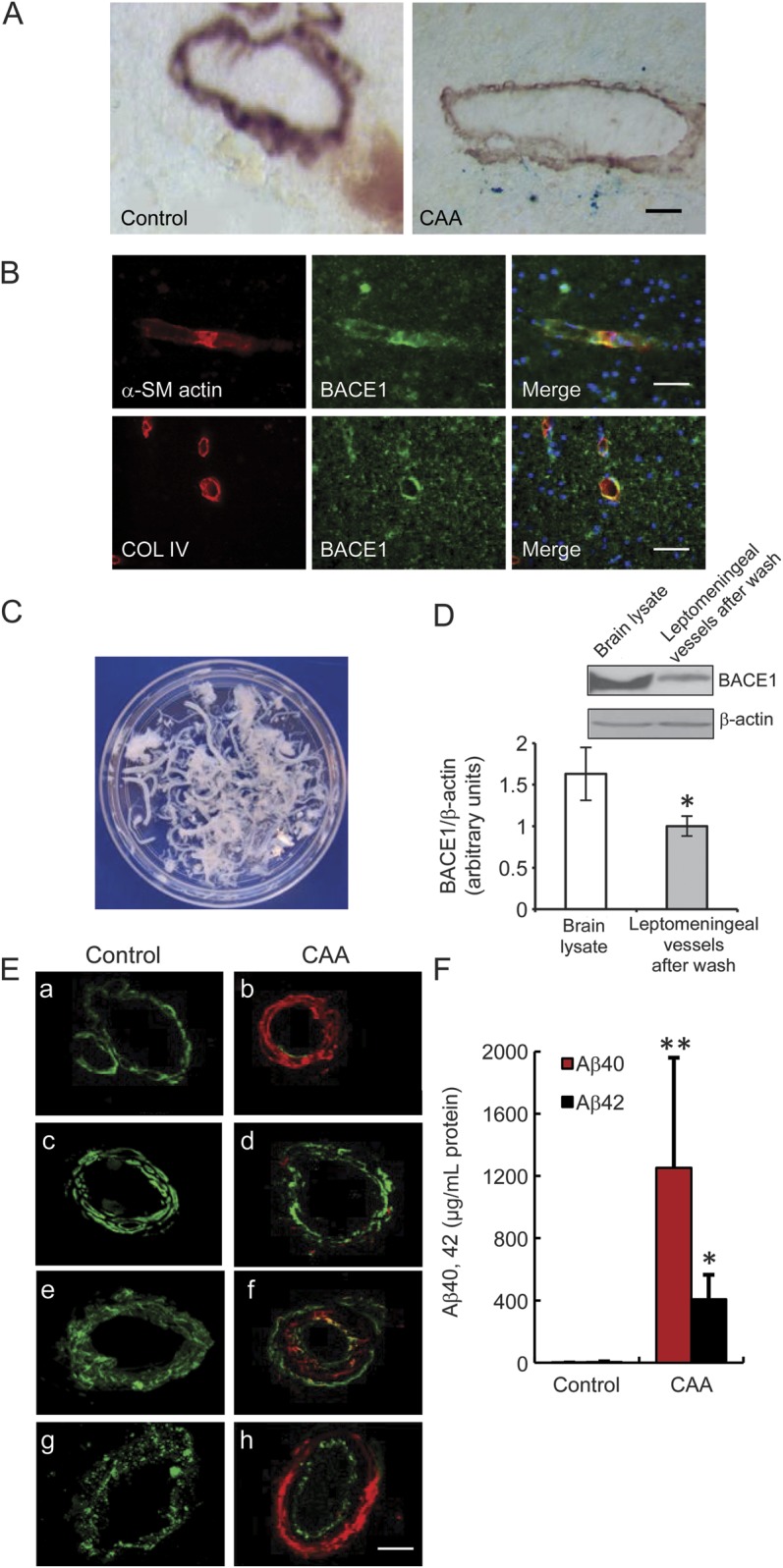
(A) Microbleeds were found in the brains of patients with CAA. The small arteries were identified with anti–α-SM actin, a smooth muscle cell marker. Cerebral microbleeds were identified by presence of hemosiderin (iron), using Prussian blue stain. A staining of ferric irons (blue) was observed in α-SM actin–positive vessels in the frontal cortex of patients with CAA, whereas it was not observed in the brains of controls without dementia. Bar: 50 μm. (B) BACE1 expression in the human cortical vessels marked by α-SM actin and collagen IV antibodies. Bar: 50 μm. (C) Leptomeninges were dissected from the brain surface in the immediate postmortem period and then each isolated vessel was washed with saline on ice until no single red blood cell could be observed, as seen in white color. (D) BACE1 expression in different tissues, including human frontal cortex and leptomeningeal vessels after wash, to confirm the expression of BACE1 in leptomeninges (*p < 0.05). (E) Vascular amyloid deposits and vascular degeneration in the leptomeningeal vessels of subjects with CAA and controls. A significant Aβ40 (red) deposition and loss of smooth muscle cells (α-SM actin, green) were observed in the leptomeningeal vessels of subjects with CAA (a, b). Aβ42 (red) deposition and loss of smooth muscle cells (α-SM actin, green) were observed in the leptomeningeal vessels of subjects with CAA (c, d). A significant Aβ (red) deposition was observed in the leptomeningeal vessels of subjects with CAA, which was labeled with antibody against the vessel basement membrane collagen IV (e, f, green), as well as the degeneration of endothelial cells, which was marked by antibody against von Willebrand factor (g, h, green). Bars: 20 μm. (F) Quantitative determination of Aβ40 and Aβ42 in the leptomeningeal vessels of patients with CAA and controls by ELISA assay. The leptomeningeal vessels of patients with CAA showed a significantly increased amount of Aβ40 and Aβ42 proteins compared with those of controls, which confirmed that the investigated leptomeningeal vessel samples showed the presence of CAA (*p < 0.05, **p < 0.01). Error bars represent SD. Aβ = β-amyloid; α-SM = α-smooth muscle; BACE1 = β-site APP-cleaving enzyme 1; CAA = cerebral amyloid angiopathy; COL = collagen.
Table.
Clinical and neuropathologic characteristics of subjects with CAA and controls
Preparation of leptomeningeal vessel samples.
Leptomeningeal tissue was prepared as described with modification.15 In brief, the larger vessels were partially transected to facilitate the release of entrapped blood. To promote hemolysis and prevent contamination, the specimens were rinsed 20 times with cold saline containing 0.01% sodium azide on a shaker at 4°C for 24 hours. The vascular network became completely colorless and transparent. Blood vessels larger than 1 mm in diameter were removed. The whole procedure was performed on ice.
Cell culture and Aβ1–42 peptide treatment.
Human aortic vascular smooth muscle cells (HA-VSMCs), human umbilical vein endothelial cells (HUVECs), and murine brain endothelial cell line bEnd.3 were purchased from American Type Culture Collection (Manassas, VA). Before treating the bEnd.3 cells, Aβ1–42 peptide (Millipore, Billerica, MA) was preaggregated at 37°C overnight and then bEnd.3 cells were exposed to 5 μM Aβ1–42 for 24 hours.
Western blot.
The samples of human frontal cortex, leptomeningeal vessels, and cells were individually homogenized in 1× RIPA buffer, and 50 to 100 μg of total protein was subjected to sodium dodecyl sulfate–polyacrylamide gel electrophoresis as previously described.7 The transferred polyvinylidene difluoride membrane blots were probed with the following antibodies: anti-BACE1 C-terminus polyclonal antibody (Calbiochem), anti-BACE1 monoclonal antibody (R&D Systems, Minneapolis, MN), anti-occludin monoclonal antibody (BD Biosciences, Franklin Lakes, NJ), anti–ZO-1 polyclonal antibody (Santa Cruz Biotechnology, Dallas, TX), anti-APP C-terminal polyclonal antibody (Calbiochem), and anti–β-actin monoclonal antibody (Sigma-Aldrich, St. Louis, MO).
BACE1 messenger RNA expression by reverse transcription PCR.
Total RNA was extracted from HA-VSMCs and HUVECs using TRIzol reagent (Invitrogen). As previously described,16 complementary DNA templates from samples were prepared from 1 µg of total RNA using the SuperScript III First-Strand Synthesis System (Invitrogen) followed by 30 PCR amplification cycles (94°C for 30 seconds, annealing at 55°C for 30 seconds, and extension at 72°C for 30 seconds). The PCR forward primer for human BACE1 was 5′-GAGGTATCGACCACTCGCTG-3′, and the reverse primer for human BACE1 was 5′-ACAGCTCCCATAACAGTGCC-3′. Fifteen microliters of each PCR product was analyzed by 1.5% agarose gel electrophoresis.
Evaluation of microvascular degeneration by immunohistochemistry and immunocytochemistry.
The fixed frontal cortex tissues were sectioned to 30 μm. Cells (1 × 105 cells/mL) were fixed with 4% paraformaldehyde on 8-well chamber slides. The sections and cells were sequentially treated with 0.15% Triton X-100 for 10 minutes and 10% goat serum for 30 minutes. Primary antibodies for BACE1 examination were applied as used in the Western blot. Anti-Aβ40 polyclonal antibody (Chemicon), anti-Aβ42 polyclonal antibody (Invitrogen), and anti-Aβ monoclonal antibody (6E10, Chemicon) were applied for Aβ detection. For microvascular degeneration, the primary antibodies for vascular smooth muscle cells, vascular endothelial cells, as well as tight junction proteins were applied as follows: anti–α-smooth muscle (α-SM) actin monoclonal antibody (Sigma-Aldrich), anti–collagen IV polyclonal antibody (Abcam, Cambridge, UK), anti–PECAM-1 (CD31) monoclonal antibody (Chemicon), anti–von Willebrand factor (vWF) polyclonal antibody (Chemicon), anti-vWF monoclonal antibody (Abcam), and anti–ZO-1 polyclonal antibody (Santa Cruz Biotechnology). Fluorescent-labeling Alexa Fluor 488- or 568-conjugated secondary antibodies against rabbit and mouse immunoglobulin G were used for detection (Invitrogen). The images were taken with a FluoView FV1000 confocal microscope (Olympus, Melville, NY).
Aβ ELISA assay.
The samples of leptomeningeal vessels were homogenized in homogenization buffer as we reported previously.7 An aliquot of the homogenate was dissolved in formic acid and neutralized with a neutralization buffer. Protein concentration was measured by protein assay (Bio-Rad Laboratories, Hercules, CA). Aβ40 and Aβ42 were measured with Aβ40 and Aβ42 ELISA kits (Invitrogen). Four technical replicates were performed for each Aβ determination.
BACE1 enzymatic activity assay.
BACE1 activity assays were performed as we described.17 In brief, samples of leptomeningeal vessels and cell lysate were lysed with a lysis buffer. BACE1 activity assays were performed by using synthetic peptide substrates containing Swedish mutant BACE1 cleavage site (Calbiochem). BACE1 substrate was dissolved in dimethylsulfoxide and mixed with 100 mM Tris-HCl and 100 mM NaCl, pH 4.5, reaction buffer. An equal amount of protein was mixed with 100 μL of substrate, and fluorescence intensity was measured with a microplate reader (BioTek, Winooski, VT) at an excitation wavelength of 430 nm and an emission wavelength of 520 nm.
Transient transfection and BACE1 inhibitor treatment.
The pcDNA3.1-BACE1 (provided by Dr. R. Yan) and APP Swedish mutation (K595N/M596L, APPsw) plasmids were prepared as described previously.18 We transfected bEnd.3 cells with different amounts of pcDNA3.1-BACE1 and/or 1 μg APPsw plasmids using Effectene (Qiagen, Venlo, Limburg). Cells were collected after transfection for Western blot analysis. The BACE1 inhibitor (1-R-45) was provided by Dr. R. Petukhov,19 BACE1 inhibitor C3 was from Calbiochem (β-secretase inhibitor IV), and cathepsin D inhibitor pepstatin A was from Millipore. All of experiments were independently replicated 3 times.
Statistical analyses.
Results were expressed as mean ± SD. All analyses were performed using SPSS version 11.5.1 (SPSS Inc., Chicago, IL). Differences between 2 groups were assessed using Student t tests. Differences between 3 or more groups were evaluated by one-way analysis of variance. The level of significance was p < 0.05.
RESULTS
BACE1 is expressed in cerebral vascular cells.
The pathologic diagnosis of CAA was consistent with the National Institute on Aging–Alzheimer's Association guidelines.20 One of the pathologic characteristics of patients with CAA was cerebral microbleeds, and ferric iron detection (figure 1A) was used to observe cerebral microbleeds for confirmation of the diagnosis of CAA (table). Results showed ferric iron staining close to small arteries, which were identified with an antibody against α-SM actin (a smooth muscle cell marker), whereas ferric iron staining was not found in the brain of the controls (figure 1A). Meanwhile, the thickness of the vessel walls was reduced in the brain of the CAA subject in comparison with that of the control subject (figure 1A). Recently, one study reported that mouse microvascular cells in cultures express BACE1.21 To examine whether BACE1 was present in human cerebral vessels in vivo, especially in the cerebral vessels from CAA, a double-labeling immunohistochemical technique was performed in the brains with CAA. We found BACE1 expression in human cortical vessels identified with 2 vessel markers: α-SM actin and collagen IV (figure 1B). Evidence has shown that leptomeningeal vessels are one of the vulnerable cerebral vessels, in both CAA and AD, and exhibited Aβ deposition (figure 1, E and F). To examine whether BACE1 is expressed in leptomeningeal vessels, leptomeningeal vessels of CAA and control subjects were dissected from the brain surface in the immediate postmortem period. Because of high levels of BACE1 protein in the blood,22 and to rule out a possibility that the presence of BACE1 in the vessels were residuals from blood BACE1, each single vessel was washed with saline on ice until no single red blood cell could be observed (figure 1C). Using Western blotting technique, we identified the presence of BACE1 in cleaned leptomeningeal vessels, although its expression levels were lower than that in neurons (figure 1D).
Because CAA often shows a patchy distribution, we performed immunohistochemistry (figure 1E) and Aβ ELISA assay (figure 1F) to confirm the presence of CAA in the investigated leptomeningeal vessel samples, and to demonstrate vascular amyloid (Aβ40) deposits (figure 1, Ea and Eb) and vascular degeneration in the leptomeningeal vessels of CAA and control subjects. Loss of smooth muscle cells (α-SM actin, green) was observed in the leptomeningeal vessels of subjects with CAA (figure 1, Ea–Ed). The antibody against the vessel basement membrane collagen IV was labeled green (figure 1, Ee and Ef), and the degeneration of endothelial cells, which were marked by antibody against vWF (figure 1, Eg and Eh, green), was observed in the leptomeningeal vessels of subjects with CAA. Quantitative determination of Aβ40 and Aβ42 in the leptomeningeal vessels of CAA and control subjects were performed using ELISA (figure 1F). The leptomeningeal vessels of subjects with CAA showed significant increases in the amount of Aβ40 and Aβ42 protein compared with those of control subjects, which confirmed that the investigated leptomeningeal vessel samples showed the presence of CAA.
To further determine what types of cerebral vascular cells express BACE1, we examined BACE1 expression in the 3 corresponding cell types: HA-VSMC, HUVEC, and bEnd.3 cells. These 2 endothelial cells exhibit the same or similar features as human cerebral endothelial cells. Of note, we found that all 3 cell types express not only BACE1 protein, which is mainly located in the cytoplasm of those cells (figure 2A), but also express BACE1 messenger RNA (mRNA) (figure 2B), which suggests that BACE1 can be synthesized in vascular cells. The presence of BACE1 protein in all 3 related vascular cells was confirmed by 2 different BACE1 monoclonal and polyclonal antibodies (data not shown).
Figure 2. BACE1 expression in different vascular cells.
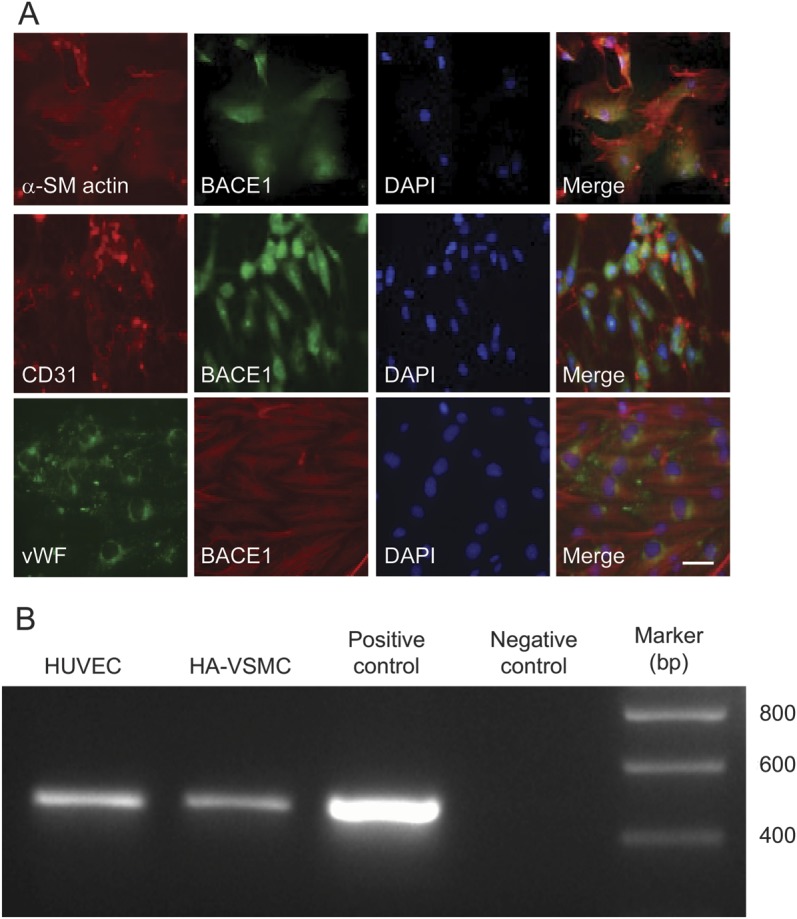
(A) BACE1 expression in different cells of the vascular walls, including HA-VSMCs, HUVECs, and murine cerebral microvessel endothelial cells (bEnd.3 cells). α-SM actin, CD31, and vWF were used as cell markers for each cell line. BACE1 is mainly located in the cytoplasm. Bar: 10 μm. (B) BACE1 mRNA expression in 2 types of human vascular cells. Reverse transcription PCR was performed to detect the BACE1 mRNA expression in HA-VSMCs and HUVECs. The 478-bp DNA band using a BACE1-specific primer set for the BACE1 mRNA was detected at both cell lines. BACE1 plasmid and water were used as positive and negative controls in PCR experiments. α-SM = α-smooth muscle; BACE1 = β-site APP-cleaving enzyme 1; bp = base pairs; DAPI = 4',6-diamidino-2-phenylindole; HA-VSMC = human aortic vascular smooth muscle cell; HUVEC = human umbilical vein endothelial cell; mRNA = messenger RNA; vWF = von Willebrand factor.
Elevated BACE1 is detected in cortical microvascular cells and isolated leptomeningeal vessels from rapidly autopsied brains of subjects with CAA.
To examine whether BACE1 levels were elevated in CAA leptomeningeal vessels where hemorrhage often occurs, we compared BACE1 protein and activity levels in isolated leptomeningeal vessels from 9 patients with CAA and 10 controls and found that BACE1 protein expression levels were significantly increased in the CAA group compared with the controls; similarly, elevated BACE1 enzyme activities were discovered in the CAA group (figure 3, B and C). To further confirm the results of high BACE1 enzyme activity found in leptomeningeal vessels, C99, the C-terminal fragment cleaved from APP by BACE1, was found to be significantly increased in the leptomeningeal vessels of subjects with CAA compared with that in control individuals (figure 3D). We have also analyzed BACE1 expression and activity levels in the leptomeningeal vessels of patients with AD without moderate-severe CAA (n = 10) (figure e-1 on the Neurology® Web site at Neurology.org).
Figure 3. Elevation of BACE1 protein levels and enzymatic activity are associated with the deficiency of tight junction proteins in the cortex and leptomeningeal vessels of patients with CAA.

(A) Reduced ZO-1 expression was observed in the cortical vessels of the CAA group compared with controls. Bar: 20 μm. (B, C) BACE1 protein expression and activities were demonstrated in the leptomeningeal vessels of controls and subjects with CAA. (D) An increased expression of C99, the C-terminal fragment cleaved from amyloid precursor protein by BACE1, was shown in leptomeningeal vessels of patients with CAA compared with those in control individuals. (E, F) The expressions of both occludin and ZO-1 were decreased in CAA leptomeningeal vessels compared with controls. (G) The BACE1 activity in the leptomeningeal vessels of subjects with CAA was negatively correlated with occludin protein levels. (H) BACE1 protein levels were negatively correlated with occludin protein levels in the leptomeningeal vessels of subjects with CAA. All data are the mean ± SD of 3 independent measurements. Error bars represent SD. *p < 0.05 vs control sample. BACE1 = β-site APP-cleaving enzyme 1; CAA = cerebral amyloid angiopathy; vWF = von Willebrand factor.
ZO-1 and occludin, 2 tight junction proteins, are deficient in cerebral vessels from CAA brains.
ZO-1 and occludin are 2 critical components of tight junction proteins for endothelial cells.23 To examine whether expression levels of ZO-1 and occludin were decreased in the cerebral vessels of CAA brains, double immunostaining of vWF (an endothelial maker) and ZO-1 (a tight junction marker) was performed, and results showed that ZO-1 expression was reduced in CAA brains with loss of vWF expression as well in the cortical microvascular cells (capillaries) (figure 3A). Furthermore, we isolated and cleaned leptomeningeal vessels from CAA brains and used Western blot methods to examine the levels of both occludin and ZO-1 expression in leptomeningeal vessels and found that levels of occludin and ZO-1 were significantly reduced in the CAA group compared with controls (figure 3, E and F). This finding is consistent with evidence that occludin and ZO-1 were lost, probably due to Aβ toxicity and BBB alteration in CAA.24,25
Elevation of BACE1 is conversely correlated with decrease of occludin in cerebral vessels with CAA.
To study whether the same leptomeningeal vessels that exhibit an increase of BACE1 activity show deficiency of occludin and whether elevation of BACE1 proteins has any converse correlation with occludin deficiency, we performed a linear regression analysis between occludin protein levels and BACE1 activity levels, and occludin and BACE1 protein levels. We found that occludin expression was significantly negatively correlated with the increased BACE1 activity (figure 3G) and protein levels (figure 3H) in the leptomeningeal vessels from patients with CAA.
Overexpression of BACE1 results in occludin deficiency.
To further study whether BACE1 increase causes occludin deficiency, we overexpressed BACE1 in endothelial cells and detected overexpressed BACE1 activity after the transfection (figure 4A). To examine whether transfection of BACE1-induced increased BACE1 activity is specific and also to rule out the possibility that other enzymes, i.e., cathepsin D, cleaves Swedish peptide in APP or APPsw-transfected cells, we transfected APPsw plus BACE1 plasmids with treatment of BACE1 inhibitor C3. We found that APPsw plus BACE1 cotransfected cells exhibited more C99, the N-terminal fragment of APP cleaved by BACE1. It is important to note that the BACE1 inhibitor C3 significantly reduced APP C99 (figure 4B), suggesting that our assay of BACE1 activity is specific. Moreover, we also examined the effects of treatment of some representative samples with pepstatin A, an inhibitor of a number of aspartyl proteases such as cathepsin D that is known to not affect BACE1 activity. To examine this possibility, we treated BACE1 stable cell lysate and human brain lysate with BACE1 inhibitor C3 and pepstatin A, respectively. As shown in figure 4C, we found that the BACE1 inhibitor C3 significantly reduced BACE1 activity as expected, but BACE1 activity had no change with pepstatin treatment, suggesting that pepstatin A indeed does not affect BACE1 and our assay of BACE1 activity is specific. We transfected different doses of BACE1 and found significantly decreased levels of occludin in endothelial cells at 48 hours (figure 4D). To explore the potential mechanistic link between elevated Aβ and aberrant expression of tight junction proteins in endothelial cells, we used Aβ1–42 peptide (5 μM) to treat the BACE1-transfected cerebral endothelial cell line, bEnd.3 cells. We observed a significant reduction of occludin protein levels in bEnd.3 cells treated with Aβ1–42 peptide (figure 4E), which supports Aβ as a potential causal toxic factor in tight junction defects. Of note, however, when the BACE1 inhibitor (1-R-45) was given at 1 and 10 pM, after 48 hours, the decline of the occludin level in endothelial cells was prevented (figure 4F). Together, these results indicate a novel mechanism by which elevation of angio-BACE1 results in occludin deficiency.
Figure 4. BACE1 overexpression results in a downregulation of occludin protein levels.

(A) BACE1 activity levels were significantly elevated in the bEnd.3 cells transfected with pcDNA-BACE1 in the BACE1 concentration of ≥0.4 μg (*p < 0.05). After adding 1 and 10 pM BACE1 inhibitor, BACE1 activity was significantly reduced (#p < 0.05). (B) BACE1 inhibitor reduces the APP C99 generation in bEnd.3 cells. bEnd.3 cells were transfected with APP Swedish mutation (K595N/M596L) and BACE1 plasmids for 24 hours in the presence of BACE1 inhibitor C3 (3 µM) and then subjected to Western blot analysis. (C) BACE1 activity was inhibited by the β-secretase inhibitor C3 (10 nM, BACE1 inhibitor IV) but not by cathepsin D inhibitor pepstatin A (1 μM) in human brain sample and BACE1 cell lysate. (D) Overexpressing BACE1 in bEnd.3 cells, occludin protein levels were significantly reduced with the transfected BACE1 amounts of 0.4 and 0.8 μg. (E) Both overexpressing BACE1 and Aβ1–42 treatment resulted in occludin deficiency. bEnd.3 cells were transfected with empty vector or pcDNA-BACE1 for 24 hours and then treated with 5 μM Aβ1–42 for 24 hours. After 48 hours, the cells were harvested and subjected to Western blot analysis. (F) After adding 1 and 10 pM BACE1 inhibitor, occludin deficiency in bEnd.3 cells with BACE1 overexpression was significantly rescued. All data are the mean ± SD of 3 independent measurements. Error bars represent SD. *p < 0.05 vs control sample. Aβ = β-amyloid; BACE1 = β-site APP-cleaving enzyme 1.
DISCUSSION
To our knowledge, this is the first report to demonstrate that human cerebral vascular cells express BACE1 in vitro and in vivo. Moreover, this is also the first report of BACE1 elevation in the leptomeningeal vessels and capillaries (microvessels) from brains of patients with CAA. We and others previously discovered significant elevation of BACE1 in AD brains,7–9 and knockout of BACE1 has resulted in reduced Aβ-mediated AD-like pathology in vivo.26,27 We noticed that BACE1 has different roles in different diseased conditions, including AD, ischemia, multiple sclerosis, and even schizophrenia via distinct mechanisms.4–6,10,28,29 Our findings suggest that overexpressing BACE1 in cerebral vessels results in their destruction via downregulation of tight junction proteins.
Although neurons are still considered a major source for BACE1 expression and Aβ production in the AD brain,30 our findings of BACE1 expression in vascular cells support the concept that BACE1 can be generated in vascular cells, and abnormal regulation of BACE1 may contribute to deficiency of BBB, especially ZO-1 and occludin. However, it is still unclear whether there is any direct link between neuronal and vascular BACE1, which needs significant study with rigorous approaches.
In the present study, analyses of BACE1 protein and enzymatic activity levels were performed using the same tissue samples, isolated leptomeningeal vessels, from CAA and nonpathologic age-matched controls that were collected, stored, and processed similarly. Notably, whether BACE1 mRNA levels change in CAA brains is not clear. However, based on our findings on BACE1 mRNA in AD brains,17 we assumed that BACE1 mRNA levels are possibly elevated in leptomeningeal vessels. This assumption is indirectly supported by our findings that an inflammatory molecule, tumor necrosis factor-α, mediates BACE1 transcriptional levels through regulation of nuclear factor κB binding sites on BACE1 promoter.17,31 Nonetheless, the findings that an increase in BACE1 activities in leptomeningeal vessels suggest more generation of Aβ and eventually contribute to CAA pathology.
The decrease of occludin and ZO-1 protein levels in patients with CAA suggests breakdown of the BBB, which is consistent with newly published evidence of BBB impairment in CAA. Moreover, we have also examined several related proteins, e.g., collagen IV, but observed little to no changes in those proteins, suggesting that the decrease of occludin and ZO-1 in CAA brains is specific. The lack of vWF and ZO-1 staining in CAA samples might be a sign of BBB damage with changes in the tight junction in endothelial cells as a part of the CAA pathology, as currently reported in human and animal models.32,33 Besides hemorrhage or microbleeds, the degradation of tight junction proteins may also contribute to the progression of white matter lesions and silent ischemic infarcts observed in CAA.34,35 The finding that the BACE1 inhibitor protects BACE1-mediated occludin degradation indicates a new possible therapeutic target for cerebral vascular degeneration. Because Aβ causes tight junction defects (figure 4E), whether a novel BACE1 mechanism beyond Aβ is involved in CAA needs to be elucidated in future studies. Moreover, it is not clear whether the roles of BACE1 in the pathogenesis of BBB disruption may exist in other, nonrelated pathologic conditions characterized by BBB opening, i.e., meningitis, brain abscess, and multiple sclerosis, and this needs further study.
Supplementary Material
ACKNOWLEDGMENT
The samples were from the Brain and Tissue Bank, Sun Health Research Institute.
GLOSSARY
- Aβ
β-amyloid
- AD
Alzheimer disease
- α-SM
α-smooth muscle
- BACE1
β-site APP-cleaving enzyme 1
- BBB
blood-brain barrier
- CAA
cerebral amyloid angiopathy
- HA-VSMC
human aortic vascular smooth muscle cell
- HUVEC
human umbilical vein endothelial cell
- mRNA
messenger RNA
- vWF
von Willebrand factor
Footnotes
Supplemental data at Neurology.org
AUTHOR CONTRIBUTIONS
Drs. Cheng, He, and Yao performed biochemical and immunochemical experiments and participated in data analyses. Dr. Cheng wrote the manuscript draft. Dr. Dong made clinical suggestions and comments. Dr. Shen and Dr. Li initiated the project and designed the experiments, participated in data analyses, overviewed manuscript drafting, and finalized the final version of the manuscript. All authors have read and approved the final manuscript.
STUDY FUNDING
Supported by grants from NIH/National Institute on Aging (NIA) 025888 (Y.S.), NIH/NIA R01AG032441 (R.L.); Alzheimer’s Association IIRG-09-61521 (Y.S.) and IIRG-07-59510 (R.L.); AHAF G2006-118 (R.L.), A2008-642 (P.H.); National Natural Science Foundation of China 81100861 (X.C.); and Specialized Research Fund for the Doctoral Program of Higher Education 20100071120081 (X.C.). The Brain and Body Donation Program is supported by the National Institute of Neurological Disorders and Stroke (U24NS072026 National Brain and Tissue Resource), NIA (P30AG19610 Arizona Alzheimer's Disease Core Center), and the Arizona Department of Health Services (contract 211002, Arizona Alzheimer's Research Center).
DISCLOSURE
The authors report no disclosures relevant to the manuscript. Go to Neurology.org for full disclosures.
REFERENCES
- 1.Greenberg SM. Cerebral amyloid angiopathy: prospects for clinical diagnosis and treatment. Neurology 1998;51:690–694 [DOI] [PubMed] [Google Scholar]
- 2.Biffi A, Anderson CD, Jagiella JM, et al. APOE genotype and extent of bleeding and outcome in lobar intracerebral haemorrhage: a genetic association study. Lancet Neurol 2011;10:702–709 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ellis RJ, Olichney JM, Thal LJ, et al. Cerebral amyloid angiopathy in the brains of patients with Alzheimer's disease: the CERAD experience, part XV. Neurology 1996;46:1592–1596 [DOI] [PubMed] [Google Scholar]
- 4.Vassar R, Bennett BD, Babu-Khan S, et al. Beta-secretase cleavage of Alzheimer's amyloid precursor protein by the transmembrane aspartic protease BACE. Science 1999;286:735–741 [DOI] [PubMed] [Google Scholar]
- 5.Sinha S, Anderson JP, Barbour R, et al. Purification and cloning of amyloid precursor protein beta-secretase from human brain. Nature 1999;402:537–540 [DOI] [PubMed] [Google Scholar]
- 6.Yan R, Bienkowski MJ, Shuck ME, et al. Membrane-anchored aspartyl protease with Alzheimer's disease beta-secretase activity. Nature 1999;402:533–537 [DOI] [PubMed] [Google Scholar]
- 7.Yang LB, Lindholm K, Yan R, et al. Elevated beta-secretase expression and enzymatic activity detected in sporadic Alzheimer disease. Nat Med 2003;9:3–4 [DOI] [PubMed] [Google Scholar]
- 8.Fukumoto H, Cheung BS, Hyman BT, Irizarry MC. Beta-secretase protein and activity are increased in the neocortex in Alzheimer disease. Arch Neurol 2002;59:1381–1389 [DOI] [PubMed] [Google Scholar]
- 9.Holsinger RM, McLean CA, Beyreuther K, Masters CL, Evin G. Increased expression of the amyloid precursor beta-secretase in Alzheimer's disease. Ann Neurol 2002;51:783–786 [DOI] [PubMed] [Google Scholar]
- 10.Tesco G, Koh YH, Kang EL, et al. Depletion of GGA3 stabilizes BACE and enhances beta-secretase activity. Neuron 2007;54:721–737 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hunter JM, Kwan J, Malek-Ahmadi M, et al. Morphological and pathological evolution of the brain microcirculation in aging and Alzheimer's disease. PLoS One 2012;7:e36893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Okazaki H, Reagan TJ, Campbell RJ. Clinicopathologic studies of primary cerebral amyloid angiopathy. Mayo Clin Proc 1979;54:22–31 [PubMed] [Google Scholar]
- 13.Beach TG, Sue LI, Walker DG, et al. The Sun Health Research Institute Brain Donation Program: description and experience, 1987–2007. Cell Tissue Bank 2008;9:229–245 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Vonsattel JP, Myers RH, Hedley-Whyte ET, Ropper AH, Bird ED, Richardson EP., Jr Cerebral amyloid angiopathy without and with cerebral hemorrhages: a comparative histological study. Ann Neurol 1991;30:637–649 [DOI] [PubMed] [Google Scholar]
- 15.Roher AE, Kuo YM, Esh C, et al. Cortical and leptomeningeal cerebrovascular amyloid and white matter pathology in Alzheimer's disease. Mol Med 2003;9:112–122 [PMC free article] [PubMed] [Google Scholar]
- 16.Li R, Lindholm K, Yang LB, et al. Amyloid beta peptide load is correlated with increased beta-secretase activity in sporadic Alzheimer's disease patients. Proc Natl Acad Sci USA 2004;101:3632–3637 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.He P, Zhong Z, Lindholm K, et al. Deletion of tumor necrosis factor death receptor inhibits amyloid beta generation and prevents learning and memory deficits in Alzheimer's mice. J Cell Biol 2007;178:829–841 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zhong Z, Ewers M, Teipel S, et al. Levels of beta-secretase (BACE1) in cerebrospinal fluid as a predictor of risk in mild cognitive impairment. Arch Gen Psychiatry 2007;64:718–726 [DOI] [PubMed] [Google Scholar]
- 19.Chirapu SR, Pachaiyappan B, Nural HF, et al. Molecular modeling, synthesis, and activity studies of novel biaryl and fused-ring BACE1 inhibitors. Bioorg Med Chem Lett 2009;19:264–274 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hyman BT, Phelps CH, Beach TG, et al. National Institute on Aging–Alzheimer's Association guidelines for the neuropathologic assessment of Alzheimer's disease. Alzheimers Dement 2012;8:1–13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Austin SA, Santhanam AV, Katusic ZS. Endothelial nitric oxide modulates expression and processing of amyloid precursor protein. Circ Res 2010;107:1498–1502 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Shen Y, He P, Cheng X, Ewers M, et al. Plasma BACE1 levels are increased in patients of mild cognitive impairment and Alzheimer's disease. Alzheimer's Association 13th International Conference on Alzheimer's Disease and Related Disorders; 2011; Paris
- 23.Zlokovic BV. The blood-brain barrier in health and chronic neurodegenerative disorders. Neuron 2008;57:178–201 [DOI] [PubMed] [Google Scholar]
- 24.Carrano A, Hoozemans JJ, van der Vies SM, Rozemuller AJ, van Horssen J, de Vries HE. Amyloid beta induces oxidative stress-mediated blood-brain barrier changes in capillary amyloid angiopathy. Antioxid Redox Signal 2011;15:1167–1178 [DOI] [PubMed] [Google Scholar]
- 25.Tai LM, Holloway KA, Male DK, Loughlin AJ, Romero IA. Amyloid-beta-induced occludin down-regulation and increased permeability in human brain endothelial cells is mediated by MAPK activation. J Cell Mol Med 2010;14:1101–1112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Cai H, Wang Y, McCarthy D, et al. BACE1 is the major beta-secretase for generation of Abeta peptides by neurons. Nat Neurosci 2001;4:233–234 [DOI] [PubMed] [Google Scholar]
- 27.Luo Y, Bolon B, Kahn S, et al. Mice deficient in BACE1, the Alzheimer's beta-secretase, have normal phenotype and abolished beta-amyloid generation. Nat Neurosci 2001;4:231–232 [DOI] [PubMed] [Google Scholar]
- 28.Hu X, Hicks CW, He W, et al. Bace1 modulates myelination in the central and peripheral nervous system. Nat Neurosci 2006;9:1520–1525 [DOI] [PubMed] [Google Scholar]
- 29.Seshadri S, Kamiya A, Yokota Y, et al. Disrupted-in-schizophrenia-1 expression is regulated by beta-site amyloid precursor protein cleaving enzyme-1-neuregulin cascade. Proc Natl Acad Sci USA 2010;107:5622–5627 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zhao J, O'Connor T, Vassar R. The contribution of activated astrocytes to Abeta production: implications for Alzheimer's disease pathogenesis. J Neuroinflammation 2011;8:150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Cheng X, He P, Lee T, Yao H, Li R, Shen Y. High activities of BACE1 in brains with mild cognitive impairment. Am J Pathol 2014;184:141–147 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Carrano A, Hoozemans JJ, van der Vies SM, van Horssen J, de Vries HE, Rozemuller AJ. Neuroinflammation and blood-brain barrier changes in capillary amyloid angiopathy. Neurodegener Dis 2012;10:329–331 [DOI] [PubMed] [Google Scholar]
- 33.Hartz AM, Bauer B, Soldner EL, et al. Amyloid-beta contributes to blood-brain barrier leakage in transgenic human amyloid precursor protein mice and in humans with cerebral amyloid angiopathy. Stroke 2012;43:514–523 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Chen YW, Gurol ME, Rosand J, et al. Progression of white matter lesions and hemorrhages in cerebral amyloid angiopathy. Neurology 2006;67:83–87 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kimberly WT, Gilson A, Rost NS, et al. Silent ischemic infarcts are associated with hemorrhage burden in cerebral amyloid angiopathy. Neurology 2009;72:1230–1235 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}