

Abstract
Palindromes are symmetrical words of DNA in the sense that they read exactly the same as their reverse complementary sequences. Representing the occurrences of palindromes in a DNA molecule as points on the unit interval, the scan statistics can be used to identify regions of unusually high concentration of palindromes. These regions have been associated with the replication origins on a few herpesviruses in previous studies. However, the use of scan statistics requires the assumption that the points representing the palindromes are independently and uniformly distributed on the unit interval. In this paper, we provide a mathematical basis for this assumption by showing that in randomly generated DNA sequences, the occurrences of palindromes can be approximated by a Poisson process. An easily computable upper bound on the Wasserstein distance between the palindrome process and the Poisson process is obtained. This bound is then used as a guide to choose an optimal palindrome length in the analysis of a collection of 16 herpesvirus genomes. Regions harboring significant palindrome clusters are identified and compared to known locations of replication origins. This analysis brings out a few interesting extensions of the scan statistics that can help formulate an algorithm for more accurate prediction of replication origins.
Keywords: DNA palindromes, Wasserstein distance, Poisson process approximation, scan statistics, replication origins
1. INTRODUCTION
DNA palindromes are words from the nucleotide base alphabet that are symmetrical in the sense that they read exactly the same as their complementary sequences in the reverse direction (see Fig. 1(a)). A DNA palindrome is necessarily even in length because the middle base in any odd-length nucleotide string cannot be identical to its complement. Palindromes are involved in a variety of biological processes. For example, the recognition sites for bacterial restriction enzymes to cut foreign DNA are mostly palindromic (Waterman, 1995, Chapter 2). Palindromes also play important roles in gene regulation and DNA replication processes (Wagner, 1991, Chapters 6, 12, 18; Kornberg and Baker, 1992, Chapter 1). It appears that palindromes have to do with DNA–protein binding. The local two-fold symmetry created by the palindrome provides a binding site for DNA-binding proteins which are often dimeric in structure. Such double binding markedly increases the strength and specificity of the binding interaction (Creighton, 1993, Chapter 8).
FIG. 1.
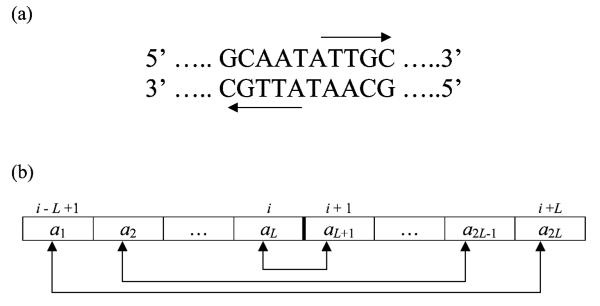
DNA palindrome. (a) This is a palindromic nucleotide sequence on the two complementary strands of DNA which are always read in opposite directions from the 5′ end to the 3′ end as shown by the arrows. The displayed segment reads exactly the same on both strands. (b) On each strand, the first base of the palindrome is complementary to the last, the second to the second last, and so on. This is a schematic representation of such complementary pairing between the bases in a 2L-palindrome centered at base i.
The herpesvirus family includes some of the well-known pathogenic viruses such as herpes simplex, varicella-zoster, Epstein-Barr, and cytomegalovirus. Some of these viruses are believed to pose major risks in immunosuppressive posttransplantation therapies, while others have been associated with life-threatening diseases such as AIDS and various cancers (Bennett et al., 2001; Biswas et al., 2001; Labrecque et al., 1995; Vital et al., 1995). A number of the animal herpesviruses are of agricultural concern. For example, the alcelaphine herpesvirus 1, indigenous to the wildebeest, is a causative agent of the fatal lymphoproliferative disease malignant catarrhal fever in cattle and deer (Bridgen, 1991).
Replication origins are places on the DNA molecules where replication processes are initiated. As DNA replication is the central step in the reproduction of many viruses, understanding the molecular mechanisms involved in DNA replication is of great importance in developing strategies to control the growth and spread of viruses (Delecluse and Hammerschmidt, 2000). For Epstein-Barr virus, one of these replication origins has been shown to associate with cellular proteins that regulate the initiation of DNA synthesis in human cells (Sugden, 2002). This suggests that these replication origins are also important locations for studying possible mechanisms of infecting human host cells. Knowledge of the locations of these replication origins will enhance the development of antiviral agents by blocking viral DNA replication or by interfering with the infection process.
As replication origins in DNA are considered major sites for regulating genome replication in general, labor-intensive laboratory procedures have been used to search for replication origins in various organisms (e.g., see Hamzeh, 1990; Zhu, 1998; Newlon and Theis, 2002). With the increasing availability of genomic DNA sequence data, the value of using computational methods to predict likely locations of replication origins before the experimental search has already been recognized although no prediction scheme that works for all DNA in general is available to date. The success of the computational prediction depends critically on the observation of the characterizing patterns in the nucleotide sequence around the replication origins of the particular kind of organisms under study. For example, the algorithm of Salzberg et al. (1998) predicted the replication origins for a number of bacterial and archaeal genomes based on the finding of seven-base and eight-base oligomers whose orientation is preferentially skewed around the replication origins. However, as pointed out by the authors, this algorithm is not suited for DNA molecules, like those in many viruses and their eukaryotic hosts, where multiple replication origins exist. In those cases, one would need to rely on other relevant sequence patterns to locate the replication origins.
The existence of high concentrations of palindromes in proximity of the replication origins of herpesviruses has been reported in some early studies (Weller et al., 1985; Reisman et al., 1985; Masse et al., 1992). This phenomenon is generally attributed to the fact that initiation of DNA replication typically requires an assembly of enzymes such as the helicases to locally unwind the helical structure of DNA and pull apart the two complementary strands. Furthermore, Masse et al. (1992) have demonstrated that by looking for palindrome clusters, among other features such as clusters of close repeats and close inversions on the nucleotide sequence, likely regions containing replication origins can be predicted.
Leung et al. (1994) describe how a statistical criterion, based on the scan statistics (Glaz, 1989; Dembo and Karlin, 1992), is developed for identifying nonrandom palindrome clusters by modeling the occurrences of palindromes in the genome as points randomly sampled from the unit interval according to the uniform distribution. Despite the fact that the criterion worked well for the cytomegalovirus genome sequence used for illustration in that article, the authors point out that the assumption of uniform distribution of palindromes has not yet been mathematically justified. In this paper, we shall justify this claim under the model that the nucleotide sequence is generated as a sequence of independent and identically distributed (i.i.d.) random variables.
The second, and more important, aim of this paper is to analyze a collection of herpesvirus genome sequences for nonrandom palindrome clusters and examine their connections with replication origins. Table 1 presents the collection of herpesvirus genomes to be analyzed. The dataset comprises all the complete genome sequences of the herpesvirus family downloaded from GenBank at the NCBI website in June 2001. Listed along with each virus name in the table are an abbreviation that will be used throughout this paper, its accession number in the GenBank database, its genome sequence length in number of bases, and the relative frequencies of the four nucleotide bases in the genome. The experimentally confirmed replication origins among the viruses in this dataset will help us assess the palindrome-based algorithm and suggest directions for improving the rate of successful prediction.
Table 1.
The List of Herpesvirus Genomes to be Analyzed
| Name | Abbrev. | Accession | Length | Base composition |
|---|---|---|---|---|
| Alcelaphine herpesvirus 1 | AHV1 | NC_002531 | 130608 | (.27, .24, .22, .26) |
| Ateline herpesvirus 3 | AtHV3 | NC_001987 | 108409 | (.32, .19, .17, .31) |
| Bovine herpesvirus 1.1 | BHV1 | NC_001847 | 135301 | (.14, .36, .37, .14) |
| Equine herpesvirus 1 | EHV1 | NC_001491 | 150223 | (.22, .29, .28, .22) |
| Equine herpesvirus 4 | EHV4 | NC_001844 | 145597 | (.25, .25, .25, .25) |
| Gallid herpesvirus 1 | MDV2 | NC_002530 | 110637 | (.24, .25, .25, .25) |
| Gallid herpesvirus 2 | MDV | NC_002229 | 138675 | (.28, .22, .21, .29) |
| Human herpesvirus 1 | HSV1 | NC_001806 | 152261 | (.16, .34, .34, .16) |
| Human herpesvirus 2 | HSV2 | NC_001798 | 154746 | (.15, .35, .35, .15) |
| Human herpesvirus 3 | VZV | NC_001348 | 124884 | (.27, .23, .23, .27) |
| Human herpesvirus 4 | EBV | NC_001345 | 172281 | (.20, .30, .29, .20) |
| Human herpesvirus 5 | HCMV | NC_001347 | 229354 | (.22, .28, .29, .21) |
| Human herpesvirus 6 | HHV6 | NC_001664 | 159321 | (.29, .22, .21, .29) |
| Human herpesvirus 7 | HHV7 | NC_001716 | 144861 | (.32, .18, .17, .32) |
| Ictalurid herpesvirus | CCV1 | NC_001493 | 134226 | (.21, .28, .28, .22) |
| Saimiriine herpesvirus 2 | HVS2 | NC_001350 | 112930 | (.33, .18, .16, .32) |
It would be of interest to ask whether our algorithm, developed for the herpesviruses, can be applied to identify replication origins in other organisms, or even to identify other functionally important regions, such as regulatory sites. While one would not anticipate that palindrome clusters will be the universal characterizing sequence pattern for all kinds of organisms and all types of functional sites, existence of palindrome clusters may serve as one possible criterion for these general purposes. For example, we have already noted that this approach adds to the skewed-oligomers method described by Salzberg et al. (1998) because it is not limited to DNA molecules with single replication origins. It is our hope that it will contribute to a general prediction tool that can be broadly applied to various domains of the tree of life.
The organization of the paper is as follows: Section 2 formulates the random process representing the palindrome occurrences on a nucleotide sequence and introduces the Wasserstein distance for measuring the difference between the palindrome process and the Poisson process. Using a general mathematical Poisson process approximation theorem, we derive an explicitly computable upper bound for this distance which approaches zero under suitable conditions. Section 3 briefly reviews how the scan statistics are used to identify nonrandom clusters of palindromes, treating them as points randomly sampled from a uniform distribution. The significant palindrome clusters obtained from the herpesviruses are presented in Section 4 where their association with replication origins is also discussed. We conclude with a few remarks about future works towards a more accurate replication origin prediction scheme in Section 5.
2. DISTRIBUTION OF PALINDROMES ON RANDOM DNA SEQUENCES
In this section, we shall see that if a DNA genome is assumed to be a sequence of nucleotide bases generated as i.i.d. random variables taking values A,C,G,T with probabilities pA,pC,pG,pT, respectively, the occurrences of palindromes above a certain minimal length can be approximated by a Poisson process. This is achieved by deriving an upper bound for the Wasserstein distance, also called the d2 metric (Barbour et al., 1992, Chapter 10), between the palindrome process and the Poisson process. Under suitable conditions, this Wasserstein distance dwindles to 0 as the sequence length increases indefinitely. Before these results can be stated, we need to first make precise the notion of the palindrome process and explain the concept of the Wasserstein distance.
2.1. The palindrome process and Wasserstein distance
Due to the complementary base pairing, it is often sufficient to represent DNA as a single nucleotide sequence. Any segment of the nucleotide sequence consisting of 2L bases will be a palindrome if its first base and its 2Lth base form a complementary pair, and so do its second and (2L − 1)st bases, the third and (2L − 2)nd, … , up to the Lth and (L + 1)st bases in the center of the segment (Fig. 1(b)). Using the center of the palindrome to indicate its position, we say that a palindrome of length 2L occurs at position i of the sequence if the bases i − j + 1 and i + j are complementary to each other for j = 1, … , L. This characterization does not preclude the possibility that a palindrome of length 2L may be extended to a longer length if the complementary pairing continues on for j > L. In this paper, it will be understood that the term “palindrome of length 2L” actually means “palindrome of length 2L or more.” For short, we shall just refer to it as a 2L-palindrome. If there are multiple 2L-palindromes centered at the same position of the sequence, only the longest one will be counted.
Because it is impossible for a nucleotide sequence of length M to have any 2L-palindrome centered at positions 1, … , L − 1 and M − L + 1, … , M, we shall represent the occurrences of palindromes as a random process on , where n = M − 2L + 1. The palindrome process is defined as
Here Ii is the indicator random variable for the occurrence of a 2L-palindrome centered at base i + L − 1 of the DNA sequence and δi/n denotes the unit point mass at i/n. In this definition, the DNA segment H of length 2L spanning bases i, … , i + L − 1, i + L, … , i + 2L − 1 is associated with the indicator Ii and the unit point mass at . For brevity, we shall say that the DNA segment H is positioned at i for the rest of the paper.
In an i.i.d. random nucleotide sequence, the success probability for the random variable Ii is
| (1) |
with θ = 2(pApT + pCpG), and the expected number of palindromes is
| (2) |
Because palindromes occurring close to each other overlap, the Ii’s are locally dependent. The neighborhood of dependence of Ii is
| (3) |
For all j outside of Ai, Ii and Ij are independent of each other.
We want to approximate the palindrome process Ξ defined on the n equally spaced discrete points in [0, 1] by the Poisson process Zλ with intensity λ on the continuous interval [0, 1]. In a number of DNA related studies (e.g., Arratia et al., 1990, 1996; Reinert et al., 2000; Reinert and Schbath, 1998; Schbath, 1995), the differences between two random processes are quantified by the total variation distance dTV between them. However, as explained in Chapter 10 of Barbour et al. (1992), the total variation distance is too strong to be useful for point processes like Ξ and Zλ. Indeed,
where the supremum is taken over all measurable subsets1 of where
is often called the configuration space of [0, 1]. From this, it can be shown that
which always equals 1 because Ξ has support only in {i/n : 1 ≤ i ≤ n} whereas Zλ has no points in {i/n : 1 ≤ i ≤ n} with probability 1.
Unlike dTV , the Wasserstein distance, also called the d2 metric, is less sensitive to small changes in the positions of points. It is more suitable for our purpose of measuring the discrepancy between the palindrome process and the Poisson process. Essentially, the Wasserstein distance between two point processes X and Y is the supremum of all expected differences between X and Y under test functions f that do not fluctuate too vigorously on the configuration space of [0, 1] with respect to the d1 metric explained below. These test functions are called Lipschitz functions. We give a concise explanation of the Wasserstein distance below. A more detailed description can be found in Barbour et al. (1992, Chapter 10, Section 2).
The Wasserstein distance between two point processes X and Y is defined as
where and
Here, d1(ξ1, ξ2) denotes the distance between two configurations ξ1 = (y11, … , y1m1) and ξ2 = (y21, … , y2m2). It is defined to be 1 if ξ1 and ξ2 have different numbers of points in [0, 1] and is defined to be the average distance between ξ1 and ξ2 under the closest matching if they have the same number of points. More precisely,
where the minimum is taken over all permutations π of (1, 2, … , m) with m being the common value of m1 and m2. Since our point processes are defined on [0, 1], without loss of generality, we can assume that y11 ≤ y12 ≤ … ≤ y1m1 and y21 ≤ y22 ≤ … ≤ y2m2. Then, when m1 = m2 = m,
The above simplified expression for d1(ξ1, ξ2) will be formally proved as Proposition 3 in the appendix.
With this background, we present the following general theorem giving an upper bound for the Wasserstein distance between a point process Ξ and the Poisson process Zλ. We will use the following notations. Let I1, … , In be indicator random variables with P(Ii = 1) = 1 − P (Ii = 0) = pi, and pij = P(Ii = Ij = 1) for 1 ≤ i, j ≤ n. For each i, Ii has a neighborhood of dependence Ai which is a collection of those indices j such that Ij may be dependent on Ii. Define a random point process
where δi/n denotes the unit point mass at i/n. We also define the point processes
We let
denote the number of points in these processes.
Theorem 1
Let Zλ denote the Poisson process on [0, 1] with intensity . Assume (Ij, j ∉ Ai) is independent of Ii and (Ik, k ∉ Ai ⋃ Aj) is independent of (Ii, Ij) for all i, j. Then we have
| (4) |
where
is the sum of products of the probabilities of observing 2L-palindromes at positions i and j within the neighborhood of dependence of each other and
is the sum of probabilities of observing overlapping 2L-palindromes at both positions i and j (∉ i) within the neighborhood of dependence of each other.
The proof of this theorem, given in the appendix, is based on an adaptation of Stein’s method (1972) for the Poisson process setting. We shall now examine how to use Theorem 1 to obtain a Poisson limit for the palindrome process. With long sequences, n is large, making the last term in the above bound negligible. We have noted in Equations (1) and (2) that for any i, pi = θL, giving λ = nθL. It is also easy to see from (3) that in the palindrome process Ξ, the neighborhood of dependence of Ii can stretch in either the forward or backward direction from base i for at most only 2L − 1 bases. This implies
| (5) |
To obtain an explicit upper bound for the Wasserstein distance between Ξ and Zλ, two things remain to be done. First, we need to examine the b2 term which involves the probabilities of overlapping palindromes. Second, we need to work out a uniform upper bound for which is independent of i, j.
The b2 term comprises the probabilities pij of observing overlapping 2L-palindromes at positions i and j that are no more than 2L − 1 bases apart. For example, the sequence ATCGATCG contains a 6-palindrome centered at position i = 3 overlapping with another 6-palindrome centered at position j = 5. The following proposition expresses the overlapping probability pij in terms of the base probabilities pA, pC, pG, and pT, the length parameter L, and the distance h = ∣i − j∣ between the centers of the two palindromes.
Proposition 1 (probability of overlapping palindromes)
Let h = ∣i − j∣ > 0. If h < 2L, the probability pij of having overlapping palindromes at both positions i and j are given by the following.
Case (a): L ≤ h ≤ 2L − 1. We have
Case (b): 0 < h < L. Here we let L = qh + r and consider two subcases according to how big the remainder r is in relation to h.
Subcase (b1): 0 ≤ r < (h + 1)/2.
Subcase (b2): (h + 1)/2 ≤ r < h.
Proof
Without loss of generality, we can assume i < j < 2L + i. Let Hi and Hj denote the DNA segments of length 2L positioned at i and j, respectively. Because of their overlap, Hi and Hj must share a common subsegment of length 2L − h at the right end of Hi and the left end of Hj. Throughout the proof, we shall use a’ to denote the complement of base a (e.g., A’ = T), w’ to denote the inverse complement of word w, and P(w) to denote the probability for w (e.g., if w = (ATC), then P(w) = pApTpC). We discuss the cases separately.
Case (a)
Let w = (a1, … , a2L−h) denote the common subsegment. For both Hi and Hj to be palindromes, we must have the arrangement as shown in Fig. 2(a). At the left end of Hi and the right end of Hj, the sequence must be w’. The center portions of Hi and Hj must be 2(h − L)-palindromes denoted, respectively, by (u’, u) and (v, v’). The probability of this arrangement is
where the sum is taken over all possible DNA words w of length 2L − h. Writing out P(w) in terms of the base probabilities, we have
where a is summed over the four bases in the alphabet .
FIG. 2.
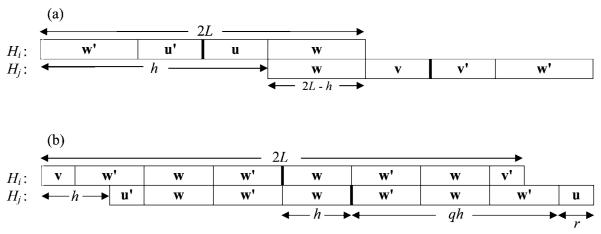
Overlapping palindromes. (a) When L ≤ h ≤ 2L − 1, the DNA segments Hi and Hj must have this structure for both of them to be 2L-palindromes centered at positions i and j, respectively. The center position of each segment is represented by a bold vertical bar. (b) Similarly, this displays the structures of Hi and Hj for both of them to be palindromes when 0 < h < L. In this illustration, q = 3.
Case (b)
This time, let w = (a1, … , ah) denote the first h bases to the right of the center of Hi and to the left of the center of Hj. Let u = (a1, … , ar) and v = (ah−r+1, … , ah), respectively, stand for the first and last r bases of w. Figure 2(b) displays the necessary structure in Hi and Hj for both of them to be palindromes when q = 3. In general, the probability of such arrangements can be computed as
Subcase (b1)
. If q is odd,
If q is even, the calculation is exactly the same except a and a’ need to be swapped, reducing to the same expression in terms of the base probabilities.
Subcase (b2)
.
when q is odd. Just like subcase (b1), a and a’ need to be swapped when q is even. Either way, the expression reduces to
The following two lemmas will facilitate proving that the palindrome process approaches a Poisson limit under the equal complementary probability (ECP) assumption of pA = pT and pC = pG.
Lemma 1
Under the ECP assumption, pij ≤ θ3L/2 for 1 ≤ i ≠ j ≤ n.
Proof
When j ∉ Ai, the inequality is trivially true because Ii and Ij are independent and pij = pipj = θ2L. We therefore only need to look at those pij’s with ∣j − i∣ < 2L. The calculations needed involve expressions of the form (0.5 − x)l + xl where x = PG = PC is a number between 0 and 0.5 and l is a positive integer. We note the following elementary algebraic properties:
A. 1/8 ≤ (0.5 − x)2 + x2 ≤ 1/4 (i.e., 1/4 ≤ θ ≤ 1/2).
B. If α > β ≥ 0, then
It follows that for any positive integer q
and
In Case (a), L ≤ h ≤ 2L − 1. Here
With θL = 2L[(0.5 − x)2 + x2]L, we have
This ratio is ≤ 1 because the numerator is ≤ 1 while the denominator is ≥ 1 by property A noted above. Similar calculations lead to
for Subcase (b1) and
for Subcase (b2). Both of these ratios are ≤ 1 again because of property A.
Remark on Lemma 1
Although not directly relevant to the analysis in this paper, it is of interest to point out that calculations similar to those in Lemma 1 will also show that if pA = pT and pC = pG, pij ≥ pipj = θ2L. Equality is obtained when one of the following special situations occur: Either all four probabilities are equal to 1/4, or one pair of probabilities is equal to 0 while the other pair is equal to 0.5. These equalities have been obtained by Ghosh and Godbole (1996). Other than these special cases, we now see that under the ECP assumption, Ii and Ij are nonnegatively correlated indicator random variables. Furthermore, we have carried out quite extensive computations, and the results indicate that the positive correlation between Ii and Ij holds even without the ECP assumption. We have yet to prove it analytically though. This positive correlation essentially says that when a palindrome occurs, it enhances the probability of having another palindrome occurring nearby that overlaps with it.
Lemma 2
Assuming ECP, then for 1 ≤ i ≤ n and j ∈ Ai, we have
| (6) |
provided that 4 ≤ L ≤ n/500.
Proof
Notice that ∣Vij∣ = ∑k∈Ai⋃Aj Ik, which is of the form ∑k∈Γij Ik. For simplicity, we suppress the notational dependence of Γij on i, j and write
Lemma 3.1 from Brown et al. (2000) states that for any random variable X ≥ 1,
| (7) |
where κ = Var (X)/E(X). Here, we let X = 1 + W. Then
For k, l ∈ Γ. where ∣k − l∣ ≥ 2L, Ik and Il are independent. So we have
Consequently, κ ≤ 5. We observe that the upper bound on E(1/X) in (7) is an increasing function in κ. Replacing κ by 5 yields
The middle inequality comes from the fact that
The last inequality follows from L ≤ n/500.
2.2. Poisson limit for the palindrome process
We shall make use of Lemmas 1 and 2 in conjunction with Theorem 1 to prove the following proposition stating that the palindrome process approaches a Poisson process in the limit under suitable conditions. This is done by showing that the Wasserstein distance between Ξ and Zλ can be made arbitrarily small provided that n → ∞ and L grows at a suitable rate proportional to log n.
Proposition 2
Assume ECP and suppose that n, L → ∞ in such a way that nθL = λ, where λ ≥ 1/32 is a fixed positive constant, then
where c is an absolute constant no greater than 131.
Proof
First note that the condition 4 ≤ L ≤ n/500 in Lemma 2 is easily satisfied when n and L become large with nθL = λ. This condition will continue to be assumed true. It has been pointed out in (5) that b1 ≤ n(4L − 1)θ2L ≤ 4LλθL. From Lemma 1, it follows that b2 ≤ n(4L − 2)θ3L/2 ≤ 4LθL/2λ. Combining the two inequalities gives
| (8) |
because θL/2 ≤ (1/2)L/2 ≤ 0.25 for L ≥ 4. From Lemma 2, it follows that
| (9) |
where the supremum is taken over 1 ≤ i ≤ n, j ∈ Ai \ {i}. Combining Inequalities (8) and (9), we see that first term in the upper bound for the Wasserstein distance in Theorem 1 is less than 130LθL/2. It can be verified that the second term 1/(2n) ≤ LθL/2 when λ ≥ 1/32. Putting the two terms together gives
Furthermore, as nθL = λ, L must be growing at the rate log n, and hence
2.3. Remarks on various assumptions in Proposition 2
Various assumptions and constraints have been made in order to prove the result in Proposition 2. How restrictive are these assumptions? Are they satisfied, at least to a reasonable extent, by the actual viral genome DNA data? We shall discuss these questions in the following remarks.
Remark 1. The ECP assumption
It should be noted that the ECP assumption is sufficient but not necessary for obtaining the Poisson process limit. The proofs of Lemma 2 and Proposition 2 require only the inequality pij ≤ θ3L/2, which can be computationally verified to hold for many base frequencies that do not satisfy the ECP assumption (e.g., pA = 0.1, pC = 0.2, pG = 0.3, pT = 0.4). In those cases, the palindrome process still approaches a limiting Poisson process.
Intuitively, the ECP assumption is quite believable, and it has been used in a number of genomic or chromosomal DNA sequence analysis studies (e.g., Burge et al., 1992; Karlin et al., 1993). Looking at the base composition of the herpesviruses in Table 1, we see that the relative frequencies of A and T are quite close to each other, and so are those of C and G. It is tempting to believe in the ECP assumption.
To examine this more objectively, one can turn to the Bayesian information criterion (BIC). For each genome, we compare the saturated multinomial model where the bases are generated with probabilities (pA, pC, pG, pT) summing to 1 against the ECP model with the extra condition that pA = pT and pC = pG. As summarized in Tavaré and Giddings (1989), the BIC of a model D can be computed by
where M is the sequence length, k is the number of free parameters in model D (k = 3 in the saturated model and k = 1 in the ECP model), and
is the log likelihood for the data, with n(a) representing the count of base a in the sequence and the maximum likelihood estimate of the base probability pa. In the saturated model, is the relative frequency of a. In the ECP model, is the averaged relative frequency of a and a’. When comparing two statistical models, the model with smaller BIC is considered superior.
It turns out from the BIC that in 8 out of the 16 herpesviruses in our dataset, the ECP model is preferred hence justifying the ECP assumption for their DNA sequences. For the rest of the viruses, we do not have such a nice statistical justification. Fortunately, one can computationally verify, by working out the values of pij and θ, that the inequality in Lemma 1 remains true for those base probabilities estimated from each of the 16 viruses, ascertaining the Poisson process limit for the palindrome process in each genome as explained above.
Remark 2. Restrictions on n, L, and λ
We have also posed the restriction of 4 ≤ L ≤ n/500 and λ ≥ 1/32 in Lemma 2 and Proposition 2. These restrictions offer no difficulty at all to our application. In each of the herpesviruses to be analyzed, n is of the order 105 and θ is close to 1/4. The values of L of interest are in the range of 4–8. One can easily verify that in each case, these requirements on n, L, and λ are satisfied.
Remark 3. The constant c
The constant c in Proposition 2 can be made much smaller than 131 in most cases of practical interest in DNA sequence analysis. The θ value calculated from real DNA base frequencies are usually close to 1/4 rather than 1/2, making the upper bound in (8) quite close to 4.25LλθL/2 and the upper bound in (9) close to 17/λ. This will reduce the constant c to about 73. There are places in Theorem 1 and Lemma 1 where the bounds can be made tighter also. However, since the main goal of Proposition 2 is to give a Poisson limit for the palindromes process, the actual value of c is not of great concern.
Remark 4. Can the Poisson limit approximate the palindrome process in real DNA?
Proposition 2 gives only an asymptotic result stating that the palindrome process will approach a Poisson process in the limit. In the proof of Proposition 2, it was further shown that the rate of convergence is of the order log . As this result is to serve as the basis of approximating the palindrome process in a real DNA sequence which is finite in length, one would naturally have to ask how large n and L have to be in order to make the Poisson process a good approximation of the palindrome process. Since the d2 distance has not been previously applied to any practical setting, at this point there is no firm basis for a reliable assessment of how good the approximation is. However, we refer to a previous work of Leung et al. (1994) where the Poisson approximation was demonstrated to be reasonable by Q-Q plots for the cytomegalovirus genome with n = 229,354 and L = 5. The d2 distance calculated for this case is 0.1644 (see Table 2). In our application to the herpesvirus genome coming up in Section 4, we shall use this value of the d2 distance as a benchmark to pick values of the parameter L for the given genome lengths and compositions.
Table 2.
Values of LθL/2 at L = 4, 5, 6, 7, 8
| Virus | L = 4 | L = 5 | L = 6 | L = 7 | L = 8 |
|---|---|---|---|---|---|
| AHV1 | 0.2523 | 0.1580 | 0.0950 | 0.0556 | 0.0318 |
| AtHV3 | 0.2867 | 0.1854 | 0.1151 | 0.0695 | 0.0411 |
| BHV1 | 0.3605 | 0.2469 | 0.1624 | 0.1038 | 0.0650 |
| EHV1 | 0.2586 | 0.1630 | 0.0986 | 0.0580 | 0.0334 |
| EHV4 | 0.2500 | 0.1563 | 0.0938 | 0.0547 | 0.0313 |
| MDV2 | 0.2500 | 0.1562 | 0.0937 | 0.0547 | 0.0312 |
| MDV | 0.2600 | 0.1641 | 0.0994 | 0.0586 | 0.0338 |
| HSV1 | 0.3213 | 0.2138 | 0.1366 | 0.0848 | 0.0516 |
| HSV2 | 0.3400 | 0.2295 | 0.1487 | 0.0937 | 0.0578 |
| VZV | 0.2531 | 0.1587 | 0.0955 | 0.0559 | 0.0320 |
| EBV | 0.2700 | 0.1720 | 0.1052 | 0.0626 | 0.0365 |
| HCMV | 0.2603 | 0.1644 | 0.0996 | 0.0587 | 0.0339 |
| HHV6 | 0.2615 | 0.1653 | 0.1003 | 0.0592 | 0.0342 |
| HHV7 | 0.2948 | 0.1920 | 0.1200 | 0.0730 | 0.0434 |
| CCV1 | 0.2577 | 0.1623 | 0.0981 | 0.0577 | 0.0332 |
| HVS2 | 0.2997 | 0.1960 | 0.1231 | 0.0751 | 0.0449 |
Remark 5. The i.i.d. sequence model
Proposition 2 is proved only for a random nucleotide sequence generated as i.i.d. random variables. While in many studies it has been pointed out that this i.i.d. sequence model does not fit well with real DNA sequences, the model is still frequently used for deriving statistical criteria to evaluate whether certain observed sequence patterns are unlikely to be observed simply by chance, mostly because of the difficulty in deriving analytical results with more elaborate models. It is also because of this limitation that the i.i.d. model is used to derive Proposition 2. The assumption of independent bases is rather hard to relax. However, one may consider allowing the base probabilities pA, pC, pG, pT to vary from one region of the sequence to another so that the local base frequencies are better reflected in the model. Indeed, there is evidence indicating that base frequencies change from one region of the genome to another. For example, Mrazrek and Karlin (1998) report a change of base preference in some herpesvirus replication origins from having more G’s than C’s on one side of the origin to having more C’s than G’s on the other. Because of the possibility of fluctuation of palindrome probabilities due to these changes in base composition, we have checked the regions around all the known replication origins in our dataset to see whether there are base compositional differences substantial enough to cause the θ value to become significantly different from that estimated from other segments of similar lengths sampled from the rest of the genome. The result in every case shows no significant difference. We have therefore chosen to simply use a value of θ estimated by the overall base frequencies in the entire genome for our analysis.
3. USE OF THE SCAN STATISTICS TO LOCATE PALINDROME CLUSTERS
In light of the mathematical result of the previous section, we can justifiably make the assumption that the midpoints of the 2L-palindromes are distributed like the events of a Poisson process on the unit interval. It then follows from the properties of a Poisson process that if the total number of 2L-palindromes is known, say, m, then these m points are distributed in the same way as m i.i.d. uniform random variables on (0, 1) (Karlin and Taylor, 1981, Chapter 4).
For a set of points X1, … , Xm distributed independently and uniformly over the unit interval (0, 1), the traditional scan statistic Nw = max1≤i≤m Nw(i), where 0 < w < 1 is a prescribed window length and Nw(i) is the number of points in the interval [X(i), X(i) + w), is a generalized likelihood ratio test statistic that has been shown to be most powerful among a class of statistics to test against the clustering alternative (Naus, 1965). More recently, Dembo and Karlin (1992) define the r-scan statistic Ar to be the minimal cumulative lengths of r consecutive distances between the ordered statistics X(1), … , X(m). Formally, let Si denote the distance between the ordered ith and (i + 1)st points, i.e., Si = X(i+1) − X(i), i = 1, … , m − 1. For any fixed integer r between 1 and m − 1, the r-scan is Ar = min{Ar(i), i = 1, … , m − r} where , i = 1, … , m − r Both the traditional and the r-scan statistics are used quite extensively in DNA sequence analysis (see Glaz et al., 2001, Chapter 6).
These two scan statistics are essentially equivalent. Consider the event {Nw(i) ≥ r + 1 for i = 1, … , m − r, which says that there are at least r + 1 points contained in the window [X(i), X(i) + w). This is equivalent to the event {Ar(i) < w} which says that there are at least r adjoining spacings, starting at X(i), whose cumulative length is less than w. See Fig. 3 for illustration with r = 3. We therefore have a simple duality relationship between Nw and Ar:
| (10) |
If either of the above probabilities is too small (say, < 0.05), then a cluster of at least r points in a window of length w is statistically significant.
FIG. 3.
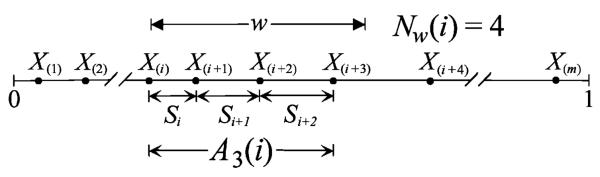
The w- and r-scan statistics. The equivalence of the events {Nw ≥ r + 1} and {Ar < w} is illustrated with r = 3.
There is a very simple asymptotic approximation for the distribution of Ar (Cressie, 1977; Dembo and Karlin, 1992): For any x > 0,
When m is large, it yields the following approximation:
| (11) |
Leung et al. (1994) make use of Equation (11) to identify nonrandom palindrome clusters for the HCMV but they observe that in some cases the approximate probabilities so computed have rather large discrepancies when compared with simulated probabilities such as those obtained by Glaz (1989). It is therefore not advisable to routinely apply this approximation to evaluate the statistical significance of palindrome clusters without first considering whether the desired accuracy can be achieved.
Leung and Yamashita (1999) review a few other Poisson type approximations to the distribution of the scan statistics with special interest in their accuracy when used on data from the palindrome occurrences in seven herpes genomes which is part of our dataset in the present paper. Their simulation results show that the compound Poisson distribution put forth by Glaz et al. (1994) based on a result of Roos (1993) produces the best approximation to Ar. We have, therefore, adopted their result to evaluate the statistical significance of palindrome clusters for the genomes in our dataset. Explicitly, this approximation is
| (12) |
where π = Q1, p = 1 − Q2/Q1, with
| (13) |
| (14) |
and . The approximation in (12) is used in the next section to assess the statistical significance of the r-clusters of palindromes observed in the herpesvirus genomes.
4. PALINDROME CLUSTERS IN HERPESVIRUS GENOMES
4.1. Choosing L
Our study of palindromes is motivated by their association with replication origins. In HCMV, the replication origin “oriLyt” is successfully located by a significant cluster of palindromes with L = 5 using the r-scan statistic The name “oriLyt” is designated to the origin of replication where DNA replication is initiated during the lytic phase of the virus life cycle. A detailed explanation of the terminology as well as the methodology is given in the article of Leung et al. (1994). The choice of L = 5 turns out to be crucial for the successful detection of the oriLyt. When the analysis is done with L = 4, too many statistically significant clusters were detected over very diverse regions of the genome. With L = 6, no significant palindrome cluster was detected at all. This indicates that the choice of L can be quite influential on the success rate of predicting a replication origin. Too small a value of L degrades the specificity while too large a value reduces the sensitivity of this method of replication origin prediction. However, apart from the knowledge that L should increase like the logarithm of the sequence length for the palindrome process to approach the Poisson limit, we do not have any guideline on what is the best length of palindromes one should examine.
We have therefore set up our algorithm in such a way that L is a parameter in the program so that its value can be easily reset by the user. The prediction accuracy is tested on our dataset with L = 4, 5, 6, 7, and 8. L = 5 is found to work best for many of the genomes, but there are several exceptions. For example, choosing L = 5 for BHV1 produced 17 significant palindrome clusters, only two of which are close to replication origins. Increasing L to 6 reduces the number of significant clusters to five, which is a more acceptable number. This is at first somewhat surprising because the BHV1 genome is much shorter than that of HCMV. On closer examination, we notice that BHV1 has a much higher C/G content than all the other genomes, yielding a higher θ, which affects the palindrome probability.
In using the scan statistics to detect palindrome clusters, the choice of L should be made so that the overall palindrome process can be approximated reasonably by a Poisson process on one hand, but it should still allow the unusual palindrome clusters to be detected on the other hand. Proposition 2 tells us that the Wasserstein distance between the palindrome process and the Poisson process is bounded above by a quantity proportional to LθL/2, which depends on the base frequencies as well as L. Table 2 contains the values of LθL/2 computed at L = 4, 5, 6, 7, 8 for the base frequencies of the 16 herpesvirus genomes. If we use the value of LθL/2 with L = 5 for HCMV as a benchmark (that is, for each virus, we choose L such that LθL/2 is closest to 0.1644), L is chosen to be 5 for most of the viruses with the exceptions of BHV1, HSV1, and HSV2 whose genomes are more C/G rich than the others. For these three viruses, L = 6 is the closest choice. The entries corresponding to the chosen L are bold-printed in Table 2.
Eventually, we hope to be able to formulate a criterion of choosing L for each genome that has a biological basis on the type of genome structure under analysis. However, this can be achieved only if we have a larger database of experimentally confirmed replication origins. For now, our hope is that by reporting these regions of significant palindrome clusters, we will be able to facilitate the experimentation to expand this database, which will in turn help improve the prediction scheme.
4.2. The palindrome clusters and replication origins
Having chosen L, we have a computer program, which is a simple adaptation of an algorithm of Leung et al. (1991), to examine each of the herpes genome nucleotide sequences and find all 2L-palindromes. It should be noted that we choose to use this particular program mainly because we have easy access to it and there are other programs for finding palindromes available in standard sequence analysis packages such as EMBOSS (Rice et al., 2000). Only the nonredundant 2L-palindromes are used for the analysis. That is, if one palindrome is completely contained in a longer one, the shorter palindrome will be discarded. The S-Plus functions developed by Leung and Yamashita (1999), based on the compound Poisson approximation to the r-scans described in Section 3, are then applied to examine the palindrome locations and identify all the nonrandom (r + 1)-clusters with r ranging from 1 to 15. Table 3 shows the spans of all the significant clusters found in HCMV, where the span of a cluster is the range of bases starting at the beginning position of the first palindrome in the cluster to the ending position of the last palindrome in the cluster. Not surprisingly, the spans of many of these clusters overlap one another. To reduce redundancy, we go through the list of clusters and join them to become one if their spans overlap. After this joining process, typically only a few nonoverlapping regions of a genome emerge. Each region contains one or more significant clusters. Table 4 lists all the regions found from the herpesvirus genomes.
Table 3.
Segments of the HCMV Genome Spanned by Significant r + 1-Clusters
| r | Clusters |
|---|---|
| 1 | None |
| 2 | None |
| 3 | 92701–92792 |
| 4 | 92526–92718, 92569–92756, 92643–92792, 92701–92868, 195029–195227, 195109–195268 |
| 5 | 92526–92756, 92569–92792, 92643–92868, 195029–195268 |
| 6 | 92526–92792, 92569–92868, 92643–93119 |
| 7 | 91953–92792, 92526–9286, 92569–93119, 92643–93260, 92701–93520, 92709–93610 AU1 |
| 8 | 91635–92792, 91953–92868, 92526–93119, 92569–93260, 92643–93520, 92701–93610 |
| 9 | 91490–92792, 91635–92868, 91953–93119, 92526–93260, 92569–93520, 92643–93610, 92701–94183 |
| 10 | 91490–92868, 91635–93119, 91953–93260, 92526–93520, 92569–93610, 92643–94183 |
| 11 | 90759–92868, 91490–93119, 91635–93260, 91953–93520, 92526–93610, 92569–94183 |
| 12 | 90251–92868, 90759–93119, 91490–93260, 91635–93520, 91953–93610, 92526–94183 |
| 13 | 90251–93119, 90759–93260, 91490–93520, 91635–93610, 91953–94183 |
| 14 | 89585–93119, 90251–93260, 90759–93520, 91490–93610, 91635–94183 |
| 15 | 89585–93260, 90251–93520, 90759–93610, 91490–94183 |
Table 4.
Regions of the Herpesvirus Genomes Containing Significant Clusters
| Genome | Region | Palindrome | Feature |
|---|---|---|---|
| BHV1 | 77155–77168 | 3 | |
| 102895–106948 | 22 | ||
| 113462–113636 | 5 | 1.75 mua from Ori | |
| 124582–124756 | 5 | 1.61 mu from Ori | |
| 131268–135221 | 21 | ||
| EHV1 | 115125–119094 | 17 | Overlaps transcriptional regulator |
| 144064–148033 | 17 | Overlaps transcriptional regulator | |
| EHV4 | None | ||
| HSV1 | None | ||
| HSV2 | None | ||
| VZV | None | ||
| EBV | 6772–11675 | 19 | Contains OriP |
| 49460–54858 | 25 | Contains OriLyt | |
| HCMV | 89585–94183 | 19 | Contains OriLyt |
| 195029–195268 | 8 | Enhancer element | |
| HHV6 | None | ||
| HHV7 | 120758–124422 | 16 | |
| AHV1 | 113456–113759 | 5 | |
| ATHV3 | 95350–100098 | 17 | |
| MDV2 | 93143–93243 | 4 | |
| 109331–110590 | 8 | ||
| MDV | None | ||
| CCV1 | None | ||
| HVS2 | None |
Here, mu stands for a map unit, which is 1% of the genome length. The distance is calculated from the midpoint of the cluster region to the midpoint of the closest replication origin.
Although our ultimate goal is to eventually make use of palindrome clusters to help predict the likely locations of replication origins, it must be recognized that at this stage, it is not yet possible to achieve much prediction accuracy. There are two main problems. First, the prediction procedure must also include information about clusters of close repeats and inversions which are also known to be characteristics of replication origins. A close repeat (respectively, inversion) is a segment of DNA with an exact (respectively, inverted complementary) copy of itself present in close vicinity, say, within 150 bases. A palindrome is actually a special case of close inversion because it is a segment of DNA followed immediately by its inverted complement. The statistical assessments of clusters for close repeats and inversions still need to be developed. Second, reports on confirmed location of replication origins is relatively scarce. We hope that the findings of the palindrome clusters in this paper will be helpful towards the experimental determination of more replication origins so that more information is available for prediction accuracy testing in the future.
Nevertheless, even with limited information, it is still of interest to examine the correspondence between these significant palindrome clusters and the actual confirmed locations of the replication origins. From various sources like the annotations in the GenBank file of these sequences and the references therein, plus published genetic maps and other biomedical articles (Farrel, 1993; Masse et al., 1992; McGeoch and Schaffer, 1993; Baumann et al., 1989), we are able to compile a list of replication origins in 10 of the 16 herpesviruses. These include one herpesvirus hosted in the cow, two in the horse, and seven in humans. It is not surprising that these viruses have been studied more than the others because of their agricultural and medical importance. The location of those origins are displayed in Table 5, and we also indicate them in the last column of Table 4 whenever they are close (within 2% of the genome length) to one of the regions found to contain significant clusters.
Table 5.
Location of Replication Origins in Ten Herpesviruses
| Virus | Replication origins |
|---|---|
| BHV1 | 111080–111300(OriS), 126918–127138(OriS) |
| EHV1 | 126187–126338 |
| EHV4 | 73900–73919(OriL), 119462–119481(OriS), 138568–138587(OriS) |
| HSV1 | 62475(OriL), 131999(OriS), 146235(OriS) |
| HSV2 | 62930(OriL), 132760(OriS), 148981(OriS) |
| VZV | 110087–110350, 119547–119810 |
| EBV | 7315–9312(OriP), 52589–53581(OriLyt) |
| HCMV | 92270–93715(OriLyt) |
| HHV6 | 67617–67993(OriLyt) |
| HHV7 | 66685–67298 |
While we see some agreements between palindrome cluster regions and replication origins in BHV1, EBV, and HCMV, many clusters regions have not been found to contain replication origins. There are two possibilities. First, there may be a replication origin which has not yet been experimentally located so that it is not documented in the biomedical literature. Second, a cluster region may correspond to a regulatory site rather than a replication origin. For example, the region 195029–195268 in HCMV is actually an enhancer element (Weston, 1988).
We also note that among the 19 replication origins compiled in Table 5, only five of them have palindrome clusters in their proximity. Palindrome clusters by themselves, therefore, will not be sufficient in terms of replication origin prediction. This is not unexpected because the sequence features of close repeat and inversion still need to be incorporated into the prediction scheme. Indeed, for the 14 replication origin sequences which do not contain any palindrome clusters, we further analyze the sequence structure around them using the program developed by Leung et al. (1991). Highly noticeable close repeats or inversions are found in all of these sequences, with only one exception.
The one exception is the oriL in the herpes Simplex I (HSV1) virus. This replication origin, located at position 62475 of the genome, is not identified by any significant palindrome clusters, nor does it have any unusual clusters of close repeats and inversions in its proximity. Instead, it has a perfect palindrome of length 144 stretching from base 62404 to base 62547. This means that it is possible to have a replication origin sequence with a highly unusual palindrome, yet our method has failed to identify it because it contains only a single long palindrome instead of a cluster of shorter ones. Upon further examination for long palindromes in the other genomes in our dataset, we find that the two replication origins of the chicken pox virus (VZV) located at bases 110087–110350 and 119547–119810 also contain two palindromes of length 36 from base 110194 to 110229 and 119668 to 119703. Again, despite the presence of a long palindrome, no significant palindrome cluster is detected.
These observations suggest the necessity of a generalization of our method of identifying palindrome clusters to take the length of the palindrome into consideration. Presently, we represent the occurrence of a palindrome by a point placed at its center. These points are considered equally as events in a Poisson process. Hence, the 144 bp palindrome carries the same weight as a 10 bp palindrome and counts as only one point. Suppose that we weigh the palindromes proportional to their lengths; then the 144 bp palindrome would count as 14 points and be deemed a significant cluster by itself. The idea of letting weights be given to events in a point process is encompassed in the theory of marked point process where each point is attached with a real-valued random variable called a mark. The Wasserstein distance between the new palindrome process and the appropriate marked Poisson process, as well as the distribution of the scan statistics will need to be developed. Such extension of the present model is under way. Together with the future incorporation of close repeats and inversions, we anticipate that a more accurate and efficient prediction scheme for replication origins can be established.
For the time being, the nonrandom palindrome clusters located by the method in this paper can be used for prioritizing which segments of DNA should be experimentally tested for replication origins first. Among the 10 herpesvirus genomes in Table 5, 12 nonrandom palindrome clusters have been identified of which 5 (over 40%) are in proximity of replication origins. One would, therefore, expect that for those herpesviruses where no replication origins have yet been identified, the chance of finding a replication origin in a segment of about 5% the length of the genome around a palindrome cluster would be much better than that of a random genome segment of similar length. We believe that, like the work of Masse et al. (1992) on HCMV, a good initial choice of genome segments for testing can expedite the experimental search for replication origins.
5. CONCLUDING REMARKS
In this paper, we have focused our analysis on herpesvirus genomes. We shall be conducting a broader analysis on other families of double-stranded DNA viruses such as the adenoviruses and papillomaviruses to gain more insight into the relationship between palindrome clusters and replication origins in other viral genomes. Moreover, we would like to point out that palindrome clusters may also be associated with the other biologically relevant functional sites (e.g., enhancer elements, transcriptional regulators as indicated in Table 4). The general statistical criterion for significant palindrome clusters will allow exploratory studies of such possible associations to be undertaken. The computer programs, implemented in C and S-Plus, for locating statistically significant clusters of palindromes using scan statistics will be accessible from the web page www.bioinformatics.utep.edu/mleung so that interested readers can adapt them for other applications.
Recently, predictions for various biological features, such as CpG islands and coding regions on the genome sequence, have been accomplished using hidden Markov models. We expect that the hidden Markov model approach can also be useful for prediction of replication origins. The difficulty, at least for now, is insufficient known replication origins that can be used for estimation of the model parameters. In cases where data is scanty, it has been suggested (Durbin et al., 1998) that Bayesian estimation should be used instead of maximum likelihood estimation. In this regard, the knowledge gained from understanding the connection between clusters of palindromes and repeats can be useful for choosing reasonable prior distributions for the model parameters.
It is a well known fact that nucleotide sequences in real DNA molecules do not fit well with the model of i.i.d. random variables (Philips et al., 1987; Prum et al., 1995; Leung et al., 1996; just to name a few). Preferably a Markov model of order at least 3 should be used. This motivates the need to generalize Theorem 1 to a Markov chain context. Even from a purely mathematical point of view, such a generalization is an interesting problem worthy of in depth investigation. Once more, this illustrates that the interaction between mathematical and biological research will prove fruitful for the advancement of both sciences.
ACKNOWLEDGMENTS
The authors gratefully acknowledge the support of the NIH MBRS program (S06GM08194-23S3), the W.M. Keck Center of Computational and Structural Biology at Rice University, and the National University of Singapore ARF Research Grant (R-146-000-013-112) and BMRC grant BMRC01/1/21/19/140. Part of this work is done at the Institute for Mathematical Sciences at the National University of Singapore in 2002 under a grant (01/1/21/19/217) from the BMRC of Singapore. The authors also wish to thank the referees for their very helpful comments.
APPENDIX: TECHNICAL PROOFS
Proposition 3
Let Γ = [0, 1] with metric d(x, y) = ∣x − y∣. If with 0 ≤ t1 ≤ ⋯ ≤ tm ≤ 1 and with 0 ≤ s1 ≤ ⋯ ≤ sm ≤ 1, then
| (15) |
for all permutations π of (1, ⋯ , m), and hence
Proof
We use mathematical induction to prove the claim. Suppose first that m = 2. Without loss of generality, we may assume t1 = min{t1, t2, s1, s2}. Then it suffices to consider the following three cases.
(i) t2 ≥ s2, then
(ii) s1 ≤ t2 < s2, then
(iii) t2 < s1, then
Now, suppose (15) holds for m ≤ k with k ≥ 2; we shall prove it holds for m = k + 1 and all permutations π of (1, ⋯ , k + 1). As a matter of fact, the claim is obvious if π(k + 1) = k + 1. Assume π(k + 1) ≠ k + 1, then follows that
Proof of Theorem 1
Define a Poisson process, , on as follows:
where ζ1, ζ2, … are independent random variables uniformly distributed on J and N is a Poisson random variable with mean λ and is independent of the ζi’s. For coupling argument below, we need to represent the usual Poisson process on [0, 1] as
where Ui’s are independent and uniformly distributed on [0, 1]. This can be seen as follows: first we pick a point from J at random and then move it to the left in a uniform manner within a distance 1/n. We shall prove Theorem 1 in two steps: first, we apply a coupling argument to show that
And in the second step, we apply Stein’s method to bound , giving the first two terms in Theorem 1. Then Theorem 1 will follow immediately by the triangle inequality:
Step 1
We see that,
Step 2
Let be an independent copy of {Ii, 1 ≤ i ≤ n}. We first derive Stein’s equation (16) follows:
where
and
Hence, we have derived Stein’s identity,
Consider Stein’s equation:
| (16) |
where and .
We need a from Brown and Xia (2001, Theorem 5.1) which gives a nonuniform bound on the solution of Stein’s equation: Suppose f satisfies and hf is the solution to Stein’s equation; then we have
| (17) |
where ∣ξ∣ denotes the number of points in the configuration ξ.
Back to the proof of Theorem 1. We have
where
In the second inequality, we write hf (Ξ) − hf (Ξ − δi/n) − hf (Vi + δi/n) + hf (Vi) as a telescoping sum and apply (17). Similarly,
Combining the bounds on the error terms R1(hf) and R2(hf) which are independent of f, we take the supremum over all f with to get the d2 distance. This completes the proof of Theorem 1.
Remark
Theorem 1 has been extended by Chen and Xia (2004) using Palm theory to a more general setting with wider applicability.
Footnotes
A subset A of is measurable if where is the smallest sigma algebra making the mappings from to R: ξ ↦ ξ(C) measurable for all sets Borel C ⊂ [0, 1].
REFERENCES
- Arratia R, Goldstein L, Gordon L. Poisson approximation and the Chen–Stein method. Statist. Sci. 1990;5:403–434. [Google Scholar]
- Arratia R, Martin D, Reinert G, Waterman MS. Poisson process approximation for sequence repeats, and sequencing by hybridization. J. Comp. Biol. 1996;3:425–463. doi: 10.1089/cmb.1996.3.425. [DOI] [PubMed] [Google Scholar]
- Barbour AD, Holst L, Janson S. Poisson Approximation. Clarendon Press; Oxford: 1992. [Google Scholar]
- Baumann RF, Yalamanchili VRR, O’Callaghan DJ. Functional mapping a DNA sequence of an equine herpesvirus 1 origin of replication. J. Virol. 1988;63(3):1275–1283. doi: 10.1128/jvi.63.3.1275-1283.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bennett JJ, Tjuvajev J, Johnson P, Doubrovin M, Akhurst T, Malholtra S, Hackman T, Balatoni J, Finn R, Larson SM, Federoff H, Blasberg R, Fong Y. Positron emission tomography imaging for herpes virus infection: Implications for oncolytic viral treatments of cancer. Nat. Med. 2001;7(7):859–863. doi: 10.1038/89991. [DOI] [PubMed] [Google Scholar]
- Biswas J, Deka S, Padmaja S, Madhavan HN, Kumarasamy N, Solomon S. Central retinal vein occlusion due to herpes zoster as the initial presenting sign in a patient with acquired immunodeficiency syndrome (AIDS) Occl. Immunol. Inflamm. 2001;9(2):103–109. doi: 10.1076/ocii.9.2.125.3976. [DOI] [PubMed] [Google Scholar]
- Bridgen A. A restriction endonuclease map for Alcelaphine herpesvirus 1 DNA. In: O’Brien SJ, editor. Genetic Maps. Book 1, Viruses. 6th ed. Cold Spring Harbor Laboratory Press; 1991. [DOI] [PubMed] [Google Scholar]
- Brown TC, Weinberg GV, Xia A. Removing logarithms from Poisson process error bounds. Stochastic Process. Appl. 2000;87:149–165. [Google Scholar]
- Brown TC, Xia A. Stein’s method and birth–death processes. Ann. Probab. 2001;29:1373–1403. [Google Scholar]
- Burge C, Campbell AM, Karlin S. Over- and under-representation of short oligonucleotides in DNA sequences. Proc. Natl. Acad. Sci. USA. 1992;89:1358–1362. doi: 10.1073/pnas.89.4.1358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen LHY, Xia A. Stein’s method, Palm theory and Poisson process approximation. To appear in Ann. Probab. 2004.
- Creighton TE. Proteins. W.H. Freeman; New York: 1993. [Google Scholar]
- Cressie N. The minimum of higher order gaps. Austral. J. Stat. 1977;19:132–143. [Google Scholar]
- Delecluse HJ, Hammerschmidt W. The genetic approach to the Epstein-Barr virus: From basic virology to gene therapy. J. Clin. Pathol. Mol. Pathol. 2000;53(5):270–279. doi: 10.1136/mp.53.5.270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dembo A, Karlin S. Poisson approximations for r-scan processes. Ann. Appl. Probab. 1992;2(2):329–357. [Google Scholar]
- Durbin R, Eddy S, Krogh A, Mitchison G. Biological Sequence Analysis—Probabilistic Models of Proteins and Nucleic Acids. Cambridge University Press; London: 1998. [Google Scholar]
- Farrell PJ. Epstein-Barr virus. In: O’Brien SJ, editor. Genetic Maps. Book 1, Viruses. 6th ed Cold Spring Harbor Laboratory Press; 1993. [Google Scholar]
- Ghosh D, Godbole AP. Palindromes in random letter generation: Poisson approximations, rates of growth and Erdös-Rényi laws. Lecture Notes in Statistics. 1996;114:99–115. [Google Scholar]
- Glaz J. Approximations and bounds for the distribution of the scan statistics. J. Am. Statist. Assoc. 1989;84(406):560–566. [Google Scholar]
- Glaz J, Naus J, Roos M, Wallenstein S. Poisson approximations for the distribution and moments of ordered m-spacings. J. Appl. Prob. 1994;31A:271–281. [Google Scholar]
- Glaz J, Naus J, Wallenstein S. Scan Statistics. Springer-Verlag; New York: 2001. [Google Scholar]
- Hamzeh FM, Lietman PS, Gibson W, Hayward GS. Identification of the lytic origin of DNA replication in human cytomegalovirus by a novel approach utilizing ganciclovir-induced chain termination. J. Virol. 1990;64:6184–6195. doi: 10.1128/jvi.64.12.6184-6195.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karlin S, Blaisdell BE, Sapolsky RJ, Cardon L, Burge C. Assessments of DNA inhomogeneities in yeast chromosome III. Nucl. Acids Res. 1993;21(3):703–711. doi: 10.1093/nar/21.3.703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karlin S, Taylor HM. A Second Course in Stochastic Processes. 2nd ed Academic Press; New York: 1981. [Google Scholar]
- Kornberg A, Baker TA. DNA Replication. 2nd ed W. Freeman; New York: 1992. [Google Scholar]
- Labrecque LG, Barnes DM, Fentiman IS, Griffin BE. Epstein-Barr virus in epithelial cell tumors: A breast cancer study. Cancer Res. 1995;55(1):39–45. [PubMed] [Google Scholar]
- Leung MY, Burge C, Blaisdell BE, Karlin S. An efficient algorithm for identifying matches with errors in multiple long molecular sequences. J. Mol. Biol. 1991;221:1367–1378. doi: 10.1016/0022-2836(91)90938-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leung MY, Marsh GM, Speed TP. Over- and underrepresentation of short DNA words in herpesvirus genomes. J. Comp. Biol. 1996;3(3):345–360. doi: 10.1089/cmb.1996.3.345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leung MY, Schachtel GA, Yu HS. Scan statistics and DNA sequence analysis: The search for an origin of replication in a virus. Nonlinear World. 1994;1:445–471. [Google Scholar]
- Leung MY, Yamashita TE. Applications of the scan statistic in DNA sequence analysis. In: Glaz J, Balakrishnan N, editors. Scan Statistics and Applications. Birkhauser Publishers; 1999. pp. 269–286. [Google Scholar]
- Masse MJ, Karlin S, Schachtel GA, Mocarski ES. Human cytomegalo-virus origin of DNA replication (oriLyt) resides within a highly complex repetitive region. Proc. Natl. Acad. Sci. USA. 1992;89:5246–5250. doi: 10.1073/pnas.89.12.5246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGeoch DJ, Schaffer PA. Herpes simplex virus. In: O’Brien SJ, editor. Genetic Maps. Book 1, Viruses. 6th ed Cold Spring Harbor Laboratory Press; 1993. [Google Scholar]
- Mrazek J, Karlin S. Strand compositional asymmetry in bacterial and large viral genomes. Proc. Natl. Acad. Sci. USA. 1998;95:3720–3725. doi: 10.1073/pnas.95.7.3720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naus JI. The distribution of the size of the maximum cluster of points on a line. J. Am. Statist. Assoc. 1965;60:532–538. [Google Scholar]
- Newlon CS, Theis JF. DNA replication joins the revolution: Whole-genome views of DNA replication in budding yeast. BioEssays. 2002;24:300–304. doi: 10.1002/bies.10075. [DOI] [PubMed] [Google Scholar]
- Phillips G, Arnold J, Ivarie R. The effect of codon usage on the oligonucleotide composition of the E. coli genome and identification of over- and underrepresented sequences by Markov chain analysis. Nucl. Acids Res. 1987;15:2627–2638. doi: 10.1093/nar/15.6.2627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prum B, Rodolphe F, De Turckheim E. Finding words with unexpected frequencies in DNA sequences. J. R. Statist. Soc. B. 1995;57(1):205–220. [Google Scholar]
- Reinert G, Schbath S. Compound Poisson and Poisson process approximations for occurrences of multiple words in Markov chains. J. Comp. Biol. 1998;5:223–253. doi: 10.1089/cmb.1998.5.223. [DOI] [PubMed] [Google Scholar]
- Reinert G, Schbath S, Waterman MS. Probabilistic and statistical properties of words: An overview. J. Comp. Biol. 2000;7:1–46. doi: 10.1089/10665270050081360. [DOI] [PubMed] [Google Scholar]
- Reisman D, Yates J, Sugden B. A putative origin of replication of plasmids derived from Epstein-Barr virus is composed of two cis-acting components. Mol. Cell. Biol. 1985;5:1822–1832. doi: 10.1128/mcb.5.8.1822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rice P, Longden I, Bleasby A. EMBOSS: The European Molecular Biology Open Software Suite. Trends Genet. 2000;16(6):276–277. doi: 10.1016/s0168-9525(00)02024-2. [DOI] [PubMed] [Google Scholar]
- Roos M. Compound Poisson approximations for the numbers of extreme spacings. Adv. Appl. Probab. 1993;25:847–874. [Google Scholar]
- Salzberg SL, Salzberg AJ, Kerlavage AR, Tomb J-F. Skewed oligomers and origins of replication. Gene. 1998;217:57–67. doi: 10.1016/s0378-1119(98)00374-6. [DOI] [PubMed] [Google Scholar]
- Schbath S. Compound Poisson approximation of word counts in DNA sequences. ESAIM: Prob. and Stat. 1995;1:1–16. [Google Scholar]
- Stein C. A bound for the error in the normal approximation to the distribution of a sum of dependent random variables. Proc. 6th Berkeley Symp. Math Statist. Probab. 1972;2:583–602. [Google Scholar]
- Sugden B. In the beginning: A viral origin exploits the cell. Trends Biochem. Sci. 2002;27(1):1–3. doi: 10.1016/s0968-0004(01)02032-1. [DOI] [PubMed] [Google Scholar]
- Tavaré S, Giddings W. Some statistical aspects of the primary structure of nucleotide sequences. In: Waterman MS, editor. Mathematical Methods for DNA Sequences. CRC Press; Boca Raton, FL: 1989. [Google Scholar]
- Vital C, Monlun E, Vital A, Martin-Negrier ML, Cales V, Leger F, Longy-Boursier M, Le Bras M, Bloch B. Concurrent herpes simplex type 1 necrotizing encephalitis, cytomegalovirus ventriculoencephalitis and cerebral lymphoma in an AIDS patient. Acta pathologica. 1995;89(1):105–108. doi: 10.1007/BF00294267. [DOI] [PubMed] [Google Scholar]
- Wagner EK, editor. Herpesvirus Transcription and its Regulation. CRC Press; Boca Raton, FL: 1991. [Google Scholar]
- Waterman MS. Introduction to Computational Biology. Chapman and Hall; London: 1995. [Google Scholar]
- Weller SK, Spadaro A, Schaffer JE, Murray AW, Maxam AM, Schaffer PA. Cloning, sequencing, and functional analysis of oriL, a herpes simplex virus type 1 origin of DNA synthesis. Mol. Cell. Biol. 1985;5:930–942. doi: 10.1128/mcb.5.5.930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weston K. An enhancer element in the short unique region of human cytomegalo virus regulates the production of a group of abundant immediate early transcripts. Virology. 1988;162:406–416. doi: 10.1016/0042-6822(88)90481-3. [DOI] [PubMed] [Google Scholar]
- Zhu Y, Huang L, Anders DG. Human cytomegalovirus oriLyt sequence requirements. J. Virol. 1998;72:4989–4996. doi: 10.1128/jvi.72.6.4989-4996.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]