

Abstract
Oxygen radical formation in mitochondria is an incompletely understood attribute of eukaryotic cells. Recently, a kinetic model was proposed, in which the ratio between electrons entering the respiratory chain via FADH2 or NADH determines radical formation. During glucose breakdown, the ratio is low; during fatty acid breakdown, the ratio is high (the ratio increasing—asymptotically—with fatty acid length to 0.5, when compared with 0.2 for glucose). Thus, fatty acid oxidation would generate higher levels of radical formation. As a result, breakdown of fatty acids, performed without generation of extra FADH2 in mitochondria, could be beneficial for the cell, especially in the case of long and very long chained ones. This possibly has been a major factor in the evolution of peroxisomes. Increased radical formation, as proposed by the model, can also shed light on the lack of neuronal fatty acid oxidation and tells us about hurdles during early eukaryotic evolution. We specifically focus on extending and discussing the model in light of recent publications and findings.
Keywords: radical formation, FADH2/NADH ratio, complex I, supercomplex, neurodegenerative disorder, peroxisome
1. Introduction
Mitochondria, derived from endosymbionts of alpha-proteobacterial origin [1,2], underwent complex evolutionary rearrangements influencing the metabolic potential of their archeal host [3]. During this process, the endosymbiont gene content was severely reduced from at least hundreds of genes [4], to present-day mitochondrial genomes encoding only up to 64 proteins in Reclinomonas americana [5] and merely 13 in humans. Thus, almost all genes encoding the endosymbiont's original metabolic pathways have been relegated to the nuclear genome, while leaving many, but not all, the endosymbiotic metabolic pathways in the organelle [6]. The proteins originally encoded by the endosymbiotic genome had to be retargeted to mitochondria. Interestingly, this losing of genes from the organelle to the nucleus and accompanying protein rerouting started immediately, in evolutionary terms, after the endosymbiosis event [4]. Recent analyses show a few endosymbiont systems retained entirely by mitochondria, despite their genes' relocalization to the nucleus. These are large protein complexes such as the respiratory chain complexes and the mitochondrial ribosome [7]. Some of the endosymbiont pathways are now found in other cellular compartments, and many mitochondrial pathways not using large protein complexes bear bacterial—not always proteobacterial—evolutionary footprints. Mitochondria also gained new functions including membrane proteins at the interface with the rest of the cell [7] with entirely new roles in, for instance, cellular apoptosis. The severely reduced genome should thus not be seen to indicate a lack of importance of the organelle.
The mitochondrion is the essential powerhouse of the cell, with more than 90% of all energy (in the form of ATP) generated by the breakdown of glucose coming from oxidative mitochondrial processes in which the electron flow through respiratory chain complexes is coupled to the establishment of a proton motive force used for ATP generation: oxidative phosphorylation, the OXPHOS system [8,9]. Most biochemists would agree that highly complex (multi) cellular organisms seem unlikely to evolve in the absence of the highly efficient ATP generation occurring in mitochondria. However, whether the role of oxygen as the final electron acceptor was crucial, is still debated. In any case, many instances of present-day anaerobic uni- or multicellular eukaryotes are known [10]. For a discussion considering which aspects of the development of mitochondria from the endosymbiont were crucial in allowing major increases in (genome) complexity, see Lane and Martin [11]. Regardless, the OXPHOS system is extremely efficient. Oxygen might also have played a more indirect role in enabling further organismal complexity, as it seems indispensable for full-fledged epigenetic signalling (via, amongst others, the marking of DNA and histone proteins, using methylation of cytosines and lysines, respectively). Getting rid of methylation in these cases seems to depend on oxygenases, and thus on oxygen [12].
There is a price to pay, however [13,14]. The use of molecular oxygen (O2) as the final electron acceptor of the respiratory chain, with the formation of H2O, inevitably leads to the formation of very toxic by-products. They are found in the form of reactive oxygen species (ROS) [15–17], such as the highly reactive free radicals (superoxide anions, .O2−, and hydroxyl radicals, HO.) and peroxides (e.g. H2O2). It is estimated that of all molecular oxygen consumed, 0.1–2.0% ends up in the form .O2−, depending on the respiratory state of the mitochondrion [17]. These form large pools of toxic molecules that the cell has to cope with. During the evolution of the two ancestors of the first eukaryotes and later on, several mechanisms evolved to deal with ROS, such as mitochondrial manganese-containing superoxide dismutase (MnSOD), cytoplasmic copper–zinc-containing superoxide dismutase (CuZnSOD), glutathione-based systems and thioredoxins. For an overview of general and mitochondrion-specific anti-oxidant measures, see Andreyev et al. [18].
Most of the present-day eukaryotes, and animals in particular, use O2 as the final electron acceptor of the respiratory chain, with the exception of organisms using mitosomes, retaining only the role the mitochondrion has in Fe–S cluster assembly, or hydrogenosomes, producing molecular hydrogen instead of water [19,20]. Even metazoans have now been found to live under conditions of continuous anoxia [10,21]. Still, most eukaryotes build up the proton motive force for ATP generation using the oxidative OXPHOS system. This means that high amounts of ATP for eukaryotes are intrinsically linked to ROS formation inside the cell: internal radicals are inevitable in complex cellular life. Eukaryotes seem to have responded in two ways: as described above, they have harnessed older mechanisms to cope with the toxic by-products of using oxygen and evolved new mechanisms of mitochondrial quality control [22]. Additionally, eukaryotes made a virtue of necessity by using mitochondria and their oxygen radical generating ability as an important part of the apoptotic process used in (multicellular) eukaryotes [23,24].
2. Challenges during early eukaryotic evolution
During the first stage of metabolic reintegration upon the uptake of the precursor of the mitochondrion, the (pre)eukaryote must have been confronted with multiple hurdles during its transition to the full eukaryotic state. We concentrate on the hurdles presumably involved in catabolic oxidative processes using the alpha-proteobacterial respiratory chain as the final route that the energy-rich electrons from different food sources have to take to end up reducing molecular oxygen, allowing optimal ATP generation. Two problems seem logical at the onset of symbiosis. First of all, toxic ROS formed in the respiratory chain becomes a bigger problem for the composite cell, than it was for its constituent alpha-proteobacterium. Radical formation on the outskirts of the bacterium (which could even have played a role in cellular defence) now occurs in the middle of the new cellular entity. Additionally, over time, the new composite organism was likely confronted with other ROS-related problems as well, as separate catabolic routes of both endosymbiont and host had to be integrated to form a functioning eukaryotic cell. The host (as reflected by the current location of glycolysis in the cytoplasm) had sugar breakdown pathways, whereas the proto-mitochondrion was equipped with aerobic catabolic pathways for lipids, amino acids and glycerol, including a complete respiratory chain, beta oxidation and at least part of the citric acid cycle [19,25,26]. Although the ability to use alternating sources of energy such as glucose or fatty acids over time is of course an enormous source of versatility, greatly extending possible ecological niches of the eukaryotes (as illustrated by yeast [27]), we can also think of intrinsic problems here.
One of the problems arising has to do with the fact that the relative amounts of FADH2 and NADH (as reflected in FADH2/NADH ratios) generated during the breakdown of glucose differ from that of the breakdown of fatty acids. A respiratory chain functioning in glucose breakdown might encounter problems upon switching to fatty acid catabolism owing to competition for fully oxidized ubiquinone, as ubiquinone is also used as an acceptor by the electron transfer flavoprotein: ubiquinone oxidoreductase (ETF-QO) oxidizing FADH2 [14,28]. This FADH2 is generated from FAD by reduction during the first recurring step of beta oxidation for every acetyl-CoA generated during fatty acid breakdown (a step catalysed by acyl-CoA dehydrogenase). This implies that especially during the breakdown of longer fatty acids the competition for ubiquinone becomes more difficult for complex I, which is known to be the major source of oxygen radicals [16]. As described before [14], large electron fluxes via both complex I on the one hand and complex II, as well as ETF, on the other hand, would probably lead to a significant increase in radical formation (see also [17,29]). We further focus on the implications of these ‘catabolic’ difficulties with regard to radical formation in (pre)eukaryotic cells for eukaryotic evolution, and for metabolic reprogramming at the cellular and multicellular (tissue) level. A schematic overview of possible solutions to the problem of increased radical formation due to catabolism of substrates associated with high FADH2/NADH ratios is given in figure 1.
Figure 1.
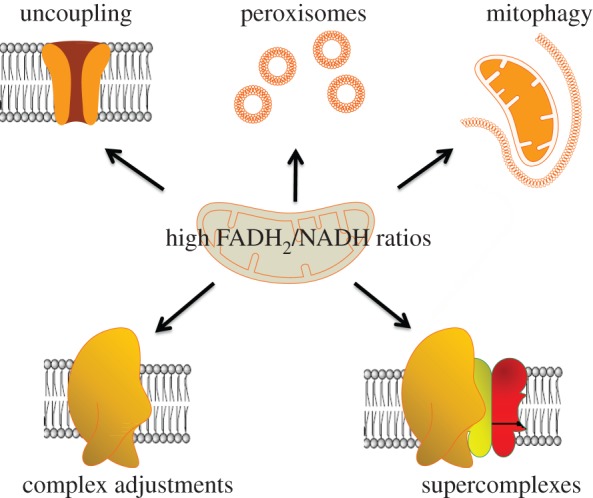
Schematic of possible adaptations to interior radical formation by respiratory chain complexes (especially complex I) upon catabolizing substrates with high FADH2/NADH ratios. Clockwise: 1. Development of mechanisms to induce (regulated) mild uncoupling (e.g. uncoupling proteins). 2. Segregation of fatty acid oxidation (completely in neurons, plants and some yeasts, limited to VLCFAs only in most mammalian cells). 3. Evolving specific mitophagy pathways. Apart from mitophagy, a mitochondrion-associated decay (MAD) pathway appeared (resembling the ERAD pathway, see text). 4. Stimulating supercomplex formation, e.g. allowing complex I ‘direct’ access to its ‘own’ ubiquinone pool. 5. Adjusting complexes (especially I) and their synthesis to lessen both radical formation and its impact. Replacing complex I completely with Ndi1, as seen in Saccharomyces cerevisiae [30], can be seen as an extreme instance of the latter. (Online version in colour.)
3. Did (proto) mitochondria create circumstances favouring the rise of peroxisomes?
A number of predictions can be derived from the hypothesis that high FADH2/NADH flux ratios lead to increased mitochondrial radical formation, especially in cells with high energy needs. First of all, eukaryotic cells would have an advantage if they evolved mechanisms to lower FADH2/NADH flux ratios during fatty acid breakdown at a minimal ATP loss. Second, all cells that are difficult to replace or very sensitive to oxidative damage should, as far as possible, forego mitochondrial catabolism in general and beta oxidation specifically. We consider the possible evolutionary solutions reflecting the first prediction next.
As already stated [14], the previous considerations give a surprisingly good explanation for the eukaryotic invention of peroxisomes. The hypothesis explaining their origin can be described as follows. Peroxisomes are known to be derived from, or coevolved with, the endoplasmic reticulum (ER) [31–36]. According to the model, the forces giving rise to the evolution of peroxisomes stem from fatty acid catabolism, because of the fact that advantage accrues from mechanisms to lower the FADH2/NADH ratio during fatty acid breakdown. This is exactly what happened in the emerging peroxisome, the oldest pathway of which is beta oxidation [36], without the generation of FADH2 which has to be taken care of by the mitochondrial respiratory chain. Beta oxidation occurs in four steps: (i) dehydrogenation (mitochondria) or direct oxidation (peroxisomes), (ii) hydration, (iii) oxidation by NAD+ and (iv) thiolysis, i.e. cleavage by the thiol group of coenzyme A. The eponymous H2O2 formed in the first reaction is dealt with by a specific peroxisomal enzyme, catalase. For more specific details, see Reddy & Hashimoto [37]. The ways in which the (partial) redistribution occurred are also telling. In mammalian peroxisomes, such as our own, the trade-off between minimal ATP loss (the ‘ATP content’ of FADH2 generated in peroxisomes is lost because of direct oxidation, forming H2O2) and overall reduction of the FADH2/NADH ratio led to the specific breakdown of very long chain fatty acids (VLCFAs) in the peroxisomes only. The VLCFAs have the highest FADH2/NADH ratios, consistent with our expectations.
In plants and most yeasts, evolution led to the migration of all beta oxidation from mitochondria to peroxisomes. This further reduces mitochondrial radical formation by lowering competition between complex I and FADH2 containing complexes, but at some energetic cost (see above). So why complete migration in plants and yeasts? Plants are able to be slightly less efficient with energy because they became autotrophs, synthesizing their own food. Plants gained this ability upon the secondary endosymbiotic event, the uptake of a cyanobacterium, which gave rise to the chloroplast. Many yeast species seem to use sugars, alcohol or organic acids as major sources of energy and in many cases have retained efficient fermentation pathways. All of this makes a complete relocation of fatty acid oxidation to the peroxisomes possible, while reducing oxygen radical generation. Reduction of radical formation by the respiratory chain can go even further, again at some energetic cost in the form of reduced proton pumping. Interestingly, mitochondria of Saccharomyces cerevisiae lack complex I, instead having a rotenone-insensitive NADH dehydrogenase (Ndi1). Ndi1 is formed by a single polypeptide which lacks all proton pumping function [30]. This points to a specific oxygen radical generating vulnerability associated with complex I even in the absence of beta oxidation (see also below). However, it should be mentioned that S. cerevisiae is not an ideal model in this context, as it survives using respiration, but thrives during fermentation.
Recently, an evolutionary force associated with fatty acid metabolism playing a role in the evolution of peroxisomes (providing a possible advantage by sequestering toxic by-products of such metabolism) has again been postulated [38,39]. However, in this case, the cellular site at which oxygen radical formation during fatty acid metabolism leads to the selective force favouring peroxisome formation is thought to be the endoplasmic reticulum, instead of the (proto)mitochondrion [38]. In addition, possible radical formation during fatty acid synthesis, instead of breakdown, is considered a driving force for peroxisomal sequestering of such reactions. There are, however, some issues that need to be resolved [39]. As mentioned, the most ancestral peroxisomal pathway is beta oxidation, the four steps of which are catalysed by only three enzymes, as step (ii) and (iii) are catalysed by only one protein: Pox2p (yeast nomenclature), an enzyme stemming from the alpha-proteobacterial heritage. This suggests a crucial contribution of mitochondrial metabolic enzymes to the evolution of peroxisome. Such a mitochondrial ‘input’ with regard to peroxisomal evolution is further strengthened by the following observations. Mammalian mitochondria and peroxisomes have been shown to communicate and cooperate intensively [40]. Quite recently, a direct relationship between both organelles has been found in HeLa cells, taking the form of cargo-selected transport from mitochondria to peroxisomes [41]. We also practically always find peroxisomes and mitochondria together. Amitochondriate, but hydrogenosome-containing species such as Giardia lamblia do not contain peroxisomes. Loss of peroxisomes goes together with extremely reduced mitochondrial function, such as lack of beta oxidation, in apicomplexans. Therefore, it appears that a likely candidate for the driving force behind peroxisome evolution is indeed reduction of mitochondrial radical formation inherent in beta oxidation of fatty acids owing to the high FADH2/NADH ratio involved. We know present-day peroxisomes are derived from the ER (see above). We also know that the peroxisomal protein import machinery is homologous to, and possibly derived from, the ER-associated degradation (ERAD) system [36]. Interestingly, a related mitochondrial system (MAD) has been discovered to operate [42–44]. This makes the reconstruction of the strict evolutionary order of events even more difficult. Although purely hypothetical, one could envisage all new endomembrane structures, such as peroxisomes, the nucleus and the ER evolving in reaction to evolutionary pressures resulting from endosymbiont entry. Both ER and Golgi formation took place over a relatively short time span, early on in eukaryotic evolution [45]. Indeed, a large number of important evolutionary events occurred rapidly between endosymbiont acquisition and subsequent eukaryotic radiation, which is consistent with early acquisition of, and adjustment to, mitochondria in eukaryotic evolution [4]. Using an adaptation to the endosymbiont to explain a general characteristic (here the peroxisome) of the eukaryotic lineage is also found in other models. One can think of Martin and Koonin's explanation for the development of a nucleus (and by extension of the ER) in response to alpha-proteobacterial group II introns migrating to the host genome [46].
4. Further adaptations to internal radical formation upon competition for ubiquinone
What other kind of eukaryotic adaptations are possibly related to suppressing radical formation during breakdown of substrates with high FADH2/NADH ratios? First of all, the widespread tendency of respiratory chain complexes to form quaternary structures called supercomplexes is in line with such a function. For instance, it was observed that all the standard respiratory chain complexes except the FADH containing complex II can associate in such supercomplexes [47–49]. Such supercomplex formation could restrict a (dedicated) ubiquinone pool to use by complex I only, reducing the chance of radical formation by reverse electron transfer (RET) from a general pool reduced by complex II. There are many indications that supercomplexes are involved in reducing radical formation [50] and that loss of supercomplex formation leads to increased superoxide formation and the mitochondrial ageing phenotype, most severely in post-mitotic tissues [51]. Supercomplex formation represents a dynamic mechanism allowing cells to organize the respiratory chain optimally in dealing with varying carbon sources (and in specific cell types [48]).
Apart from supercomplex formation, the appearance of so-called uncoupling proteins (UCPs) could be linked to the dangers of radical formation by the mitochondrial respiratory chain inside the cell. Extensive radical formation via RET at complex I is found to be more pronounced when electrons are supplied to ubiquinone from succinate, alpha-glycerophosphate or fatty acid oxidation [17]. This radical production comes from electrons entering complex I through the ubiquinone-binding site(s), but is only observed if Δp is high (e.g. during extensive oxidation of both NADH and FADH2). All the observations above are consistent with the kinetic model of radical formation owing to competition for ubiquinone. The precise molecular mechanisms playing a role are unclear. However, a high proton motive force (Δp) and large electron flows via FADH containing enzymes seem to be essential. In isolated mitochondria, RET-induced radical formation is abolished upon slight decreases of Δp [17]. This could mean that lowering Δp by the expression of UCPs would efficiently reduce radical formation in the cell. The UCPs form a family of proton transporters in the mitochondrial inner membrane which have been implicated in thermoregulation and protection against oxidative damage by their ability to induce uncoupling [52–54]. UCPs belong to the mitochondrial carriers family, an inner membrane protein family involved in transporting keto acids, amino acids, nucleotides, inorganic ions and cofactors across the mitochondrial inner membrane [55]. Mitochondrial carriers are part of the organellar interface—and as such arose with the early eukaryote [7]. More recent evolution of UCPs indicates multiple duplications in vertebrates [56]. Five different UCPs have been identified [57]. They are found in highly variable amounts in various tissues in all modern mammals [52], as well as in plants and unicellular eukaryotes. UCP1–3 have been studied in some detail, but UCP4 and 5 (interestingly, both mainly expressed in the brain, see below) have only been characterized more recently. UCP1 in mammalian brown adipose tissue has a clear role in heat production, whereas UCP1, UCP2 and UCP3, in other species, are implicated in ROS reduction, though their precise roles are still heavily debated. Interestingly, when mammalian UCP1 was expressed in yeast mitochondria, proton transport was increased significantly in the presence of the (saturated C-16) fatty acid palmitate or an oxidized fatty acid (4-hydroxy-2-nonenal) only. Together, they showed a clear synergistic effect [58]. 4-Hydroxy-2-nonenal can be formed by the activity of superoxide, a well-known ROS species, which can also activate UCPs on its own. So it seems that, as high ROS formation owing to RET in complex I driven by oxidation of fatty acids is acutely sensitive to Δp, it could thus be controlled via slight uncoupling of oxidative phosphorylation. This is accomplished by fatty acids via activation of UCPs. That activation would be even stronger in the simultaneous presence of oxidized fatty acids (see also [59,29]). In conclusion, the data regarding most members of the UCP protein family seem to support a role in dealing with specific problems of internal radical formation during fatty acid oxidation.
5. The ‘special’ position of neurons
Above, we predicted that all cells which are ‘irreplaceable’ or extraordinarily sensitive to oxidative damage should have reduced levels of mitochondrial fatty acid oxidation. A prime example of such cells are neurons. Neurons have very high energy demands. It has been calculated that the brain only accounts for approximately 2% of body mass, while it consumes 20% of total oxygen taken up by the entire body. In the brain, neurons have the maximum energy consumption, whereas astrocytes are only responsible for approximately 5–15% of its energy requirements [60]. This immense metabolic demand of neurons results from the fact that they are highly differentiated cells needing large amounts of ATP for maintenance of ionic gradients across cell membranes for neurotransmission [61]. We should also point out that neurons are completely dependent on the OXPHOS system for their ATP needs, as they are not able to switch to glycolysis when OXPHOS becomes limited [62]. In an apparent conflict with these high energy demands, it is beneficial for organisms if neurons are long lived, because they carry essential information for the organism. This poses a dilemma, as oxidative catabolism generates toxic oxygen radicals. Our considerations regarding FADH2/NADH ratios explain why neurons do not use beta oxidation [14,63,64], instead relying on glucose (and ketone bodies) for their energy supply, even though fatty acids have about twice the energy content of sugars. Despite the banishment of fatty acids from the menu, neurons of course still need complete oxidative catabolism of glucose, with full mitochondrial involvement. This still results in radical damage, albeit reduced, that neurons accumulate owing to their high energy needs.
Mitochondrial diseases have extremely inconsistent clinical presentations, affecting any organ in isolation or combination at any age with variable severity. The genetic basis of these diseases is complex. Mutations in more than 100 genes encoded by the nucleus, inherited in a Mendelian manner, or on the mitochondrial genome inherited maternally, have been found so far [65,66]. Organs with high energy demand are especially afflicted, most prominently the brain [65–67]. Neurodegeneration, in which neurons are specifically steadily destroyed, leads to a progressive loss of nervous system structure and function [68,69]. Again, the pathological picture in neurodegenerative disorders is heterogeneous, affecting unique areas of the nervous system. In the brain, Purkinje cells and neurons of the dentate nuclei of the cerebellum as well as neurons of the inferior olivary nuclei of the medulla are considered to be most energy demanding, and thus most susceptible to damage [70]. Symptoms range from acute and rapid in progression to subtle and chronic [71]. The critical function of the neuronal OXPHOS system in maintaining bioenergetic needs is illustrated by the fact that OXPHOS dysfunction leads to defects in growth and trafficking as well as to apoptosis and neuronal death [72]. This, in turn, can lead to microglial activation, a pathophysiological consequence that can trigger catastrophic events such as further oxidative and nitrosative stress, ultimately leading to further neuronal damage and death [73]. OXPHOS dysfunction seems also involved in the occurrence of non-functional synapses and axonal degeneration [74].
6. The possible role of mitochondrial reactive oxygen species production in neurodegenerative disorders
Despite not using beta oxidation, neuronal radical formation by complex I is still involved in pathogenesis. Increasing evidence shows that disrupted mitochondrial function, especially at complexes I and III, enhances ROS production in neurodegenerative disorders. Here, we briefly review the role of ROS in Parkinson's disease (PD), amyotrophic lateral sclerosis (ALS) and Alzheimer's disease (AD) [75–77].
In PD, neurons in the motor centres such as the cerebellum and the striatum are decreased. Substantial decreases in dopaminergic neurons, containing proteinaceous Lewy bodies, of the substantia nigra pars compactica are observed [78]. PD patients not only show depletion of dopaminergic neurons, but also decreases in Purkinje cell numbers in the cerebellar cortex, explaining motor skill losses. It is still mostly unknown what causes PD; however, genetic mutations in Parkin [79] and PINK1 [80], both implicated in the removal of damaged mitochondria via autophagy, have been observed. Defects in mitochondrial respiration, particularly at complex I [79,81], also result in elevated ROS production which further leads to increased oxidative stress and dopaminergic neuronal degeneration, all strongly correlating with development of PD [77]. Interestingly, in the context of our model regarding high FADH2/NADH ratios leading to ROS production because of competition for the acceptor ubiquinone, reduced complex I activity can be compensated by increased complex II activity in PD fibroblasts. Indeed, the contribution of complex II to ATP generation is hardly diminished when compared with control fibroblasts [79]. Moreover, 50% of late onset sporadic PD patient derived fibroblasts even were able to restore complex I activity upon incubation with ubiquinone [82]. These findings are paralleled in Drosophila mitochondria: parkin and PINK1 mutant phenotypes were found to be enhanced by mutations in the UBIAD1/Heix gene, involved in making the alternative electron acceptor vitamin K2. Overexpressing wildtype Heix or supplying vitamin K2 ‘rescued’ Drosophila from PD-like symptoms [83].
The motor neuron disease ALS shows loss of motor neurons in cortex, brain stem and spinal cord with aggregated neurofilaments in proximal axons of motor neurons. ALS presents Bunina bodies, eosinophilic aggregates found in anterior horn cells [84]. Apart from rare predisposing mutations in the mitochondrial superoxide dismutase gene (MnSOD; see above), ALS has no known causes. Surprisingly, in MnSOD mutant rats, binding of the misfolded mutant protein to VDAC1 was shown, resulting in reduced VDAC1 activity and loss of motor neurons [85]. This could imply that loss of MnSOD function in ROS protection is not important in the development of ALS.
AD is characterized by significant decreases in intellectual ability and memory, as well as personality changes [86]. AD gives rise to neurofibrillary tangles, constituted of hyper phosphorylated tau proteins. Extracellular senile plaques of β-amyloid (Aβ) are most prominently found on cholinergic neurons supplying the hippocampus [87]. Increasing evidence points to a role of OXPHOS dysfunction in the development of AD. In a transgenic mouse model study, decreased mitochondrial membrane potential and decreased ATP levels in neurons could be observed and degree of cognitive impairment correlated with the amount of synaptic mitochondrial dysfunction [88]. Interestingly, Aβ seems to inhibit several key mitochondrial enzymes in the brain, complex IV looking most relevant [89]. This last observation nicely illustrates one of the key problems in describing a neurodegenerative disease such as AD from a perspective of mitochondrial dysfunction and concomitant radical formation: is it cause or effect? [90].
7. Complex I as the most important site of radical formation
As seen from the description of the most clear example of the involvement of ROS generation and a dysfunctional OXPHOS in the aetiology of neurodegenerative disease, PD, complex I plays a crucial role. It is known to be the major source of oxygen radicals [16]. Complex I has grown considerably in size during evolution, gathering so-called supernumerary subunits. It grew from its 14 bacterial subunits, to encompass 30 in algae and plants, 37 in fungi and at least 44 in human beings [91]. The process of complexification was most likely already (almost) complete in the very last eukaryotic common ancestor [92–94]. It is tempting to think, that apart from performing chaperone functions during assembly and membrane insertion, as well as giving stability to this huge protein complex, some supernumerary subunits are specifically involved in minimizing the occurrence of, and combating, radical formation. This however has to await further research into the specific roles of ‘extra’ subunits.
Interestingly, at the origin of the vertebrate lineage, acyl-CoA dehydrogenase 9 (ACAD9), closely resembling very long-chain acyl-CoA dehydrogenase (VLCAD), lost its function in mitochondrial oxidation of fatty acids. Instead, it became a required factor for complex I assembly [95]. This could be another indication for a functional link between expressing enzymes involved in fatty acid oxidation and stress for complex I [14]. Very recently, pluripotent stem cells from patients carrying a common mutation in mitochondrial DNA, m.3243A>G, mutating the tRNA LEU gene, were generated. The most pronounced problems were associated with complex I. Upon differentiation to neurons, complex I seemed to be actively and specifically (!) degraded via mitophagy (involving PINK1 and Parkin-positive autophagosomes). After the concentration of complex I is thus diminished, complex IV becomes slightly impaired, possibly owing to extensive radical formation, and complex II is upregulated, presumably to compensate for the loss in ATP generation, as previously observed in patients carrying this mitochondrial DNA mutation [96]. Although it is known that oxidative stress can induce autophagy [24,97], such specific targeting of malfunctioning complex I points to its crucial position in the balancing act between ATP generation and radical formation.
8. Conclusion
Aerobic eukaryotic life has to perform a balancing act between efficient ATP generation and oxygen radical formation. If the cell is easily replaceable in a multicellular organism and it has high energy needs, then it will use mitochondrial beta oxidation of fatty acids, with the exception of one class, as explained by the model presented here. Such cells will only relegate VLCFAs to the peroxisomes, giving high ATP yields, while only somewhat reducing radical formation via complex I. Neurons have a different position along the line of mammalian cells as characterized by the trade-off between efficient ATP generation and amount of ROS generated. Neurons will not oxidize any fatty acids, but consume only less energy-rich glucose. However, breakdown has to be performed efficiently, with full mitochondrial involvement. Stem cells and some cancer cells (consider the Warburg effect) also only consume glucose, but can rely on permanent glycolysis with resulting lactate being recycled to glucose by other cells in the body (such as the hepatocytes), even further reducing their mitochondrial contribution and amount of ROS generated, especially by complex I. An impressive array of cellular mechanisms evolved partially to cope with the arrival of an internal oxygen radical generator in the form of the (pre)mitochondrion, among others: uncoupling mechanisms, partial or total segregation of fatty acid oxidation, mitophagy, the MAD pathway, supercomplex formation, adjustment of the respiratory chain complexes themselves and even apoptosis when possible. The results described here are fully consistent with the model we focused on, one in which FADH2/NADH ratios, as encountered during breakdown of different metabolites, are crucial to ROS formation. However, in the light of all the cellular adaptations just mentioned, proof will not be easy to come by. We have to await future experiments.
Acknowledgements
This work was supported by Horizon grant (no. 050-71-053) from the Netherlands Organization for Scientific Research (NWO) and the CSBR (Centres for Systems Biology Research) initiative from NWO (no: CSBR09/013V).
References
- 1.Thrash JC, et al. 2011. Phylogenomic evidence for a common ancestor of mitochondria and the SAR11 clade. Sci. Rep. 1, 13 ( 10.1038/srep00013) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Yang D, Oyaizu Y, Oyaizu H, Olsen GJ, Woese CR. 1985. Mitochondrial origins. Proc. Natl Acad. Sci. USA 82, 4443–4447. ( 10.1073/pnas.82.13.4443) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Yutin N, Makarova KS, Mekhedov SL, Wolf YI, Koonin EV. 2008. The deep archaeal roots of eukaryotes. Mol. Biol. Evol. 25, 1619–1630. ( 10.1093/molbev/msn108) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Huynen MA, Duarte I, Szklarczyk R. 2013. Loss, replacement and gain of proteins at the origin of the mitochondria. Biochim. Biophys. Acta 1827, 224–231. ( 10.1016/j.bbabio.2012.08.001) [DOI] [PubMed] [Google Scholar]
- 5.Lang BF, Burger G, O'Kelly CJ, Cedergren R, Golding GB, Lemieux C, Sankoff D, Turmel M, Gray MW. 1997. An ancestral mitochondrial DNA resembling a eubacterial genome in miniature. Nature 387, 493–497. ( 10.1038/387493a0) [DOI] [PubMed] [Google Scholar]
- 6.Gabaldon T, Huynen MA. 2005. Lineage-specific gene loss following mitochondrial endosymbiosis and its potential for function prediction in eukaryotes. Bioinformatics 21(Suppl. 2), ii144–ii150. ( 10.1093/bioinformatics/bti1124) [DOI] [PubMed] [Google Scholar]
- 7.Szklarczyk R, Huynen MA. 2010. Mosaic origin of the mitochondrial proteome. Proteomics 10, 4012–4024. ( 10.1002/pmic.201000329) [DOI] [PubMed] [Google Scholar]
- 8.Smeitink J, van den Heuvel L, DiMauro S. 2001. The genetics and pathology of oxidative phosphorylation. Nat. Rev. Genet. 2, 342–352. ( 10.1038/35072063) [DOI] [PubMed] [Google Scholar]
- 9.Mitchell P. 1961. Coupling of phosphorylation to electron and hydrogen transfer by a chemi-osmotic type of mechanism. Nature 191, 144–148. ( 10.1038/191144a0) [DOI] [PubMed] [Google Scholar]
- 10.Muller M, et al. 2012. Biochemistry and evolution of anaerobic energy metabolism in eukaryotes. Microbiol. Mol. Biol. Rev. 76, 444–495. ( 10.1128/MMBR.05024-11) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lane N, Martin W. 2010. The energetics of genome complexity. Nature 467, 929–934. ( 10.1038/nature09486) [DOI] [PubMed] [Google Scholar]
- 12.Jeltsch A. 2013. Oxygen, epigenetic signaling, and the evolution of early life. Trends Biochem. Sci. 38, 172–176. ( 10.1016/j.tibs.2013.02.001) [DOI] [PubMed] [Google Scholar]
- 13.Harman D. 1981. The aging process. Proc. Natl Acad. Sci. USA 78, 7124–7128. ( 10.1073/pnas.78.11.7124) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Speijer D. 2011. Oxygen radicals shaping evolution: why fatty acid catabolism leads to peroxisomes while neurons do without it: FADH(2)/NADH flux ratios determining mitochondrial radical formation were crucial for the eukaryotic invention of peroxisomes and catabolic tissue differentiation. Bioessays 33, 88–94. ( 10.1002/bies.201000097) [DOI] [PubMed] [Google Scholar]
- 15.Turrens JF. 2003. Mitochondrial formation of reactive oxygen species. J. Physiol. 552, 335–344. ( 10.1113/jphysiol.2003.049478) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Brand MD. 2010. The sites and topology of mitochondrial superoxide production. Exp. Gerontol. 45, 466–472. ( 10.1016/j.exger.2010.01.003) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Murphy MP. 2009. How mitochondria produce reactive oxygen species. Biochem. J. 417, 1–13. ( 10.1042/BJ20081386) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Andreyev AY, Kushnareva YE, Starkov AA. 2005. Mitochondrial metabolism of reactive oxygen species. Biochemistry (Mosc.) 70, 200–214. ( 10.1007/s10541-005-0102-7) [DOI] [PubMed] [Google Scholar]
- 19.Hackstein JH, Tjaden J, Huynen M. 2006. Mitochondria, hydrogenosomes and mitosomes: products of evolutionary tinkering! Curr. Genet. 50, 225–245. ( 10.1007/s00294-006-0088-8) [DOI] [PubMed] [Google Scholar]
- 20.Lindmark DG, Muller M. 1973. Hydrogenosome, a cytoplasmic organelle of the anaerobic flagellate Tritrichomonas foetus, and its role in pyruvate metabolism. J. Biol. Chem. 248, 7724–7728. [PubMed] [Google Scholar]
- 21.Danovaro R, Dell'Anno A, Pusceddu A, Gambi C, Heiner I, Kristensen RM. 2010. The first metazoa living in permanently anoxic conditions. BMC Biol. 8, 30 ( 10.1186/1741-7007-8-30) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Szklarczyk R, Nooteboom M, Osiewacz HD. 2014. Control of mitochondrial integrity in ageing and disease. Phil. Trans. R. Soc. B 369, 20130439 ( 10.1098/rstb.2013.0439) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wang C, Youle RJ. 2009. The role of mitochondria in apoptosis. Annu. Rev. Genet. 43, 95–118. ( 10.1146/annurev-genet-102108-134850) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Korsmeyer SJ, Yin XM, Oltvai ZN, Veis-Novack DJ, Linette GP. 1995. Reactive oxygen species and the regulation of cell death by the Bcl-2 gene family. Biochim. Biophys. Acta 1271, 63–66. ( 10.1016/0925-4439(95)00011-R) [DOI] [PubMed] [Google Scholar]
- 25.Gabaldon T, Huynen MA. 2003. Reconstruction of the proto-mitochondrial metabolism. Science 301, 609 ( 10.1126/science.1085463) [DOI] [PubMed] [Google Scholar]
- 26.Gabaldon T, Huynen MA. 2004. Shaping the mitochondrial proteome. Biochim. Biophys. Acta 1659, 212–220. ( 10.1016/j.bbabio.2004.07.011) [DOI] [PubMed] [Google Scholar]
- 27.Landry CR, Townsend JP, Hartl DL, Cavalieri D. 2006. Ecological and evolutionary genomics of Saccharomyces cerevisiae. Mol. Ecol. 15, 575–591. ( 10.1111/j.1365-294X.2006.02778.x) [DOI] [PubMed] [Google Scholar]
- 28.Watmough NJ, Frerman FE. 2010. The electron transfer flavoprotein: ubiquinone oxidoreductases. Biochim. Biophys. Acta 1797, 1910–1916. ( 10.1016/j.bbabio.2010.10.007) [DOI] [PubMed] [Google Scholar]
- 29.Brand MD, et al. 2004. Mitochondrial superoxide and aging: uncoupling-protein activity and superoxide production. Biochem. Soc. Symp. 71, 203–213. [DOI] [PubMed] [Google Scholar]
- 30.Iwata M, Lee Y, Yamashita T, Yagi T, Iwata S, Cameron AD, Maher MJ. 2012. The structure of the yeast NADH dehydrogenase (Ndi1) reveals overlapping binding sites for water- and lipid-soluble substrates. Proc. Natl Acad. Sci. USA 109, 15 247–15 252. ( 10.1073/pnas.1210059109) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.van der Zand A, Braakman I, Tabak HF. 2010. Peroxisomal membrane proteins insert into the endoplasmic reticulum. Mol. Biol. Cell 21, 2057–2065. ( 10.1091/mbc.E10-02-0082) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Tabak HF, Hoepfner D, van der Zand A, Geuze HJ, Braakman I, Huynen MA. 2006. Formation of peroxisomes: present and past. Biochim. Biophys. Acta 1763, 1647–1654. ( 10.1016/j.bbamcr.2006.08.045) [DOI] [PubMed] [Google Scholar]
- 33.Tabak HF, van der Zand A, Braakman I. 2008. Peroxisomes: minted by the ER. Curr. Opin. Cell. Biol. 20, 393–400. ( 10.1016/j.ceb.2008.05.008) [DOI] [PubMed] [Google Scholar]
- 34.van der Zand A, Gent J, Braakman I, Tabak HF. 2012. Biochemically distinct vesicles from the endoplasmic reticulum fuse to form peroxisomes. Cell 149, 397–409. ( 10.1016/j.cell.2012.01.054) [DOI] [PubMed] [Google Scholar]
- 35.Tabak HF, Braakman I, van der Zand A. 2013. Peroxisome formation and maintenance are dependent on the endoplasmic reticulum. Annu. Rev. Biochem. 82, 723–744. ( 10.1146/annurev-biochem-081111-125123) [DOI] [PubMed] [Google Scholar]
- 36.Gabaldon T, Snel B, van Zimmeren F, Hemrika W, Tabak H, Huynen MA. 2006. Origin and evolution of the peroxisomal proteome. Biol. Direct. 1, 8 ( 10.1186/1745-6150-1-8) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Reddy JK, Hashimoto T. 2001. Peroxisomal beta-oxidation and peroxisome proliferator-activated receptor alpha: an adaptive metabolic system. Annu. Rev. Nutr. 21, 193–230. ( 10.1146/annurev.nutr.21.1.193) [DOI] [PubMed] [Google Scholar]
- 38.Gabaldón T. In press A metabolic scenario for the evolutionary origin of peroxisomes from the endomembranous system. Cell. Mol. Life Sci. ( 10.1007/s00018-013-1424-z) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Speijer D. In press Reconsidering ideas regarding the evolution of peroxisomes: the case for a mitochondrial connection. Cell. Mol. Life Sci. ( 10.1007/s00018-013-1507-x) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Schrader M, Yoon Y. 2007. Mitochondria and peroxisomes: are the ‘big brother’ and the ‘little sister’ closer than assumed? Bioessays 29, 1105–1114. ( 10.1002/bies.20659) [DOI] [PubMed] [Google Scholar]
- 41.Neuspiel M, Schauss AC, Braschi E, Zunino R, Rippstein P, Rachubinski RA, Andrade-Navarro MA, McBride HM. 2008. Cargo-selected transport from the mitochondria to peroxisomes is mediated by vesicular carriers. Curr. Biol. 18, 102–108. ( 10.1016/j.cub.2007.12.038) [DOI] [PubMed] [Google Scholar]
- 42.Margineantu DH, Emerson CB, Diaz D, Hockenbery DM. 2007. Hsp90 inhibition decreases mitochondrial protein turnover. PLoS ONE 2, e1066 ( 10.1371/journal.pone.0001066) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Heo JM, et al. 2010. A stress-responsive system for mitochondrial protein degradation. Mol. Cell 40, 465–480. ( 10.1016/j.molcel.2010.10.021) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Taylor EB, Rutter J. 2011. Mitochondrial quality control by the ubiquitin-proteasome system. Biochem. Soc. Trans. 39, 1509–1513. ( 10.1042/BST0391509) [DOI] [PubMed] [Google Scholar]
- 45.Dacks JB, Field MC. 2007. Evolution of the eukaryotic membrane-trafficking system: origin, tempo and mode. J. Cell. Sci. 120, 2977–2985. ( 10.1242/jcs.013250) [DOI] [PubMed] [Google Scholar]
- 46.Martin W, Koonin EV. 2006. Introns and the origin of nucleus-cytosol compartmentalization. Nature 440, 41–45. ( 10.1038/nature04531) [DOI] [PubMed] [Google Scholar]
- 47.Barrientos A, Ugalde C. 2013. I function, therefore I am: overcoming skepticism about mitochondrial supercomplexes. Cell Metab. 18, 147–149. ( 10.1016/j.cmet.2013.07.010) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Lapuente-Brun E, et al. 2013. Supercomplex assembly determines electron flux in the mitochondrial electron transport chain. Science 340, 1567–1570. ( 10.1126/science.1230381) [DOI] [PubMed] [Google Scholar]
- 49.Moreno-Lastres D, Fontanesi F, Garcia-Consuegra I, Martin MA, Arenas J, Barrientos A, Ugalde C. 2012. Mitochondrial complex I plays an essential role in human respirasome assembly. Cell Metab. 15, 324–335. ( 10.1016/j.cmet.2012.01.015) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Winge DR. 2012. Sealing the mitochondrial respirasome. Mol. Cell. Biol. 32, 2647–2652. ( 10.1128/MCB.00573-12) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Gomez LA, Hagen TM. 2012. Age-related decline in mitochondrial bioenergetics: does supercomplex destabilization determine lower oxidative capacity and higher superoxide production? Semin. Cell. Dev. Biol. 23, 758–767. ( 10.1016/j.semcdb.2012.04.002) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Jastroch M, Divakaruni AS, Mookerjee S, Treberg JR, Brand MD. 2010. Mitochondrial proton and electron leaks. Essays Biochem. 47, 53–67. ( 10.1042/bse0470053) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Azzu V, Brand MD. 2010. The on-off switches of the mitochondrial uncoupling proteins. Trends Biochem. Sci. 35, 298–307. ( 10.1016/j.tibs.2009.11.001) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Nicholls DG, Bernson VS, Heaton GM. 1978. The identification of the component in the inner membrane of brown adipose tissue mitochondria responsible for regulating energy dissipation. Experientia Suppl. 32, 89–93. ( 10.1007/978-3-0348-5559-4_9) [DOI] [PubMed] [Google Scholar]
- 55.Kunji ER, Robinson AJ. 2010. Coupling of proton and substrate translocation in the transport cycle of mitochondrial carriers. Curr. Opin. Struct. Biol. 20, 440–447. ( 10.1016/j.sbi.2010.06.004) [DOI] [PubMed] [Google Scholar]
- 56.Hughes J, Criscuolo F. 2008. Evolutionary history of the UCP gene family: gene duplication and selection. BMC Evol. Biol. 8, 306 ( 10.1186/1471-2148-8-306) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Ricquier D, Bouillaud F. 2000. The uncoupling protein homologues: UCP1, UCP2, UCP3, StUCP and AtUCP. Biochem. J. 345, 161–179. ( 10.1042/0264-6021:3450161) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Esteves TC, Parker N, Brand MD. 2006. Synergy of fatty acid and reactive alkenal activation of proton conductance through uncoupling protein 1 in mitochondria. Biochem. J. 395, 619–628. ( 10.1042/BJ20052004) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Brand MD, Affourtit C, Esteves TC, Green K, Lambert AJ, Miwa S, Pakay JL, Parker N. 2004. Mitochondrial superoxide: production, biological effects, and activation of uncoupling proteins. Free Radic. Biol. Med. 37, 755–767. ( 10.1016/j.freeradbiomed.2004.05.034) [DOI] [PubMed] [Google Scholar]
- 60.Attwell D, Laughlin SB. 2001. An energy budget for signaling in the grey matter of the brain. J. Cereb. Blood Flow Metab. 21, 1133–1145. ( 10.1097/00004647-200110000-00001) [DOI] [PubMed] [Google Scholar]
- 61.Kann O, Kovacs R. 2007. Mitochondria and neuronal activity. Am. J. Physiol. Cell Physiol. 292, C641–C657. ( 10.1152/ajpcell.00222.2006) [DOI] [PubMed] [Google Scholar]
- 62.Knott AB, Perkins G, Schwarzenbacher R, Bossy-Wetzel E. 2008. Mitochondrial fragmentation in neurodegeneration. Nat. Rev. Neurosci. 9, 505–518. ( 10.1038/nrn2417) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Yang SY, He XY, Schulz H. 1987. Fatty acid oxidation in rat brain is limited by the low activity of 3-ketoacyl-coenzyme A thiolase. J. Biol. Chem. 262, 13 027–13 032. [PubMed] [Google Scholar]
- 64.Schonfeld P, Reiser G. 2013. Why does brain metabolism not favor burning of fatty acids to provide energy? Reflections on disadvantages of the use of free fatty acids as fuel for brain. J. Cereb. Blood Flow Metab. 33, 1493–1499. ( 10.1038/jcbfm.2013.128) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Schapira AH. 2012. Mitochondrial diseases. Lancet 379, 1825–1834. ( 10.1016/S0140-6736(11)61305-6) [DOI] [PubMed] [Google Scholar]
- 66.Tucker EJ, Compton AG, Thorburn DR. 2010. Recent advances in the genetics of mitochondrial encephalopathies. Curr. Neurol. Neurosci. Rep. 10, 277–285. ( 10.1007/s11910-010-0112-8) [DOI] [PubMed] [Google Scholar]
- 67.Kirby DM, Thorburn DR. 2008. Approaches to finding the molecular basis of mitochondrial oxidative phosphorylation disorders. Twin Res. Hum. Genet. 11, 395–411. ( 10.1375/twin.11.4.395) [DOI] [PubMed] [Google Scholar]
- 68.Deuschl G, Elble R. 2009. Essential tremor: neurodegenerative or nondegenerative disease towards a working definition of ET. Mov. Disord. 24, 2033–2041. ( 10.1002/mds.22755) [DOI] [PubMed] [Google Scholar]
- 69.Przedborski S, Vila M, Jackson-Lewis V. 2003. Neurodegeneration: what is it and where are we? J. Clin. Invest. 111, 3–10. ( 10.1172/JCI17522) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Breuer ME, Koopman WJ, Koene S, Nooteboom M, Rodenburg RJ, Willems PH, Smeitink JA. 2013. The role of mitochondrial OXPHOS dysfunction in the development of neurologic diseases. Neurobiol. Dis. 51, 27–34. ( 10.1016/j.nbd.2012.03.007) [DOI] [PubMed] [Google Scholar]
- 71.Koopman WJ, Distelmaier F, Smeitink JA, Willems PH. 2013. OXPHOS mutations and neurodegeneration. EMBO J. 32, 9–29. ( 10.1038/emboj.2012.300) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Fukui H, Moraes CT. 2008. The mitochondrial impairment, oxidative stress and neurodegeneration connection: reality or just an attractive hypothesis? Trends Neurosci. 31, 251–256. ( 10.1016/j.tins.2008.02.008) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.DiFilippo M, Chiasserini D, Tozzi A, Picconi B, Calabresi P. 2010. Mitochondria and the link between neuroinflammation and neurodegeneration. J. Alzheimers Dis. 20(Suppl. 2), S369–S379. ( 10.3233/JAD-2010-100543) [DOI] [PubMed] [Google Scholar]
- 74.Dutta R, et al. 2006. Mitochondrial dysfunction as a cause of axonal degeneration in multiple sclerosis patients. Ann. Neurol. 59, 478–489. ( 10.1002/ana.20736) [DOI] [PubMed] [Google Scholar]
- 75.Lin MT, Beal MF. 2006. Mitochondrial dysfunction and oxidative stress in neurodegenerative diseases. Nature 443, 787–795. ( 10.1038/nature05292) [DOI] [PubMed] [Google Scholar]
- 76.Scaglia F. 2010. The role of mitochondrial dysfunction in psychiatric disease. Dev. Disabil. Res. Rev. 16, 136–143. ( 10.1002/ddrr.115) [DOI] [PubMed] [Google Scholar]
- 77.de Moura MB, dos Santos LS, Van Houten B. 2010. Mitochondrial dysfunction in neurodegenerative diseases and cancer. Environ. Mol. Mutagen. 51, 391–405. ( 10.1002/em.20575) [DOI] [PubMed] [Google Scholar]
- 78.Tansey MG, Goldberg MS. 2010. Neuroinflammation in Parkinson's disease: its role in neuronal death and implications for therapeutic intervention. Neurobiol. Dis. 37, 510–518. ( 10.1016/j.nbd.2009.11.004) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Mortiboys H, et al. 2008. Mitochondrial function and morphology are impaired in parkin-mutant fibroblasts. Ann. Neurol. 64, 555–565. ( 10.1002/ana.21492) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Heeman B, et al. 2011. Depletion of PINK1 affects mitochondrial metabolism, calcium homeostasis and energy maintenance. J. Cell. Sci. 124, 1115–1125. ( 10.1242/jcs.078303) [DOI] [PubMed] [Google Scholar]
- 81.Mounsey RB, Teismann P. 2010. Mitochondrial dysfunction in Parkinson's disease: pathogenesis and neuroprotection. Parkinsons Dis. 2011, 617472 ( 10.4061/2011/617472) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Winkler-Stuck K, Wiedemann FR, Wallesch CW, Kunz WS. 2004. Effect of coenzyme Q10 on the mitochondrial function of skin fibroblasts from Parkinson patients. J. Neurol. Sci. 220, 41–48. ( 10.1016/j.jns.2004.02.003) [DOI] [PubMed] [Google Scholar]
- 83.Vos M, et al. 2012. Vitamin K2 is a mitochondrial electron carrier that rescues pink1 deficiency. Science 336, 1306–1310. ( 10.1126/science.1218632) [DOI] [PubMed] [Google Scholar]
- 84.Goodall EF, Morrison KE. 2006. Amyotrophic lateral sclerosis (motor neuron disease): proposed mechanisms and pathways to treatment. Expert Rev. Mol. Med. 8, 1–22. ( 10.1017/S1462399406010854) [DOI] [PubMed] [Google Scholar]
- 85.Israelson A, Arbel N, Da CS, Ilieva H, Yamanaka K, Shoshan-Barmatz V, Cleveland DW. 2010. Misfolded mutant SOD1 directly inhibits VDAC1 conductance in a mouse model of inherited ALS. Neuron 67, 575–587. ( 10.1016/j.neuron.2010.07.019) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Perry RJ, Hodges JR. 1999. Attention and executive deficits in Alzheimer's disease. A critical review. Brain 122, 383–404. ( 10.1093/brain/122.3.383) [DOI] [PubMed] [Google Scholar]
- 87.Kar S, Slowikowski SP, Westaway D, Mount HT. 2004. Interactions between beta-amyloid and central cholinergic neurons: implications for Alzheimer's disease. J. Psychiatry Neurosci. 29, 427–441. [PMC free article] [PubMed] [Google Scholar]
- 88.Dragicevic N, Mamcarz M, Zhu Y, Buzzeo R, Tan J, Arendash GW, Bradshaw PC. 2010. Mitochondrial amyloid-beta levels are associated with the extent of mitochondrial dysfunction in different brain regions and the degree of cognitive impairment in Alzheimer's transgenic mice. J. Alzheimers Dis. 20(Suppl. 2) S535–S550. ( 10.3233/JAD-2010-100342) [DOI] [PubMed] [Google Scholar]
- 89.Reddy PH, Beal MF. 2008. Amyloid beta, mitochondrial dysfunction and synaptic damage: implications for cognitive decline in aging and Alzheimer's disease. Trends Mol. Med. 14, 45–53. ( 10.1016/j.molmed.2007.12.002) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Morais VA, De Strooper B. 2010. Mitochondria dysfunction and neurodegenerative disorders: cause or consequence. J. Alzheimers Dis. 20(Suppl. 2), S255–S263. ( 10.3233/JAD-2010-100345) [DOI] [PubMed] [Google Scholar]
- 91.Balsa E, Marco R, Perales-Clemente E, Szklarczyk R, Calvo E, Landazuri MO, Enriquez JA. 2012. NDUFA4 is a subunit of complex IV of the mammalian electron transport chain. Cell Metab. 16, 378–386. ( 10.1016/j.cmet.2012.07.015) [DOI] [PubMed] [Google Scholar]
- 92.Gabaldon T, Rainey D, Huynen MA. 2005. Tracing the evolution of a large protein complex in the eukaryotes, NADH:ubiquinone oxidoreductase (complex I). J. Mol. Biol. 348, 857–870. ( 10.1016/j.jmb.2005.02.067) [DOI] [PubMed] [Google Scholar]
- 93.Cardol P. 2011. Mitochondrial NADH:ubiquinone oxidoreductase (complex I) in eukaryotes: a highly conserved subunit composition highlighted by mining of protein databases. Biochim. Biophys. Acta 1807, 1390–1397. ( 10.1016/j.bbabio.2011.06.015) [DOI] [PubMed] [Google Scholar]
- 94.Huynen MA, de Hollander M, Szklarczyk R. 2009. Mitochondrial proteome evolution and genetic disease. Biochim. Biophys. Acta 1792, 1122–1129. ( 10.1016/j.bbadis.2009.03.005) [DOI] [PubMed] [Google Scholar]
- 95.Nouws J, et al. 2010. Acyl-CoA dehydrogenase 9 is required for the biogenesis of oxidative phosphorylation complex I. Cell Metab. 12, 283–294. ( 10.1016/j.cmet.2010.08.002) [DOI] [PubMed] [Google Scholar]
- 96.Hamalainen RH, Manninen T, Koivumaki H, Kislin M, Otonkoski T, Suomalainen A. 2013. Tissue- and cell-type-specific manifestations of heteroplasmic mtDNA 3243A>G mutation in human induced pluripotent stem cell-derived disease model. Proc. Natl Acad. Sci. USA 110, E3622–E3630. ( 10.1073/pnas.1311660110) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Gurusamy N, Das DK. 2009. Autophagy, redox signaling, and ventricular remodeling. Antioxid. Redox Signal. 11, 1975–1988. ( 10.1089/ARS.2009.2524) [DOI] [PMC free article] [PubMed] [Google Scholar]