

Abstract
The tumour suppressor p53 is activated to induce cell-cycle arrest or apoptosis in the DNA damage response (DDR). p53 phosphorylation at Ser46 by HIPK2 (homeodomain-interacting protein kinase 2) is a critical event in apoptosis induction. Interestingly, HIPK2 is degraded by Mdm2 (a negative regulator of p53), whereas Mdm2 is downregulated by HIPK2 through several mechanisms. Here, we develop a four-module network model for the p53 pathway to clarify the role of interplay between Mdm2 and HIPK2 in the DDR evoked by ultraviolet radiation. By numerical simulations, we reveal that Mdm2-dependent HIPK2 degradation promotes cell survival after mild DNA damage and that inhibition of HIPK2 degradation is sufficient to trigger apoptosis. In response to severe damage, p53 phosphorylation at Ser46 is promoted by the accumulation of HIPK2 due to downregulation of nuclear Mdm2 in the later phase of the response. Meanwhile, the concentration of p53 switches from moderate to high levels, contributing to apoptosis induction. We show that the presence of three mechanisms for Mdm2 downregulation, i.e. repression of mdm2 expression, inhibition of its nuclear entry and HIPK2-induced degradation, guarantees the apoptosis of irreparably damaged cells. Our results agree well with multiple experimental observations, and testable predictions are also made. This work advances our understanding of the regulation of p53 activity in the DDR and suggests that HIPK2 should be a significant target for cancer therapy.
Keywords: HIPK2 degradation, downregulation of nuclear Mdm2, p53 phosphorylation, cell-fate decision
1. Introduction
Upon DNA damage, the tumour suppressor p53 is stabilized and activated to induce cell-cycle arrest or apoptosis by regulating expression of target genes [1]. Apoptosis can also be induced by p53 through transcription-independent mechanisms [2–4]. How p53 guides the cell-fate decision after DNA damage is still incompletely understood although it has been investigated extensively.
The posttranslational modifications of p53 markedly influence the cellular outcome [5]. For example, the phosphorylation of p53 at Ser46 is induced only in response to severe damage, promoting cell death [6]. In human cells, HIPK2 (homeodomain-interacting protein kinase 2) specifically phosphorylates p53 at Ser46 after lethal damage caused by ultraviolet (UV) radiation or chemotherapeutic agents such as adriamycin (ADR) and cisplatin [7]. p53DINP1 (p53-dependent damage inducible nuclear protein 1) acts as a cofactor for HIPK2-mediated p53 phosphorylation [8,9]. However, such phosphorylation can be prevented due to Mdm2-dependent degradation of HIPK2 [10]. This raises the issue of how HIPK2 accumulates to promote apoptosis following severe damage.
On the other hand, HIPK2 can modulate Mdm2 activity through several mechanisms. Once p53 is phosphorylated by HIPK2 at Ser46, transcription of the mdm2 gene is reduced [11,12]. HIPK2 also interacts with Mdm2, promoting its nuclear export and proteasomal degradation [11,13]. Moreover, phosphatase and tensin homologue (PTEN), which is induced by p53 phosphorylated at Ser46 [12], represses the nuclear entry of Mdm2 by inhibiting the phosphorylation of Akt [14]. Therefore, HIPK2 and Mdm2 regulate each other in a complicated manner. It is intriguing to explore the significance of their interplay during the p53-mediated DNA damage response (DDR).
A series of experimental and theoretical studies have recently probed the dynamics and function of p53 in the DDR [15–17]. Although the phosphorylation of p53 at Ser46 is considered a critical event in apoptosis induction, how it is modulated by the interplay between Mdm2 and HIPK2 is largely unknown. Moreover, the p53 dynamics depend heavily on cellular context and stress type, exhibiting pulsatile or switch-like behaviours [18]. For example, a series of fixed pulses was observed in p53 levels upon ionizing radiation (IR) [19]. By contrast, p53 shows a graded behaviour with its concentration in proportion to the dose of UV radiation [20]. How the UV-irradiated cells make a decision between life and death remains unclear.
In this study, we aim to clarify the mechanism for cell-fate decision in response to UV radiation, focusing on the interplay between Mdm2 and HIPK2. We proposed an integrated model for the p53 signalling network, characterizing DNA repair, ATR (ataxia telangiectasia mutated (ATM) and Rad3-related) activation, p53 activation and determination of cellular output. We found that p53 undergoes one- or two-phase dynamics, depending on the severity of DNA damage. Upon mild damage, the concentration of p53 reaches a moderate level, and a transient cell-cycle arrest is induced; the cell resumes proliferation after the damage is fixed. Following severe damage, the concentration of p53 rises from a moderate to high level, and apoptosis is evoked. The interplay between Mdm2 and HIPK2 is crucial to a reliable cell-fate decision. The degradation of HIPK2 is important for survival of reparably damaged cells, whereas the HIPK2-dependent downregulation of nuclear Mdm2 promotes apoptosis of severely damaged cells. Our findings suggest a critical role for Mdm2 downregulation in p53-dependent tumour suppression.
2. Model and method
The p53 signalling network responding to DNA damage is rather complicated and can be considered as an information processing network [21]. We develop an integrated model of the p53 network, composed of four modules: the DNA repair module, the ATR sensor, the p53-centred feedback control module and the cell-fate decision module (figure 1). A large difference from our previous models [16,17] is that the current model characterizes the cellular response to UV radiation as well as the interplay between HIPK2 and Mdm2. To this end, we consider several new aspects including the detection of UV-induced DNA damage by ATR, repression of mdm2 transcription in apoptosis induction, degradation of Mdm2 by HIPK2, Mdm2-dependent HIPK2 degradation and regulation of p53Ser46 phosphorylation by HIPK2 and p38MAPK. The key points of the model are addressed as follows.
Figure 1.

Schematic diagram of the integrated model. The model is composed of four modules, separately characterizing the repair of DNA damage caused by UV, ATR activation, regulation of p53 activity and cell-fate decision. Based on its phosphorylation status, active p53 is further divided into p53-arrester and p53-killer. We focus on the interplay between Mdm2 and HIPK2: Mdm2 targets HIPK2 for proteasomal degradation, whereas HIPK2 promotes the accumulation of p53-killer followed by the repression of mdm2 transcription and sequestration of Mdm2 in the cytoplasm by PTEN. Dashed lines denote gene expression, while arrow-headed solid lines signify the transitions between different states. The promotion and inhibition of state transition are denoted by circle- and bar-headed lines, respectively. The implications of acronyms in the figure are listed in the electronic supplementary material, table S1.
2.1. Detection and repair of DNA damage
When cells are exposed to UV radiation, DNA single-strand breaks (SSBs) are produced [22]. The level of DNA damage is denoted by LD. For simplicity, the damage is assumed to be repaired at a constant rate (electronic supplementary material, equations (1) and (2)). SSBs are mainly detected by the ATR kinase, which is activated by phosphorylation [22]. Phosphorylated ATR (ATRp) indirectly promotes the further activation of ATR [23], thereby enclosing a positive-feedback loop.
The total level of ATR is assumed to be constant, because ATR is mainly regulated posttranslationally [24]. The dynamics of ATR are characterized by equations (3) and (4) of the electronic supplementary material. The phosphorylation and dephosphorylation of ATR are taken as enzyme-catalysed reactions and assumed to follow the Michaelis–Menten kinetics [25]. The activation rate of ATR is positively associated with the ATRp level and the extent of DNA damage [23]. In simulations, we set 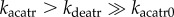 to ensure that ATR is inactive in unstressed cells but is activated strongly upon DNA damage.
to ensure that ATR is inactive in unstressed cells but is activated strongly upon DNA damage.
2.2. Regulation of p53 activity and cell-fate decision
Upon UV-induced DNA damage, p53 is activated by ATRp and regulated by several feedback loops. ATRp activates p53 in two manners, blocking the interaction between p53 and Mdm2 via phosphorylating p53 and inhibiting the ubiquitin-ligase activity of Mdm2 via phosphorylating Mdm2 in the nucleus [26,27]. Note that the c-Abl kinase is activated by ATM in response to IR and some DNA-damaging agents [28,29]. Active c-Abl activates p53 by phosphorylating Mdm2 [30]. However, c-Abl cannot be activated following UV irradiation [31]. Thus, we do not include c-Abl in the model.
Four forms of Mdm2 are included here: Mdm2c (dephosphorylated cytoplasmic Mdm2), Mdm2cp (phosphorylated cytoplasmic Mdm2), Mdm2n (nuclear Mdm2 with ubiquitin-ligase activity) and Mdm2np (phosphorylated nuclear Mdm2 without ubiquitin-ligase activity). The mdm2 mRNA, mdm2m, is also included in the model. Only nuclear p53 is considered, comprising non-phosphorylated p53 (p53, inactive form), primarily phosphorylated p53 (p53-arrester, primarily active form) and further phosphorylated p53 (p53-killer, fully activated form). Here, p53-arrester refers to p53 primarily phosphorylated at Ser15, whereas p53-killer is p53 further phosphorylated at Ser46, i.e. the doubly phosphorylated form; they promote cell-cycle arrest and apoptosis, respectively [6]. Phosphorylated p53 is collectively symbolized by p53p.
The production rate of p53 is assumed to be constant (electronic supplementary material, equation (6)), because p53 is mainly regulated by posttranslational modifications [32]. The conversion between inactive p53 and p53-arrester is controlled by its reversible phosphorylation [33]. It is assumed that the rate constants of p53 activation and inactivation, kacp53 and kdep53, depend on the levels of ATRp and Wip1 (wild-type p53-induced phosphatase 1), respectively (electronic supplementary material, equations (9) and (10)) [27,34]. As the E3-ligase activity of Mdm2n is inhibited after it is phosphorylated by ATR [26], p53 is targeted for proteasomal degradation by Mdm2n rather than Mdm2np, which is characterized by the Michaelis–Menten kinetics [25]. We set 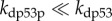 because the weak binding affinity of Mdm2n for p53-arrester and p53-killer prevents their degradation [33]. We consider only the degradation of p53 in the nucleus, whereas the Mdm2-dependent nuclear export and cytoplasmic degradation of p53 are ignored.
because the weak binding affinity of Mdm2n for p53-arrester and p53-killer prevents their degradation [33]. We consider only the degradation of p53 in the nucleus, whereas the Mdm2-dependent nuclear export and cytoplasmic degradation of p53 are ignored.
p53-arrester transactivates Wip1, p21 and p53DINP1, whereas p53-killer transactivates p53DINP1, p53AIP1 (p53-regulated apoptosis-inducing protein 1) and PTEN [6,12]. p53-killer is more efficient in inducing p53DINP1 [8], i.e. 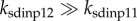 in equation (31) of the electronic supplementary material. p53DINP1 promotes the accumulation of p53-killer by acting as a partner of HIPK2 [8]. Wip1 promotes the reversion from p53-killer to p53-arrester by inhibiting p38MAPK, which is another kinase for p53 phosphorylation at Ser46 [35]. Following UV-induced DNA damage, p38MAPK is activated through the ATR–MAP3K–MAP2K–MAPK phosphorylation cascade [36]. Here, MAP3K mainly refers to TAO kinases [37], and MAP2K could be MKK3/6 [38]. The characterization of MAPK activation is derived from a recent modelling study on MAPK cascade [39]. All p53-mediated expression of genes is characterized by the Hill function, and the Hill coefficient is set to 4 given the cooperativity of the tetrameric form of p53 as a transcription factor [40].
in equation (31) of the electronic supplementary material. p53DINP1 promotes the accumulation of p53-killer by acting as a partner of HIPK2 [8]. Wip1 promotes the reversion from p53-killer to p53-arrester by inhibiting p38MAPK, which is another kinase for p53 phosphorylation at Ser46 [35]. Following UV-induced DNA damage, p38MAPK is activated through the ATR–MAP3K–MAP2K–MAPK phosphorylation cascade [36]. Here, MAP3K mainly refers to TAO kinases [37], and MAP2K could be MKK3/6 [38]. The characterization of MAPK activation is derived from a recent modelling study on MAPK cascade [39]. All p53-mediated expression of genes is characterized by the Hill function, and the Hill coefficient is set to 4 given the cooperativity of the tetrameric form of p53 as a transcription factor [40].
The mdm2 gene is transactivated by both p53-arrester and p53-killer. Because mdm2 transcription is repressed after p53 phosphorylation at Ser46 [11,12], the rate constant of mdm2 transcription by p53-killer is much smaller than that by p53-arrester, i.e. 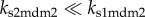 in equation (11) of the electronic supplementary material. Such a reduction in mdm2 transcription is termed transcriptional suppression in the following text. After the synthesis of mdm2 transcripts, Mdm2 proteins are produced. Mdm2c can be phosphorylated by Akt, promoting its nuclear entry [41]. For simplicity, we assume that only Mdm2cp can enter the nucleus. Akt is activated by phosphatidylinositol-3,4,5-trisphosphate (PIP3) through phosphorylation [42], whereas it is deactivated indirectly by PTEN dephosphorylating PIP3 into phosphatidylinositol-4,5-bisphosphate [43]. Thus, there exists a double-negative feedback loop among p53, PTEN, Akt and Mdm2. Mdm2n also targets HIPK2 for proteasomal degradation [10], whereas HIPK2 promotes the degradation of Mdm2 [11]. Taken together, our model includes the double-negative feedback loops between Mdm2 and HIPK2 at both the transcriptional and posttranslational levels.
in equation (11) of the electronic supplementary material. Such a reduction in mdm2 transcription is termed transcriptional suppression in the following text. After the synthesis of mdm2 transcripts, Mdm2 proteins are produced. Mdm2c can be phosphorylated by Akt, promoting its nuclear entry [41]. For simplicity, we assume that only Mdm2cp can enter the nucleus. Akt is activated by phosphatidylinositol-3,4,5-trisphosphate (PIP3) through phosphorylation [42], whereas it is deactivated indirectly by PTEN dephosphorylating PIP3 into phosphatidylinositol-4,5-bisphosphate [43]. Thus, there exists a double-negative feedback loop among p53, PTEN, Akt and Mdm2. Mdm2n also targets HIPK2 for proteasomal degradation [10], whereas HIPK2 promotes the degradation of Mdm2 [11]. Taken together, our model includes the double-negative feedback loops between Mdm2 and HIPK2 at both the transcriptional and posttranslational levels.
As downstream effectors of p53, p21 and p53AIP1 induce cell-cycle arrest and apoptosis, respectively [6]. p53AIP1 promotes the release of cytochrome c (CytoC) from mitochondria [44]. Subsequently, CytoC activates the caspase cascade in the cytoplasm [45]. Finally, caspase-3 (Casp3) is activated and apoptosis ensues. We consider the positive feedback between CytoC release and Casp3 activation, where Casp3 cleaves the inhibitors of CytoC release [46].The enhancement of CytoC release by Casp3 and that of Casp3 activation by CytoC release are characterized by Hill functions. To ensure the irreversibility of apoptosis induction, the Hill functions should have high cooperativity [47], and thus the Hill coefficients are set to 4 (electronic supplementary material, equations (35) and (36)).
2.3. Remarks on the model
Although we include various proteins related to regulation of p53-dependent apoptosis in the model, some others that influence p53Ser46 phosphorylation are still ignored for simplicity. It was suggested that HIPK2-mediated p53 phosphorylation at Ser46 facilitates p53 acetylation at K382 [7,48]. Li et al. [49] showed that p53 with mutations at three lysine sites in the central DNA-binding domain is deficient in inducing cell-cycle arrest, senescence and apoptosis, but retains its tumour suppressor function. How the phosphorylation and acetylation of p53 influence each other is not well understood and remains to be investigated. Several other kinases (like PKCδ and DYRK2) are also involved in the phosphorylation of p53 at Ser46 [7,50]. It would be intriguing to investigate whether there exists mutual regulation between Mdm2 and those kinases in apoptosis induction. The proteins such as PML, Axin and Daxx act as cofactors of HIPK2 in p53Ser46 phosphorylation [7,51], and their dynamic regulation of HIPK2 is worth considering. We do not explicitly characterize the activation of HIPK2 kinase activity; HIPK2 per se becomes catalytically active after its production [52], and its stability is critical to its activation. We consider the repression of Akt activity by PTEN in the cytoplasm, which contributes to the sequestration of Mdm2 in the cytoplasm. Moreover, PTEN can translocalize into the nucleus and form a complex with p53 after DNA damage, enhancing its transcriptional activity [53]. As how the subcellular localization of PTEN is regulated remains unclear, we do not consider the direct interaction in the nucleus. Overall, it is important to add more factors to the p53 signalling network and evaluate their roles when enough data are available.
2.4. Methods
The concentration of each species is represented by a dimensionless state variable in rate equations ([…] denotes the concentration of species throughout the paper). These ordinary differential equations are presented in the electronic supplementary material. All the initial values of variables are their lower steady-state values under unstressed conditions. The standard parameter values together with notes on parameter setting are listed in the electronic supplementary material, table S2. The robustness of the network dynamics to parameter variations is analysed in the electronic supplementary material, Text, and the corresponding data are presented in the electronic supplementary material, table S3. The unit of time is minutes, and the units of parameters are determined such that the concentrations of proteins are dimensionless. The rate equations are numerically solved using Oscill8 (http://oscill8.sourceforge.net). Experimentally, p53Ser46 phosphorylation and apoptosis begin to appear in response to UV radiation of 20–25 J m−2 [6]. Here, the minimum LD capable of inducing apoptosis is about 4.8 with the standard parameter setting, and thus one unit of LD roughly corresponds to the damage caused by 5 J m−2 UV radiation.
3. Results
3.1. Cell-fate determination by the p53 signalling network
DNA SSBs are induced upon UV irradiation [22]. The initial level of DNA damage, LD0, is taken as the input to the p53 signalling network. Our model can characterize the whole process from the detection and repair of DNA damage to the choice of cell fate. We associate the network dynamics with cellular outcome. To illustrate the typical dynamics of the p53 network, we show the temporal evolution of the output of each module under typical stress conditions (figure 2).
Figure 2.
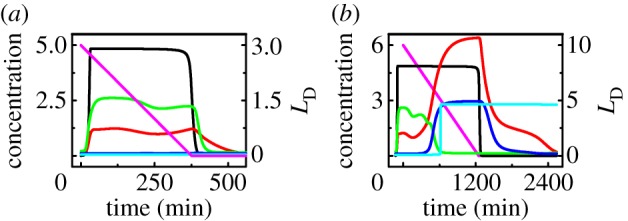
Dynamics of the p53 signalling network in response to UV. Temporal evolution of the levels of DNA damage (magenta), ATRp (black), p53p (red), p21 (green), p53AIP1 (blue) and Casp3 (cyan) with the initial extent of DNA damage being 3 (a) or 10 (b).
At LD0 = 3, the concentration of phosphorylated ATR rises quickly to a high level and remains there until the damage is fixed (figure 2a). Accordingly, the concentration of phosphorylated p53 is maintained at a moderate level until [ATRp] drops to a basal level. p21 is induced by p53p to arrest the cell cycle, whereas both [p53AIP1] and [Casp3] are kept at basal levels. Consequently, the cell recovers to normal proliferation, surviving mild DNA damage.
At LD0 = 10, [ATRp] remains at a high level for a long time (figure 2b). [p53p] exhibits two-phase dynamics: it stays at a moderate level in the first phase and switches to a high level in the second phase. p21 and p53AIP1 are induced in the first and second phases, respectively. Consequently, Casp3 is activated to evoke apoptosis. The timing of Casp3 activation (about 11 h) is consistent with the observation that p53Ser46 phosphorylation becomes marked between 8 and 12 h post-irradiation [6]. Here, the irreversible activation of Casp3 is considered a marker of apoptosis induction, and the events following apoptosis induction are not represented (as we concern only the determination of cell fate). Thus, [p53p] and [p53AIP1] still return to their basal levels after the damage is fixed. In fact, DNA repair proteins will be cleaved by Casp3, and the repair process will stop after Casp3 activation. Together, apoptosis is triggered before the damage can be entirely repaired in response to severe DNA damage.
The irreversibility of Casp3 activation results from the positive feedback between CytoC release and Casp3 activation. The relationship between the steady-state levels of Casp3 and p53AIP1 is shown in the electronic supplementary material, figure S1. The activation of Casp3 behaves like a bistable switch. [Casp3] switches to the ‘ON’ state when [p53AIP1] ≥ 1.41 and remains there until [p53AIP1] < 0.003. The switching is actually irreversible because the basal level of p53AIP1 is 0.1. Therefore, Casp3 activation requires a sufficient amount of p53AIP1, but can be sustained robustly.
3.2. Detecting DNA damage by ATR
Here, we explore the mechanism for ATR activation. As shown above, [ATRp] exhibits a switch-like behaviour upon DNA damage. In the bifurcation diagram of [ATRp] versus LD (ignoring DNA repair), there exists a saddle-node bifurcation (figure 3a). [ATRp] remains in the lower state when LD is less than the upper threshold (0.139). When LD exceeds this threshold, [ATRp] switches to the upper state. Reversely, [ATRp] returns to the lower state only when LD is less than the lower threshold (0.065). Thus, ATR is competent and reliable as a sensor of DNA damage [54].
Figure 3.
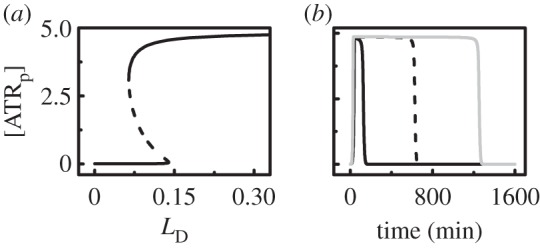
Sensing DNA damage by ATR. (a) Bifurcation diagram of the steady-state level of ATRp as a function of LD (without DNA repair). The stable and unstable steady states are represented by solid and dashed lines, respectively. (b) Time courses of [ATRp] at LD0 = 1 (black solid), 5 (dashed) or 10 (grey).
Figure 3b displays the temporal evolution of [ATRp] at different initial levels of DNA damage. Given the repair of DNA damage, ATR can be activated when LD0 ≥ 0.7. Notably, the duration of the activation state extends with increasing LD0, and ATRp is deactivated only after the damage is effectively fixed. Collectively, ATR is sensitive to DNA damage, and the activation of ATR signifies the presence of DNA damage.
3.3. Dynamics of p53 and Mdm2
ATRp transmits the stress signal by activating p53. Upon sublethal damage (e.g. LD0 = 3), [p53p] accumulates to a moderate level (figure 4a). Although [p53-killer] is greater than [p53-arrester] after a transient, it is too low to induce apoptosis before the damage is fixed. On the other hand, [mdm2m] evidently rises following p53 activation (figure 4b). As most Mdm2 molecules enter the nucleus, [Mdm2c] is close to zero. The majority of nuclear Mdm2 is phosphorylated by ATRp and loses its E3-ligase activity [26]. Thus, [Mdm2n] rises mildly. Together, ATRp activates p53 and inhibits Mdm2 by phosphorylating them.
Figure 4.
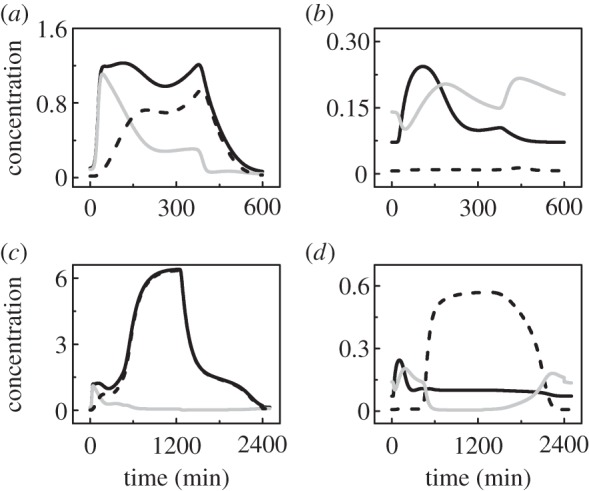
Dynamics of p53 and Mdm2. Time courses of [p53p] (black solid), [p53-arrester] (grey) and [p53-killer] (dashed) at LD0 = 3 (a) or 10 (c), and those of [mdm2m] (black solid), [Mdm2c] (dashed) and [Mdm2n] (grey) at LD0 = 3 (b) or 10 (d).
Following lethal damage (e.g. LD0 = 10), [p53p] first reaches a moderate level and then rises to a high level in the second phase, where p53-killer is absolutely dominant over p53-arrester (figure 4c). In contrast to p53p, [mdm2m] drops evidently in the second phase (figure 4d), consistent with the experimental observations that mdm2 transcription is repressed in the later phase of the cellular response to lethal damage [6,11]. As most of Mdm2 is sequestered in the cytoplasm, [Mdm2c] is much higher than [Mdm2n] in the second phase. These results indicate the connection between marked downregulation of nuclear Mdm2 and higher levels of p53p.
3.4. Dynamics of Wip1 and the p38MAPK cascade
In addition to HIPK2, p38MAPK is another kinase for p53Ser46 phosphorylation, and its activity can be inhibited by Wip1 via dephosphorylation. We show the temporal evolution of the levels of MAP3Kp, MAP2Kpp, p38MAPKpp, Wip1 and p53-killer in the electronic supplementary material, figure S2. Upon DNA damage, [MAP3Kp] and [MAP2Kpp] quickly rise to high platforms and remain there until the damage is fixed, whereas the dynamics of [p38MAPKpp] depend on the extent of DNA damage.
At LD0 = 3, enough Wip1 is induced by p53-arrester following DNA damage, and thus [p38MAPKpp] is at a low level (electronic supplementary material, figure S2a). At LD0 = 10, the concentrations of these proteins also exhibit two-phase dynamics (electronic supplementary material, figure S2b). Wip1 is expressed only in the first phase, and its inhibition of p38AMPK activity is relieved in the second phase. Consequently, p38MAPK is activated to promote the accumulation of p53-killer in the second phase. Therefore, Wip1 represses p53Ser46 phosphorylation by inhibiting the activation of p38MAPK. The presence of two kinases for p53Ser46 phosphorylation may confer robustness to p53-dependent apoptosis induction. In the following, we will focus on the interplay between Mdm2 and HIPK2 in the regulation of p53-mediated cell-fate decision.
3.5. Repression of p53 phosphorylation at Ser46 by Mdm2-regulated HIPK2 degradation
It was reported that HIPK2 is degraded by Mdm2 to promote cell survival after sublethal DNA damage [10]. In our model, the drop in HIPK2 levels originates from its degradation by nuclear Mdm2. Upon mild damage, [HIPK2] falls slightly because [Mdm2n] rises mildly (figure 5a, upper). When the damage is severe, [HIPK2] remains lower than its basal level in the first phase but rises to a high level in the second phase, where [Mdm2n] drops to a rather low level (figure 5a, lower). These results agree with the observation that HIPK2 is differently expressed following distinct levels of DNA damage [10].
Figure 5.
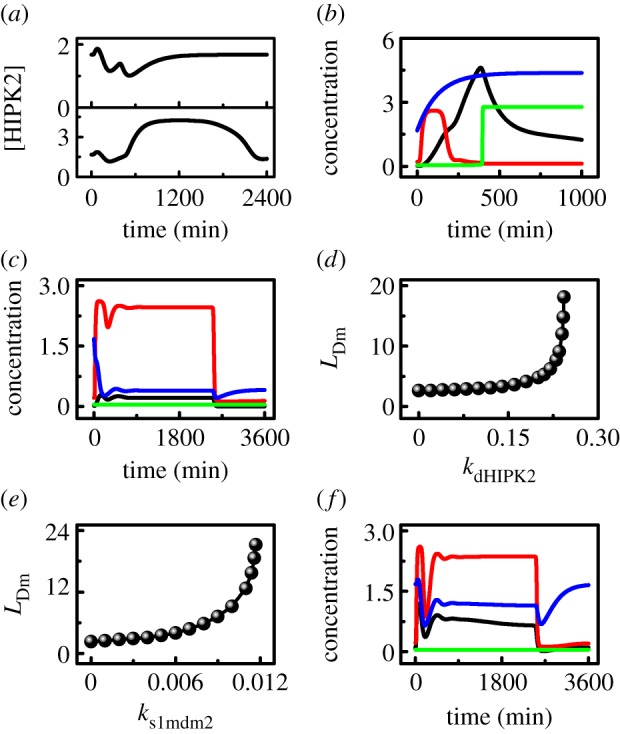
Mdm2-regulated HIPK2 degradation is critical for cell survival. (a) Time courses of [HIPK2] at LD0 = 3 (upper) or 10 (lower). (b,c) Temporal evolution of [p21] (red), [HIPK2] (blue), [p53-killer] (black) and [Casp3] (green) with kdHIPK2 = 0.0 and LD0 = 3 (b), or kdHIPK2 = 0.4 and LD0 = 20 (c). (d,e) Minimum of LD0 required for apoptosis induction as a function of kdHIPK2 (d) or ks1mdm2 (e). (f) Time courses of [p53p] (black), [p21] (red), [HIPK2] (blue) and [Casp3] (green) at ks1mdm2 = 0.015 and LD0 = 20.
We further probe the influence of the rate constant of HIPK2 degradation by Mdm2, kdHIPK2, on cellular outcome. With the standard parameter setting (kdHIPK2 = 0.2), p53-killer accumulates slowly and becomes remarkable after about 11 h poststimulus at LD0 = 10, whereas p21 is induced only in the first phase (cf. figures 2b and 4c). At kdHIPK2 = 0, [HIPK2] rises quickly towards a high level even at LD0 = 3 because of weakened degradation (figure 5b); the duration of p21-induced cell-cycle arrest is shortened significantly, and Casp3 is activated around 6.5 h poststimulation. These results agree with the experimental observation that the HIPK2K1182R mutant, which is resistant to Mdm2-regulated degradation, is more competent than the wild-type one in apoptosis induction [10]. At kdHIPK2 = 0.4, [HIPK2] drops very fast to a low level even at LD0 = 20 (figure 5c); p21 is induced persistently throughout the response, whereas Casp3 is inactivated. Consequently, the severely damaged cell may undergo senescence when apoptosis is inhibited [55]. Together, the degradation rate of HIPK2 markedly affects the cell fate.
The minimum level of initial DNA damage required for apoptosis induction, LDm, reflects the sensitivity of the p53 pathway to DNA damage. LDm becomes larger with increasing kdHIPK2 (figure 5d). When kdHIPK2 is less than 0.15, LDm is lower than 3.5. This means that the damaged cell becomes rather sensitive to DNA damage when the degradation of HIPK2 by Mdm2 is inefficient. Differently, LDm rises quickly with increasing kdHIPK2 when it is greater than 0.15. At kdHIPK2 = 0.24, for example, apoptosis is triggered only after very severe damage with LD0 ≥ 12 (compared with 4.8 under the standard parameter setting). Together, the degradation rate of HIPK2 can markedly influence the sensitivity of the cell to DNA damage. These results suggest that inhibition of HIPK2 degradation is an efficient way to convert a cytostatic stimulus into a pro-apoptotic one [10].
Following severe DNA damage, the degradation of HIPK2 mainly occurs in the first phase and is affected markedly by the level of Mdm2n, which depends on the rate constant ks1mdm2. Thus, LDm also varies as a function of ks1mdm2. LDm is rather low if ks1mdm2 < 0.008 (figure 5e), in agreement with the finding that Mdm2 depletion results in apoptosis even in response to sublethal damage [10]. On the other hand, it becomes very difficult to induce apoptosis when Mdm2 is overexpressed. With further increasing ks1mdm2, LDm rises quickly and equals 21.2 at ks1mdm2 = 0.0117. At ks1mdm2 = 0.015, [p53p] remains at a moderate level and cannot be fully activated even at LD0 = 20 (figure 5f). p21 is induced persistently, whereas Casp3 remains inactive. Thus, the cell may become senescent, consistent with the observation that Mdm2 overexpression markedly inhibits p53-mediated apoptosis by enhancing HIPK2 degradation [10]. This provides another explanation for the carcinogenic role of Mdm2 in many tumours [56].
The ATR-induced Mdm2 phosphorylation only inhibits its E3-ligase activity rather than leads to its degradation [26]. The dephosphorylation of Mdm2np contributes to Mdm2 reactivation and HIPK2 degradation. To explore the effect of Mdm2np dephosphorylation on cell-fate decision, the rate constant kdphm2n is set to different values in the electronic supplementary material, figure S3. At kdphm2n = 0 and LD0 = 10, [Mdm2n] drops to a low level after a transient (electronic supplementary material, figure S3a). With increasing kdphm2n, the downregulation of Mdm2n occurs later. When kdphm2n is large enough, [Mdm2n] instead remains relatively high. Thus, it takes a longer time for [HIPK2] to reach its peak with increasing kdphm2n, or [HIPK2] stays at low levels when kdphm2n is sufficiently large (electronic supplementary material, figure S3b). Consequently, Casp3 is activated later with increasing kdphm2n, or it cannot be activated at all (electronic supplementary material, figure S3c). These results reveal that the reactivation of nuclear Mdm2 via dephosphorylation promotes the cellular resistance to apoptosis. This is also manifested in the electronic supplementary material, figure S3d, where LDm rises markedly with increasing kdphm2n. Only when the extent of DNA damage exceeds some threshold can the anti-apoptotic effect of Mdm2 reactivation be overcome by downregulation of Mdm2, leading to apoptosis.
3.6. Mdm2 downregulation promotes p53 phosphorylation at Ser46
We have shown that the Mdm2-dependent HIPK2 degradation prevents phosphorylation of p53 at Ser46 and contributes to cell survival. Thus, downregulation of Mdm2 is essential for HIPK2-mediated p53Ser46 phosphorylation and apoptosis after severe DNA damage. Indeed, nuclear Mdm2 can be downregulated at both the transcriptional and posttranslational levels: repressed expression of mdm2 by p53-killer, sequestration of Mdm2 in the cytoplasm by p53-killer-induced PTEN, and HIPK2-dependent degradation of Mdm2 [11–13]. In this sense, there exists a positive-feedback mechanism in the induction of p53-killer, i.e. p53-killer promotes its own accumulation by downregulating Mdm2. The feedback strength is related to the rate constants of mdm2 and PTEN expression by p53-killer and HIPK2-dependent degradation of nuclear Mdm2, i.e. ks2mdm2, ksPTEN and 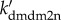 . In the following, we investigate how the mechanisms for Mdm2 downregulation affect p53Ser46 phosphorylation, whose extent is represented by the accumulation of p53-killer.
. In the following, we investigate how the mechanisms for Mdm2 downregulation affect p53Ser46 phosphorylation, whose extent is represented by the accumulation of p53-killer.
Figure 6a shows the bifurcation diagrams of the steady-state level of p53-killer versus ksPTEN under different conditions of Mdm2 downregulation at fixed LD0 = 10 (without DNA repair). With suppression of mdm2 expression and HIPK2-dependent Mdm2 degradation, [p53-killer] rises monotonically with increasing ksPTEN; it remains relatively high even at ksPTEN = 0. Without transcriptional repression (i.e. ks2mdm2 = ks1mdm2 = 0.007), [p53-killer] shows bistability over some range and switches to the upper state only when ksPTEN > 0.093. That is, a sufficient amount of PTEN is required for the induction of p53-killer to trigger apoptosis in the absence of repression of mdm2 expression in the second phase. Without both transcriptional repression and HIPK2-dependent degradation (ks2mdm2 = 0.007 and 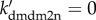 ), [p53-killer] is always at low levels. Thus, PTEN induction alone cannot ensure marked accumulation of p53-killer.
), [p53-killer] is always at low levels. Thus, PTEN induction alone cannot ensure marked accumulation of p53-killer.
Figure 6.
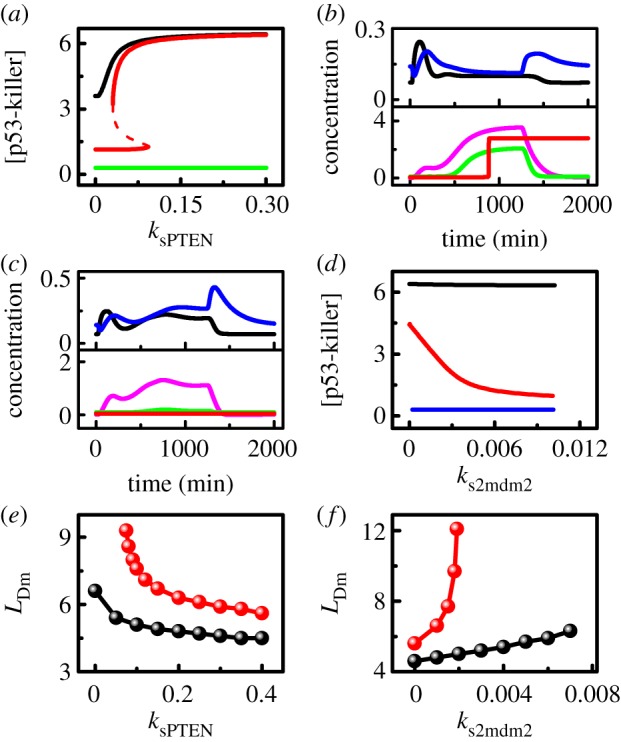
Effects of downregulation of nuclear Mdm2 on the phosphorylation of p53 at Ser46. (a) Bifurcation diagrams of the steady-state level of p53-killer versus ksPTEN at fixed LD = 10. Different lines correspond to different conditions of Mdm2 downregulation: normal case (black), without repression of mdm2 transcription (red), without both transcriptional repression and HIPK2-dependent Mdm2 degradation (green). (b,c) Time courses of the levels of mdm2m (black), Mdm2n (blue), p53-killer (magenta), p53AIP1 (green) and Casp3 (red) at LD0 = 10 with ksPTEN = 0 and ks2mdm2 = 0.001 (b) or ks2mdm2 = 0.007 (c). (d) The steady-state level of p53-killer versus ks2mdm2 at fixed LD = 10 under different conditions of Mdm2 downregulation: normal case (black), without PTEN induction (red) and without both PTEN induction and HIPK2-dependent Mdm2 degradation (blue). (e) Minimum extent of DNA damage required for apoptosis induction as a function of ksPTEN under the same conditions corresponding to the red and black lines in (a). (f) Minimum of LD0 required for apoptosis induction as a function of ks2mdm2 under the same conditions corresponding to the red and black lines in (d).
With ksPTEN = 0 and ks2mdm2 = 0.001, [Mdm2n] drops in the second phase due to a marked reduction in [mdm2m] at LD0 = 10 despite PTEN deficiency (figure 6b). Consequently, [p53-killer], [p53AIP1] and [Casp3] reach high levels in the late phase. Therefore, the marked downregulation of nuclear Mdm2 by transcriptional repression and HIPK2-dependent degradation is sufficient to induce p53-killer and apoptosis. On the other hand, with ksPTEN = 0.2 and ks2mdm2 = 0.007, enough PTEN is induced at LD0 = 10, and most of Mdm2 is sequestered in the cytoplasm despite abundant Mdm2 produced in the second phase (electronic supplementary material, figure S4a). Whereas [mdm2m] rises mildly in the second phase, [Mdm2n] falls markedly. Casp3 is still activated to trigger apoptosis (electronic supplementary material, figure S4b). Thus, suppression of mdm2 transcription and PTEN-induced sequestration of Mdm2 in the cytoplasm can complement each other in downregulating nuclear Mdm2.
In the absence of both transcriptional repression and PTEN induction (e.g. ks2mdm2 = 0.007 and ksPTEN = 0), the amount of mdm2 mRNA in the second phase is comparable to that in the first phase at LD0 = 10, and most of Mdm2 enters the nucleus to degrade p53 and HIPK2 (figure 6c). Accordingly, the levels of p53-killer and p53AIP1 in the second phase are much lower than those in figure 6b, and Casp3 cannot be activated to induce apoptosis. This may account for both the PTEN deficiency and Mdm2 overexpression in tumour cells [57].
We also show the diagrams of the steady-state level of p53-killer versus ks2mdm2 for three cases in figure 6d. With both PTEN induction and HIPK2-dependent degradation of Mdm2, the change of ks2mdm2 almost has no influence on [p53-killer]. Without PTEN induction (ksPTEN = 0), [p53-killer] drops monotonically with increasing ks2mdm2. The repression of mdm2 transcription becomes critical for the induction of p53-killer in PTEN-deficient cells. When both the two mechanisms are impaired (i.e. ksPTEN = 0 and 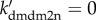 ), [p53-killer] settles at basal levels even at ks2mdm2 = 0. That is, suppression of mdm2 transcription alone cannot drive [p53-killer] to high levels. These results suggest that both transcriptional repression and HIPK2-dependent degradation of Mdm2 are required for remarkable p53Ser46 phosphorylation in PTEN-deficient cells.
), [p53-killer] settles at basal levels even at ks2mdm2 = 0. That is, suppression of mdm2 transcription alone cannot drive [p53-killer] to high levels. These results suggest that both transcriptional repression and HIPK2-dependent degradation of Mdm2 are required for remarkable p53Ser46 phosphorylation in PTEN-deficient cells.
We display how the minimum extent of DNA damage required for apoptosis induction varies with ksPTEN in figure 6e. Generally, LDm falls with increasing ksPTEN because enhancing the expression of PTEN facilitates apoptosis induction. At ks2mdm2 = 0.001, only a small amount of Mdm2 could be induced in the second phase, and apoptosis can be triggered in response to DNA damage with intermediate intensities even at ksPTEN = 0. At ks2mdm2 = 0.007, a sufficient amount of cytoplasmic Mdm2 could be induced in the second phase; to induce apoptosis, abundant PTEN is required to sequester Mdm2 in the cytoplasm. Here, apoptosis cannot be induced when ksPTEN ≤ 0.07, and LDm drops markedly with increasing ksPTEN. Therefore, a sufficient amount of PTEN is required for apoptosis induction in the absence of marked repression of mdm2 transcription following p53Ser46 phosphorylation.
We also present the curves of LDm versus ks2mdm2 for different values of ksPTEN in figure 6f. At ksPTEN = 0.2, LDm rises moderately from 4.6 to 6.3 when ks2mdm2 is increased from 0 to 0.007. Clearly, the presence of PTEN induction compensates for a deficiency in transcriptional repression. At ksPTEN = 0, apoptosis can be triggered only when ks2mdm2 is below 0.002, and LDm rises quickly from 5.6 to 12.1 as ks2mdm2 is increased from 0 to 0.002. Thus, marked transcriptional repression is required for apoptosis induction in the absence of PTEN induction. Together, these two mechanisms can cooperate to downregulate Mdm2, and the malfunction in one mechanism can be compensated for by the other when the latter is strong enough. The significance of HIPK2-dependent Mdm2 degradation in apoptosis induction is further analysed in the electronic supplementary material, Text (see also the electronic supplementary material, figure S5).
4. Discussion and conclusion
It is assumed here that p53-arrester and p53-killer induce pro-arrest and pro-apoptotic genes, respectively. This assumption is based on several facts: p53 phosphorylation at Ser46 is closely related to the induction of pro-apoptotic genes including PTEN and p53AIP1 [6,12]; p21 is induced transiently, and its level significantly drops later in response to lethal damage [6]. The downregulation of p21 may result from either transcriptional inhibition or degradation [58,59]. Most recently, Kracikova et al. [60] reported that p53 transactivates both pro-arrest and pro-apoptotic genes proportionally to its levels and apoptosis is induced after p53 levels reach some threshold. These phenomena were observed in B5/589 cells, whereas the selective expression of p53-targeted genes was observed in MCF-7 and several other cell lines. Notably, p53 levels should also exceed some threshold to induce apoptosis in our work. It would be intriguing to clarify how to make a decision between cell-cycle arrest and apoptosis when p53 induces pro-arrest and pro-apoptotic genes simultaneously.
The degradation of HIPK2 by Mdm2 promotes cell survival. Upon sublethal damage, Mdm2 degrades HIPK2 to inhibit p53Ser46 phosphorylation, while the cell cycle is arrested by p21 to allow for DNA repair. HIPK2 can also be degraded by other E3-ligases such as p53-inducible Siah-1 [61]. However, Siah-1 becomes unstable after it is phosphorylated by ATM or ATR, leading to accumulation of HIPK2. It still remains unclear how the degradation of HIPK2 by Siah-1 is differently modulated based on the severity of DNA damage. Moreover, the E3-ligase for HIPK2 degradation may depend on cellular context and stress type.
The downregulation of nuclear Mdm2 in three manners represents a robust mechanism for p53 activation and apoptosis induction. Specifically, when one mechanism is impaired, apoptosis can still be triggered provided that the others function normally. Their action modes may be cell- and stress-type dependent. In U2OS cells, for example, PTEN induction and repression of mdm2 transcription promote the full activation of p53 in the DDR [12]. In PTEN-deficient MCF-7 cells, suppression of mdm2 transcription may play a major role in p53Ser46 phosphorylation; a remarkable reduction in mdm2 transcription was observed following lethal DNA damage caused by UV radiation [6]. By contrast, mdm2 transcription is repressed mildly in human embryonic kidney 293 cells after ADR treatment, and HIPK2-dependent Mdm2 degradation may contribute to apoptosis induction [11]. Collectively, downregulation of nuclear Mdm2 provides an effective manner to induce apoptosis.
In this study, we mainly explored how the interplay between Mdm2 and HIPK2 affects the p53-mediated cellular response to UV-induced DNA damage. We found that p53 is activated progressively: upon repairable damage, it reaches a moderate level and becomes primarily activated to induce cell-cycle arrest; it further rises to a high level and gets fully activated to trigger apoptosis when the damage is irreparable. HIPK2 can be degraded by nuclear Mdm2 to prevent apoptosis induction after sublethal damage. Following lethal damage, nuclear Mdm2 can be downregulated via three manners: suppression of gene transcription by p53-killer, inhibition of its nuclear entry by PTEN and degradation by HIPK2. As a result, HIPK2 accumulates and promotes p53 phosphorylation at Ser46. Our results suggest that inhibition of the E3-ligase activity of Mdm2 may make cells more sensitive to cancer treatment such that cancer cells can be killed more easily through p53-dependent apoptosis. Moreover, reactivation of PTEN in some tumours may improve the efficacy of cancer therapy.
Funding statement
This work was supported by the 973 programme (no. 2013CB834104), National Natural Science Foundation of China (nos. 11175084, 11204126 and 31361163003) and the Priority Academic Program Development of Jiangsu Higher Education Institutions.
References
- 1.Kruse JP, Gu W. 2009. Modes of p53 regulation. Cell 137, 609–622. ( 10.1016/j.cell.2009.04.050) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Speidel D. 2009. Transcription-independent p53 apoptosis: an alternative route to death. Trends Cell Biol. 20, 14–24. ( 10.1016/j.tcb.2009.10.002) [DOI] [PubMed] [Google Scholar]
- 3.Pu T, Zhang XP, Liu F, Wang W. 2010. Coordination of the nuclear and cytoplasmic activities of p53 in response to DNA damage. Biophys. J. 99, 1696–1705. ( 10.1016/j.bpj.2010.07.042) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Tian XJ, Liu F, Zhang XP, Li J, Wang W. 2012. A two-step mechanism for cell fate decision by coordination of nuclear and mitochondrial p53 activities. PLoS ONE 7, e38164 ( 10.1371/journal.pone.0038164) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Meek DW, Anderson CW. 2009. Posttranslational modification of p53: cooperative integrators of function. Cold Spring Harb. Perspect. Biol. 1, a000950 ( 10.1101/cshperspect.a000950) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Oda K, et al. 2000. p53AIP1, a potential mediator of p53-dependent apoptosis, and its regulation by Ser-46-phosphorylated p53. Cell 102, 849–862. ( 10.1016/S0092-8674(00)00073-8) [DOI] [PubMed] [Google Scholar]
- 7.Puca R, Nardinocchi L, Givol D, D'Orazi G. 2010. Regulation of p53 activity by HIPK2: molecular mechanisms and therapeutical implications in human cancer cells. Oncogene 29, 4378–4387. ( 10.1038/onc.2010.183) [DOI] [PubMed] [Google Scholar]
- 8.Okamura S, Arakawa H, Tanaka T, Nakanishi H, Ng CC, Taya Y, Monden M, Nakamura Y. 2001. p53DINP1, a p53-inducible gene, regulates p53-dependent apoptosis. Mol. Cell 8, 85–94. ( 10.1016/S1097-2765(01)00284-2) [DOI] [PubMed] [Google Scholar]
- 9.Tomasini R, et al. 2003. TP53INP1s and homeodomain-interacting protein kinase-2 (HIPK2) are partners in regulating p53 activity . J. Biol. Chem. 278, 37 722–37 729. ( 10.1074/jbc.M301979200) [DOI] [PubMed] [Google Scholar]
- 10.Rinaldo C, Prodosmo A, Mancini F, Iacovelli S, Sacchi A, Moretti F, Soddu S. 2007. MDM2-regulated degradation of HIPK2 prevents p53Ser46 phosphorylation and DNA damage-induced apoptosis. Mol. Cell 25, 739–750. ( 10.1016/j.molcel.2007.02.008) [DOI] [PubMed] [Google Scholar]
- 11.Di Stefano V, Mattiussi M, Sacchi A, D'Orazi G. 2005. HIPK2 inhibits both MDM2 gene and protein by, respectively, p53-dependent and independent regulations. FEBS Lett. 579, 5473–5480. ( 10.1016/j.febslet.2005.09.008) [DOI] [PubMed] [Google Scholar]
- 12.Mayo LD, Seo YR, Jackson MW, Smith ML, Guzman JR, Korgaonkar CK, Donner DB. 2005. Phosphorylation of human p53 at serine 46 determines promoter selection and whether apoptosis is attenuated or amplified. J. Biol. Chem. 280, 25 953–25 959. ( 10.1074/jbc.M503026200) [DOI] [PubMed] [Google Scholar]
- 13.Di Stefano V, Blandino G, Sacchi A, Soddu S, D'Orazi G. 2004. HIPK2 neutralizes MDM2 inhibition rescuing p53 transcriptional activity and apoptotic function. Oncogene 23, 5185–5192. ( 10.1038/sj.onc.1207656) [DOI] [PubMed] [Google Scholar]
- 14.Mayo LD, Dixon JE, Durden DL, Tonks NK, Donner DB. 2002. PTEN protects p53 from Mdm2 and sensitizes cancer cells to chemotherapy. J. Biol. Chem. 277, 5484–5489. ( 10.1074/jbc.M108302200) [DOI] [PubMed] [Google Scholar]
- 15.Zhang T, Brazhnik P, Tyson JJ. 2007. Exploring mechanisms of the DNA-damage response: p53 pulses and their possible relevance to apoptosis. Cell Cycle 6, 85–94. ( 10.4161/cc.6.1.3705) [DOI] [PubMed] [Google Scholar]
- 16.Zhang XP, Liu F, Cheng Z, Wang W. 2009. Cell fate decision mediated by p53 pulses. Proc. Natl Acad. Sci. USA 106, 12 245–12 250. ( 10.1073/pnas.0813088106) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zhang XP, Liu F, Wang W. 2011. Two-phase dynamics of p53 in the DNA damage response. Proc. Natl Acad. Sci. USA 108, 8990–8995. ( 10.1073/pnas.1100600108) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Murray-Zmijewski F, Slee EA, Lu X. 2008. A complex barcode underlies the heterogeneous response of p53 to stress. Nat. Rev. Mol. Cell Biol. 9, 702–712. ( 10.1038/nrm2451) [DOI] [PubMed] [Google Scholar]
- 19.Lahav G, Rosenfeld N, Sigal A, Geva-Zatorsky N, Levine AJ, Elowitz MB, Alon U. 2004. Dynamics of the p53-Mdm2 feedback loop in individual cells. Nat. Genet. 36, 147–150. ( 10.1038/ng1293) [DOI] [PubMed] [Google Scholar]
- 20.Batchelor E, Loewer A, Mock C, Lahav G. 2011. Stimulus-dependent dynamics of p53 in single cells. Mol. Syst. Biol. 7, 488 ( 10.1038/msb.2011.20) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Batchelor E, Loewer A, Lahav G. 2009. The ups and downs of p53: understanding protein dynamics in single cells. Nat. Rev. Cancer 9, 371–377. ( 10.1038/nrc2604) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Latonen L, Laiho M. 2005. Cellular UV damage responses: functions of tumor suppressor p53. Biochim. Biophys. Acta 1755, 71–89. ( 10.1016/j.bbcan.2005.04.003) [DOI] [PubMed] [Google Scholar]
- 23.Liu S, Shiotani B, Lahiri M, Maréchal A, Tse A, Leung CC, Glover JN, Yang XH, Zou L. 2011. ATR autophosphorylation as a molecular switch for checkpoint activation. Mol. Cell 43, 192–202. ( 10.1016/j.molcel.2011.06.019) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Cimprich KA, Cortez D. 2008. ATR: an essential regulator of genome integrity. Nat. Rev. Mol. Cell Biol. 9, 616–627. ( 10.1038/nrm2450) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kholodenko BN. 2006. Cell-signalling dynamics in time and space. Nat. Rev. Mol. Cell Biol. 7, 165–176. ( 10.1038/nrm1838) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Shinozaki T, Nota A, Taya Y, Okamoto K. 2003. Functional role of Mdm2 phosphorylation by ATR in attenuation of p53 nuclear export. Oncogene 22, 8870–8880. ( 10.1038/sj.onc.1207176) [DOI] [PubMed] [Google Scholar]
- 27.Tibbetts RS, Brumbaugh KM, Williams JM, Sarkaria JN, Cliby WA, Shieh SY, Taya Y, Prives C, Abraham RT. 1999. A role for ATR in the DNA damage-induced phosphorylation of p53. Genes Dev. 13, 152–157. ( 10.1101/gad.13.2.152) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Shafman T, et al. 1997. Interaction between ATM protein and c-Abl in response to DNA damage. Nature 387, 520–523. ( 10.1038/387520a0) [DOI] [PubMed] [Google Scholar]
- 29.Baskaran R, et al. 1997. Ataxia telangiectasia mutant protein activates c-Abl tyrosine kinase in response to ionizing radiation. Nature 387, 516–519. ( 10.1038/387516a0) [DOI] [PubMed] [Google Scholar]
- 30.Waning DL, Lehman JA, Batuello CN, Mayo LD. 2011. c-Abl phosphorylation of Mdm2 facilitates Mdm2–Mdmx complex formation. J. Biol. Chem. 286, 216–222. ( 10.1074/jbc.M110.183012) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Liu ZG, et al. 1996. Three distinct signalling responses by murine fibroblasts to genotoxic stress. Nature 384, 273–276. ( 10.1038/384273a0) [DOI] [PubMed] [Google Scholar]
- 32.Lavin MF, Gueven N. 2006. The complexity of p53 stabilization and activation. Cell Death Differ. 13, 941–950. ( 10.1038/sj.cdd.4401925) [DOI] [PubMed] [Google Scholar]
- 33.Ma L, Wagner J, Rice JJ, Hu W, Levine AJ, Stolovitzky GA. 2005. A plausible model for the digital response of p53 to DNA damage. Proc. Natl Acad. Sci. USA 102, 14 266–14 271. ( 10.1073/pnas.0501352102) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lu X, Nannenga B, Donehower LA. 2005. PPM1D dephosphorylates Chk1 and p53 and abrogates cell cycle checkpoints. Genes Dev. 19, 1162–1174. ( 10.1101/gad.1291305) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Takekawa M, Adachi M, Nakahata A, Nakayama I, Itoh F, Tsukuda H, Taya Y, Imai K. 2000. p53-inducible Wip1 phosphatase mediates a negative feedback regulation of p38 MAPK-p53 signaling in response to UV radiation. EMBO J. 19, 6517–6526. ( 10.1093/emboj/19.23.6517) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Cuadrado A, Nebreda AR. 2010. Mechanisms and functions of p38 MAPK signalling. Biochem. J. 429, 403–417. ( 10.1042/BJ20100323) [DOI] [PubMed] [Google Scholar]
- 37.Raman M, Earnest S, Zhang K, Zhao Y, Cobb MH. 2007. TAO kinases mediate activation of p38 in response to DNA damage. EMBO J. 26, 2005–2014. ( 10.1038/sj.emboj.7601668) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Coulthard LR, White DE, Jones DL, McDermott MF, Burchill SA. 2009. p38MAPK: stress responses from molecular mechanisms to therapeutics. Trends Mol. Med. 15, 369–379. ( 10.1016/j.molmed.2009.06.005) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sun J, et al. 2014. Enhancement of tunability of MAPK cascade due to coexistence of processive and distributive phosphorylation mechanisms. Biophys. J. 106, 1215–1226. ( 10.1016/j.bpj.2014.01.036) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Jeffrey PD, Gorina S, Pavletich NP. 1995. Crystal structure of the tetramerization domain of the p53 tumor suppressor at 1.7 angstroms. Science 267, 1498–1502. ( 10.1126/science.7878469) [DOI] [PubMed] [Google Scholar]
- 41.Mayo LD, Donner DB. 2001. A phosphatidylinositol 3-kinase/Akt pathway promotes translocation of Mdm2 from the cytoplasm to the nucleus. Proc. Natl Acad. Sci. USA 98, 11 598–11 603. ( 10.1073/pnas.181181198) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Manning BD, Cantley LC. 2007. AKT/PKB signaling: navigating downstream. Cell 129, 1261–1274. ( 10.1016/j.cell.2007.06.009) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Stambolic V, MacPherson D, Sas D, Lin Y, Snow B, Jang Y, Benchimol S, Mak TW. 2001. Regulation of PTEN transcription by p53. Mol. Cell 8, 317–325. ( 10.1016/S1097-2765(01)00323-9) [DOI] [PubMed] [Google Scholar]
- 44.Matsuda K, Yoshida K, Taya Y, Nakamura K, Nakamura Y, Arakawa H. 2002. p53AIP1 regulates the mitochondrial apoptotic pathway. Cancer Res. 62, 2883–2889. [PubMed] [Google Scholar]
- 45.Ow YLP, Green DR, Hao Z, Mak TW. 2008. Cytochrome c: functions beyond respiration. Nat. Rev. Mol. Cell Biol. 9, 532–542. ( 10.1038/nrm2434) [DOI] [PubMed] [Google Scholar]
- 46.Kirsch DG, Doseff A, Chau BN, Lim DS, de Souza-Pinto NC, Hansford R, Kastan MB, Lazebnik YA, Hardwick JM. 1999. Caspase-3-dependent cleavage of Bcl-2 promotes release of cytochrome c. J. Biol. Chem. 274, 21 155–21 161. ( 10.1074/jbc.274.30.21155) [DOI] [PubMed] [Google Scholar]
- 47.Angeli D, Ferrell JE, Jr, Sontag ED. 2004. Detection of multistability, bifurcations, and hysteresis in a large class of biological positive-feedback systems. Proc. Natl Acad. Sci. USA 101, 1822–1827. ( 10.1073/pnas.0308265100) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Puca R, et al. 2009. HIPK2 modulates p53 activity towards pro-apoptotic transcription. Mol. Cancer 8, 85 ( 10.1186/1476-4598-8-85) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Li T, et al. 2012. Tumor suppression in the absence of p53-mediated cell-cycle arrest, apoptosis, and senescence. Cell 149, 1269–1283. ( 10.1016/j.cell.2012.04.026) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Taira N, Nihira K, Yamaguchi T, Miki Y, Yoshida K. 2007. DYRK2 is targeted to the nucleus and controls p53 via Ser46 phosphorylation in the apoptotic response to DNA damage. Mol. Cell 25, 725–738. ( 10.1016/j.molcel.2007.02.007) [DOI] [PubMed] [Google Scholar]
- 51.Li QX, et al. 2007. Daxx cooperates with the Axin/HIPK2/p53 complex to induce cell death. Cancer Res. 67, 66–74. ( 10.1158/0008-5472.CAN-06-1671) [DOI] [PubMed] [Google Scholar]
- 52.Saul VV, de la Vega L, Milanovic M, Krüger M, Braun T, Fritz-Wolf K, Becker K, Schmitz ML. 2013. HIPK2 kinase activity depends on cis-autophosphorylation of its activation loop. J. Mol. Cell Biol. 5, 27–38. ( 10.1093/jmcb/mjs053) [DOI] [PubMed] [Google Scholar]
- 53.Li AG, Piluso LG, Cai X, Wei G, Sellers WR, Liu X. 2006. Mechanistic insights into maintenance of high p53 acetylation by PTEN. Mol. Cell 23, 575–587. ( 10.1016/j.molcel.2006.06.028) [DOI] [PubMed] [Google Scholar]
- 54.Ünsal-Kaçmaz K, Makhov AM, Griffith JD, Sancar A. 2002. Preferential binding of ATR protein to UV-damaged DNA. Proc. Natl Acad. Sci. USA 99, 6673–6678. ( 10.1073/pnas.102167799) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Leontieva OV, Gudkov AV, Blagosklonny MV. 2010. Weak p53 permits senescence during cell cycle arrest. Cell Cycle 9, 4323–4327. ( 10.4161/cc.9.21.13584) [DOI] [PubMed] [Google Scholar]
- 56.Momand J, Jung D, Wilczynski S, Niland J. 1998. The MDM2 gene amplification database. Nucleic Acids Res. 26, 3453–3459. ( 10.1093/nar/26.15.3453) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Song MS, Salmena L, Pandolfi PP. 2012. The functions and regulation of the PTEN tumour suppressor. Nat. Rev. Mol. Cell Biol. 13, 283–296. ( 10.1038/nrm3330) [DOI] [PubMed] [Google Scholar]
- 58.Das S, et al. 2007. Hzf determines cell survival upon genotoxic stress by modulating p53 transactivation. Cell 130, 624–637. ( 10.1016/j.cell.2007.06.013) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Stoyanova T, Roy N, Kopanja D, Bagchi S, Raychaudhuri P. 2009. DDB2 decides cell fate following DNA damage. Proc. Natl Acad. Sci. USA 106, 10 690–10 695. ( 10.1073/pnas.0812254106) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Kracikova M, Akiri G, George A, Sachidanandam R, Aaronson SA. 2013. A threshold mechanism mediates p53 cell fate decision between growth arrest and apoptosis. Cell Death Differ. 20, 576–588. ( 10.1038/cdd.2012.155) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Winter M, Sombroek D, Dauth I, Moehlenbrink J, Scheuermann K, Crone J, Hofmann TG. 2008. Control of HIPK2 stability by ubiquitin ligase Siah-1 and checkpoint kinases ATM and ATR. Nat. Cell Biol. 10, 812–824. ( 10.1038/ncb1743) [DOI] [PubMed] [Google Scholar]