

Abstract
Mapping genome-wide data to human subtelomeres has been problematic due to the incomplete assembly and challenges of low-copy repetitive DNA elements. Here, we provide updated human subtelomere sequence assemblies that were extended by filling telomere-adjacent gaps using clone-based resources. A bioinformatic pipeline incorporating multiread mapping for annotation of the updated assemblies using short-read data sets was developed and implemented. Annotation of subtelomeric sequence features as well as mapping of CTCF and cohesin binding sites using ChIP-seq data sets from multiple human cell types confirmed that CTCF and cohesin bind within 3 kb of the start of terminal repeat tracts at many, but not all, subtelomeres. CTCF and cohesin co-occupancy were also enriched near internal telomere-like sequence (ITS) islands and the nonterminal boundaries of subtelomere repeat elements (SREs) in transformed lymphoblastoid cell lines (LCLs) and human embryonic stem cell (ES) lines, but were not significantly enriched in the primary fibroblast IMR90 cell line. Subtelomeric CTCF and cohesin sites predicted by ChIP-seq using our bioinformatics pipeline (but not predicted when only uniquely mapping reads were considered) were consistently validated by ChIP-qPCR. The colocalized CTCF and cohesin sites in SRE regions are candidates for mediating long-range chromatin interactions in the transcript-rich SRE region. A public browser for the integrated display of short-read sequence–based annotations relative to key subtelomere features such as the start of each terminal repeat tract, SRE identity and organization, and subtelomeric gene models was established.
Subtelomeric DNA is crucial for telomere (TTAGGG)n tract length regulation and telomeric chromatin integrity. A telomeric repeat–containing family of RNAs (TERRA) is transcribed from subtelomeres into the (TTAGGG)n tracts (Azzalin et al. 2007; Schoeftner and Blasco 2008; Porro et al. 2010) and forms an integral component of a functional telomere; perturbation of its abundance and/or localization causes telomere dysfunction and genome instability (Azzalin et al. 2007; Deng et al. 2009). Telomere dysfunction caused by critically short telomere DNA sequence or by disruption of telomeric chromatin integrity induces DNA damage response pathways that cause cellular senescence or apoptosis (depending on the cellular context) in the presence of a functional p53 tumor suppressor pathway (Palm and de Lange 2008). Only one or a few critically short telomeres in a cell are sufficient to induce DDR-mediated senescence or apoptosis (Zou et al. 2004; Meier et al. 2007). Senescence or apoptosis of somatic cells can disrupt tissue microenvironments, and senescence or apoptosis of stem cell populations can prevent proper replenishment of rapidly dividing cellular lineages, both impacting aging phenotypes and age-related diseases, including cancer (Coppé et al. 2010; Davalos et al. 2010; Jaskelioff et al. 2010; Sahin and Depinho 2010).
Subtelomeric DNA elements regulate both TERRA levels and haplotype-specific (TTAGGG)n tract length and stability (Graakjaer et al. 2003, 2006; Britt-Compton et al. 2006; Deng et al. 2009; Nergadze et al. 2009), with accumulating evidence for specific epigenetic modulation of these effects (Yehezkel et al. 2008; Caslini et al. 2009; Deng et al. 2009; Nergadze et al. 2009; Arnoult et al. 2012). Heterogeneously sized TERRA transcripts with as yet ill-defined transcription start sites and potential splice patterns originate in many, perhaps all, human subtelomere regions (Azzalin et al. 2007; Porro et al. 2010; Deng et al. 2012), with the sizes of the larger transcripts (>15 kb) suggesting structural overlap with some transcribed subtelomeric gene families (Riethman 2008a,b). While many details of the dynamic interplay between shelterin, telomere chromatin structure, TERRA expression, and telomere biology remain unclear, recent work from our group indicates that CTCF and cohesin are integral components of most human subtelomeres, and important for the regulation of TERRA transcription and telomere end protection (Deng et al. 2012).
The chromatin organizing factor CTCF has been implicated in numerous aspects of chromosome biology, including chromatin insulator, enhancer blocker, transcriptional activator and repressor, DNA methylation–sensitive parental imprinting, and DNA-loop formation between transcriptional control elements (Bushey et al. 2008; Phillips and Corces 2009; Ohlsson et al. 2010). In addition to its role in TERRA regulation, CTCF has been implicated in the transcriptional repression of a subtelomeric D4Z4 macrosatellite repeat transcript ∼30 kb from the telomere repeats of chromosome 4q (Ottaviani et al. 2011). At D4Z4, CTCF interacts with lamin A and tethers the chromosome 4q telomere to the nuclear periphery (Ottaviani et al. 2009a,b). A more general role for CTCF has been found in its ability to colocalize with cohesin subunits at many chromosomal positions (Parelho et al. 2008; Rubio et al. 2008; Stedman et al. 2008; Wendt et al. 2008). Cohesin is a multiprotein complex consisting of core subunits SMC1, SMC3, RAD21, and STAG1 or STAG2, which can form a ring-like structure capable of encircling or embracing two DNA molecules (Nasmyth and Haering 2005; Hirano 2006). Cohesin was originally identified as a regulator of sister chromatid cohesion, but subsequent studies in higher eukaryotes indicate functions in mediating long-distance interactions between DNA elements required for transcription regulation (Kagey et al. 2010; Dorsett 2011). Cohesin subunit STAG1 is recruited to telomere repeats by the shelterin protein TINF2, and this interaction is required for telomeric sister chromatid cohesion and efficient telomere replication (Canudas and Smith 2009; Remeseiro et al. 2012). STAG1 binds directly to telomere repeat DNA through a unique AT hook, and overexpression of STAG1 alone is sufficient to induce cohesion at telomeres independently of cohesin ring components (Bisht et al. 2013). In contrast, colocalized cohesin ring components and CTCF both contribute to subtelomeric TERRA transcriptional regulation and telomere end protection (Deng et al. 2012).
In humans, telomere regulation occurs in the context of subtelomeric DNA segmental duplications known as subtelomeric repeat elements (SREs), which comprise ∼80% of the most distal 100 kb and 25% of the most distal 500 kb in human DNA (The International Human Genome Sequencing Consortium 2004; Riethman et al. 2004). SRE regions of human chromosomes contain mosaic patchworks of duplicons (Der-Sarkissian et al. 2002; Mefford and Trask 2002; Ambrosini et al. 2007) apparently generated by translocations involving the tips of chromosomes, followed by transmission of unbalanced chromosomal complements to offspring (Linardopoulou et al. 2005). Along with highly elevated sister chromatid exchange (SCE) rates in subtelomeres (Rudd et al. 2007), these studies indicate that human subtelomeres are duplication-rich hotspots of DNA breakage and repair.
Here, we have generated improved human subtelomere assemblies by sequencing additional subtelomeric clones and revising the reference sequence of distal subtelomere regions. A bioinformatic pipeline for annotation of the updated subtelomere assemblies using short-read data sets is developed and implemented. A public browser for the integrated display of short-read–based annotations relative to key subtelomere features such as the start of each terminal repeat tract, SRE identity and organization, and subtelomeric gene models is established and used to investigate cohesin and CTCF binding in SRE regions.
Results
Gap-filling and detection of distal telomeric structural variants
In order to fill remaining telomere-adjacent gaps from our previous reference subtelomere assembly (Ambrosini et al. 2007), we sampled telomere-adjacent DNA from deep fosmid clone libraries prepared from sheared genomic DNA samples (The International Human Genome Sequencing Consortium 2004; Kidd et al. 2008, 2010). Since each fosmid from these libraries had been end-sequenced using Sanger methods, we computationally searched for (CCCTAA)n sequence (the DNA sequence and orientation expected from fosmids ends located within telomere terminal repeat tracts) and selected the (CCCTAA)n-positive group of clones for further analysis. Each mate-pair read associated with a (CCCTAA)n read was mapped to our laboratory’s previous assembly (Ambrosini et al. 2007) to create a deep-coverage resource of mapped fosmid clones containing telomere-adjacent DNA. Using this mapping information, representative single clones that spanned gaps in the assembly were selected and sequenced (Table 1). Included in this group of clones were two structural variants identified in the mapping studies that, while capturing telomere-adjacent DNA for these chromosome ends, removed some SRE sequence from our previous assembly (analogous to the sequenced 16p allele relative to the longer mapped variant 16p alleles) (Flint et al. 1997). A second allele for the distal 4q subtelomere, which shared high sequence similarity with distal 10q (Lemmers et al. 2007), was also sequenced, as was a yeast artificial clone (YAC)–derived sequence we identified which filled a 12q gap. Finally, the mapped telomere fosmid resource was used to complete 8q and 18q telomere-adjacent sequences that contained sequence ambiguities and misassemblies immediately adjacent to the (TTAGGG)n tract in the previous assembly (Ambrosini et al. 2007); these errors were retained in hg19. Further details relating to fosmid library screening and characterization, the mapped telomere fosmid resources available from this work, and direct sequencing from distal telomere fosmids are provided in the Supplemental Material (Mapped Telomere Fosmid Resource; Supplemental Figs. 1, 2; Supplemental Tables 1–4).
Table 1.
Subtelomeric sequences from telomeric clones
Updated subtelomere assemblies
Rather than simply extending our previous assembly, we combined our new sequences with all other available fully sequenced subtelomere clones in NCBI to create an updated clone-based assembly of human subtelomere regions (Supplemental Table 5). We used, to the extent possible, contiguous segments of the existing hg19 assembly for the preparation of our 500-kb-sized subtelomere assemblies, only altering regions where our data indicated substantial change was required. The subtelomere regions that changed relative to hg19 are shown in Figure 1; 18 telomere-adjacent regions were altered: 15 by addition to or replacement of hg19 sequence and three by truncation of hg19 sequence. For all telomeres not showing change relative to hg19 in Fig 1, the distal-most telomere gaps and clone gaps (where they existed immediately adjacent to telomere gaps), represented in hg19 by a long string of N’s, were removed. Distal telomere tract sequence was also removed, so that coordinate 1 of each assembly corresponds to the start of the terminal repeat tract on the strand oriented toward the centromere (to maintain a consistent starting coordinate for subtelomere annotation). For the seven telomeres whose reference sequences do not extend to the terminal repeat (6p, 8p, 1p, 11p, 3q, 9q, 20p), coordinate 1 corresponds to the most distal base of the subtelomere assembly. The five acrocentric short arm telomeres are not represented in our assemblies; while they are known to contain a characteristic SRE organization closely related to distal 4p (Youngman et al. 1992), they cannot be distinguished from each other and assemblies adjacent to them are unavailable. Thirty-five of the telomere assemblies extend to the start of the terminal telomere repeat tract, and those that do not can be defined relative to the start of the terminal repeat tract by comparison with known SRE organizations and independent mapping data (Supplemental Table 5; Riethman et al. 2004; Linardopoulou et al. 2005; Ambrosini et al. 2007).
Figure 1.

Sequence organization of updated subtelomere sequence assemblies. The assemblies are oriented with the telomere on the left and aligned to maximize paralogous blocks of SREs following the methods described in Linardopoulou et al. (2005). Regions of the assemblies differing from hg19 are indicated by the black brackets above the altered region of the assembly. An internal gap in the 1q assembly is indicated by the magenta line segment. The pseudoautosomal region of Xq and Yq shares the same reference sequence and is indicated by the thick gray line distal to the dotted line. Blocks 43 and 44 are shown as subtelomere paralogs because they are duplicated at the 2q site of an ancestral telomere fusion; other internal paralogies are not shown or analyzed here. A selection of named transcripts mapping primarily to the indicated blocks is listed; a much larger number of uncharacterized transcripts and ncRNAs is not shown here but is annotated on the subtelomere browser. The average percentage of identity shared by copies of paralogous blocks is indicated by the groupings to the left of the color key. The positions of telomeres, ITSs, and CTCF/cohesion colocalization sites in the three cell types examined in detail are as indicated in the figure.
Figure 1 shows the distal parts of the assemblies, encompassing all SRE regions. The one-copy DNA at the centromeric end of each assembly corresponded to and was connected to hg19 at the coordinates shown in Supplemental Table 6. In a few cases, large segments of hg19 subtelomeric sequence were removed in our assemblies (e.g., removal of ∼520 kb of distal hg19 sequence at the 1p subtelomere), but in most cases, the updated assemblies were similar to those in hg19 with the exception of the most distal DNA segments. The resulting “hybrid genome,” composed mostly of hg19 sequence but modified by incorporation of our new subtelomere assemblies, allowed consistent genome-wide annotation that takes into account the entire reference sequence. The subtelomere browser described below displays only the first 500 kb of each chromosome arm from the annotated hybrid genome. It is important to note that the subtelomere assemblies are not from single haplotypes. The hg19 genome assembly is composed of clones from the DNA of many individuals, and the sequences we have added are from four additional individual genomes (Table 1; for description of the mapped telomere fosmid resource, see Supplemental Information); it is important to consider these limitations in the interpretation of read-mapping results (see Discussion).
Subtelomere annotation
The hybrid genome was used to annotate subtelomeric sequence features as described in the study by Ambrosini et al. (2007) and to map several ChIP (chromatin immunoprecipitation)-seq data sets of particular interest to subtelomere function (Deng et al. 2012). Figure 2 illustrates these annotations for the first 250 kb of the 19p subtelomere. Both coding and noncoding transcripts are abundant in SRE regions; while some are clearly functional, most are not well characterized (Linardopoulou 2001; Riethman et al. 2004; Linardopoulou et al. 2007; Riethman 2008a).
Figure 2.
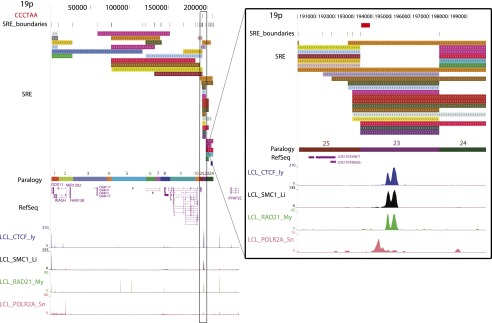
Subtelomere annotation features. The first 250 kb of the 19p subtelomere assembly is shown to illustrate key features of subtelomere sequence organization annotated on our browser. Coordinate 1 on the browser corresponds to the centromeric end of the terminal repeat tract [i.e., the last (CCCTAA)n repeat unit before subtelomere DNA starts]. The 207-kb-long SRE region on 19p is subdivided into duplication modules (“duplicons”) defined by segments of similarity (>90% nucleotide identity, >1 kb in length) between 19p and other subtelomeres (Ambrosini et al. 2007). Each rectangle represents a separate duplicon. Duplicated segments are identified by chromosome (color) as described previously (Ambrosini et al. 2007); additional details included on the live browser but omitted for the sake of clarity include the subject subtelomere identity, starting and ending coordinates of the duplicon in the subject subtelomere sequence, and the percentage of nucleotide sequence similarity of non-RepeatMasked sequences from the duplicon segment of the subject subtelomere to 19p (vader.wistar.upenn.edu/humansubtel). Each SRE boundary is indicated on a single track (SRE_boundaries), as are the internal telomere-like sequence (ITS) islands as defined in Methods (red ticks in the CCCTAA track). Gene models for transcripts included in the RefSeq (shown) (Pruitt et al. 2012) and Ensembl (hidden in this figure) (Flicek et al. 2012) transcript databases were mapped using Spidey (Wheelan et al. 2001). The paralogy track corresponds to the blocks, as shown in Figure 1. Enrichment profiles for four ChIP-seq data sets originally mapped only to subterminal DNA sequences (Deng et al. 2012) are displayed. (Inset) Close-up view of an internal SRE boundary region showing the association of the boundaries with an ITS (red rectangle on top line) and enrichment peaks for CTCF, cohesin subunits SMC1A and RAD21, and RNA polymerase II large subunit (POLR2A).
A paralogy map for SRE regions was prepared based upon the paralogy blocks defined previously (Linardopoulou et al. 2005) to facilitate graphic visualization of similar sequence segments occurring in multiple telomeres (see Fig. 1; Methods). Previously defined paralogy blocks covered most SRE regions, but we identified five new blocks and divided block 19 into two sub-blocks because of subtelomeric sequence not available to Linardopoulou et al. (2005). Paralogy blocks as defined by Linardopoulou et al. (2005) were developed as graphic visualization tools and have inexact borders with lower boundary resolution than the duplicons defined by Ambrosini et al. (2007). In addition, the paralogy blocks share slightly higher percentages of nucleotide sequence similarity than the duplicons defined by Ambrosini et al. (2007), because the paralogy blocks include high copy repeat sequence for this analysis, whereas the duplicon analysis of Ambrosini et al. (2007) uses only non-repeat-masked sequence for sequence comparisons.
The mapping of short-read data sets to human subtelomere regions requires special consideration because of the recent segmental duplication content. To deal with this challenge, we used a strategy of assigning a mapping likelihood (mL tag) to reads equal to the inverse of its genome-wide mapping positions; in effect, splitting up a read and mapping an equal portion of it to all of its possible sites of true mapping (Wang et al. 2010; Chung et al. 2011). By using this alternative mapping strategy, we then build fragment densities to display on enrichment tracks and to call peaks (see Methods). Concurrently, a track for each sample was built using only uniquely mapping reads (with an mL tag of 1) for comparison with the multiread track. The multiread tracks are shown in the figures; tracks for uniquely mapping reads can be found in the subtelomere browser (vader.wistar.upenn.edu/humansubtel).
By using this pipeline, enrichment profiles for four of the ChIP-seq data sets originally mapped only to telomere-adjacent DNA sequences (Deng et al. 2012) are displayed in Figure 2 on the subtelomere browser after mapping to the entire hybrid genome using the multiread mapping approach and then displaying the distal 500 kb on the subtelomere browser (see Methods). The same subterminal binding enrichments for CTCF, SMC1A, RAD21, and RNA polymerase II large subunit (POLRA2), which were found and validated by ChIP-qPCR (quantitative PCR) in our previous work (Deng et al. 2012), are evident in the current annotation (<3 kb from the telomere tract at 19p in Fig. 2; for other telomeres, see Supplemental Fig. 4). In addition, enrichment peaks for these proteins throughout the 19p subtelomere region are shown in Figure 2 (for other subtelomeres, see Supplemental Fig. 4). The inset highlights an internal SRE boundary region shared by many duplicons, showing the proximity of these boundaries with an ITS (red rectangle on top line) and enrichment peaks for CTCF, cohesin subunits SMC1 and RAD21, and POLR2A. Interestingly, the sequences adjacent to this ITS share similar but nonidentical features with sequences adjacent to terminal (TTAGGG)n repeat tracts. The POLR2A peak is positioned over a degenerate version of the subterminal 29-mer element (Deng et al. 2012); this ITS-adjacent binding site corresponds to a 23-mer element that, like the 29-mer repeat, is CpG rich. The CTCF/cohesin peaks span an extended 61-mer repeat array (7.3 copies in the ITS-adjacent sequence, vs. between two and four copies at most subterminal sites), but only 44 of 61 bases on the consensus 61-mer sequences are shared between subterminal and internal copies. The pattern of CTCF, cohesin, and POLR2A binding to these internal sequences is nearly identical to that found adjacent to terminal repeats (Deng et al. 2012), even though the sequences have diverged substantially. In fact, the sequences adjacent to this ITS are more similar to several other subtelomeric ITS-adjacent sequences (90%) than they are to any subterminal copies (85%).
SRE boundary enrichments
Publicly available CTCF and cohesin subunit ChIP-seq data sets from human ES cells and primary diploid fibroblasts (IMR90) were mapped in the same fashion and compared with the LCL data. All of the data sets used in this study and their mapping characteristics are summarized in Supplemental Table 7. Broadly speaking, similar patterns of CTCF and cohesin binding to the terminal boundary regions [defined as within 3 kb of the (TTAGGG)n repeat tract] were observed in LCL, ES, and primary fibroblast (IMR90) cell types, although the relative peak heights sometimes varied substantially (Fig. 3; Supplemental Fig. 4). For example, terminal boundary RAD21 enrichment peaks were almost always visible at some level in the bedGraphs of the expected subtelomeres, but for some data sets, many of the peaks did not reach the MACS significance threshold set for peak-calling subtelomere-wide (P < 1.0 × 10−4) (Supplemental Table 8). Many, but not all CTCF and cohesin sites across the SRE regions in LCLs were also detectable in the ES cell lines and in IMR90 (Fig. 3; Supplemental Fig. 4). Differences in library quality and depth, as well as differences in the antibodies used for ChIP-seq (Supplemental Table 7), are expected to have an effect on binding enrichments. However, easily discernible proportional differences in peak heights as well as clear instances of differential peak presence/absence between cell types may be indicative of true differential binding. These candidate differentially binding sites are easily detectable visually (e.g., compare relative CTCF peak heights and relative RAD21 peak heights on distal 6q in Fig. 3, and in all subtelomeres in Supplemental Fig. 4 and on the subtelomere browser; vader.wistar.upenn.edu/humansubtel). Most CTCF binding sites have been thought to be invariant between cell types, but a recent study suggested significant plasticity in CTCF occupancy at a majority of sites genome-wide, with 41% of the variable occupancy sites linked to differential CpG methylation (Wang et al. 2012). Similarly, a subset of cohesin binding sites are known to display cell-type specificity, colocalizing with tissue-specific transcription factors (Merkenschlager and Odom 2013). In this context, it will be intriguing to follow up our initial annotations here with detailed studies of the differential CTCF and cohesin occupancy of binding sites in the telomere-adjacent regions, and their potential implications for telomere length and stability.
Figure 3.

Example of an annotated subtelomere with CTCF and cohesin binding enrichment peaks from multiple cell types. The first 160 kb of 6q is shown in our browser. The PCR assay track marks the primer sites used for ChIP-qPCR (see Fig. 4). In addition to the ChIP-seq data sets shown in Figure 2 for LCLs (Deng et al. 2012), enrichment profiles for CTCF and RAD21 are shown following mapping of the ENCODE Project ChIP-seq data sets from the pluripotent human embryonic stem cell line H1-hESC and the primary fibroblast cell line IMR90.
Visually noted apparent association of CTCF and cohesin peaks with some SRE boundaries was analyzed systematically using only significant peaks called for each data set by MACS (Table 2; Supplemental Table 8; Zhang et al. 2008). The terminal SRE boundary was defined as the start of the terminal (TTAGGG)n tract; since the CTCF and cohesin binding sites associated with terminal repeat tracts are consistently <3 kb from this boundary (Deng et al. 2012), we initially used a 3-kb window to scan all SRE boundaries for CTCF and cohesin subunit peaks. Peak association enrichments are the observed ratio of peaks in the boundary window regions to the expected peak number within these windows if the total number of peaks in the SRE regions were distributed evenly. Some boundaries are within the allowable window of each other; in these instances, a peak can be associated with more than one boundary, although no additional weighting is added to the boundary association of these peaks. To calculate a P-value for the enrichment of peaks in boundary regions, a one-sided binomial test was performed.
Table 2.
SRE boundary enrichments
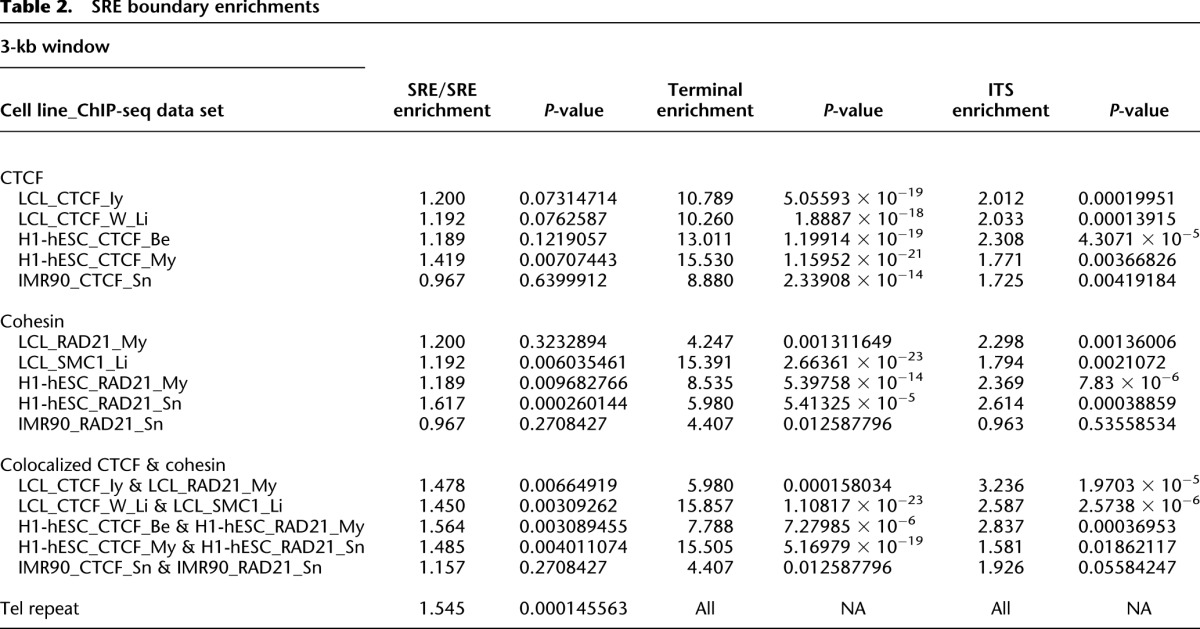
This analysis confirmed the strong association of CTCF and cohesin sites with the terminal boundaries in the cell types examined and also revealed a strong association of CTCF and cohesin sites with ITSs in all data sets except for IMR90 RAD21 (Table 2). There were weaker and often statistically insignificant associations of CTCF and cohesin sites with internal SRE/SRE boundaries in the individual data sets from these cell types (Table 2). However, boundary analysis of just the strictly colocalized peaks for CTCF and cohesin subunits showed significant associations with SRE/SRE boundaries for LCLs and ES cells, but not for the primary fibroblast cell line IMR90 (Table 2). The positions of all colocalized CTCF and cohesin peaks occurring in at least one of these three cell types are shown relative to SRE organization in Figure 1.
Experimental validation of ChIP-seq peaks by ChIP-qPCR
Several recent reports have suggested that some human ITSs bind the shelterin components TERF1 and TERF2 (Simonet et al. 2011; Yang et al. 2011), which seems plausible given the demonstrated ability of TERF1 and TERF2 to interact with the very short TTAGGGTT motif in some contexts (Deng et al. 2003; Zhou et al. 2005). This could have important functional implications and suggest potential long-range interaction of ITSs with telomeres. We therefore mapped TERF1 and TERF2 ChIP-seq data sets we prepared from LCLs, as well as publically available TERF1 and TERF2 ChIP-seq data sets from a transformed BJ fibroblast cell line (Simonet et al. 2011). Enrichment peaks localizing to many subtelomeric ITSs were initially found for both TERF1 and TERF2, in both cell types. However, in each case the mapped reads contributing to the peak did not have a normal distribution (Supplemental Fig. 3A), the consequence of a pile-up of reads mapped on both strands underneath a central peak region being extended to the ChIP fragment length, resulting in peak shoulders that do not correspond to true fragment ends (see Methods). The reads mapping to ITSs were composed of telomere-like repeat arrays. While these reads map “uniquely” according to sequence aligners, this is only in relation to the rest of the reference genome. Neither hg19 nor our hybrid genome includes proximal regions of terminal repeat tracts, known to contain extended regions of telomere-like sequences interspersed with pure (TTAGGG)n repeats (Baird et al. 1995). When telomere and telomere-like sequences were specifically removed from the data sets, peaks at all subtelomeric ITSs disappeared (Supplemental Fig. 3A). Examination of read orientations underneath a typical ITS peak compared with a true CTCF enrichment peak shows that reads responsible for ITS peaks are piled up in random orientation, whereas a true enrichment peak has reads oriented nonrandomly toward the peak of the enrichment (Supplemental Fig. 3B,C). In addition, true binding sites should be marked by noticeable enrichments in sequences flanking the central binding sites, but these enrichments were not found.
To test experimentally the computationally predicted subtelomeric CTCF and RAD21 colocalization sites in SRE regions and whether the called TERF1/TERF2 ChIP-seq peaks described above correlate with TERF1 and TERF2 binding, we carried out a series of ChIP-qPCR experiments summarized in Figure 4 and in Supplemental Figure 5. In Figure 4A, the colocalized CTCF and RAD21 sites in segments of the 6q and 16q SRE regions were examined; each of these sites were not called as peaks when only the uniquely mapping read sets were considered, but peaks were called at these positions using our multiread mapping pipeline. Each of the CTCF and RAD21 binding sites predicted by ChIP-seq mappings (primer positions 2, 4–6, 8–10) show the expected enrichments upon ChIP-qPCR relative to the control primer sets (3 and 7). In addition, the telomere-adjacent sites at primer positions 1 and 2 show the expected TERF1 and TERF2 enrichment very close to the terminal repeat tracts (Deng et al. 2012), whereas more distant subtelomeric sites at positions 3–10 show only background TERF1 and TERF2 levels. In Figure 4B, the expected CTCF and RAD21 enrichments are also seen in assays corresponding to colocalized CTCF and RAD21 sites predicted by ChIP-seq (positions 2, 4–6, 8). The telomere-adjacent Xq sites at positions 1 and 2 detect the expected TERF1 and TERF2 enrichments (Deng et al. 2012), but the position at 17p corresponding to an ITS with called TERF1 and TERF2 ChIP-seq peaks (position 5) shows only background levels of TERF1 and TERF2 binding. The Xq ITS adjacent to position 3 lacked a ChIP-seq enrichment peak in the TERF1 and TERF2 data sets, yet the ChIP-qPCR showed slight enrichment for TERF2, possibly because it is relatively close (9 kb) to the Xq telomere. Additional ChIP-qPCR assays from 19p and 11p show no correlation between ITS-associated ChIP-seq peaks called in the TERF1 and TERF2 data sets and binding enrichment by ChIP-qPCR, while showing anticipated ChIP-qPCR enrichments at CTCF and RAD21 colocalization sites predicted by ChIP-seq (Supplemental Fig. 5). Thus, we conclude that CTCF and RAD21 binding sites in SRE regions predicted by ChIP-seq multiread mappings are true binding sites, but that the ITS-associated ChIP-seq peaks called in the TERF1 and TERF2 data sets cannot be used to predict true TERF1 and TERF2 binding.
Figure 4.

ChIP-qPCR analysis of subtelomeric DNA protein binding sites predicted by ChIP-seq data set mappings. Candidate sites of CTCF, cohesin, TERF1, and TERF2 binding were analyzed by ChIP-qPCR. Segments of the 6q and 16q (A) and the Xq and 17p (B) subtelomeres are shown, with the coordinates (in bp) shown at the top and the subtelomere paralogy regions indicated on the respective segments. The positions of ITSs are indicated by red rectangles extending from the segments; an ITS with called TERF1 and TERF2 ChIP-seq enrichment peaks is marked with a red asterisk. The positions of colocalized CTCF and cohesin (RAD21) peaks called in LCLs are shown as green dots (if not called in other cell types) and as blue dots (if also called in ES and/or IMR90 cells). A diamond beneath a dot indicates a site where no ChIP-seq peak was called when only uniquely mapping reads were considered. Numbered ticks show the positions of primer sets used in the ChIP-qPCR experiments, and correspond to the numbered ChIP-qPCR results shown for CTCF, RAD21, and TERF1 and TERF2 graphed as the percentage of input DNA. The bar graphs represent the average of percentage input (mean ± SD) for each ChIP from three independent ChIP experiments. Ticks numbered 1 and 2 are qPCR assays for DNA immediately adjacent to the telomere, used here as positive controls for TERF1 and TERF2 binding (primer positions 1 and 2) and a positive control for a previously validated subtelomeric CTCF/RAD21 colocalization site (primer position 2).
CTCF data sets from additional primary and cancer cell lines
To test whether the terminal boundary and the ITS CTCF peak associations seen in the cell types described above are also seen in additional cell types, we mapped publically available CTCF ChIP-seq data sets from four primary cell lines (HMECs [human mammary epithelial cells], SAECs [small airway epithelial cells], HREs [human renal cortical epitheliums], and HRPEpiC [retinal pigment epithelial cells]) and four immortal cell lines (MCF-7 [mammary gland adenocarcinoma], A549 [lung carcinoma], HEK 293 [embryonic kidney cells transformed by Adenovirus 5 DNA], and WERI-Rb-1 [a retinoblastoma line]). Boundary analysis indicated a similar number of subtelomeric CTCF binding sites and a similar range of P-values for terminal boundaries and ITS associations with peaks (Supplemental Table 9) as were found in ChIP-seq data sets for LCLs, human ES cells, and IMR90 (Supplemental Table 8). As with the individual CTCF data sets for LCLs, ES cells, and IMR90, nonterminal SRE/SRE boundary associations with just CTCF peaks were usually not significant in the cell lines. Cohesin ChIP-seq data sets were not available for most of these cell lines, so we could not determine colocalized CTCF and cohesin binding sites and test their boundary associations. While most of the same CTCF peaks were called near the terminal boundary and the ITSs, visual comparison of peaks showed clear differences in relative levels of peak enrichments between the cell lines (vader.wistar.upenn.edu/humansubtel), as well as some differentially called peaks. These preliminary observations merit follow-up with much larger data sets as well as experimental validation.
Discussion
With this work, we have revised and updated human subtelomere assemblies such that 34 of the 41 genetically distinct chromosome ends extend to the start of terminal repeat tracts (Fig. 1). This represents a significant advance over the previous human subtelomere assemblies (Riethman et al. 2004; Ambrosini et al. 2007). We also provide a multiread mapping pipeline that enables the systematic analysis of distal chromosome regions using short-read sequencing–based methods, leveraging the wealth of public genome-wide data sets available to help understand subtelomere and telomere function. We have also established a public browser (vader.wistar.upenn.edu/humansubtel) that integrates novel aspects of subtelomere sequence organization with short-read sequence–based annotations and displays this information in a manner optimized for understanding potential functional properties associated with the annotations relative to the telomere terminal repeat tract as well as subtelomeric sequence features. As additional annotation is added, we believe it will become an increasingly valuable resource for the telomere and chromosome biology communities.
The updated subtelomere reference assemblies are subject to caveats as are all regions of the reference human genome sequence; they are composed of DNA segments derived from multiple individuals, and for any sequenced clone, only one allele is represented. This means that the depicted reference allele sequences may not completely match that of corresponding subtelomere alleles from other source genomes. Much of the natural variation in human subtelomeres is due to differential placement of SRE regions at specific subsets of subtelomeres (Linardopoulou et al. 2005; Riethman 2008a,b), and this may complicate interpretation of ChIP-seq signal strengths at specific high-similarity SRE sites when comparing data sets from nonisogenic source genomes. For example, a CTCF peak predicted by multiread mapping in a high-similarity SRE segment of the reference assembly is expected to have a higher enrichment level in a data set from a genome with more copies of the SRE segment than a data set with fewer copies of the SRE segment. Copy numbers of all known highly similar SRE blocks vary by a factor of two or less in the human population, although most vary by considerably less than twofold (Linardopoulou et al. 2005, 2007); depending on the SRE segment in question, a doubling or halving of an enrichment value at a peak may not be meaningful for a given data set. Prior knowledge of SRE copy number in the respective source genomes would help to mitigate this issue. Even with these limitations, the more complete sequence representation of our assemblies, especially in the distal subtelomere regions, has already permitted novel annotation leading to experimental validation and functional insights into telomere biology (Deng et al. 2012), which we have extended here. As new technologies capable of adding complete alternative long-range subtelomere haplotypes to the reference assemblies are developed, these sequences will be annotated and incorporated into our browser.
The use of our multiread mapping approach for ChIP-seq short-read data sets had a very large impact on the annotation of candidate binding sites in SRE regions; most candidate CTCF and RAD21 binding sites in SRE regions were missed (between 70% and 90% of called peaks, depending on the data set) when only uniquely mapping reads were considered. This is illustrated dramatically in Figure 4, where all of the sites predicted by the multiread mapping in the SRE regions were missed in the analysis considering only uniquely mapping reads. Comparison of the multiread mapping tracks and the unique read mapping tracks on the bedGraphs in the subtelomere browser for the same experiment often revealed a small unique read peak corresponding to a much larger and robust multiread peak for SRE sites, indicating that some fraction of the reads were mapping uniquely to the site but that the unique enrichment peak was too weak to be called statistically significant. However, as we showed previously (Deng et al. 2012), in SRE regions with very high sequence similarity to paralogs, there was no detectable enrichment in the uniquely mapping data sets.
Because peaks detected using the multiread mapping method represent an average of enrichments over all genomic sites to which the reads map, there is the potential for prediction of false-positive peaks called due to extremely high true binding at one or a few sites, causing called peaks at all of them. This is a limitation of the approach and an important caveat to consider in the interpretation of the results. Short-read–based annotations in SRE regions or, for that matter, any region of the genome, are models. While perhaps revealing valuable insights into subtelomere biology, they ultimately require independent validation. The ChIP-qPCR results of the predicted CTCF and RAD21 peaks shown in Figure 4 provide strong validation of the ChIP-seq binding predictions in SRE regions; however, even here ChIP-qPCR primer sets in very high similarity duplicated regions sometimes cannot distinguish all individual copies (see Supplemental Table 10).
Somewhat to our surprise, we did not find evidence for specific TERF1 or TERF2 binding to ITS sites. Interestingly, however, we found evidence for enrichment of CTCF and cohesin subunit binding adjacent to ITS boundaries, in addition to the binding sites near terminal (TTAGGG)n sites noted previously (Table 2; Deng et al. 2012). When we considered only the CTCF and cohesin subunit peaks that colocalized exactly (see Fig. 1), the significance of association with telomere-adjacent DNA and ITSs typically increased, while the colocalized peak association with SRE/SRE boundaries reached significance for the ES and LCL lines but not for IMR90 (Table 2). Strong cohesin sites colocalizing with CTCF have been implicated in long-range chromosomal interactions (Merkenschlager and Odom 2013), suggesting colocalized cohesin/CTCF sites may mediate DNA looping and long-range DNA interactions as well as regulate transcription (Chien et al. 2011; Lee and Iyer 2012; Merkenschlager and Odom 2013). Even in the potential absence of direct shelterin interactions between ITSs and telomeres, it is possible that CTCF/cohesin interactions between binding sites associated with the terminal boundaries and internal binding sites such as the ITS-associated ones could mediate events impacting telomeres as well as the regulation of subtelomeric gene families. For example, long-range cohesin/CTCF-mediated interactions involving the telomere-adjacent cohesin/CTCF colocalization sites implicated in TERRA regulation (Deng et al. 2012) may provide a means to coordinate the regulated transcription of TERRA from subtelomeric loci, similar in principle to the coordinated regulation of other complex loci and multigene families by cohesin and CTCF (Merkenschlager and Odom 2013). Using our subtelomere browser and bioinformatics pipeline to leverage the rich public resource of additional short-read data sets for further annotation of these regions may point to focused experiments to test this hypothesis and help to tease out candidate functional sequences involved in subtelomere biology.
Methods
Fosmid library screening; fosmid end sequence mapping, gap-filling, and detection of telomeric structural variants; and directed sequencing of distal ends of terminal fosmids
Methods and materials for these experiments are described in text associated with Supplemental Tables 1 through 4 and Supplemental Figures 1 and 2.
Updated subtelomere assemblies
Supplemental Table 5 describes the complete clone-based subtelomere assemblies as well as their relationship to current clone-based tiling path files (TPFs) being used to update the human reference sequence. The hybrid genome was built by tying the updated subtelomere assemblies into hg19 at their connection point. These points were found by using BLAST (Altschul 1997) to align the most centromeric 10 kb of sequence from each subtelomere assembly with hg19 sequence. The BLAST results produced one perfect 10-kb hit in the expected orientation, forward for p arm subtelomeres and reverse for q arm subtelomeres. The positions of these hits were then used to extract the nonsubtelomeric portion of the hybrid genome using BEDTools (Quinlan and Hall 2010). The sequence of each 500-kb subtelomere assembly is provided as a concatenated FASTA file in the Supplemental Material. The joining coordinates for connecting hg19 to the subtelomere assemblies are listed in Supplemental Table 6.
Sequence feature annotation
SRE and SD annotation were carried out as described previously (Ambrosini et al. 2007). Duplicon boundaries were defined as the end positions of duplicon blocks. Boundaries within 40 bp of each other were combined at a position corresponding to the weighted average of the number of boundaries they incorporate and were declared a single boundary for analysis purposes. Paralogy tracks were generated by first comparing the representative blocks identified by Linardopoulou et al. (2005) with the updated assemblies and then adding blocks corresponding to new SRE segments shared in the manner described by Linardopoulou et al. (2005). Existing Block 19 was broken into two separate blocks based upon the SRE/1-copy boundary generated by 17q sequence, which was not available to Linardopoulou et al. (2005). Representative sequences for paralogy blocks 19a, 19b, 45, 46, 47, 48, and 49 are provided as a concatenated FASTA file in Supplemental File S2. Subtelomere sequence assemblies were analyzed with RepeatMasker (Smit et al. 1996 at http://repeatmasker.org) and Tandem Repeats Finder (Benson 1999). Ensembl transcripts (Flicek et al. 2012) and RefSeq genes (Pruitt et al. 2012) were aligned to subtelomeres using Spidey (Wheelan et al. 2001).
Short-read–based annotation pipeline
Data sets analyzed in this study are listed with their specific sample and control GEO accessions, as well as the specific antibodies used and their sources, in Supplemental Table 7. The LCL-associated data sets for CTCF, RAD21, and SMC1 were the same as described previously (Deng et al. 2012). Additional data sets were downloaded as raw data FASTQ files from the ENCODE Project (The ENCODE Project Consortium 2011) through the UCSC portal. H1-hESC_CTCF_Be and HMEC_CTCF_Be are from the Bernstein laboratory (GSE29611 series) at the Broad Institute. IMR90_CTCF_Sn, IMR90_RAD21_Sn, IMR90_POLR2A_Sn, and H1-hESC_RAD21_Sn correspond to the Snyder laboratory data from Stanford (GSE31477 series). H1-hESC_RAD21_My, H1-hESC_CTCF_My, and H1-hESC_POLR2A_My correspond to the Myers data from HudsonAlpha (GSE32465 series).
Reads were aligned to the hybrid genome using BWA 0.6.2 (Li and Durbin 2009), allowing multimapping up to 101 locations (-n 101). BWA does not prioritize multimapping reads, and alternate mapping locations are not included as reads but instead are listed in an XA tag. Alternate positions were then expanded from the XA tag to one mapping position per line. A mapping likelihood (mL) tag was added as the inverse of the number of mapping locations. It is still possible to only consider uniquely mapping reads by analyzing only those reads with an mL tag equal to one. Fragment length was estimated by cross-correlation implemented in the SPP ChIP-seq mapping program (Kharchenko et al. 2008). bedGraph coverage files were created from the mapping positions by extending read mappings to the estimated fragment size. Fragment coverage for each position was calculated as the sum of mL values of fragments overlapping that position and then averaged over a 20-bp sliding window. Adjacent positions were given the same value if the coverage was within 0.1. To simplify fold change calculations, values less than one were given a pseudo count to be equal to one. Fold enrichment tracks were built between control (Input or IgG) and sample to be used as a signal track, normalizing the control data set to the size of the sample. Negative values were used to show stronger signal in the control. A pseudo count of one was used in locations where there was no mapping for the sample or control. A smoothing window of 500 bases was used on all control data sets. Peak calls were made using MACS 2.0.10 using the sample and control bedGraphs. First, bdgcmp –m ppois was called, setting ppois as the method and calculating P-value tracks. Peaks were called using bdgpeakcall –l 50 –c 4, setting minimum peak length to 50 and a P-value significance cut of 4 (10−4) (Zhang et al. 2008). Overall quality and mapping metrics for the data sets were determined as previously described (Landt et al. 2012) and are included in Supplemental Table 7.
TERF1 and TERF2 data sets
Publicly available data sets from Simonet et al. 2011 (GSE26005) were downloaded and analyzed. These are color space reads mapped on the AB SOLiD System 3.0. The color space reads were mapped using SHRiMP 2.2.3 (Rumble et al. 2009), allowing for reads mapping up to 101 mapping positions (-o 102). Once mapping positions were determined, the pipeline followed was the same as other ChIP-seq data sets. However, cross-correlation analysis failed at finding a fragment size, so the selected fragment size of 200 bases was used (Supplemental Table 7). Additional TERF1 and TERF2 ChIP-seq data sets were generated for LCL as described previously (Lu et al. 2012), using rabbit antibodies to TERF1 and TERF2, which were generated against recombinant protein and affinity purified. The 100-bp Illumina reads in these data sets were trimmed from both the 3′ and 5′ ends up to the first high-quality base (>Phred 30). Telomere and telomere-like simple repeats were identified by RepeatMasker (Smit et al. 1996).
Subtelomere browser
The subtelomere browser can be found on a mirror site of the UCSC Genome Browser maintained by the Wistar Bioinformatics Facility (vader.wistar.upenn.edu/humansubtel). The entire subtelomere region of interest is displayed by typing it in the format chrNp:1-500000 or chrNq:1-500000. The subtelomere browser has similar navigation and mapped data set selection functionalities as the UCSC Genome Browser (Kent et al. 2002). The updated subtelomere assemblies in FASTA format are found in Supplemental File S2 and can be found on the Riethman laboratory website (http://www.wistar.org/sites/default/files/protected/htel_1-500K_1_10_12_v4_3_12fasta.TXT).
Peak/boundary association enrichment calculation
Peak/boundary association enrichments were defined as the ratio of the number of peaks observed in defined boundary window regions (across all SRE sequence space) to the expected number of peaks within these window regions if the total number of peaks in the SRE sequence space were distributed evenly. Some boundaries were within the allowable window of each other; in these instances, a peak was associated with more than one boundary, although no additional weighting was added to the boundary association of these peaks. To calculate a P-value, a one-sided binomial test was performed, using the expected percentage as the probability of success, the associated number of peaks as the number of successes, and the total number of peaks in the SRE as the number of trials. Terminal boundaries and their associated peaks were excluded when calculating P-values for peak association with ITSs.
ChIP assay
ChIP assays were performed with the protocol provided by Millipore with minor modifications as described previously (Deng et al. 2009). Briefly, LCLs were crosslinked in 1% formaldehyde with shaking for 15 min, and DNA was sheared to between 200- and 400-bp fragments by sonication with a Diagenode Bioruptor. Quantification of ChIP DNA at subtelomeric regions was determined using quantitative PCR (qPCR) with the ABI 7900 sequence detection system (Applied Biosystems). qPCR was performed in triplicates from three independent ChIP experiments, and PCR data were normalized to input values. Primer sequences used for qPCR were designed using Primer Express (Applied Biosystems), and listed in Supplemental Table 10. Each primer sets was validated by using melting curve analysis, in which one major dissociation peak was observed. ChIP DNA at telomeres was assayed by dot blotting with γ-[32P]ATP-labeled probes specific for telomere (4 × TTAGGG) or Alu repeats (cggagtctcgctctgtcgcccaggctggagtgcagtggcgcga). After hybridization, the blot was developed with a Typhoon 9410 imager (GE Healthcare) and quantified with ImageQuant 5.2 software (Molecular Dynamics). Antibodies used in ChIP assay include rabbit polyclonal antibodies to CTCF (Millipore 07-729) and RAD21 (abcam ab992). Rabbit antibodies to TERF1 and TERF2 were generated against recombinant protein and affinity purified.
Data access
DNA sequence for gap-filling clones and clone fragments were submitted to the NCBI GenBank (https://www.ncbi.nlm.nih.gov/genbank/) under accession numbers AC215217, AC215219, AC213859, AC213860, AC215218, AC213861, AC215220, AC215221, AC225782, AC225782, AC215524, AC215522, AC226150, KF477190, KF477189, KF477188, KF477185, KF477187, KF477186, and KF477184 (see Table 1). The TERF1 and TERF2 ChIP-seq data sets generated as part of this study were submitted to the NCBI Genome Expression Omnibus (GEO; http://www.ncbi.nlm.nih.gov/gds) under accession numbers GSM1328844 and GSM1328845. Each of the 500-kb subtelomere reference assemblies are available as a concatenated FASTA file in Supplemental File S1. New SRE paralogy blocks 45–49, 19a, and 19b are available as a concatenated FASTA file in Supplemental File S2. The subtelomere browser link is vader.wistar.upenn.edu/humansubtel.
Acknowledgments
We acknowledge contributions from the Wistar Cancer Center Core facilities in Bioinformatics and Genomics, especially Priyankara Wickramasinghe for maintenance and updating of the subtelomere browser in the Wistar Bioinformatics Core facility, and Brett Taylor for assistance with the computational resources in the Wistar Center for Systems and Computational Biology. This work was supported by the Wistar Cancer Center core grant (P30 CA10815) and the Commonwealth Universal Research Enhancement Program, PA Department of Health. Additional support was provided by the Philadelphia Health Care Trust and a predoctoral NRSA F31 Diversity award (N.S.) and by NIH grants to H.R. (R21CA143349 and R21HG007205) and P.L. (RO1CA140652). Z.D. was supported by an American Heart Association grant (11SDG5330017) and R.V.D. by R01LM011297. E.E.E. was supported by HG004120 and thanks Maika Malig and Jeff Kidd for technical/bioinformatics assistance. Work done by T.G., C.C.F., L.C., and R.K.W. was supported by U54 HG003079.
Author contributions: N.S. and H.R. designed the experiments for the project, and S.H. and S.P. carried out the mapping and sequencing experiments in H.R.’s laboratory. Fosmid clones were provided by E.E.E., and full Sanger sequencing of selected clones was done by C.C.F., L.C., and T.G. R.K.W., R.G., A.K.W., and R.V.D. assisted with the initial analysis of ChIP-seq data. Z.D. and P.L. carried out the ChIP-qPCR experiments and assisted with interpretation of the data. N.S. developed the bionformatics pipeline for multimapping ChIP-seq analysis. N.S. and H.R. led the analysis and interpretation of the data, assembled the figures, and wrote the manuscript.
Footnotes
[Supplemental material is available for this article.]
Article published online before print. Article, supplemental material, and publication date are at http://www.genome.org/cgi/doi/10.1101/gr.166983.113.
References
- Altschul S 1997. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res 25: 3389–3402 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ambrosini A, Paul S, Hu S, Riethman H 2007. Human subtelomeric duplicon structure and organization. Genome Biol 8: R151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arnoult N, Van Beneden A, Decottignies A 2012. Telomere length regulates TERRA levels through increased trimethylation of telomeric H3K9 and HP1α. Nat Struct Mol Biol 19: 948–956 [DOI] [PubMed] [Google Scholar]
- Azzalin CM, Reichenbach P, Khoriauli L, Giulotto E, Lingner J 2007. Telomeric repeat containing RNA and RNA surveillance factors at mammalian chromosome ends. Science 318: 798–801 [DOI] [PubMed] [Google Scholar]
- Baird DM, Jeffreys AJ, Royle NJ 1995. Mechanisms underlying telomere repeat turnover, revealed by hypervariable variant repeat distribution patterns in the human Xp/Yp telomere. EMBO J 14: 5433–5443 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benson G 1999. Tandem repeats finder: a program to analyze DNA sequences. Nucleic Acids Res 27: 573–580 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bisht KK, Daniloski Z, Smith S 2013. SA1 binds directly to DNA through its unique AT-hook to promote sister chromatid cohesion at telomeres. J Cell Sci 126: 3493–3503 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Britt-Compton B, Rowson J, Locke M, Mackenzie I, Kipling D, Baird DM 2006. Structural stability and chromosome-specific telomere length is governed by cis-acting determinants in humans. Hum Mol Genet 15: 725–733 [DOI] [PubMed] [Google Scholar]
- Bushey AM, Dorman ER, Corces VG 2008. Chromatin insulators: regulatory mechanisms and epigenetic inheritance. Mol Cell 32: 1–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Canudas S, Smith S 2009. Differential regulation of telomere and centromere cohesion by the Scc3 homologues SA1 and SA2, respectively, in human cells. J Cell Biol 187: 165–173 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caslini C, Connelly JA, Serna A, Broccoli D, Hess JL 2009. MLL associates with telomeres and regulates telomeric repeat-containing RNA transcription. Mol Cell Biol 29: 4519–4526 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chien R, Zeng W, Ball AR, Yokomori K 2011. Cohesin: a critical chromatin organizer in mammalian gene regulation. Biochem Cell Biol 89: 445–458 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chung D, Kuan PF, Li B, Sanalkumar R, Liang K, Bresnick EH, Dewey C, Keleş S 2011. Discovering transcription factor binding sites in highly repetitive regions of genomes with multi-read analysis of ChIP-Seq data. PLoS Comput Biol 7: e1002111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coppé J-P, Desprez P-Y, Krtolica A, Campisi J 2010. The senescence-associated secretory phenotype: the dark side of tumor suppression. Annu Rev Pathol 5: 99–118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davalos AR, Coppe J-P, Campisi J, Desprez P-Y 2010. Senescent cells as a source of inflammatory factors for tumor progression. Cancer Metastasis Rev 29: 273–283 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng Z, Atanasiu C, Burg JS, Broccoli D, Lieberman PM 2003. Telomere repeat binding factors TRF1, TRF2, and hRAP1 modulate replication of Epstein-Barr virus OriP. J Virol 77: 11992–12001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng Z, Norseen J, Wiedmer A, Riethman H, Lieberman PM 2009. TERRA RNA binding to TRF2 facilitates heterochromatin formation and ORC recruitment at telomeres. Mol Cell 35: 403–413 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng Z, Wang Z, Stong N, Plasschaert R, Moczan A, Chen H-S, Hu S, Wikramasinghe P, Davuluri RV, Bartolomei MS, et al. 2012. A role for CTCF and cohesin in subtelomere chromatin organization, TERRA transcription, and telomere end protection. EMBO J 31: 4165–4178 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Der-Sarkissian H, Vergnaud G, Borde Y-M, Thomas G, Londoño-Vallejo J-A 2002. Segmental polymorphisms in the proterminal regions of a subset of human chromosomes. Genome Res 12: 1673–1678 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dorsett D 2011. Cohesin: genomic insights into controlling gene transcription and development. Curr Opin Genet Dev 21: 199–206 [DOI] [PMC free article] [PubMed] [Google Scholar]
- The ENCODE Project Consortium. 2011. A user’s guide to the encyclopedia of DNA elements (ENCODE). PLoS Biol 9: e1001046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flicek P, Amode MR, Barrell D, Beal K, Brent S, Carvalho-Silva D, Clapham P, Coates G, Fairley S, Fitzgerald S, et al. 2012. Ensembl 2012. Nucleic Acids Res 40: D84–D90 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flint J, Bates G, Clark K, Dorman A, Willingham D, Roe B, Micklem G, Higgs D, Louis E 1997. Sequence comparison of human and yeast telomeres identifies structurally distinct subtelomeric domains. Hum Mol Genet 6: 1305–1313 [DOI] [PubMed] [Google Scholar]
- Graakjaer J, Bischoff C, Korsholm L, Holstebroe S, Vach W, Bohr VA, Christensen K, Kølvraa S 2003. The pattern of chromosome-specific variations in telomere length in humans is determined by inherited, telomere-near factors and is maintained throughout life. Mech Ageing Dev 124: 629–640 [DOI] [PubMed] [Google Scholar]
- Graakjaer J, Der-Sarkissian H, Schmitz A, Bayer J, Thomas G, Kolvraa S, Londoño-Vallejo J-A 2006. Allele-specific relative telomere lengths are inherited. Hum Genet 119: 344–350 [DOI] [PubMed] [Google Scholar]
- Hirano T 2006. At the heart of the chromosome: SMC proteins in action. Nat Rev Mol Cell Biol 7: 311–322 [DOI] [PubMed] [Google Scholar]
- The International Human Genome Sequencing Consortium. 2004. Finishing the euchromatic sequence of the human genome. Nature 431: 931–945 [DOI] [PubMed] [Google Scholar]
- Jaskelioff M, Muller FL, Paik J-H, Thomas E, Jiang S, Adams AC, Sahin E, Kost-Alimova M, Protopopov A, Cadiñanos J, et al. 2010. Telomerase reactivation reverses tissue degeneration in aged telomerase-deficient mice. Nature 469: 102–106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kagey MH, Newman JJ, Bilodeau S, Zhan Y, Orlando DA, van Berkum NL, Ebmeier CC, Goossens J, Rahl PB, Levine SS, et al. 2010. Mediator and cohesin connect gene expression and chromatin architecture. Nature 467: 430–435 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kent WJ, Sugnet CW, Furey TS, Roskin KM, Pringle TH, Zahler AM, Haussler D 2002. The Human Genome Browser at UCSC. Genome Res 12: 996–1006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kharchenko PV, Tolstorukov MY, Park PJ 2008. Design and analysis of ChIP-seq experiments for DNA-binding proteins. Nat Biotechnol 26: 1351–1359 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kidd JM, Cooper GM, Donahue WF, Hayden HS, Sampas N, Graves T, Hansen N, Teague B, Alkan C, Antonacci F, et al. 2008. Mapping and sequencing of structural variation from eight human genomes. Nature 453: 56–64 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kidd JM, Graves T, Newman TL, Fulton R, Hayden HS, Malig M, Kallicki J, Kaul R, Wilson RK, Eichler EE 2010. A human genome structural variation sequencing resource reveals insights into mutational mechanisms. Cell 143: 837–847 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Landt SG, Marinov GK, Kundaje A, Kheradpour P, Pauli F, Batzoglou S, Bernstein BE, Bickel P, Brown JB, Cayting P, et al. 2012. ChIP-seq guidelines and practices of the ENCODE and modENCODE consortia. Genome Res 22: 1813–1831 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee B-K, Iyer VR 2012. Genome-wide studies of CCCTC-binding factor (CTCF) and cohesin provide insight into chromatin structure and regulation. J Biol Chem 287: 30906–30913 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lemmers RJLF, Wohlgemuth M, van der Gaag KJ, van der Vliet PJ, van Teijlingen CMM, de Knijff P, Padberg GW, Frants RR, van der Maarel SM 2007. Specific sequence variations within the 4q35 region are associated with facioscapulohumeral muscular dystrophy. Am J Hum Genet 81: 884–894 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Durbin R 2009. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25: 1754–1760 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Linardopoulou E 2001. Transcriptional activity of multiple copies of a subtelomerically located olfactory receptor gene that is polymorphic in number and location. Hum Mol Genet 10: 2373–2383 [DOI] [PubMed] [Google Scholar]
- Linardopoulou EV, Williams EM, Fan Y, Friedman C, Young JM, Trask BJ 2005. Human subtelomeres are hot spots of interchromosomal recombination and segmental duplication. Nature 437: 94–100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Linardopoulou EV, Parghi SS, Friedman C, Osborn GE, Parkhurst SM, Trask BJ 2007. Human subtelomeric WASH genes encode a new subclass of the WASP family. PLoS Genet 3: e237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu F, Tsai K, Chen H-S, Wikramasinghe P, Davuluri RV, Showe L, Domsic J, Marmorstein R, Lieberman PM 2012. Identification of host-chromosome binding sites and candidate gene targets for Kaposi’s sarcoma-associated herpesvirus LANA. J Virol 86: 5752–5762 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mefford HC, Trask BJ 2002. The complex structure and dynamic evolution of human subtelomeres. Nat Rev Genet 3: 91–102 [DOI] [PubMed] [Google Scholar]
- Meier A, Fiegler H, Muñoz P, Ellis P, Rigler D, Langford C, Blasco MA, Carter N, Jackson SP 2007. Spreading of mammalian DNA-damage response factors studied by ChIP-chip at damaged telomeres. EMBO J 26: 2707–2718 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merkenschlager M, Odom DT 2013. CTCF and cohesin: linking gene regulatory elements with their targets. Cell 152: 1285–1297 [DOI] [PubMed] [Google Scholar]
- Nasmyth K, Haering CH 2005. The structure and function of SMC and kleisin complexes. Annu Rev Biochem 74: 595–648 [DOI] [PubMed] [Google Scholar]
- Nergadze SG, Farnung BO, Wischnewski H, Khoriauli L, Vitelli V, Chawla R, Giulotto E, Azzalin CM 2009. CpG-island promoters drive transcription of human telomeres. RNA 15: 2186–2194 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohlsson R, Lobanenkov V, Klenova E 2010. Does CTCF mediate between nuclear organization and gene expression? Bioessays 32: 37–50 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ottaviani A, Rival-Gervier S, Boussouar A, Foerster AM, Rondier D, Sacconi S, Desnuelle C, Gilson E, Magdinier F 2009a. The D4Z4 macrosatellite repeat acts as a CTCF and A-type lamins-dependent insulator in facio-scapulo-humeral dystrophy. PLoS Genet 5: e1000394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ottaviani A, Schluth-Bolard C, Rival-Gervier S, Boussouar A, Rondier D, Foerster AM, Morere J, Bauwens S, Gazzo S, Callet-Bauchu E, et al. 2009b. Identification of a perinuclear positioning element in human subtelomeres that requires A-type lamins and CTCF. EMBO J 28: 2428–2436 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ottaviani A, Schluth-Bolard C, Gilson E, Magdinier F 2011. D4Z4 as a prototype of CTCF and lamins-dependent insulator in human cells. Nucleus 1: 30–36 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palm W, de Lange T 2008. How shelterin protects mammalian telomeres. Annu Rev Genet 42: 301–334 [DOI] [PubMed] [Google Scholar]
- Parelho V, Hadjur S, Spivakov M, Leleu M, Sauer S, Gregson HC, Jarmuz A, Canzonetta C, Webster Z, Nesterova T, et al. 2008. Cohesins functionally associate with CTCF on mammalian chromosome arms. Cell 132: 422–433 [DOI] [PubMed] [Google Scholar]
- Phillips JE, Corces VG 2009. CTCF: master weaver of the genome. Cell 137: 1194–1211 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Porro A, Feuerhahn S, Reichenbach P, Lingner J 2010. Molecular dissection of TERRA biogenesis unveils the presence of distinct and multiple regulatory pathways. Mol Cell Biol 30: 4808–4817 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pruitt KD, Tatusova T, Brown GR, Maglott DR 2012. NCBI reference sequences (RefSeq): current status, new features and genome annotation policy. Nucleic Acids Res 40: D130–D135 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quinlan AR, Hall IM 2010. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics 26: 841–842 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Remeseiro S, Cuadrado A, Carretero M, Martínez P, Drosopoulos WC, Cañamero M, Schildkraut CL, Blasco MA, Losada A 2012. Cohesin-SA1 deficiency drives aneuploidy and tumourigenesis in mice due to impaired replication of telomeres. EMBO J 31: 2076–2089 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riethman H 2008a. Human subtelomeric copy number variations. Cytogenet Genome Res 123: 244–252 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riethman H 2008b. Human telomere structure and biology. Annu Rev Genomics Hum Genet 9: 1–19 [DOI] [PubMed] [Google Scholar]
- Riethman H, Ambrosini A, Castaneda C, Finklestein J, Hu X-L, Mudunuri U, Paul S, Wei J 2004. Mapping and initial analysis of human subtelomeric sequence assemblies. Genome Res 14: 18–28 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rubio ED, Reiss DJ, Welcsh PL, Disteche CM, Filippova GN, Baliga NS, Aebersold R, Ranish JA, Krumm A 2008. CTCF physically links cohesin to chromatin. Proc Natl Acad Sci 105: 8309–8314 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rudd MK, Friedman C, Parghi SS, Linardopoulou EV, Hsu L, Trask BJ 2007. Elevated rates of sister chromatid exchange at chromosome ends. PLoS Genet 3: e32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rumble SM, Lacroute P, Dalca AV, Fiume M, Sidow A, Brudno M 2009. SHRiMP: accurate mapping of short color-space reads. PLoS Comput Biol 5: e1000386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sahin E, Depinho RA 2010. Linking functional decline of telomeres, mitochondria and stem cells during ageing. Nature 464: 520–528 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schoeftner S, Blasco MA 2008. Developmentally regulated transcription of mammalian telomeres by DNA-dependent RNA polymerase II. Nat Cell Biol 10: 228–236 [DOI] [PubMed] [Google Scholar]
- Simonet T, Zaragosi L-E, Philippe C, Lebrigand K, Schouteden C, Augereau A, Bauwens S, Ye J, Santagostino M, Giulotto E, et al. 2011. The human TTAGGG repeat factors 1 and 2 bind to a subset of interstitial telomeric sequences and satellite repeats. Cell Res 21: 1028–1038 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smit AFA, Hubley R, Green P 1996. RepeatMasker Open-3.0. http://www.repeatmasker.org
- Stedman W, Kang H, Lin S, Kissil JL, Bartolomei MS, Lieberman PM 2008. Cohesins localize with CTCF at the KSHV latency control region and at cellular c-myc and H19/Igf2 insulators. EMBO J 27: 654–666 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J, Huda A, Lunyak VV, Jordan IK 2010. A Gibbs sampling strategy applied to the mapping of ambiguous short-sequence tags. Bioinformatics 26: 2501–2508 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H, Maurano MT, Qu H, Varley KE, Gertz J, Pauli F, Lee K, Canfield T, Weaver M, Sandstrom R, et al. 2012. Widespread plasticity in CTCF occupancy linked to DNA methylation. Genome Res 22: 1680–1688 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wendt KS, Yoshida K, Itoh T, Bando M, Koch B, Schirghuber E, Tsutsumi S, Nagae G, Ishihara K, Mishiro T, et al. 2008. Cohesin mediates transcriptional insulation by CCCTC-binding factor. Nature 451: 796–801 [DOI] [PubMed] [Google Scholar]
- Wheelan SJ, Church DM, Ostell JM 2001. Spidey: a tool for mRNA-to-genomic alignments. Genome Res 11: 1952–1957 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang D, Xiong Y, Kim H, He Q, Li Y, Chen R, Songyang Z 2011. Human telomeric proteins occupy selective interstitial sites. Cell Res 21: 1013–1027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yehezkel S, Segev Y, Viegas-Péquignot E, Skorecki K, Selig S 2008. Hypomethylation of subtelomeric regions in ICF syndrome is associated with abnormally short telomeres and enhanced transcription from telomeric regions. Hum Mol Genet 17: 2776–2789 [DOI] [PubMed] [Google Scholar]
- Youngman S, Bates GP, Williams S, McClatchey AI, Baxendale S, Sedlacek Z, Altherr M, Wasmuth JJ, MacDonald ME, Gusella JF, et al. 1992. The telomeric 60 kb of chromosome arm 4p is homologous to telomeric regions on 13p, 15p, 21p, and 22p. Genomics 14: 350–356 [DOI] [PubMed] [Google Scholar]
- Zhang Y, Liu T, Meyer CA, Eeckhoute J, Johnson DS, Bernstein BE, Nusbaum C, Myers RM, Brown M, Li W, et al. 2008. Model-based analysis of ChIP-Seq (MACS). Genome Biol 9: R137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou J, Chau CM, Deng Z, Shiekhattar R, Spindler M-P, Schepers A, Lieberman PM 2005. Cell cycle regulation of chromatin at an origin of DNA replication. EMBO J 24: 1406–1417 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zou Y, Sfeir A, Gryaznov SM, Shay JW, Wright WE 2004. Does a sentinel or a subset of short telomeres determine replicative senescence? Mol Biol Cell 15: 3709–3718 [DOI] [PMC free article] [PubMed] [Google Scholar]