

Abstract
Signal transducer and activator of transcription 3 (Stat3) is constitutively activated in a number of human cancers and cancer cell lines. Via its Src homology 2 (SH2) domain, Stat3 is recruited to phosphotyrosine residues on intracellular domains of cytokine and growth factor receptors, whereupon it is phosphorylated on Tyr705, dimerizes, translocates to the nucleus and is reported to participate in the expression of genes related to angiogenesis, metastasis, growth and survival. To block this process, we are developing cell-permeable, phosphatase-stable phosphopeptide mimics, targeted to the SH2 domain of Stat3, that inhibit the phosphorylation of Tyr705 of Stat3 in cultured tumor cells (Mandal et al., J. Med. Chem. 54, 3549–5463, 2011). At concentrations that inhibit tyrosine phosphorylation, these materials were not cytotoxic, similar to recent reports on JAK inhibitors. At higher concentrations, cytotoxicity was accompanied by off-target effects. We report that treatment of MDA-MB-468 human breast cancer xenografts in mice with peptidomimetic PM-73G significantly inhibited tumor growth, which was accompanied by reduction in VEGF production and microvessel density. No evidence of apoptosis or changes in the expression of the canonical genes cyclin D1 or survivin were observed. Thus selective inhibition of Stat3 Tyr705 phosphorylation may be a novel anti-angiogenesis strategy for the treatment of cancer.
INTRODUCTION
Signal transducer and activator of transcription 3 (Stat3) has received considerable attention due to reports that it is constitutively activated in a number of human cancers and cancer cell lines (reviewed in 1–4). Stat3 was originally described as the acute phase response factor in the innate immune response, mediating signals from interleukin-6 (IL-6) and its receptor (5). Subsequently it was reported to transmit signals from growth factor receptors such as those of epidermal growth factor (EGF), vascular endothelial growth factor (VEGF), as well as other IL-6-famly cytokines (reviewed in 2, 3). In the canonical mechanism, cytokine or growth factor binding leads to tyrosine phosphorylation on the intracellular regions of the corresponding receptor and Stat3 is recruited to these sites via its Src homology 2 (SH2) domain. Associated Janus kinases (JAK), Src kinase, or the kinase activity of the receptor phosphorylates Stat3 on Tyr705 (pStat3), termed activation. pStat3 dimerizes via reciprocal SH2 domain-pTyr705 interactions and is translocated to the nucleus where it participates in the transcription of a variety of genes. Antisense, dominant negative, siRNA, and decoy oligonucleotides targeting Stat3 have been reported to induce apoptosis in tumor cell lines. Taken together, these results support the hypothesis that Tyr705 phosphorylation of Stat3 plays a major role in proliferation, cell cycling, angiogenesis, and survival of cancer cells.
Recent reports, however, have described alternative activities of Stat3. Unphosphorylated Stat3 (U-Stat3) complexes with unphosphorylated NF-κB resulting in the transcription of κB-dependent genes.(6) In non-transcriptional roles, Ser727-phosphorylated Stat3 has been found in electron transport complexes in mitochondria (7) and in this capacity supports the growth of Ras transformed cells by sustaining glycolytic and oxidative phosphorylation (8). Thus, the reported cytotoxicity cited above and alterations in gene transcription ensuing from Stat3 knockdown and dominant-negative over-expression may, in part, be due to mechanisms not related to pTyr705-driven transcription. Highly potent and selective inhibitors of Stat3 Tyr705 phosphorylation are needed both to better understand the role(s) of Tyr705 phosphorylation in cancer cell growth and as leads for drug development. Our strategy is to develop phosphopeptide mimetics targeting the SH2 domain to block recruitment to receptors, thereby preventing phosphorylation and subsequent transcriptional activity.
SH2 domains recognize pTyr and the first 2–5 residues to its C-terminus and are involved in protein complexes that transmit signals from growth factors, cytokines, and integrins (reviewed in 9). SH2 domains are involved in several dysregulated signaling pathways that contribute to the cancer phenotype including the PI3K/Akt axis, Bcr-Abl and its downstream effectors, as well as the JAK (or Src)-STAT pathways. Since the discovery of this target in the early 1990’s research groups from industry, academia, and government laboratories have developed phosphotyrosyl peptide-based inhibitors of a variety of SH2 domains (reviewed in 10–16). Due to the difficulties in delivering compounds with the required high negative charge density of a phosphate to cells and tissues, the pharmaceutical industry abandoned this supposedly undruggable target.
We and others (17–30) have been developing phosphopeptide mimetics targeting the SH2 domain of Stat3 with the goal of blocking recruitment to cytokine or growth factor receptors which would lead to inhibition of Tyr705 phosphorylation and subsequent Stat3 dimerization, nuclear translocation and transcriptional activity. Starting with Ac-pTyr-Leu-Pro-Gln-Thr-Val-NH2, which contains the Tyr-Xaa-Yaa-Gln Stat3 recognition motif (31, 32), we have studied peptide-protein interactions with both structure-affinity relationship (33–37) and molecular modeling approaches (38–40). This work has led to the development of phosphatase-stable, cell-permeable prodrugs that inhibit both constitutive and IL-6-stimulated Tyr705 phosphorylation in a variety of tumor cell lines (41, 42). We recently reported the design and synthesis of a highly selective and potent phosphopeptidomimetic prodrug, PM-73G (Figure 1) (41). In this inhibitor, glutamine was replaced with Apa (4-aminopentamide), Leu-Pro was substituted with the tricyclic mimic Haic ((2S,5S)-5-amino-1,2,4,5,6,7-hexahydro-4-oxo-azepino[3,2,1-hi]indole-2-carboxylic acid), and phosphotyrosine was replaced with 4-phosphoryldifluoromethyl-β-methyl cinnamate. The phosphonodifluoromethyl group was employed to prevent cleavage of the phosphate by phosphatases. To block the negative charges of the phosphonate oxygens which prevent cell penetration (41), esterase-labile pivaloyloxymethyl (POM) groups were added. As we reported (41) in MDA-MB-468 breast cancer cells in vitro, PM-73G inhibited constitutive phosphorylation of Stat3 with an IC50 of 100–500 nM. As a measure of selectivity, at 5 μM, the prodrug did not inhibit the constitutive phosphorylation of Akt or Tyr861 of focal adhesion kinase (FAK), or EGF-stimulated phosphorylation of Stat5, processes that rely on the SH2 domains of p85, Src, and Stat5, respectively. Of note, at this concentration PM-73G was not cytotoxic to either MDA-MB-468 (breast), HCC-827 (lung), or SKOV3-ip (ovarian) cells, which all harbor constitutively active pStat3. However, at the higher concentration of 25 μM, cytotoxicity was noted with concomitant reduction in Akt and FAK phosphorylation, suggesting off-target effects. These results call into question the role of Stat3 phosphorylation in tumor cell viability both in vitro and in vivo.
Figure 1.

The chemical structure of PM-73G, the cell-permeable, phosphatase-stable phosphopeptide mimic prodrug that targets the SH2 domain of Stat3.
In this communication, we demonstrate that treatment with PM-73G, as a single agent, inhibits the growth of orthotopic MDA-MB-468 human breast tumor xenografts in nude mice. Inhibition of Stat3 phosphorylation and VEGF expression were observed, along with reductions in CD31/microvessel density. However, no inhibition of either cyclin D1 or survivin was noted and no apoptosis was observed, suggesting that these are indeed not down-stream targets regulated by pStat3.
RESULTS
PM-73G inhibits Stat3 phosphorylation in breast tumor xenografts in nude mice
To initially asses the ability of the prodrug to inhibit phosphorylation of Stat3 in vivo, established orthotopic MDA-MB-468 breast tumor xenografts (~100 mm3) in female nude mice were treated intratumorally (i.t.) with 100 microliters of PM-73G, formulated in 20% hydroxypropyl-β-cyclodextrin (Trappsol) in PBS to facilitate solubility. Pilot studies had demonstrated that pStat3 levels were quenched following i.t. injections of 5 mM PM-73G, its highest level of aqueous solubility (data not shown). To establish a context for subsequent systemic administration of PM-73G, we employed a dose-de-escalating approach with i.t. administration to determine whether lower tumor tissue levels that might be achievable systemically were also effective. Concentrations of PM-73G in the injectate ranged from 5 mM to as low as 0.3 μM. Tumors were harvested at 2 hr post injection and PM-73G levels were assessed by immunohistochemistry. An injectate concentration of 8 μM was found to result in significant inhibition (Figure 2).
Figure 2.

Intratumoral administration of PM-73G inhibits Tyr705 phosphorylation of Stat3 in vivo. Established tumors were injected intratumorally with decreasing doses of PM73G in Trappsol and two hr later tumors were harvested, preserved in formalin, sectioned, and pStat3 was monitored with immunohistochemistry.
PM-73G inhibits growth of breast tumor xenografts in nude mice
Having demonstrated that PM-73G could inhibit Stat3 phosphorylation in tumors via i.t. injection, the prodrug was next evaluated for its ability to inhibit tumor growth in orthotopic MDA-MB-468 xenografts. Mice were injected in the mammary fat pad with MDA-MB-468 cells, and after 15 days they were separated into three groups, all receiving i.t. injections. Group 1 received PBS, group 2 received 20% Trappsol in PBS and group 3 received 5 mM PM-73G in 20% Trappsol/PBS (100 μL injections). On day 1, mice were started on daily treatment for 5 days, then given 2 days of rest; they were treated again for 5 days, and given 2 days rest. On day 15, mice were treated again and 2 hours later tumors were harvested and divided into two parts. The first part was frozen and the other was fixed in formalin for immunohistochemistry.
Average tumor volumes increased ~2.3- and ~2.5-fold over 15 days in the PBS and Trappsol/PBS vehicle control groups, respectively. However, tumors treated with PM-73G increased only ~1.6-fold in volume (Figure 3). In addition, PM-73G strongly inhibited the phosphorylation of Stat3 in treated tumors, as compared to vehicle controls (Figure 4, 3rd Row). There was no apparent difference in CD31 staining in the tumor tissue isolated from mice injected with PBS compared with Trappsol/PBS vehicle, suggesting that the Trappsol vehicle alone had no effect on angiogenesis during growth of the xenografts. In contrast, tumors treated with PM-73G were avascular, with a significant decrease in the number and size of microvessels, as determined by the lack of CD31 staining (Figure 4, 1st Row). These results suggested that PM-73G has anti-angiogenic activity, targeting angiogenic growth factors such as VEGF A (Figure 4, 2nd Row), known to be stimulated through Stat3 signaling pathways (43).
Figure 3.
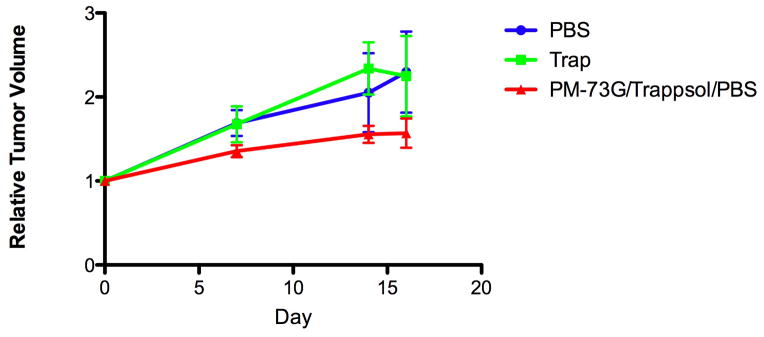
Inhibition of growth of established MDA-MB-468 tumor xenografts with i.t. administration of 5 mM PM-73G.
Figure 4.

Intratumoral treatment with PM-73G inhibits microvessel density, VEGF expression, and phosphorylation of Tyr705 of Stat3.
Immunohistochemical analysis showed no reduction in cyclin D1 or survivin with no evidence of apoptosis (data not shown). Thus, inhibition of Stat3 phosphorylation had no effect on expression of the “canonical” downstream genes, which is consistent with our in vitro results (41).
The next phase of the in vivo studies was undertaken to determine whether systemic administration of PM-73G would result in an anti-tumor response. Using the results of the dose-response characteristics of PM-73G in blocking Stat3 phosphorylation following i.t. administration as a guide, a pilot efficacy study using intraperitoneal (i.p.) administration of PM-73G was conducted. Mice with MDA-MB-468 tumor xenografts were treated i.p. with 170 mg/kg PM-73G in the same vehicle as above, according to a schedule of 2 weekly cycles of 5-days-per week injection, followed by a last injection three days later to allow for assessment of labile pStat3 inhibition. Tumor volumes of mice treated with vehicle (n=3) increase an average of 2.5-fold, whereas those treated with PM-73G (n=2) increased 1.5-fold. The experiment was repeated for 4 weekly cycles with the same dose (treated, n=8; vehicle control, n=6) again followed by a last injection three days later to allow for assessment of labile pStat3 inhibition. Tumor volumes of mice treated with vehicle increased an average of 3.3-fold, whereas those treated with PM-73G increased 2.1-fold (Figure 5). Immunohistochemical analyses verified that pStat3 levels in tumors were reduced in the PM-73G treated arm, as was CD31/MVD (Figure 6). Full necroscopy examination of organs harvested after this treatment schedule did not reveal any toxicity. Complete blood counts were normal for both treatment and control groups. Treated animals exhibited no weight loss or altered behavioral characteristics. Taken together, these observations indicate that systemic monotherapy with PM-73G is both highly efficacious and apparently not toxic on this schedule.
Figure 5.
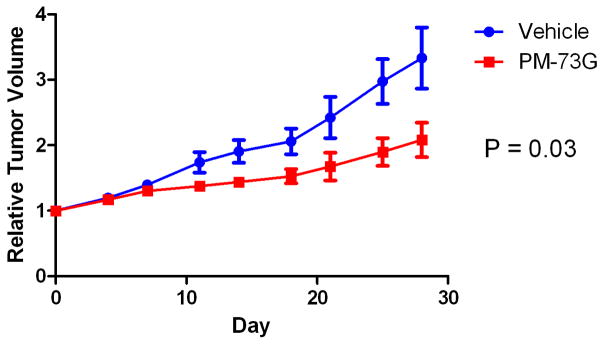
Inhibition of growth of MDA-MB-468 tumor xenografts by i.p. treatment with PM-73G.
Figure 6.

Inhibition of Stat3 phosphorylation and microvessel density in MDA-MB-468 xenografts treated with PM-73G i.p.
DISCUSSION
Targeting SH2 domains is challenging in that the targets recognize highly negatively charged phosphopeptides which prevents diffusion of phosphopeptide-based inhibitors through cell membranes. Furthermore, phosphatase cleavage of phosphates renders ligands unrecognizable by SH2 domains. Based on rationale that takes these constraints into account, we developed a lead candidate that embodies a novel combination of structural features. In addition to mimicking the Tyr-Xaa-Yaa-Gln recognition motif for Stat3 (31), PM-73G possesses the non-hydrolyzable difluoromethylphosphonate group, and the esterase-labile POM groups block the negative charges thus allowing passive diffusion across cell membranes (41, 42). Although other phosphopeptide-based inhibitors incorporating free phosphate surrogates (44–46) or bioreversible esters (47–52) have been reported to inhibit their targets in cells, PM-73G is the first example of a phosphopeptide targeted to an SH2 domain that inhibits its target in vivo following systemic administration. These results suggest that SH2 domains are indeed druggable.
There are three known activities of Stat3. Ser727-phosphorylated Stat3 has been found in electron transport complexes in mitochondria (7, 8). In the nucleus, unphosphorylated Stat3 interacts with NF-κB and is involved with gene transcription (6). The canonical Tyr705 phosphorylation mechanism has been reported to result in the transcription of several “canonical” downstream genes: Bcl2-family proteins, Cyclin D1, survivin, and others (reviewed in (2, 3, 53, 54). Perturbation of Stat3 levels with antisense RNA, dominant negative, or siRNA techniques led to apoptosis in cultured tumor cells. These knockdown methods are unable to discriminate between “non-pTyr705” and the pTyr705 activities because they deplete STAT3 from the cell. Our findings demonstrate that selective inhibition of Tyr705 phosphorylation either in vitro (41) or in vivo does not result in apoptosis nor in inhibition of the expression of canonical downstream genes. This suggests that mitochondrial or unphosphorylated Stat3, or an as yet undiscovered activity, is essential for tumor cell survival.
Recent reports show that inhibition of JAK2 kinase activity with pyridone P6 (melanoma) (55), AZD 1840 (breast, ovarian, prostate) (56, 57) and ruxolitinib (lung) (58) is also not cytotoxic to tumor cells of epithelial origin in culture at concentrations that inhibit Tyr705 phosphorylation. These reports and our in vitro results suggest that 1, these models do not depend on Stat3 phosphorylation for survival and 2, if a compound inhibits Stat3 phosphorylation, and it results in cell death, the cytotoxicity is due to perturbations of off-target pathways.
Despite the lack of in vitro cytotoxicity, the results of evaluation of the effects of in vivo, intratumoral and intraperitoneal administration of PM-73G to mice bearing MDA-MB-468 tumor xenografts demonstrate significant inhibition of tumor growth. The reduction in tumor volume was accompanied by decreased microvessel density (MVD) as well as a reduction in VEGF protein production. Thus, in human tumor xenografts, VEGF production and angiogenesis appear to depend on Stat3 Tyr705 phosphorylation. Stat3 has previously been reported to both transmit signals from and to regulate the expression of VEGF (reviewed in 43). CHIP analysis indicated that there is a Stat3 binding site on the VEGF promoter (59, 60). JAK inhibitor AZD1840 also reduced growth of human tumor xenografts, accompanied by reduction in angiogenesis. Taken together, it appears that angiogenesis is dependent on Stat3 Tyr705 phosphorylation. We propose that SH2 domain-targeted Stat3 inhibition is a potential new anti-angiogenesis strategy.
MATERIALS AND METHODS
Reagents
PM-73G was synthesized and purity verified as described by Mandal et al. (41)
Cell lines
The triple-negative phenotype MDA-MB-468 human mammary adenocarcinoma cell line was from the American Type Tissue Collection (ATCC, Manassas, VA, U.S.A.).
Tumor Model
MDA-MB-468 cells in log-phase growth were harvested and single cell suspensions prepared. Two million cells in 100 mliters of matrigel were injected orthotopically in one of the lower right fat pads of 6–8 week old female nude mice. Once tumors grew to ~ 70–110 mm3 volume (V = w x l x w/2), the mice were distributed into groups with approximately equivalent average tumor volumes and treatments were initiated.
Pilot Toxicity Study
Female nude mice (Harlan Laboratories; 4–5 weeks of age upon arrival) were injected intraperitoneally (i.p.) with either 50, 150 or 500 μl of a 10 mM solution of PM-73G dissolved in PBS/Trappsol. Injections were administered daily for 4 consecutive days, the mice were rested for 2 days, and injections resumed again for 5 consecutive days. Bodyweights were taken on every day of injection, and finally at 3 days after the last injection.
Anti-tumor Efficacy Studies
Preparation of PM-73G solutions
Due to the stability characteristics of PM-73G, solutions were freshly prepared on each day of treatment. The drug was dissolved in 20% Trappsol/PBS at maximum apparent solubility, ~ 5 mM, with vortexing to enhance the rate of dissolution and minimize the time prior to injection.
Intra-tumoral (i.t.) injections
In initial studies with both therapeutic intent and for biomarker evaluation, mice with orthotopic MDA-MB-468 xenografts were injected i.t. with 50 μliters of 10 mM PM-73G (containing 425 μg of prodrug), and control mice received equivalent volumes of Trappsol/PBS or PBS alone. Injections were repeated daily for 5 days, mice were rested for 2 days, and the set of 5 injections were repeated. Tumor measurements were made prior to the first and sixth injections, three days after the tenth injection, and then again prior to the final injection. Due to the transient nature of STAT3 phosphorylation, a final injection of PM73G was made 3 days after the tenth injection, and mice were sacrificed within 1 hr later. Tumors were excised and placed in formalin or OCT for subsequent immunohistochemical analyses.
To further examine the dose-response effect on pSTAT3 levels, tumor bearing mice were given 100 μL i.t. injections of either 200 μM, 40 μM, 8 μM, 1.6 μM, or 0.3 μM PM-73G. Tumors were harvested 1.5 hr post-injection, and pStat3 levels analyzed by immunohistochemistry
Intra-peritoneal (i.p.) injections
In an initial study based on the pilot toxicity study that demonstrated that PM-73G was well-tolerated, mice bearing MDA-MB-468 tumors were injected i.p. with 1 ml of a 5 mM solution of PM-73G dissolved in PBS/Trappsol. Injections were repeated daily for 5 consecutive days, the mice were rested for 2 days, and injections resumed again for 5 consecutive days, followed by a final injection 3 days later and subsequent tumor harvest on that day. Tumor measurements were made prior to the first, fifth, sixth and tenth injections, and then again two days prior to and on the day of the final (11th) injection, five days later. Harvested tumors were analyzed by IHC for CD31, and pStat3 expression.
In an expanded anti-tumor efficacy and pilot toxicology study with systemic administration, the protocol was modified to allow four weeks of five-days-per-week of i.p. injections, followed by a last injection three days later. Two hr later, the groups of mice were split and tumors prepared for immunohistochemical analyses or the intact mice sent for histopathological analyses. A subset of mice from the PM-73G treatment group was given either PBS/Trappsol or PM-73G on the last injection day and then sacrificed 2 hr later and prepared for immunohistochemical and histopathological analyses, as before; this would allow determination of chronic effects (four weeks) of PM-73G, either four days or 2 hr after the last injection. In addition, the control mice were split and either given a final injection of PBS/Trappsol or PM-73G and sacrificed 2 hr later for immunohistochemical analyses. Each tumor was measured and its percentage growth was tabulated. For each group (treated, n = 8; and vehicle, n = 6) the mean and standard error of the mean of the percentage growth was plotted for each time point. The unpaired Student’s t test was used to calculate the significance of the difference in growth.
Acknowledgments
This work was supported by the National Cancer Institute (CA096652, JSM, PKM, JK) and the CTT/TI-3D Chemistry & Molecularly-Targeted Therapeutic Development Grant Program (JSM, FAM). We also acknowledge the NCI Cancer Center Support Grant CA016672 for the support of our NMR facility, the Research Histology Laboratory, and the Translational Chemistry Core Facility (mass spectrometry).
References
- 1.Costantino L, Barlocco D. STAT 3 as a Target for Cancer Drug Discovery. Curr Med Chem. 2008;15:834–843. doi: 10.2174/092986708783955464. [DOI] [PubMed] [Google Scholar]
- 2.Darnell JE., Jr Transcription factors as targets for cancer therapy. Nat Rev Cancer. 2002;2:740–749. doi: 10.1038/nrc906. [DOI] [PubMed] [Google Scholar]
- 3.Yu H, Jove R. The STATs of cancer--new molecular targets come of age. Nat Rev Cancer. 2004;4:97–105. doi: 10.1038/nrc1275. [DOI] [PubMed] [Google Scholar]
- 4.Yue P, Turkson J. Targeting STAT3 in cancer: how successful are we? Expert Opin Investig Drugs. 2009;18:45–56. doi: 10.1517/13543780802565791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Akira S, Nishio Y, Inoue M, Wang XJ, Wei S, Matsusaka T, Yoshida K, Sudo T, Naruto M, Kishimoto T. Molecular cloning of APRF, a novel IFN-stimulated gene factor 3 p91-related transcription factor involved in the gp130-mediated signaling pathway. Cell. 1994;77:63–71. doi: 10.1016/0092-8674(94)90235-6. [DOI] [PubMed] [Google Scholar]
- 6.Yang J, Stark GR. Roles of unphosphorylated STATs in signaling. Cell Res. 2008;18:443–451. doi: 10.1038/cr.2008.41. [DOI] [PubMed] [Google Scholar]
- 7.Wegrzyn J, Potla R, Chwae YJ, Sepuri NB, Zhang Q, Koeck T, Derecka M, Szczepanek K, Szelag M, Gornicka A, Moh A, Moghaddas S, Chen Q, Bobbili S, Cichy J, Dulak J, Baker DP, Wolfman A, Stuehr D, Hassan MO, Fu XY, Avadhani N, Drake JI, Fawcett P, Lesnefsky EJ, Larner AC. Function of mitochondrial Stat3 in cellular respiration. Science. 2009;323:793–797. doi: 10.1126/science.1164551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gough DJ, Corlett A, Schlessinger K, Wegrzyn J, Larner AC, Levy DE. Mitochondrial STAT3 supports Ras-dependent oncogenic transformation. Science. 2009;324:1713–1716. doi: 10.1126/science.1171721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Waksman G, Kumaran S, Lubman O. SH2 domains: role, structure and implications for molecular medicine. Expert Reviews in Molecular Medicine. 2004;6:1–18. doi: 10.1017/S1462399404007331. [DOI] [PubMed] [Google Scholar]
- 10.Sawyer TK. Src homology-2 domains: structure, mechanisms, and drug discovery. Biopolymers. 1998;47:243–261. doi: 10.1002/(SICI)1097-0282(1998)47:3<243::AID-BIP4>3.0.CO;2-P. [DOI] [PubMed] [Google Scholar]
- 11.Cody WL, Lin Z, Panek RL, Rose DW, Rubin JR. Progress in the development of inhibitors of SH2 domains. Curr Pharm Des. 2000;6:59–98. doi: 10.2174/1381612003401532. [DOI] [PubMed] [Google Scholar]
- 12.Muller G. Peptidomimetic SH2 domain antagonists for targeting signal transduction. Topics in Current Chemistry. 2001;211:17–59. [Google Scholar]
- 13.Shakespeare WC. SH2 domain inhibition: a problem solved? Curr Opin Chem Biol. 2001;5:409–415. doi: 10.1016/s1367-5931(00)00222-2. [DOI] [PubMed] [Google Scholar]
- 14.Metcalf CA, III, Sawyer T. Src homology-2 domains and structure-based, small-molecule library approaches to drug discovery. In: Makriyannis A, Biegel D, editors. Drug Discovery Strategies and Methods. Marcel Dekker, Inc; New York, NY: 2004. pp. 23–59. [Google Scholar]
- 15.Garcia-Echeverria C. Antagonists of the homology 2 (SH2) domains of Grb2, Src, Lck and ZAP-70. Curr Med Chem. 2001;8:1589–1604. doi: 10.2174/0929867013371905. [DOI] [PubMed] [Google Scholar]
- 16.Garcia-Echeverria C. Inhibitors of signaling interfaces: targeting Src homology 2 domains in drug discovery. Protein Tyrosine Kinases. 2006:31–52. [Google Scholar]
- 17.Fletcher S, Singh J, Zhang X, Yue P, Page BD, Sharmeen S, Shahani VM, Zhao W, Schimmer AD, Turkson J, Gunning PT. Disruption of transcriptionally active Stat3 dimers with non-phosphorylated, salicylic acid-based small molecules: potent in vitro and tumor cell activities. Chembiochem. 2009;10:1959–1964. doi: 10.1002/cbic.200900172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gunning PT, Katt WP, Glenn M, Siddiquee K, Kim JS, Jove R, Sebti SM, Turkson J, Hamilton AD. Isoform selective inhibition of STAT1 or STAT3 homo-dimerization via peptidomimetic probes: structural recognition of STAT SH2 domains. Bioorg Med Chem Lett. 2007;17:1875–1878. doi: 10.1016/j.bmcl.2007.01.077. [DOI] [PubMed] [Google Scholar]
- 19.Shahani VM, Yue P, Fletcher S, Sharmeen S, Sukhai MA, Luu DP, Zhang X, Sun H, Zhao W, Schimmer AD, Turkson J, Gunning PT. Design, synthesis, and in vitro characterization of novel hybrid peptidomimetic inhibitors of STAT3 protein. Bioorg Med Chem. 2011;19:1823–1838. doi: 10.1016/j.bmc.2010.12.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Shahani VM, Yue P, Haftchenary S, Zhao W, Lukkarila JL, Zhang X, Ball D, Nona C, Gunning PT, Turkson J. Identification of Purine-Scaffold Small-Molecule Inhibitors of Stat3 Activation by QSAR Studies. ACS Med Chem Lett. 2011;2:79–84. doi: 10.1021/ml100224d. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 21.Siddiquee KA, Gunning PT, Glenn M, Katt WP, Zhang S, Schrock C, Sebti SM, Jove R, Hamilton AD, Turkson J. An oxazole-based small-molecule Stat3 inhibitor modulates Stat3 stability and processing and induces antitumor cell effects. ACS Chem Biol. 2007;2:787–798. doi: 10.1021/cb7001973. [DOI] [PubMed] [Google Scholar]
- 22.Turkson J, Kim JS, Zhang S, Yuan J, Huang M, Glenn M, Haura E, Sebti S, Hamilton AD, Jove R. Novel peptidomimetic inhibitors of signal transducer and activator of transcription 3 dimerization and biological activity. Mol Cancer Ther. 2004;3:261–269. [PubMed] [Google Scholar]
- 23.Turkson J, Ryan D, Kim JS, Zhang Y, Chen Z, Haura E, Laudano A, Sebti S, Hamilton AD, Jove R. Phosphotyrosyl peptides block Stat3-mediated DNA binding activity, gene regulation, and cell transformation. The Journal of biological chemistry. 2001;276:45443–45455. doi: 10.1074/jbc.M107527200. [DOI] [PubMed] [Google Scholar]
- 24.Zhang X, Yue P, Fletcher S, Zhao W, Gunning PT, Turkson J. A novel small-molecule disrupts Stat3 SH2 domain-phosphotyrosine interactions and Stat3-dependent tumor processes. Biochem Pharmacol. 2010;79:1398–1409. doi: 10.1016/j.bcp.2010.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Shao H, Cheng HY, Cook RG, Tweardy DJ. Identification and characterization of signal transducer and activator of transcription 3 recruitment sites within the epidermal growth factor receptor. Cancer research. 2003;63:3923–3930. [PubMed] [Google Scholar]
- 26.Shao H, Xu X, Mastrangelo MA, Jing N, Cook RG, Legge GB, Tweardy DJ. Structural requirements for signal transducer and activator of transcription 3 binding to phosphotyrosine ligands containing the YXXQ motif. The Journal of biological chemistry. 2004;279:18967–18973. doi: 10.1074/jbc.M314037200. [DOI] [PubMed] [Google Scholar]
- 27.Xu X, Kasembeli MM, Jiang X, Tweardy BJ, Tweardy DJ. Chemical probes that competitively and selectively inhibit Stat3 activation. PloS one. 2009;4:e4783. doi: 10.1371/journal.pone.0004783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Chen J, Bai L, Bernard D, Nikolovska-Coleska Z, Gomez C, Zhang J, Yi H, Wang S. Structure-Based Design of Conformationally Constrained, Cell-Permeable STAT3 Inhibitors. ACS Med Chem Lett. 2010;1:85–89. doi: 10.1021/ml100010j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Chen J, Nikolovska-Coleska Z, Yang CY, Gomez C, Gao W, Krajewski K, Jiang S, Roller P, Wang S. Design and synthesis of a new, conformationally constrained, macrocyclic small-molecule inhibitor of STAT3 via ‘click chemistry’. Bioorg Med Chem Lett. 2007;17:3939–3942. doi: 10.1016/j.bmcl.2007.04.096. [DOI] [PubMed] [Google Scholar]
- 30.Dourlat J, Valentin B, Liu WQ, Garbay C. New syntheses of tetrazolylmethylphenylalanine and O-malonyltyrosine as pTyr mimetics for the design of STAT3 dimerization inhibitors. Bioorg Med Chem Lett. 2007;17:3943–3946. doi: 10.1016/j.bmcl.2007.04.107. [DOI] [PubMed] [Google Scholar]
- 31.Stahl N, Farruggella TJ, Boulton TG, Zhong Z, Darnell JE, Jr, Yancopoulos GD. Choice of STATs and other substrates specified by modular tyrosine-based motifs in cytokine receptors. Science. 1995;267:1349–1353. doi: 10.1126/science.7871433. [DOI] [PubMed] [Google Scholar]
- 32.Gerhartz C, Heesel B, Sasse J, Hemmann U, Landgraf C, Schneider-Mergener J, Horn F, Heinrich PC, Graeve L. Differential activation of acute phase response factor/STAT3 and STAT1 via the cytoplasmic domain of the interleukin 6 signal transducer gp130. I Definition of a novel phosphotyrosine motif mediating STAT1 activation. The Journal of biological chemistry. 1996;271:12991–12998. doi: 10.1074/jbc.271.22.12991. [DOI] [PubMed] [Google Scholar]
- 33.Coleman DRI, Kaluarachchi K, Ren Z, Chen X, McMurray JS. Solid phase synthesis of phosphopeptides incorporating 2,2-dimethyloxazolidine pseudoproline analogs: evidence for trans Leu-Pro peptide bonds in Stat3 inhibitors. Int J Pept Res Ther. 2008;14:1–9. [Google Scholar]
- 34.Coleman DRI, Ren Z, Mandal PK, Cameron AG, Dyer GA, Muranjan S, Campbell M, Chen X, McMurray JS. Investigation of the binding determinants of phosphopeptides targeted to the SRC homology 2 domain of the signal transducer and activator of transcription 3. Development of a high-affinity peptide inhibitor. J Med Chem. 2005;48:6661–6670. doi: 10.1021/jm050513m. [DOI] [PubMed] [Google Scholar]
- 35.Mandal PK, Heard PA, Ren Z, Chen X, McMurray JS. Solid-phase synthesis of Stat3 inhibitors incorporating O-carbamoylserine and O-carbamoylthreonine as glutamine mimics. Bioorg Med Chem Lett. 2007;17:654–656. doi: 10.1016/j.bmcl.2006.10.099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Mandal PK, Ren Z, Chen X, Xiong C, McMurray JS. Structure-affinity relationships of glutamine mimics incorporated into phosphopeptides targeted to the SH2 domain of signal transducer and activator of transcription 3. J Med Chem. 2009;52:6126–6141. doi: 10.1021/jm901105k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ren Z, Cabell LA, Schaefer TS, McMurray JS. Identification of a high-affinity phosphopeptide inhibitor of Stat3. Bioorg Med Chem Lett. 2003;13:633–636. doi: 10.1016/s0960-894x(02)01050-8. [DOI] [PubMed] [Google Scholar]
- 38.Mandal PK, Limbrick D, Coleman DR, Dyer GA, Ren Z, Birtwistle JS, Xiong C, Chen X, Briggs JM, McMurray JS. Conformationally constrained peptidomimetic inhibitors of signal transducer and activator of transcription 3: evaluation and molecular modeling. J Med Chem. 2009;52:2429–2442. doi: 10.1021/jm801491w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.McMurray JS. Structural basis for the binding of high affinity phosphopeptides to Stat3. Biopolymers. 2008;90:69–79. doi: 10.1002/bip.20901. [DOI] [PubMed] [Google Scholar]
- 40.Dhanik A, McMurray JS, Kavraki L. On modeling peptidomimetics in complex with the SH2 domain of Stat3. Conf Proc IEEE Eng Med Biol Soc. 2012;2011:3229–3232. doi: 10.1109/IEMBS.2011.6090878. [DOI] [PubMed] [Google Scholar]
- 41.Mandal PK, Gao F, Lu Z, Ren Z, Ramesh R, Birtwistle JS, Kaluarachchi KK, Chen X, Bast RC, Liao WS, McMurray JS. Potent and Selective Phosphopeptide Mimetic Prodrugs Targeted to the Src Homology 2 (SH2) Domain of Signal Transducer and Activator of Transcription 3. J Med Chem. 2011;54:3549–5463. doi: 10.1021/jm2000882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Mandal PK, Liao WS, McMurray JS. Synthesis of phosphatase-stable, cell-permeable peptidomimetic prodrugs that target the SH2 domain of Stat3. Org Lett. 2009;11:3394–3397. doi: 10.1021/ol9012662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Chen Z, Han ZC. STAT3: a critical transcription activator in angiogenesis. Med Res Rev. 2008;28:185–200. doi: 10.1002/med.20101. [DOI] [PubMed] [Google Scholar]
- 44.Atabey N, Gao Y, Yao ZJ, Breckenridge D, Soon L, Soriano JV, Burke TR, Jr, Bottaro DP. Potent blockade of hepatocyte growth factor-stimulated cell motility, matrix invasion and branching morphogenesis by antagonists of Grb2 Src homology 2 domain interactions. The Journal of biological chemistry. 2001;276:14308–14314. doi: 10.1074/jbc.M010202200. [DOI] [PubMed] [Google Scholar]
- 45.Giubellino A, Gao Y, Lee S, Lee MJ, Vasselli JR, Medepalli S, Trepel JB, Burke TR, Jr, Bottaro DP. Inhibition of tumor metastasis by a growth factor receptor bound protein 2 Src homology 2 domain-binding antagonist. Cancer research. 2007;67:6012–6016. doi: 10.1158/0008-5472.CAN-07-0022. [DOI] [PubMed] [Google Scholar]
- 46.Soriano JV, Liu N, Gao Y, Yao ZJ, Ishibashi T, Underhill C, Burke TR, Jr, Bottaro DP. Inhibition of angiogenesis by growth factor receptor bound protein 2-Src homology 2 domain bound antagonists. Mol Cancer Ther. 2004;3:1289–1299. [PubMed] [Google Scholar]
- 47.Garrido-Hernandez H, Moon KD, Geahlen RL, Borch RF. Design and synthesis of phosphotyrosine peptidomimetic prodrugs. J Med Chem. 2006;49:3368–3376. doi: 10.1021/jm060142p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Gay B, Suarez S, Weber C, Rahuel J, Fabbro D, Furet P, Caravatti G, Schoepfer J. Effect of potent and selective inhibitors of the Grb2 SH2 domain on cell motility. The Journal of biological chemistry. 1999;274:23311–23315. doi: 10.1074/jbc.274.33.23311. [DOI] [PubMed] [Google Scholar]
- 49.Gay B, Suarez S, Caravatti G, Furet P, Meyer T, Schoepfer J. Selective GRB2 SH2 inhibitors as anti-Ras therapy. Int J Cancer. 1999;83:235–241. doi: 10.1002/(sici)1097-0215(19991008)83:2<235::aid-ijc15>3.0.co;2-b. [DOI] [PubMed] [Google Scholar]
- 50.Liu WQ, Vidal M, Mathe C, Perigaud C, Garbay C. Inhibition of the ras-dependent mitogenic pathway by phosphopeptide prodrugs with antiproliferative properties. Bioorg Med Chem Lett. 2000;10:669–672. doi: 10.1016/s0960-894x(00)00077-9. [DOI] [PubMed] [Google Scholar]
- 51.Liu WQ, Vidal M, Olszowy C, Million E, Lenoir C, Dhotel H, Garbay C. Structure-activity relationships of small phosphopeptides, inhibitors of Grb2 SH2 domain, and their prodrugs. J Med Chem. 2004;47:1223–1233. doi: 10.1021/jm031005k. [DOI] [PubMed] [Google Scholar]
- 52.Stankovic CJ, Surendran N, Lunney EA, Plummer MS, Para KS, Shahripour A, Fergus JH, Marks JS, Herrera R, Hubbell SE, Humblet C, Saltiel AR, Stewart BH, Sawyer TK. The role of 4-phosphonodifluoromethyl- and 4-phosphono-phenylalanine in the selectivity and cellular uptake of SH2 domain ligands. Bioorg Med Chem Lett. 1997;7:1909–1914. [Google Scholar]
- 53.Bromberg J, Darnell JE., Jr The role of STATs in transcriptional control and their impact on cellular function. Oncogene. 2000;19:2468–2473. doi: 10.1038/sj.onc.1203476. [DOI] [PubMed] [Google Scholar]
- 54.Levy DE, Darnell JE., Jr Stats: transcriptional control and biological impact. Nat Rev Mol Cell Biol. 2002;3:651–662. doi: 10.1038/nrm909. [DOI] [PubMed] [Google Scholar]
- 55.Kreis S, Munz GA, Haan S, Heinrich PC, Behrmann I. Cell density dependent increase of constitutive signal transducers and activators of transcription 3 activity in melanoma cells is mediated by Janus kinases. Mol Cancer Res. 2007;5:1331–1341. doi: 10.1158/1541-7786.MCR-07-0317. [DOI] [PubMed] [Google Scholar]
- 56.Hedvat M, Huszar D, Herrmann A, Gozgit JM, Schroeder A, Sheehy A, Buettner R, Proia D, Kowolik CM, Xin H, Armstrong B, Bebernitz G, Weng S, Wang L, Ye M, McEachern K, Chen H, Morosini D, Bell K, Alimzhanov M, Ioannidis S, McCoon P, Cao ZA, Yu H, Jove R, Zinda M. The JAK2 inhibitor AZD1480 potently blocks Stat3 signaling and oncogenesis in solid tumors. Cancer Cell. 2009;16:487–497. doi: 10.1016/j.ccr.2009.10.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Xin H, Herrmann A, Reckamp K, Zhang W, Pal S, Hedvat M, Zhang C, Liang W, Scuto A, Weng S, Morosini D, Cao ZA, Zinda M, Figlin R, Huszar D, Jove R, Yu H. Antiangiogenic and antimetastatic activity of JAK inhibitor AZD1480. Cancer Res. 2011;71:6601–6610. doi: 10.1158/0008-5472.CAN-11-1217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Looyenga BD, Hutchings D, Cherni I, Kingsley C, Weiss GJ, Mackeigan JP. STAT3 Is Activated by JAK2 Independent of Key Oncogenic Driver Mutations in Non-Small Cell Lung Carcinoma. PLoS One. 2012;7:e30820. doi: 10.1371/journal.pone.0030820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Niu G, Wright KL, Huang M, Song L, Haura E, Turkson J, Zhang S, Wang T, Sinibaldi D, Coppola D, Heller R, Ellis LM, Karras J, Bromberg J, Pardoll D, Jove R, Yu H. Constitutive Stat3 activity up-regulates VEGF expression and tumor angiogenesis. Oncogene. 2002;21:2000–2008. doi: 10.1038/sj.onc.1205260. [DOI] [PubMed] [Google Scholar]
- 60.Gray MJ, Zhang J, Ellis LM, Semenza GL, Evans DB, Watowich SS, Gallick GE. HIF-1alpha, STAT3, CBP/p300 and Ref-1/APE are components of a transcriptional complex that regulates Src-dependent hypoxia-induced expression of VEGF in pancreatic and prostate carcinomas. Oncogene. 2005;24:3110–3120. doi: 10.1038/sj.onc.1208513. [DOI] [PubMed] [Google Scholar]