

Abstract
Estrogen receptor α (ERα) can be phosphorylated at various residues, one of which is serine 212 in the DNA binding domain. The majority of human nuclear receptors conserves, as a motif, this serine residue within their DNA binding domain. Among these nuclear receptors, phosphorylation of the corresponding threonine 38 in the nuclear receptor CAR is essential for determining its activity [9]. Here, we have investigated the role of phosphorylated serine 212 in the regulation of ERα activity by comparing it with serine 236, another potential phosphorylation site within the DNA binding domain, and demonstrated that phosphorylation of serine 212 confers upon ERα a distinct activity regulating gene expression in Huh-7 cells. In Western blot analysis, wild type ERα and mutants ERα S212A, ERα S212D, ERα S236A and ERα S236D were equally expressed in the nucleus, thus indicating that phosphorylation does not determine nuclear localization of ERα. ERα S212D, but not ERα S236D, retained its capability of activating an ERE-reporter gene in luciferase assays. Similar results were also obtained for human ERβ; the ERβ S176D mutant retained its trans-activation activity, but the ERβ S200D mutant did not. cDNA microarray and Ingenuity Pathway Analysis, employed on Huh-7 cells ectopically expressing either ERα S212A or ERα S212D, revealed that phosphorylation of serine 212 enabled ERα to regulate a unique set of genes and cellular functions.
Keywords: Estrogen receptor, Phosphorylation, DNA binding domain, Gene expression, Nuclear receptor CAR
1. Introduction
Estrogen receptor, a member of a group of transcription factors that binds small lipophilic compounds, is activated by estradiol as well as estrogenic chemicals such as diethylstilbestrol (DES) and bisphenol A (BPA), thereby regulating the transcription of genes involved in many aspects of physiology, development and diseases. In addition to these agonistic ligands, phosphorylation of specific residues within ER has been a major subject of investigations as it may significantly impact ER signaling [1]. A number of phosphorylation sites have been identified in the AF-1 domain of ERα, among which phosphorylation of serine 167 has been recently implicated as a marker for therapeutic efficacy and post relapse survival of metastatic breast cancer patients [2]. In the DNA binding domain (DBD), phosphorylation of serine 236 is known to result in the inactivation of the ERα trans-activation activity [3,4]. A recent study using mass spectroscopy identified 14 phosphorylation sites of endogenous ERα in MCF7 cells, which included these serine residues, and in addition to serine 236, serine 212 was also identified as a phosphorylation site within the DNA binding domain [5]. Since phosphorylation of ERα has been suggested to regulate the actions of estrogen and disease development, we wanted to assign a functional role to phosphorylation of serine 212, which should provide us with an experimental basis to implicate this phosphorylation in the biological consequences of estrogen actions.
The serine 212 residue of ERα is located in the region between two zinc fingers, which forms a helix that contains the C-terminal part of the first zinc finger and extends towards the second zinc finger within the DBD. Nearly all hepatic genes regulated by estradiol require the DBD to directly bind to them [6]. The serine 212 residue, which is conserved in the DBD of 40 out of 45 human nuclear receptors, is analogous to threonine 38 of nuclear receptor CAR (NR1I3) which was first characterized as a phenobarbital-activated transcription factor [7]. CAR, a xenobiotic-sensing nuclear receptor, regulates hepatic xenobiotic and energy metabolism in response to exposures, exhibiting similarities with and differences from ERα in their structures and functions [8]. Threonine 38 phosphorylation suppresses the constitutive activation of CAR and sequesters it in the cytoplasm of hepatocytes. Phenobarbital, a CAR activator, induces de-phosphorylation of threonine 38 by protein phosphatase 2A to activate CAR and translocate it into the nucleus [9]. Given this key role of phosphorylation of threonine 38 in regulating the activity of CAR, the objective of the present work was to determine the functional consequences of phosphorylation of serine 212 in the regulation of ERα activity using hepatocarcinoma Huh 7 cells as a model system; these cells have extremely low or no endogenous ERα and no phosphorylation of ERα has been reported in Huh 7 cells. To this end, we generated the ERα S212D mutant, which can mimic the phosphorylated form of the receptor, and the ERα S212A mutant, which mimics the non-phosphorylated form, and compared their activities with those of the ERα S236D and ERα S236A mutants in luciferase reporter assays. In addition, cDNA microarray analysis was employed to measure their effects on global gene expression patterns in human hepatoma-derived Huh-7 cells. Here we present experimental results to demonstrate that the ERα S212D mutant, but not the ERα S236D mutant, retains the trans-activation activity of ERα and regulates a distinct set of genes and cell signaling pathways. The implications of phosphorylation of serine 212 in providing ERα with novel functionality will be discussed.
2. Experimental
2.1. Construction of ER mutants
Using the pcDNA3.1 human ERα and ERβ plasmids as templates, the pcDNA3.1 ERα S212D, pcDNA3.1 ERα S212A, pcDNA3.1 ERα S236D, pcDNA3.1 ERα S236A, the pcDNA3.1 ERβ S176D, pcDNA3.1 ERβ S176A, pcDNA3.1 ERβ S200D and pcDNA3.1 ERβ S200A mutants were generated using the QuickChange site-directed mutagenesis system (Stratagene, La Jolla, Calif.) with appropriate primers. The pEYFP-C1 human ERβ plasmid was produced by cloning human ERβ cDNA into the pEYFP-C1 plasmid and this was utilized as a template to generate the pEYFP-C1 ERβ S176D, pEYFP-C1 ERβ S176A, pEYFP-C1 ERβ S200D and pEYFP-C1 ERβ S200A mutants. These mutations were verified by DNA sequencing.
2.2. Cell culture and reporter assays
Huh-7 cells were cultured in minimal essential medium (MEM, Gibco BRL) supplemented with 10% FBS (Atlanta Biologicals), 2 mM l-glutamine, penicillin (100 U/ml) and streptomycin (100 μ/ml) in an atmosphere of 5% CO2 at 37 °C. For luciferase reporter assays, Huh 7 cells were harvested and suspended in MEM media without phenol red, containing 10% dextran charcoal stripped serum, 2 mM l-glutamine, penicillin (100 U/ml) and streptomycin (100 μg/ml), and plated in 24 well plates at 0.75 × 105 cells/well. Twenty-four hours later, the cells were transfected with either 3×-ERE-TATA-firefly luciferase reporter (0.1 μg/well), pRL-TK Renilla luciferase for transfection control (Promega) (0.05 μg/well) and pcDNA3.1 ERα (ERβ) or one of its mutants (0.05 μg/well), using FuGene 6 (Roche Applied Science, Indianapolis, IN) in Opti-MEM (Gibco) according to the manufacturer’s instructions. Twenty four hours after transfection, the media was replaced with media containing various concentrations of ligand, produced by 500-fold dilutions of stock solutions of ligands in ethanol with media. After an additional 24 h, the cells were washed with PBS, treated with lysis buffer and cell extracts were used for luciferase assays using the dual luciferase reporter assay system (Promega) according to the manufacturer’s instructions. Luciferase activities were measured using a Veritas Microplate Luminometer with dual injectors (Turner Biosystems, Sunnyvale, CA) and reported as a ratio of firefly luciferase/renilla luciferase.
2.3. Real-time PCR
Real-time PCR was performed using TaqMan® probes for human RAB27B and CCR7 mRNAs (Hs01072206_m1* and Hs0103469_m1 for RAB27B and CCR7, respectively), the TaqMan PCR Master Mix (Life Technologies) and an ABI prism 7700 sequence detection system (Life Technologies). The TaqMan human β-actin probe (Life Technologies) was used as an internal control.
2.4. Western blot
Huh-7 cells were scraped, collected by centrifugation, washed with PBS and homogenized in 10 mM HEPES buffer, pH 7.6, containing 10 mM KCl, 1.5 mM MgCl2, 0.3% NP-40 and Complete Protease Inhibitor Cocktail Tablets (Roche, Indianapolis, IN). The homogenates were centrifuged at 2000g for 5 min to obtain nuclei. The resulted nuclei were lysed by incubating them at 4 °C for 30 min in 10 mM HEPES buffer, pH 7.6, containing 0.4 M NaCl, 0.1 M KCl, 3 mM MgCl2, 0.1 mM EDTA, 1 mM Na3VO4, 20% glycerol and Complete Protease Inhibitor Cocktail Tablets. Subsequently, the lysate was centrifuged at 38,000 rpm for 30 min to obtain nuclear extracts. Nuclear extracts (5 μg) from the Huh-7 cells that were transfected with pcDNA3.1 ERα, pEYFP-C1 ERβ or one of their mutants (1 μg/well) using Lipofectamine 2000 (Invitrogen) were subjected to electrophoresis on a 10% SDS–polyacrylamide gel under reducing conditions, and proteins were transferred to Hybond™-P membrane (GE Healthcare). After blocking with 5% skim milk in PBS for 1 h at room temperature, blots were incubated overnight at 4 °C with rabbit polyclonal antibody raised against the estrogen receptor α (sc-542), TFIID (TBP) (sc-273) (Santa Cruz Biotechnology) or an anti-GFP goat antibody-conjugated HRP (Abcam). After washing, the blots were incubated for 1 h at room temperature with donkey anti-rabbit immunoglobulin G conjugated with horseradish peroxidase (Santa Cruz Biotechnology), and then visualized with an ECL Plus Western Blotting Detection Reagent (GE Healthcare) following by exposure on Kodak BioMax MR film (Kodak).
2.5. Microarrays and data analysis
Huh-7 cells (3 × 106 cells in 10 ml of MEM supplemented with 10% FBS, 2 mM l-glutamine, penicillin (100 U/ml) and streptomycin (100 μg/ml)) were transfected in suspension with pcDNA3.1 ERα S212D, pcDNA3.1 ERα S212A or empty pcDNA3.1 vector (6 μg) using Fugene 6 according to the manufacturer’s instructions. Cells were then plated in 10 cm dishes, with six dishes for each receptor treatment and empty vector. Twenty four hours later, the media was replaced with media containing 100 nM estradiol for three dishes of each transfection treatment and 0.2% ethanol for the other three dishes of each transfection treatment. Twenty four hour later, the cells were lysed in Trizol reagent (Invitrogen) and RNA was extracted according to the manufacturer’s instructions. The RNA was then purified using the RNeasy kit (Quiagen) with DNase treatment and quantified using an Implen Nanophotometer Version 2.2. The RNA quality was confirmed by Agilent Bioanalyser analysis.
Microarray analysis was conducted to compare the effects of transfection and activation of the S212A and S212D mutants of ERα on gene expression using Agilent Human 4×44k genome arrays (Agilent Technologies). Fifty nanograms of total RNA was amplified and labeled as directed in the NuGEN Ovation Pico WTA System protocol for Agilent microarrays. For each sample, 3 μg of Cy3 labeled cRNAs were fragmented and hybridized for 17 h in a rotating hybridization oven. Slides were washed and then scanned with an Agilent Scanner. Data was obtained using the Agilent Feature Extraction software (v9.5), using the 1-color defaults for all parameters. The Agilent Feature Extraction Software performed error modeling, adjusting for additive and multiplicative noise. The resulting data were processed using the Rosetta Resolver® system (version 7.2) (Rosetta Biosoftware, Kirkland, WA). Significant differences in gene expression were estimated by ANOVA analysis. The significant gene lists were analyzed using Ingenuity Pathway Analysis to identify canonical pathways that are significantly represented in the individual data sets (Ingenuity Systems, www.ingenuity.com). This procedure considers the number of genes in the data set that is involved in a metabolic pathway compared to the total number of genes in the reference set for that pathway. Significant pathways were identified using P values obtained using the right-tailed Fisher Exact test.
3. Results and Discussion
3.1. Role of phosphorylation in trans-activation activity
Serine 212 of ERα is located in the region between the 1st and 2nd zinc fingers and serine 236 is located in the 2nd zinc finger of the DNA binding domain (Fig. 1). The amino acid sequence of the region that encompasses these serine residues was aligned with the corresponding sequences of ERβ and CAR (Fig. 1). Serine 212 is aligned with serine 176 and threonine 38 in ERβ and CAR, respectively. Serine 236 corresponds to serine 200 in ERβ but is not conserved in CAR. Serine 212 and serine 236 of human ERα in pcDNA were mutated to alanine and aspartic acid to produce the mutants ERα S212A, ERα S212D, ERα S236A and ERα S236D; the D mutants can mimic the phosphorylated form and the A mutants mimic the non-phosphorylated form of ERα These pcDNA plasmids were then transfected into Huh-7 cells, from which nuclear extracts were prepared. Western blot analysis of these nuclear extracts with an anti-ERα antibody demonstrated nuclear expression of these mutants was similar to that of wild-type ERα, although ERα S212D levels were slightly lower compared with those of the others (Fig. 2A). These results indicated that the nuclear accumulation of ERα in Huh-7 cells was not affected by the phosphorylation status of serine 212 or serine 236.
Fig. 1.
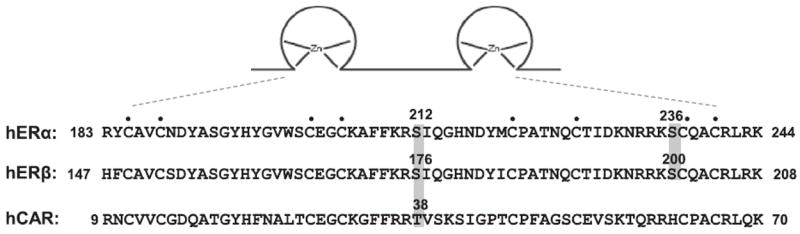
Intra-molecular localization of serine 212 in ERα and its alignment with ERβ and CAR. Amino acid sequences encompassing the two zinc fingers of ERα, ERβ and CAR were aligned manually. Numbers indicate the residues at the start and end of the aligned sequences. Black circles mark the eight cysteine residues which form zinc fingers. The left gray box shows the conserved serine residues in ERα and ERβ and threonine residue in CAR, while the right box showed conserved serine 236 and 200 in ERα and ERβ, respectively.
Fig. 2.
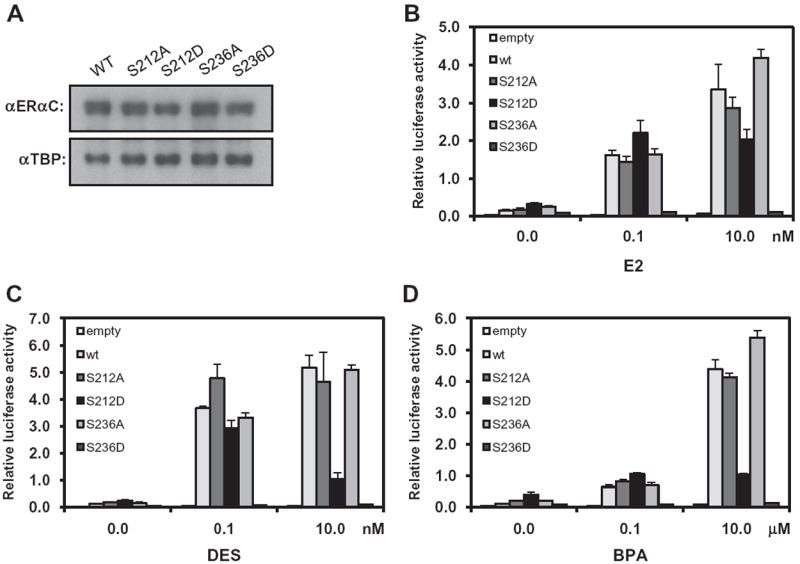
Activation of ERE reporter by ERα and its mutants in Huh-7 cells. (A) Nuclear extracts were prepared from Huh-7 cells that were transfected with expression plasmids for ERα and its A and D mutants and were subjected to Western blot analysis with an anti-ERα (αERαC) and an anti-TBP antibody. (B) A given expression plasmid was co-transfected with (ERE)3-luciferase reporter plasmid in Huh-7 cells and cells were treated with estradiol. (C) Co-transfected cells were treated with DES. (D) Co-transfected cells were treated with BPA. The results of luciferase reporter assays are reported as the mean of triplicate analysis with each experiment repeated three times.
The A and D mutants and wild type ERα were co-expressed with the (ERE)3-Luc reporter gene in Huh-7 cells. Cells were then treated with ERα activators at various concentrations and their luciferase activities were determined. Estradiol treatment at 0.1 and 10 nM induced the transcriptional activation of wild-type ERα and of both ERα S212A and ERα S236A (Fig. 2B). While ERα S236D lost activity, ERα S212D retained its capability to activate the ERE reporter following treatment with estradiol. The activation by ERα S212D was highest at 1.0 nM of estradiol and was slightly decreased at 10 and 100 nM of estradiol (Supplemental Fig. 1). This decreased activation of ERα S212D at higher dose of ligand was more prominent when DES was used. DES at 0.1 nM activated ERα S212D, but not ERα S236D, as effectively as wild type ERα (Fig. 2C). DES treatment at 0.1 and 10 nM induced the transcriptional activation of wild-type ERα by 35-fold and 50-fold, respectively, whereas the transcriptional activation of ERα S212D was induced by 30-fold and 10-fold, respectively, at 0.1 and 10 nM DES. ERα S236D was not activated by DES, even when a concentration of 10 nM was used. DES activated ERα S212A, ERα S236A and wild type ERα to similar levels. BPA, an estrogenic environmental contaminant, weakly activated ERα S212D by only 5-fold, a much weaker level of activation when compared to the over 30-fold activation conferred by estradiol and DES (Fig. 2D). No activation by BPA was observed with ERα S236D, but the A mutants of ERα were activated by BPA in a similar fashion to wild type ERα. Thus, phosphorylation of S212, which retains ERE activation activity, has dramatically different effects on the activity of ERα from the phosphorylation of S236, even though both of these serine residues are located within the DNA binding domain. However, how well the activity is retained appeared to be dependent on the type and dosage of the activators.
We next investigated the nuclear localization and the activity of different ERβ mutants in (ERE)3-luciferase reporter assays: ERβ S176A, ERβ S176D, ERβ S200A and ERβ S200D. Neither the phosphorylation of these residues of ERβ nor an investigation of the activity of these mutants has been previously reported. Similar to ERα mutants, all of these ERβ mutants were expressed in the nucleus as equally as wild type ERβ (Fig. 3A). Estradiol activated ERβ S176D, but not ERβ S200D, to a degree similar to its activation of wild type ERβ and the A mutants (Fig. 3B). Dose-dependent activation by DES was similar among wild type ERβ and the A mutants, whereas ERβ S200D was not activated (Fig. 3C). DES at 10 nM exhibited diminished activation of ERα S212D (Fig. 2C), but this effect was not observed with ERβ S176D (Fig. 3C). Activation of ERβ and its mutants by BPA was similar to that of estradiol and DES; ERβ S176D, but not ERβ S200D, was activated (Fig. 3D). Thus, the role of serine 212 phosphorylation in ERα appeared to be conserved in ERβ in the form of phosphorylation of serine 176.
Fig. 3.
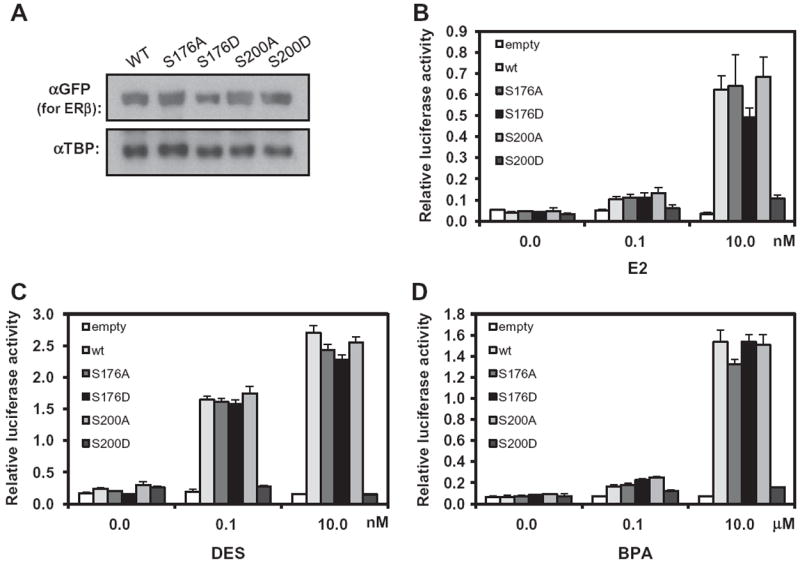
Activation of ERE reporter by ERβ and its mutants in Huh-7 cells. (A) Nuclear extracts were prepared from Huh-7 cells that were transfected with expression plasmids for YFP-tagged ERβ and its A and D mutants and were subjected to Western blot analysis with an anti-GFP antibody conjugated with HRP or an anti-TBP antibody. (B) A given expression plasmid was co-transfected with (ERE)3-luciferase reporter plasmid in Huh-7 cells and cells were treated with estradiol. (C) Co-transfected cells were treated with DES. (D) Co-transfected cells were treated with BPA. The results of luciferase reporter assays are reported as the mean of triplicate analysis with each experiment repeated three times.
3.2. Regulation of endogenous genes by ERα S212D
Given our findings that the ERα S212D mutant retained its capability to activate the ERE and its type and dose-dependent activation differed dramatically from that of ERα S212A, we employed cDNA microarray analysis to determine if these mutants would differentially affect the expression of endogenous genes. To this end, ERα S212D and ERα S212A were ectopically expressed by transfecting their expression plasmids into Huh-7 cells, with an empty plasmid vector transfected as a control. Cells were treated with estradiol or ethanol vehicle, from which total RNA was prepared for microarray analysis. After removing genes that were affected by estradiol treatment in cells transfected with empty vector, 1864 genes were found to be affected specifically by ERα S212D, 786 genes were affected by ERα S212A and 118 genes were affected by both mutants following estradiol treatment (Fig. 4A). These numbers decreased to 262, 272 and 4 when a 1.7-fold cutoff was imposed on the analysis. These results clearly indicated that these mutants of ERα regulated unique sets of genes in Huh7 cells. The effects on gene expression by activation of S212A ranged from +13.5 to −2.8-fold, while the range in gene expression by activation of S212D was +3.7 to −2.6-fold. Gene ontology analysis (IPA) was also performed to identify significant canonical signal pathways that were regulated specifically by these mutants (Fig. 4B). ERα S212A appeared to regulate known estrogen-elicited cell signals, such as arylhydrocarbon receptor (AhR) signaling, LPS/IL-1 and IL-6 signaling. ERα is known to modulate AhR-mediated transcription [10]. An ERα knock-in mouse with abolished DNA binding ability of ERα resulted in the loss of LPS-induced signaling [6]. ERα S212D, on the other hand, elicited signals that down-regulate immune function, in addition to an OX40 (TNFRS4)-mediated apoptotic signal. Thus, ERα S212D was found to regulate a distinct group of genes and cell signals in Huh-7 cells. However, the molecular mechanism by which ERα S212D regulates these genes and cell signals remains a major question for future investigations. CCR7 and RAB27B were selected from our microarray experiments as genes specifically regulated by ERα S212A and ERα S212D, respectively, and their mRNA levels were determined by real-time PCR. It was found that CCR7 mRNA levels only increased with ERα S212A, while RAB27B mRNA levels increased only with ERα S212D (Fig. 5) We also performed additional co-transfection experiments using wild-type ERα and 10 nM E2 (Supplemental Fig. 2). The ERα S212D mutant, but not the wild type ERα or the ERα S212A mutant, activated RAB27B at 10 nM E2. CCR7 was activated by wild type ERα and the ERα S212A mutant, but not ERα S212D at 10 nM E2. RAB27B is a small GTPase regulating exocytosis; a recent study with knock-out mice has demonstrated that it regulates exocytosis of granules in neutrophils [11]. CCR7 is known to regulate lymphocyte migration [12].
Fig. 4.
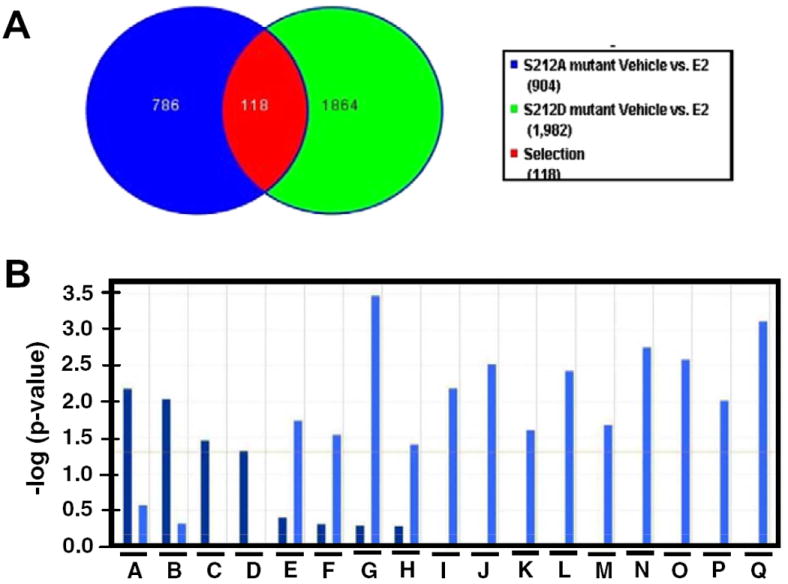
Differential gene regulation by ERα S212A and ERα S212D. (A) Venn diagram illustrating the number of specific and common genes regulated by the two ERα mutants. (B) Gene ontology analysis (IPA) for significant canonical pathways. A, arylhydrocarbon receptor signaling; B, tumoricidal function of hepatic natural killer cells; C, LPS/IL-1 mediated inhibition of RXR function; D, IL-6 signaling; E, dendritic cell maturation; F, communication between innate and adaptive immune cells; G, OX40 signaling pathway; H, regulation of IL-2 expression in activated and anergic T lymphocytes; I, CTLA4 signaling in cytotoxic T lymphocytes; J, T helper cell differentiation; K, IL-9 signaling; L, CD28 signaling in T helper cells; M, ICOs-ICOSL signaling in T helper cells; N, Crosstalk between dendritic cells and natural killer cells; O, PKCδ signaling in T lymphocytes; P, MSP-ROH signaling pathway; Q, antigen presentation pathway. Bars in darker blue and in lighter blue represent signaling pathways that were regulated by ERα S121A and ERα S212D, respectively.
Fig. 5.
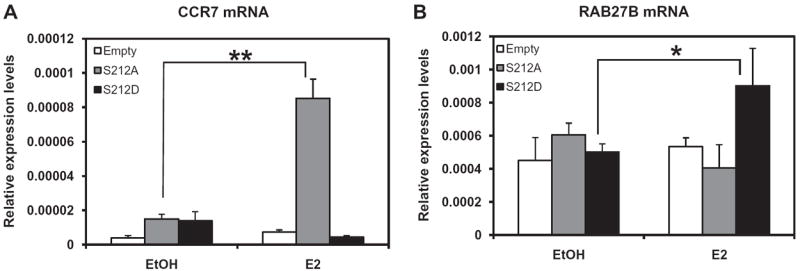
Mutation-specific increase of CCR7 and RAB27B mRNAs. Huh-7 cells were transfected with pcDNA3.1 ERα S212D, pcDNA3.1 ERα S212A or empty pcDNA3.1 for 24 h prior to treatment with 100 nM estradiol for an additional 24 h, from which total RNAs were prepared and subjected to real-time PCR and were subjected to real time PCR: CCR7 mRNA (A) and RAB27B mRNA (B). The mRNA levels are expressed relative to the levels of β-actin mRNA. Bars represent the mean ± SD. **significantly different at P < 0.01 (A) and * at P < 0.05 (B).
3.3. Conclusion
Our present study has demonstrated that phosphorylation of the DNA binding domain affects ERα activity in different ways, depending on which residue is phosphorylated; phosphorylation of serine 212 retains ERα in the nucleus and activates the ERE, whereas phosphorylation of serine 236 abolishes activity. This differential effect of phosphorylation is also observed with ERβ. Upon activation by estrogen, ERα S212D elicited cell signaling that regulates immune cells, raising an interesting question whether some immune cells, in fact, express phosphorylated ERα and are regulated by it. Phosphorylation of serine 212 of ERα is conserved in the form of the corresponding residue threonine 38 in the nuclear receptor CAR. While phosphorylation is conserved in ERα and CAR, it regulates their activities differently. Phosphorylation of threonine 38 inactivates the constitutive activity of CAR and sequesters it in the cytoplasm of hepatocytes, while de-phosphorylation activates CAR and translocates it into the nucleus [9]. In addition to CAR and ERβ, Serine 212 is also conserved as either a serine or threonine residue in 48 out of the 54 total human nuclear receptors, and phosphorylation of this residue may also be conserved. The possibility exists that phosphorylation of this single residue in the DNA binding domain can play diverse roles in the regulation of nuclear receptors, thereby impacting the research of nuclear receptor biology.
Supplementary Material
Acknowledgments
We thank the laboratory of Dr. Ken Korach at NIEHS for kindly providing us with human ERα and ERβ plasmids as well as for useful suggestions. We also thank Dr. Kosuke Saito for ANOVA analysis and the DNA sequencing and cDNA microarray cores of NIEHS for their assistance. This research was supported by the Intramural Research Program of National Institute of Environmental Health Sciences, Z01ES1005-01.
Footnotes
Appendix A. Supplementary data
Supplementary data associated with this article can be found, in the online version, at doi:10.1016/j.steroids.2012.01.001.
References
- 1.Murphy LC, Seekallu SV, Watson PH. Clinical significance of estrogen phosphorylation. Endocr-Relat Cancer. 2011;18:R1–R14. doi: 10.1677/ERC-10-0070. [DOI] [PubMed] [Google Scholar]
- 2.Yamashita H, Nishio M, Toyama T, Sugiura H, Kondo N, Kobayashi S, et al. Low phosphorylation of estrogen receptor a (ERα) serine 118 and high phosphorylation of ERα serine 167 improve survival in ER-positive breast cancer. Endocr-Relat Cancer. 2008;15:755–63. doi: 10.1677/ERC-08-0078. [DOI] [PubMed] [Google Scholar]
- 3.Chen D, Pace PE, Coombes RC, Ali S. Phosphorylation of human estrogen receptor α by protein kinase A regulates dimerization. Mol Cell Biol. 1999;19:1002–15. doi: 10.1128/mcb.19.2.1002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lannigan DA. Estrogen receptor phosphorylation. Steroids. 2003;68:1–9. doi: 10.1016/s0039-128x(02)00110-1. [DOI] [PubMed] [Google Scholar]
- 5.Atsriku C, Britton DJ, Held JM, Schilling B, Scott GK, Gibson BW, et al. Systematic mapping of posttranslational modifications in estrogen receptor-α with emphasis on novel phosphorylation sites. Mol Cell Prot. 2009;83:467–80. doi: 10.1074/mcp.M800282-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ahlbory-Dieker DL, Stride BD, Leder G, Schkoldow J, Trolenberg S, Seidel H, et al. DNA binding by estrogen receptor-α is essential for the transcriptional response to estrogen in the liver and the uterus. Mol Endocrinol. 2009;23:1544–55. doi: 10.1210/me.2009-0045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Honkakoski P, Zelko I, Sueyoshi T, Negishi M. The nuclear orphan receptor CAR-retinoid X receptor heterodimer activates the phenobarbital-responsive enhancer module of the CYP2B gene. Mol Cell Biol. 1998;18:5652–8. doi: 10.1128/mcb.18.10.5652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Timsit YE, Negishi M. CAR and PXR: the xenobiotic-sensing receptor. Steroids. 2007;72:231–46. doi: 10.1016/j.steroids.2006.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Mutoh S, Osabe M, Inoue K, Moore R, Pedersen L, Perera L, et al. Dephosphorylation of threonine 38 is required for nuclear translocation and activation of human xenobiotic receptor CAR (NR1I3) J Biol Chem. 2009;284:34785–92. doi: 10.1074/jbc.M109.048108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wihlen B, Ahmed S, Inzunza J, Matthews J. Estrogen receptor subtype- and promoter-specific modulation of arylhydrocarbon receptor-dependent transcription. Mol Cancer Res. 2009;7:977–86. doi: 10.1158/1541-7786.MCR-08-0396. [DOI] [PubMed] [Google Scholar]
- 11.Johnson J, Brzezinska AA, Tolmachova T, Munafo DB, Ellis BA, Sebra MC, et al. Rab27a and Rab27b regulate neutrophil azuophilic granule exocytosis and NADPH oxidase activity by independent mechanisms. Traffic. 2010;11:533–47. doi: 10.1111/j.1600-0854.2009.01029.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Calpe E, Codony C, Baptista MJ, Abrisqueta P, Del Carpio C, Purroy N, et al. ZAP-70 enhances migration of malignant B lymphocytes towards CCL21 by inducing CCR7 expression via IgM-ERK1/2 activation. Blood. 2011 doi: 10.1182/blood-2011-01-333682. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.