

Abstract
The insulin minisatellite (INS VNTR) has been intensively analyzed due to its associations with diseases including diabetes. We have previously used patterns of variant repeat distribution in the minisatellite to demonstrate that genetic diversity is unusually great in Africans compared to non-Africans. Here we analyzed variation at 56 single nucleotide polymorphisms (SNPs) flanking the minisatellite in individuals from six populations, and we show that over 40% of the total genetic variance near the minisatellite is due to differences between Africans and non-Africans, far higher than seen in most genomic regions and consistent with differential selection acting on the insulin gene region, most likely in the non-African ancestral population. Linkage disequilibrium was lower in African populations, with evidence of clustering of historical recombination events. Analysis of haplotypes from the relatively nonrecombining region around the minisatellite revealed a star-shaped phylogeny with lineages radiating from an ancestral African-specific haplotype. These haplotypes confirmed that minisatellite lineages defined by variant repeat distributions are monophyletic in origin. These analyses provide a framework for a cladistic approach to future disease association studies of the insulin region within both African and non-African populations, and they identify SNPs which can be rapidly analyzed as surrogate markers for minisatellite lineage.
The insulin minisatellite, located within the promoter of the human insulin gene, has been intensely investigated for nearly two decades due to its associations with diseases such as diabetes (Bell et al. 1984; Bennett and Todd 1996). Most studies have analyzed populations of European descent where low diversity at the minisatellite combined with strong linkage disequilibrium in flanking regions make it difficult to distinguish between etiological and associated variants (Bennett and Todd 1996; Doria et al. 1996). The identification of etiological polymorphisms in the insulin region may therefore require analysis of a range of different populations, in particular those showing a greater range of haplotype diversity than that seen in Europeans.
As a first step in these studies, we used a system of minisatellite variant repeat mapping by PCR (MVR-PCR; Jeffreys et al. 1991) to analyze variation at the insulin minisatellite both in allele size (number of tandem repeats) and structure (distribution of different variant repeats) in a range of populations from Africa, Asia, and Europe (Stead and Jeffreys 2000, 2002). Pairwise comparisons between all alleles showed that structures were either very different from each other, indicating that they had diverged through multiple complex mutational rearrangements, or displayed very similar patterns of variant repeat distribution, resulting in these closely related structures having similar overall sizes. In this way, alleles could be readily divided into lineage groups with low levels of variation within each lineage both in allele size and structure (Stead and Jeffreys 2000, 2002). Analysis of three African populations from the Ivory Coast (156 alleles), Zimbabwe (138), and Kenya (84) revealed substantial structural diversity with minisatellite alleles assigned to one of 22 different lineages. In contrast, all alleles within non-African populations from the U.K. (1080 alleles), Japan (118), and Kazakhstan (80) belong to just three lineages. These lineages, termed I, IIIA, and IIIB, are all present in Africa. This reduction in diversity from 22 lineages in Africans to a subset of just three lineages in non-Africans is consistent with a wealth of genetic evidence from mtDNA, Y chromosome polymorphisms, and autosomal markers which demonstrates a greater diversity in Africans than non-Africans and supports an African origin for anatomically modern humans (for review, see Tishkoff and Williams 2002). Although other explanations are possible, an African origin is further supported by the presence at multiple loci of ancestral sequences seen exclusively in Africa (Takahata et al. 2001).
Although most studies of genetic variation have identified greater diversity in Africans compared with non-Africans, the difference in lineage frequencies at the insulin minisatellite between these population groups is unusually large, with 28% of the total genetic variance being due to differences between Africans and non-Africans (Stead and Jeffreys 2002). This is substantially greater than that at most other loci (average 11%; Barbujani et al. 1997). Although this increased population differentiation is consistent with differential selection acting in the past between Africans and non-Africans, it is unclear whether the differentiation is simply an artifact of using minisatellite lineages rather than haplotypes defined by SNPs.
We now extend these minisatellite structural studies by analyzing SNP variation flanking the minisatellite in the three African and three non-African populations previously analyzed. If the 22 minisatellite lineages are truly monophyletic and deeply diverged from each other, then the unusually great differentiation between Africans and non-Africans should be reflected by elevated haplotype differentiation. SNP-based haplotypes could also give further clues about the nature, timing, and intensity of selective pressures in this region of DNA.
Haplotype data are also highly relevant to disease-association studies. Lineage diversity is too great in Africans for each lineage to be tested independently for association, and the use of MVR-PCR is too labor-intensive for lineage identification in large cohorts. Haplotype analysis should identify SNPs that can act as surrogate markers for minisatellite lineages, allowing lineage identification in large cohorts. Furthermore, the identification of closely related haplotypes will allow different but related lineages to be pooled, thus providing a framework for a hierarchical cladistic approach to disease-association studies (Templeton et al. 2000).
RESULTS
Global SNP Diversity in the Insulin Gene Region
Previous analysis of variant repeat structures of 1278 non-African and 378 African alleles at the insulin minisatellite identified 22 lineages in Africans, with a subset of just three lineages present in non-Africans (Stead and Jeffreys 2000, 2002). We used this information to select three non-Africans and 13 Africans for SNP discovery who between them contained 21 of these 22 lineages; this strategy helped maximize the chance of detecting SNPs and gaining a global view of DNA diversity. Analysis of a 7.8-kb region including the minisatellite identified 53 polymorphisms, with an additional three polymorphisms determined from the literature (Table 1). To the best of our knowledge, only 22 of these 56 SNPs have been described previously (Elbein et al. 1985; Kelsoe et al. 1988; Julier et al. 1991; Lucassen et al. 1993; Owerbach and Gabbay 1993; Johnson et al. 2001). Nucleotide diversity across the 32 sequences was estimated as π = 0.00124. Although this estimate will be biased upwards, as individuals were selected for sequencing based on maximal diversity at the minisatellite, similar estimates were derived from haplotype data (described below) on unselected chromosomes, which will be biased downwards because only known variants were typed on most chromosomes (data not shown). These measures of the average difference per nucleotide between two randomly chosen sequences selected from a population are similar to estimates from elsewhere in the genome (Jorde et al. 2001). The 56 polymorphisms include 40 transitions and 14 transversions, plus a 4-bp insertion and a 5-bp deletion. Twenty-seven polymorphisms (48%) were at CpG doublets (24 transitions and three transversions), four were within 3 bp of polymerase α-arrest sites (TGRRGA), and three were within mononucleotide runs of ≥5 bp (Templeton et al. 2000). The higher frequency of polymorphism at CpG doublets compared with estimates from elsewhere in the genome (Templeton et al. 2000) is perhaps unsurprising given the high GC content of the region (65% GC excluding the minisatellite) and evidence of imprinting in this region (Moore et al. 2001). Indeed, levels of polymorphism are known to increase with GC content, at least over short genomic regions (Sachidanandam et al. 2001; Smith and Lercher 2002).
Table 1.
Polymorphisms in the Insulin Gene Region
|
Allele frequency
|
|||||||||
|---|---|---|---|---|---|---|---|---|---|
|
Africans
|
Non-Africans
|
||||||||
| Identity | Position | Ancestral allele | Derived allele | Ancestral | Derived | Ancestral | Derived | Sequence context | Nomenclature (ref.) |
| INS-1 | -4217 | T | C | 0.820 | 0.180 | 0.489 | 0.511 | Ti | -4217 PstI (b,d) |
| INS-2 | -4069 | G | A | 0.997 | 0.003 | 1 | 0 | Ti (CpG,α) | |
| INS-3 | -4041 | C | T | 0.968 | 0.032 | 1 | 0 | Ti | |
| INS-4 | -3602 | T | C | 0.892 | 0.108 | 1 | 0 | Ti (CpG) | |
| INS-5 | -3440 | C | T | 0.910 | 0.090 | 1 | 0 | Ti (CpG) | |
| INS-6 | -3071 | G* | A* | 0.992 | 0.008 | 0.997 | 0.003 | Ti (CpG) | |
| INS-7 | -3012 | G | C | 0.701 | 0.299 | 1 | 0 | Tv | |
| INS-9 | -2733 | C | A | 0.817 | 0.183 | 0.196 | 0.804 | Tv | -2733 A/C (d) |
| INS-10 | -2250 | C | T | 0.796 | 0.204 | 1 | 0 | Ti (CpG) | |
| INS-11 | -2221 | C | T | 1 | 0 | 0.860 | 0.140 | Ti (CpG) | -2221 MspI (d) |
| INS-12 | -1986 | C | T | 0.979 | 0.021 | 1 | 0 | Ti (r) | |
| INS-14 | -1851 | T* | C* | 0.013 | 0.987 | 0 | 1 | Ti (CpG) | |
| INS-16 | -1743 | G | A | 0.995 | 0.005 | 1 | 0 | Ti | |
| INS-17 | -1467 | T | A | 0.981 | 0.019 | 1 | 0 | Tv | |
| INS-18 | -1333 | A | G | 0.960 | 0.040 | 1 | 0 | Ti (CpG) | |
| INS-19 | -1319 | C | T | 0.997 | 0.003 | 1 | 0 | Ti (CpG) | |
| INS-20 | -1265 | C | T | 0.968 | 0.032 | 1 | 0 | Ti (α) | |
| INS-21 | -1148 | A | C | 0.563 | 0.437 | 1 | 0 | Tv (r) | |
| INS-24 | -293 | C | G | 0.513 | 0.487 | 1 | 0 | Tv | -293 HincII (a) |
| INS-25 | -286 | G | A | 0.995 | 0.005 | 1 | 0 | Ti | |
| INS-69 | -192 | — | TTGC | 0.746 | 0.254 | 1 | 0 | ID | |
| INS-26 | -158 | G | A | 0.950 | 0.050 | 1 | 0 | Ti | |
| INS-27 | -23 | T | A | 0.817 | 0.183 | 0.188 | 0.812 | Tv | -23 HphI (c) |
| INS-72 | -9 | C | T | 1 | 0 | 0.995 | 0.005 | Ti | dbSNP rs5505 |
| INS-70 | +35 | G | A | 0.968 | 0.032 | 1 | 0 | Ti (CpG) | |
| INS-28 | +197 | C | T | 0.728 | 0.272 | 1 | 0 | Ti (CpG) | dbSNP rs5506 |
| INS-31 | +431 | C | T | 0.989 | 0.011 | 1 | 0 | Ti (CpG) | |
| INS-32 | +573 | T | C | 0.997 | 0.003 | 1 | 0 | Ti (CpG) | |
| INS-34 | +805 | G | C | 0.746 | 0.254 | 0.148 | 0.852 | Tv | +805 DraIII (c) |
| INS-35 | +862 | C | T | 0.926 | 0.074 | 0.960 | 0.040 | Ti (CpG) | +862 Tsp45I (f) |
| INS-36 | +934 | G | A | 0.989 | 0.011 | 1 | 0 | Ti (CpG) | |
| INS-37 | +957 | C | T | 0.976 | 0.024 | 1 | 0 | Ti (CpG) | dbSNP rs5507 |
| INS-38 | +1127 | C | T | 0.955 | 0.045 | 0.852 | 0.148 | Ti (CpG) | +1127 PstI (c) |
| INS-39 | +1140 | A | C | 0.820 | 0.180 | 0.188 | 0.812 | Tv (r) | +1140 A/C (c) |
| INS-40 | +1355 | C | T | 1 | 0 | 0.852 | 0.148 | Ti (CpG) | +1355 C/T (c) |
| INS-41 | +1404 | G | T | 0.939 | 0.061 | 0.854 | 0.146 | Tv | +1404 Fnu4HI (c) |
| INS-42 | +1428 | G | A | 0.939 | 0.061 | 0.852 | 0.148 | Ti | +1428 FokI (c) |
| INS-43 | +1973 | G | C | 0.966 | 0.034 | 1 | 0 | Tv (CpG) | |
| INS-45 | +2003 | C | T | 0.989 | 0.011 | 1 | 0 | Ti | |
| INS-49 | +2331 | A | T | 0.741 | 0.259 | 0.664 | 0.336 | Tv | +2331 A/T (d) |
| INS-71 | +2336 | CTGGG | — | 0.741 | 0.259 | 0.664 | 0.336 | ID (α) | +2336 INDEL (d) |
| INS-51 | +2844 | G* | C* | 0.500 | 0.500 | 0.336 | 0.664 | Tv (CpG) | |
| INS-52 | +2883 | G | A | 0.892 | 0.108 | 0.966 | 0.034 | Ti | +2883 A/G (e) |
| INS-53 | +2992 | C | A | 0.743 | 0.257 | 0.664 | 0.336 | Tv (α) | +2992 DdeI (f) |
| INS-54 | +3006 | T | C | 0.757 | 0.243 | 1 | 0 | Ti | |
| INS-73 | +3078 | T* | C* | 0 | 1 | 0.024 | 0.976 | Ti (CpG) | +3078 (f) |
| INS-56 | +3131 | G | A | 0.987 | 0.013 | 1 | 0 | Ti | |
| INS-74 | +3201 | G | A | 0.997 | 0.003 | 0.952 | 0.048 | Ti (CpG) | +3201 HaeII (d) |
| INS-57 | +3256 | A | G | 0.950 | 0.050 | 1 | 0 | Ti (CpG) | |
| INS-60 | +3305 | G | A | 0.979 | 0.021 | 1 | 0 | Ti | |
| INS-63 | +3526 | C | T | 0.757 | 0.243 | 1 | 0 | Ti | |
| INS-64 | +3562 | G | C | 0.950 | 0.050 | 1 | 0 | Tv (CpG) | |
| INS-65 | +3565 | T* | C* | 0.757 | 0.243 | 1 | 0 | Ti | |
| INS-66 | +3579 | C* | T* | 0.794 | 0.206 | 1 | 0 | Ti (CpG) | |
| INS-67 | +3580 | G* | A* | 0.968 | 0.032 | 0.728 | 0.272 | Ti (CpG) | +3580 MspI (d) |
| INS-68 | +3614 | G | A | 0.984 | 0.016 | 1 | 0 | Ti (CpG) | |
Details of 56 polymorphisms within a 7.8-kb region surrounding the insulin minisatellite are presented. Locations in bp are consistent with terminology of Julier et al. (1991). Ancestral states were determined by comparing sequence data between humans, chimpanzee, gorilla, and orangutan. * indicates polymorphisms for which ancestral states were ambiguous as both human alleles were found in different primate species. Allele frequencies of ancestral and derived alleles are presented and are combined for either all African or all non-African populations. The type of polymorphism is indicated as Ti (transition), Tv (transversion), and ID (insertion/deletion). Over half of the polymorphisms are located at or near mutational hotspots (Templeton et al. 2000). (CpG) denotes polymorphism at a CpG doublet, (α) denotes polymorphisms within 3 bp from a TGRRGA polymerase α-arrest site, and (r) denotes polymorphisms located within mononucleotide run of ≥5 bases. Nomenclature of polymorphisms published elsewhere is indicated with reference source as follows: (a) Elbein et al. (1985); (b) Kelsoe et al. (1988); (c) Julier et al. (1991); (d) Lucassen et al. (1993); (e) Owerbach and Gabbay (1993); (f) Johnson et al. (2001).
The ancestral states of 49 of the 56 polymorphisms could be determined unambiguously by comparison with chimpanzee, gorilla, and orangutan sequences. The remaining seven ambiguous sites showed evidence of recurrent mutation in primates, with five occurring at CpG doublets. One CpG doublet was polymorphic in humans for both TpG and CpA variants (SNPs INS-66 and INS-67) with the gorilla showing the likely ancestral CpG form, the orangutan the TpG form, and the chimpanzee the CpA form. The only amino acid substitutional changes among the four species were within the signal peptide of preproinsulin, with a conservative Ala→Val switch at position –13 in chimpanzees (Seino et al. 1992) plus an Ala/Gln/Ser three-way substitution at position –2 which is also polymorphic in humans, resulting in an Ala→Thr substitution (Oda et al. 2001).
Sequence comparisons between humans and chimpanzees generated a sequence divergence estimate of 2.11%. By random concatenation of human and chimpanzee sequence data from 53 noncoding autosomal loci described by Chen and Li (2001), we generated a distribution of divergences between these sequences. Divergence estimates as high as 2.11% were not observed in this distribution, allowing us to show that sequence divergence at the insulin region was significantly (P < 0.05) higher than at most other loci (see Methods). Furthermore, sequence comparisons with other great apes revealed high divergence between all species analyzed (Supplemental Information Table S1, available online at www.genome.org; Chen and Li 2001). This elevated divergence may reflect the high GC content near the insulin gene (65%) and the abundance of segregating sites at CpG dinucleotides. A relative rates test performed on these sequence data showed that the insulin gene region was evolving in a clock-like fashion (P > 0.05).
Genotype and Haplotype Diversity
To determine the level of diversity around the insulin gene, we typed all of the 56 polymorphisms in 189 non-Africans from the U.K. (102 individuals), Kazakhstan (28), and Japan (59), plus 189 Africans from the Ivory Coast (78 individuals), Zimbabwe (69), and Kenya (42). All of these individuals had been previously characterized for insulin minisatellite structures using MVR-PCR (Stead and Jeffreys 2002). Fifty-two of these SNPs were polymorphic in Africans, compared with only 21 in non-Africans (Table 1).
To investigate haplotype diversity around the insulin gene, we inferred haplotypes from diploid SNP data in silico using the PHASE algorithm (Table 2; Stephens et al. 2001). A total of 77 different haplotypes were identified (70 with >95% confidence). Africans contained 66 of these haplotypes (61 with >95% confidence), compared to only 20 in Europeans and Asians (18 with >95% confidence). These differences in haplotype diversity prompted further investigations of population differentiation by Fst and analysis of molecular variance (AMOVA) analyses (Table 3; data not shown). Both approaches revealed unusually high differentiation between Africans and non-Africans, compared to modest differentiation between Europe and Asia and very little differentiation within Asia or between the three African populations tested. This picture is very similar quantitatively to the levels of population differentiation seen using minisatellite lineage data (Table 3). This demonstrates that the unexpectedly large reduction in minisatellite lineage diversity seen in non-Africans (Stead and Jeffreys 2002) is not an artifact of using data from groups of related minisatellite alleles but instead reflects a genuine major shift in lineage frequencies not only at the minisatellite but also at flanking SNPs and haplotypes.
Table 2.
Inferred Haplotypes at the Insulin Region

Table 3.
Fst Estimates Between Populations From Minisatellite and Haplotype Diversity
| UK | Kazakhstan | Japan | Ivory Coast | Zimbabwe | Kenya | |
|---|---|---|---|---|---|---|
| UK | 0.051 | 0.124 | 0.231 | 0.238 | 0.263 | |
| Kazakhstan | 0.039 | 0.016 | 0.271 | 0.271 | 0.312 | |
| Japan | 0.085 | 0.001 | 0.372 | 0.376 | 0.448 | |
| Ivory Coast | 0.245 | 0.281 | 0.347 | 0.001 | 0.000 | |
| Zimbabwe | 0.250 | 0.273 | 0.341 | 0.001 | 0.001 | |
| Kenya | 0.241 | 0.298 | 0.383 | 0.003 | 0.011 |
Pairwise Fst values were estimated either from SNP genotype data (bottom left) or from minisatellite lineage data (top right, taken from Stead and Jeffreys 2002). Bold indicates P < 0.01. All calculations were performed with Arlequin v.2.000 (Schneider et al. 2000).
As noted previously (Stead and Jeffreys 2002), this extreme population differentiation between Africans and non-Africans is consistent with differential and directional selection acting to increase genetic differentiation between population groups (Cavalli-Sforza et al. 1994), whereas the similar lineage composition in the three non-African populations suggests that selection acted on a population which was ancestral to all three. If selection is acting at or near the minisatellite, then markers closest to the minisatellite should show the highest levels of population differentiation, with recombination releasing more distal markers from these selective pressures and reducing the differentiation between populations. This prediction was tested using a sliding window AMOVA analysis on blocks of 16 consecutive SNPs across the insulin region (Fig. 1). This analysis showed that the component of variance attributed to differences between Africans and non-Africans rose to a peak of 43.5% near the minisatellite, far higher than the genome average of 10.8% (Barbujani et al. 1997) and fell to 4.2% towards the 5′ end of IGF2. In contrast, neither the HKA test (Hudson et al. 1987; comparing the insulin region with the LPL locus; Clark et al. 1998) nor estimates of Tajima's D (–1.027; Tajima 1989) found evidence of selection at the insulin locus (P > 0.05; data not shown). Although this appears to contradict the Fst estimates, the high Fst values reflect the very low lineage and haplotype diversity in non-Africans, suggesting that selection may have occurred since the MRCA and specifically in these populations. If so, then the HKA test, which we used to compare human/great ape sequence divergence with human sequence diversity, would have little power to detect this recent selection. Similarly, the Tajima test, which compares nucleotide diversity with the number of segregating sites in humans, would have little power to detect selection in our sequence data set established primarily (81%) from Africans.
Figure 1.

Differentiation between Africans and non-Africans across the insulin region. Components of genetic variance attributed to differences in haplotype frequencies between African and non-African populations were determined by sliding-window AMOVA analysis (Excoffier et al. 1992) using the program SWAPS (see Methods) from overlapping blocks of 16 SNPs across the insulin region. Gene structure and SNP locations are represented below. Gray boxes are exons, black boxes introns, and the striped box is the minisatellite.
Patterns of Linkage Disequilibrium
D′ is a measure of complete disequilibrium where |D′| = 1 if at most three of the four possible haplotypes are present (Lewontin 1964). To investigate patterns of linkage disequilibrium (LD) at the insulin gene region, D′ was estimated between all pairwise combinations of markers in each of the six populations tested (Fig. 2). High LD was observed in each of the three non-African populations across the 7.8-kb region. The only exception is the 5′ most distal SNP INS-1, though LD has been reported to extend over 6 kb 5′ of this position (Doria et al. 1996), suggesting that LD breakdown at INS-1 may instead have resulted from recurrent mutation or localized gene conversion at this marker rather than from crossover. The main difference between the non-African populations was reduced statistical power supporting LD in the Japanese, a consequence of 95% of chromosomes within this population belonging to lineage I.
Figure 2.

Linkage disequilibrium (LD) across 7.8 kb of the insulin gene region. (A) LD patterns in U.K. and Ivory Coast populations. D′ measures of complete LD between each pair of markers (bottom right) and the likelihood ratio vs. free association (top left) are shown for the largest non-African population (102 individuals from the U.K.) and the largest African population (78 from the Ivory Coast) included in this study. LD measures were estimated from diploid genotype data, after excluding all markers with minor allele frequencies <0.15. Regions of high LD appear as red blocks. Each colored block is centered on the two SNPs being compared and extends halfway to the adjacent marker. Positions of SNPs are shown below and to the right of each plot, with SNP identities indicated below. (B) Schematic of the insulin gene region, as in Fig. 1. Regions of high absolute association (Δ) are indicated in yellow, with regions of high complete association (D′) in pink. Checked pink boxes show four African SNPs within a block of complete LD which display evidence of obligate recombination. (C) Decay of absolute disequilibrium (Δ) with distance. Δ was estimated for each pair of markers with minor allele frequency ≥0.15 and plotted against physical distance between markers for each of the six populations. Lines show the least-squares best-fit curves calculated for the theoretical relationship 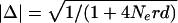 where Ne is the effective population size, r is the recombination frequency per Mb, and d is the intermarker distance in Mb (Sved 1971).
where Ne is the effective population size, r is the recombination frequency per Mb, and d is the intermarker distance in Mb (Sved 1971).
To investigate haplotype structure, we analyzed levels of absolute association in these populations using the LD measure Δ for which |Δ| = 1 where at most only two of the possible four haplotypes are present (Hill and Robertson 1968). Two blocks of absolute association were apparent in non-Africans, indicating the presence of only two major haplotypes per block (Table 2A). These blocks of very low haplotype diversity, one covering the INS gene and 5′ flanking region and the other extending over the first exon of the IGF2 gene, correlate strongly with the heavily reduced minisatellite lineage diversity seen in Europeans and Asians. Thus, the two main haplotypes in the INS block, which corresponds to the region of disequilibrium previously described by Lucassen et al. (1993), are associated with minisatellite lineages I and IIIA in all three non-African populations (Table 2A). Similarly, one haplotype in the IGF2 block corresponds to sublineage IC, with the other haplotype being associated with ID and IIIA lineages. Haplotypes associated with lineage IIIB differ from the two main haplotypes in each block, but are at low frequency in these populations and so do not dramatically reduce levels of absolute association. We found no evidence of historical recombination between the two blocks of association.
The degree of LD in the insulin gene region is lower in Africans compared to non-Africans (Fig. 2). Patterns of complete disequilibrium (D′) do show evidence for possible LD blocks that broadly correspond to the two domains of absolute disequilibrium (Δ) identified in non-Africans (Table 2B). Application of the four-gamete test to these populations found evidence of recombination between the two blocks of disequilibrium. Furthermore, there was evidence of homoplasy at four markers within the INS LD block (INS-21, INS-24, INS-69, and INS-28) which cluster in an interval from 50 bp 5′ of the minisatellite to 800 bp 3′ of the minisatellite (Fig. 2B, Table 2B). This clustering, plus the observation that two of the four markers are not located at sites prone to increased mutation, argue against recurrent mutation and instead provide evidence of a minisatellite-associated increase in local recombination frequency. Interestingly there is little evidence that linkage phase has been disrupted between markers distal to these apparent recombinants, suggesting that either crossing-over occurred between similar haplotypes or that recombination may be generating conversions in this region. The lack of observable recombinants in this region in non-Africans may simply reflect the fact that these markers are monomorphic in Europeans and Asians.
Historical recombination rates in the insulin gene region were investigated by analyzing the rate of decay of absolute disequilibrium with distance (Fig. 2C). LD decays more rapidly in Africans than non-Africans, with extended LD greatest in the Japanese population, followed by Kazakhstan, then the U.K. Assuming an effective population size of 10,000 (Jorde et al. 1998), the decay of LD suggests a mean recombination activity of 5.3 cM/Mb across the region (ranging from 0.4 to 12.8 cM/Mb depending on the population considered), somewhat higher than the genome average rate of 1 cM/Mb (Yu et al. 2001). This is likely to be an overestimate given the evidence of gene conversion and recurrent mutation in this region, both of which will contribute to LD decay. Assuming that the recombination activity is the same in all populations, the LD decay data suggest a ratio of effective population sizes among Africa, Asia, and Europe of 10:1:3. This is similar to estimates of 7:1:2 based on simple tandem repeat (STR) data (Relethford and Jorde 1999) and 3:1:1 based on craniometric and genetic data sets (Relethford and Harpending 1994).
Phylogenetic Network Analysis
We used median joining network analysis (Bandelt et al. 1999) of the insulin haplotypes to explore the phylogenetic relationships between minisatellite lineages (Fig. 3A). To minimize reticulation arising from historical recombination events, we focused on a 3.6-kb region spanning the minisatellite, from markers INS-11 to INS-40, that is two SNPs (512 bp) shorter than the largest block of complete disequilibrium found in African populations. This analysis revealed a star-like phylogeny in which the residual reticulation involved only markers INS-21, INS-24, INS-69, and INS-28 near the minisatellite which had been previously identified as being prone to recombination.
Figure 3.

Median-joining (MJ) network analysis of the insulin gene region. (A) MJ network analysis of all haplotypes. MJ networks were based on 26 polymorphisms within a 3.6-kb region around the insulin minisatellite and including SNPs INS-11 to INS-40. Haplotypes are represented by circles, with areas reflecting the frequency of each haplotype group within the total African plus non-African data set. A reference circle representing n = 5 chromosomes is shown. African haplotypes are marked in black and non-African in white. The lengths of lines connecting haplotype nodes are proportional to the number of mutation steps separating lineages, with the identity of the SNPs mutated along each branch indicated. SNPs displaying homoplasy are underlined. The network was rooted by comparison with primate sequences at the African-specific haplotypes S, V, and H (indicated by stars). (B) MJ network analysis of sublineages of lineage I chromosomes. Relationships between sublineages were resolved by network analysis using all markers across the 7.8-kb haplotype.
Most nodes on the haplotype phylogeny contain only one minisatellite lineage, confirming that these lineages defined by repeat DNA structure are genuinely monophyletic and that many of them are highly diverged, in particular lineages I and IIIA that account for the majority of non-African chromosomes. Discounting singletons, only three lineages shared the same flanking haplotype (lineages S/V, lineages IIIB/X, and lineage K with a subset of lineage J). Minisatellite alleles can therefore on occasion become highly diverged in structure without accumulating flanking mutations. Conversely, three lineages were spread over similar haplotypes related by mutation (lineages IIIA and Y) or by either mutation or recombination (lineage J), indicating that major rearrangements in the minisatellite must occur sufficiently rarely to allow flanking mutations to sometimes accumulate within a minisatellite lineage. It therefore follows that major rearrangements must occur at a rate similar to base substitution over this 3.6-kb region (c.7×10–5 per gamete). Interestingly, this rate is similar to the frequency of major complex rearrangements at the minisatellite detected in human sperm (c.2×10–5; Stead and Jeffreys 2000).
To investigate whether closely related haplotypes had minisatellite lineages of similar sizes, pairwise comparisons were performed between the most common haplotype from each of the 22 lineages. There was no correlation between the distance between haplotypes and the size difference between minisatellite allele lineages, either with the shortened haplotype used for the phylogenetic studies (Kendall τ = 0.069, P = 0.12) or with full haplotypes (Kendall τ = 0.026, P = 0.56). This implies that major rearrangements occurring in the minisatellite must involve radical changes in allele lengths, as seen in complex sperm mutants (Stead and Jeffreys 2000).
Comparison of human and primate sequences located the root of the network at or near the central node of the star phylogeny (lineages S/V or H, respectively). Haplotypes near the root were uncommon in Africans and absent from non-Africans. Two of the three lineages found outside Africa (lineages I and IIIB) lie on the same haplotypes in Africans and non-Africans (Fig. 3A,B). In contrast, lineage IIIA alleles in Europeans and Asians have diverged from their African counterparts by mutation at INS-11 and/or INS-40, suggesting diversification following the founding of non-African populations.
DISCUSSION
Previous studies of lineage diversity at the insulin minisatellite revealed an unusually high level of genetic differentiation between Africans and non-Africans, considerably greater than that seen at most other genomic regions analyzed using SNPs or microsatellites (Barbujani et al. 1997; Jorde et al. 1998). We now show that this hyperdifferentiation is not an artifact of using minisatellite lineages as markers, but instead correlates with a remarkable degree of population differentiation in SNP-based haplotypes near the minisatellite. Furthermore, this differentiation does not appear to affect the whole insulin gene region uniformly; rather, it extends across a region previously implicated in susceptibility to type 1 diabetes (Lucassen et al. 1993), reaching a peak at and near the minisatellite. These data are consistent with directional selection acting on the minisatellite or a nearby polymorphism, with more distal markers being released from selective pressures by historical recombination. As previously noted (Stead and Jeffreys 2002), the substantial over-representation of lineage I chromosomes in Europeans and Asians raises the possibility that positive selection has acted to increase the frequency of class I chromosomes specifically in non-African populations, as a result for example of adaptation to different environments. The finding of the same three lineages in all non-African populations suggests that this selection acted early during the migration out of Africa, and that shared evolutionary history accounts for the similar lineage composition of the three non-African populations. Finally, we found no evidence of differences in selective environments between Europe and Japan, as Fst values between these populations are no higher at the insulin locus than at other autosomal loci (Cavalli-Sforza et al. 1994). We are not aware of comparative data for Kazakhstan.
It will be of considerable interest to extend these analyses more broadly in the INS region, to see whether other peaks of population hyperdifferentiation can be detected that may mark regions that have undergone prehistoric selection. If selective environments have remained sufficiently stable over time, such sites may colocate with regions containing variants underlying current disease etiologies. These studies might also prove more informative as to the nature of the putative selection that has acted on this locus. For example, analyses of extended haplotypes have recently been used to find signatures of recent selection at two genes implicated in resistance to malaria, G6PD, and the CD40 ligand (Sabeti et al. 2002). However, this approach may not be fruitful at the insulin locus, where selection is likely to be much older and therefore harder to detect; also, neutral `core haplotypes' at the same locus in the same populations are not available for comparison.
There is substantial linkage disequilibrium across the insulin gene region, most noticeably in non-Africans but still present in African populations; for example, 57% of comparisons between SNPs with minor allele frequencies >0.15 yielded D′ values >0.9 in the Ivory Coast sample. Despite these associations, there is evidence that historical recombination events have contributed to haplotype diversity. The indirectly estimated recombination activity of this region is roughly fivefold higher than the mean rate of 1 cM/Mb in the human genome (Yu et al. 2001). However, historical recombination events appear not to be randomly scattered but rather cluster into two regions, one between INS and IGF2, and another spanning a region from just upstream of the insulin minisatellite to 800 bp 3′ of the minisatellite. There is growing evidence that minisatellites are generated from errors in meiotic recombination centered at recombination hotspots (Jeffreys et al. 1998). It is therefore possible that the insulin minisatellite may also be associated with a weak hotspot perhaps 3′ to the minisatellite. Consistent with this, sperm mutation studies have revealed a low frequency recombination-based mode of repeat DNA instability in the germline, with some evidence that these events occur preferentially towards the 3′ end of the minisatellite (Stead and Jeffreys 2000).
Variant repeat analysis at the insulin minisatellite identified 22 lineages with highly diverged structures, most of which are apparently present only in Africa (Stead and Jeffreys 2002). Network analysis of haplotype data has now confirmed that most minisatellite lineages are indeed monophyletic in origin (Fig. 3). Although the structural divergence between minisatellite lineages prevents the construction of phylogenies from MVR data, haplotype networks show that some closely related lineages do share common motifs within the minisatellite. For example, lineages IIIB, X, and Y all share a motif of consecutive F, A, C, and E variant repeats, whereas lineages L, M, and W display similarities at both 5′ and 3′ ends. This indicates that motif sharing between lineages can be due to a relatively recent common ancestry as opposed to recombination or mutational convergence. However, neither these motifs nor minisatellite allele lengths can serve as reliable phylogenetic markers for identifying related lineages, and lineage phylogeny can only be established by analysis of flanking haplotypes.
These data provide a framework for future disease association studies at the insulin region in a range of populations. Most previous studies analyzed populations of European descent in which low minisatellite and haplotype diversity combined with high LD near the minisatellite make it difficult to distinguish etiological from associated variants (Bennett and Todd 1996; Stead et al. 2000). Our data suggest that this limitation would apply to the analysis of any non-African population, given that Asians and Europeans share the same few lineages and associated haplotypes. However, increased lineage diversity and reduced LD in African populations open up the possibility of a second phase of higher-resolution studies with the potential to be able to dissect out etiological variants which in non-African populations cannot be uniquely identified due to strong associations with other markers. The present study allowed us to identify either lineage-specific SNPs or combinations of SNPs for every known minisatellite lineage; these could be rapidly analyzed as surrogate markers of minisatellite lineage in every population (Table 2, Suppl. Information S2). Furthermore, although many African lineages are found at too low a frequency to allow their effects on disease susceptibility to be independently analyzed, our haplotype phylogenetic analysis describes relationships between different lineages, allowing different closely related lineages to be combined into a single group of sufficient size for association analysis. This allows for a hierarchical cladistic approach to association studies in Africans in which analysis of a small number of SNPs will allow closely related lineages to be initially analyzed as a single group (Templeton et al. 2000). The effects of specific lineages within individual groups could then be further investigated by analyzing additional polymorphisms.
Finally, there is considerable interest in defining haplotype blocks in the human genome and identifying haplotype-tag (ht) SNPs that can be used to capture efficiently most or all of the haplotype information within a block (Johnson et al. 2001; Gabriel et al. 2002). There is evidence that common haplotypes within a block can to some extent at least be global, shared by most or all major population groups, and raising the possibility of defining a set of universal ht-SNPs for an LD block applicable to any population (Gabriel et al. 2002). The present data show that this approach may face substantial difficulties. For example, the haplotypes carrying lineage IIIA alleles in non-Africans (IIIAa,b) are absent from Africans, with allele INS-40T which uniquely identifies this lineage in Europeans and Asians (Table 2) being absent from Africans. Similarly, marker INS-74 used elsewhere to define one of the most common haplotypes within the U.K. population (Johnson et al. 2001) was not detectably polymorphic in other non-African populations. The implications are that, even if LD blocks are shared across populations, the underlying haplotypes and ht-SNPs may show substantial population specificity.
METHODS
DNA Source
Genomic DNA was extracted from blood or sperm from individuals sampled at random with respect to disease status from six populations: U.K. (102 individuals), Kazakhstan (28), Japan (59), Ivory Coast (78), Zimbabwe (69), and Kenya (42). The use of human samples was approved by the Leicestershire Local Research Ethics Committee.
Nomenclature
This study describes variation both at the insulin minisatellite and in the flanking haplotype. We reserve the terms “lineage” and “sublineage” exclusively for minisatellite diversity. Briefly, structures of all alleles within a lineage can be aligned to each other, whereas different lineages cannot be aligned. Some lineages contained subgroups of alleles that were more similar to each other than to other alleles within that lineage. These subgroups are termed `sublineages.' Our nomenclature incorporates terminology used in previous publications (Bell et al. 1984; Stead and Jeffreys 2000). Originally, minisatellite alleles were divided into three classes on the basis of size: class I (small), II (intermediate), and III (large) (Bell et al. 1984; Rotwein et al. 1986). In Europeans, class I alleles form a single lineage called lineage I, class II alleles are rare, and class III alleles divide into two lineages termed IIIA and IIIB (Stead and Jeffreys 2000). Other lineages are denoted with capital letters, and sublineages are indicated by Roman numerals; thus IIIAii is the second sublineage of lineage IIIA. The only exceptions are sublineages IC, ID, and IE of lineage I, named prior to this study (Stead and Jeffreys 2000, 2002). The names of specific minisatellite alleles reflect minisatellite lineage/sublineage, allele size (number of repeats), and a further discriminator; for example, allele ID42.4 is the fourth allele of 42 repeats identified from the ID sublineage of lineage I.
Haplotypes were inferred from combined SNP and minisatellite lineage data and named according to minisatellite lineage, with different haplotype variants indicated in lowercase letters; thus Wa and Wb are the first and second haplotypes identified in chromosomes carrying the minisatellite from lineage W.
SNP Discovery and Genotyping
PCR primers designed from the genomic sequence of the insulin region (accession number L15440; Lucassen et al. 1993) were used to amplify a 3.163-kb region 5′ of the minisatellite (primers 5F1 to 5R1) and two overlapping amplicons covering a 4.237-kb region 3′ of the minisatellite (primers 3F1–3R3 and 3F6–3R1). To screen for polymorphisms, these amplicons were resequenced using BigDye terminators (ABI) on an ABI 377 Automated Sequencer, and SNPs were identified using ABI AutoAssembler software. Diploid PCR products from 16 individuals were analyzed. Their sample references (italics) and genotypes at the insulin minisatellite are as follows: 11 individuals from Zimbabwe (Z-10 IC35.8/F17.1; Z-14 N82.2/U119.1; Z-15 Q86.1/X144.1; Z-16 T107.1/Y422.1; Z-20 G30.1/K44.1; Z-21 J43.1/W145.1; Z-46 L51.1/K52.2; Z-51 IIIB142.2/W148.1; Z-58 M86.1/V216.1; Z-67 K44.2/Z191.1; Z-86 IC31.8/IE39.1), two from Kenya (K-3I H29.1/S110.1; K-5B P79.1/IIIA211.1), and three from the U.K. (UK-101 ID40.2/ID44.1; UK-102 IIID144.1/IIIA147.2; UK-103 IC30.1/IIIA159.2). Orthologous regions from one chimpanzee, one gorilla, and one orangutan were also amplified and sequenced using the same primers (GenBank acc. nos. for human AY138589, AY138590; chimpanzee AY137496, AY137497; gorilla AY137498, AY137499, AY137500; orangutan AY137501, AY137502, AY137503). We identified 53 polymorphisms in the 16 human samples, with one additional SNP (INS-72) from dbSNP plus two (INS-73 and INS-74) described by Johnson et al. (2001). We genotyped all 56 SNPs using genomic DNA from our panel of unrelated individuals from six populations by allele-specific oligonucleotide (ASO) hybridization to dotblots of the three PCR-generated amplicons described above (Wood et al. 1985; Jeffreys et al. 1998). Each dotblot included amplicons used for sequence-based SNP-discovery resulting in genotypes being determined by both ASO hybridization and sequence analysis for 16 subjects (896 genotypes). No genotyping discrepancies between the two techniques were observed. Furthermore, there was no evidence for ambiguity at any of the resulting 21,168 genotypes. After Bonferroni correction, none of the markers showed deviation from Hardy-Weinberg equilibrium in any of the populations tested. Details of SNPs, ASOs, PCR primers, primate sequences, genotypes, and allele frequencies, plus the full MVR data from 1985 alleles analyzed in this and other studies (Stead et al. 2000; Stead and Jeffreys 2000, 2002; J.D.H. Stead, M.I. McCarthy, and A.J. Jeffreys, unpubl.) can be found on our Web site (http://www.le.ac.uk/ge/ajj/insulin).
In Silico Analyses
Haplotypes were inferred from genotype data in silico using PHASE software (Stephens et al. 2001) available at http://www.stats.ox.ac.uk/mathgen/software.html which can analyze multi-allelic loci including minisatellite lineages and which assigns a probability of the correct inference of haplotype phase at every heterozygous position. Each of the 22 minisatellite lineages were given a unique identifier prior to analysis except for lineage I alleles, which were subdivided into the three main sublineages IC, ID, and IE. PHASE simulations were repeated five times. Differences between outputs were minimal (<2%) and only involved haplotypes determined with <95% confidence. In these cases, the most common output was accepted. PHASE inferred haplotypes with >95% confidence for 186 of the 189 non-Africans genotyped, and 171 of the 189 Africans. Incorporation of data from specific alleles at the minisatellite (as opposed to minisatellite lineage) allowed haplotypes to be inferred with confidence from a further two non-Africans and eight Africans. PHASE-inferred haplotypes for all 378 individuals can be found on our Web site (http://www.le.ac.uk/ge/ajj/insulin).
Sequence divergence between hominoids was estimated using Jukes-Cantor distances (Jukes and Cantor 1969) from an alignment of a 6774-bp region surrounding the insulin locus for which full sequence data were available. Human values were averaged over the 32 complete sequences, inferred using PHASE software from the 16 diploid sequences described above. To compare our estimates of sequence divergence between humans and chimpanzees at the insulin locus (2.11%) with other autosomal loci, we randomly (with replacement) concatenated sequence data from 53 noncoding autosomal loci described by Chen and Li (2001) until total sequence length matched that which was available for the insulin region, after which sequence divergence was determined. Replication of this process 10,000 times generated a divergence distribution of 0.96%–1.51% (between 2.5 and 97.5 centiles). A sequence divergence as high as 2.11% was never observed, despite the insulin region containing coding regions under selective constraint.
To determine whether these great ape and human sequences were evolving in a clock-like fashion, a relative rates test was conducted on the aligned sequences using TREE-PUZZLE software (Strimmer and von Haeseler 1996) available at http://www.tree-puzzle.de/. This generated a likelihood test statistic of 4.67 which fails to reject the molecular clock for this data (P > 0.05 assuming a χ2 distribution with two degrees of freedom).
We applied two tests of neutrality to the sequence data. Tajima's D statistic (Tajima 1989) compares estimates of θ derived either from the average pairwise difference between sequences, or from the number of segregating sites. Under neutrality and mutation/drift equilibrium these measures should be equal, giving a Tajima's D value of 0. The Hudson/Kreitman/Aguade (HKA) test (Hudson et al. 1987) compares levels of intraspecific polymorphism with interspecific divergence between the locus being investigated and a neutral reference locus. The ratio of polymorphism against divergence should be equal between loci if both are evolving neutrally, as both measures depend on the underlying neutral mutation rate. In contrast, directional selection will cause a reduction in this ratio in the locus under selection. The lipoprotein lipase gene (LPL [MIM 238600]; Clark et al. 1998), which has been used elsewhere as a neutral control for HKA analysis (Fullerton et al. 2000), was selected as our reference locus. Tajima's D and the HKA test were both analyzed using the program DnaSP (Rozas and Rozas 1999) available at http://www.ub.es/dnasp.
Fst calculations were performed on all SNP genotype data excluding the insulin minisatellite, or on minisatellite lineage data excluding flanking SNPs, from each population using Arlequin ver. 2.000 (Schneider et al. 2000) available at http://lgb.unige.ch/arlequin. Analysis of molecular variance (AMOVA; Excoffier et al. 1992) was calculated from pairwise distances between SNP haplotypes and was performed using Arlequin ver. 2.000 (Schneider et al. 2000). To investigate variation in the degree of population subdivision across the insulin region, we used a program which performs a sliding window analysis of population subdivision (SWAPS), written by M.E. Hurles, which will be described in detail elsewhere. Here, the program performs an AMOVA analysis (Excoffier et al. 1992) on 41 overlapping windows, each composed of 16 informative sites, shifting in position by a single SNP across the region analyzed. Larger windows reduce resolution, whereas smaller windows produce more peaks and troughs by exaggerating the effects of individual population-specific SNPs.
Linkage disequilibrium (LD) was analyzed from diploid genotype data (excluding the minisatellite) and plotted using software described elsewhere (Jeffreys et al. 2001) which employs LD measures of both complete association (D′) and absolute association (Δ), and estimates the likelihood ratio (LR) in favor of significant linkage disequilibrium. Markers with minor allele frequencies below 0.15 were excluded from LD analysis; these markers tend to lack statistical power to detect LD and are also likely to be the result of relatively recent mutations and will not therefore report on ancient recombination events.
Indirect estimates of recombination activity were derived from the LD data using markers with minor allele frequencies ≥0.15. Assuming that crossovers are randomly distributed and that haplotypes are selectively neutral and at crossover/drift equilibrium, the expected rate of decay of absolute disequilibrium (|Δ|) with physical distance is given by 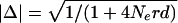 where Ne is the effective population size, r is the recombination frequency per Mb, and d is the intermarker distance in Mb (Sved 1971).
where Ne is the effective population size, r is the recombination frequency per Mb, and d is the intermarker distance in Mb (Sved 1971).
Ancestral relationships between inferred haplotypes were investigated using the Median-Joining (MJ) network algorithm (Bandelt et al. 1999) within NETWORK 2.0 software available at http://www.fluxus-engineering.com/sharenet.htm. Haplotypes which could not be inferred with confidence were excluded. Inclusion of all full-length haplotypes generated highly reticulated networks due to historical recombination events. To reduce reticulation, singleton haplotypes were excluded except for haplotypes of minisatellite lineages F and Z, as these lineages were each identified only once in our data set. Haplotype lengths were restricted to a 3.6-kb block of linkage disequilibrium surrounding the minisatellite and including all 26 SNPs between INS-11 and INS-40. Although networks were based on SNP data and not minisatellite lineage, lineage data were included in haplotype inference. SNP and minisatellite lineage data are therefore both represented within the network. The network was rooted at lineages H, S, and V by comparison with the ancestral states of these 26 SNPs determined from primate sequence analysis. The ancestral state of INS-14 was ambiguous, so this site was discounted. Minisatellite structures in primates bear no similarity to human minisatellite lineages and are uninformative regarding ancestral state (Stead and Jeffreys 2002).
Acknowledgments
We thank J. Clegg and Y. Dubrova for kindly providing DNA samples and M. Jobling and colleagues for invaluable discussions. This work was supported by grants to A.J.J. from the Wellcome Trust (ref. 061869/Z/00/Z) and the Royal Society, and by the McDonald Institute for Archaeological Research.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC section 1734 solely to indicate this fact.
[Supplemental material is available online at www.genome.org and at the authors' Web site; http://www. leicester.ac.uk/genetics/ajj/insulin. The sequence data from this study have been submitted to GenBank under accession nos.: human AY138589, AY138590; chimpanzee AY137496, AY137497; gorilla AY137498, AY137499, AY137500; orangutan AY137501, AY137502, AY137503. The SNP data from this study have been submitted to dbSNP. The following individuals kindly provided reagents, samples, or unpublished information as indicated in the paper: J.Clegg and Y.Dubrova.]
Article and publication are at http://www.genome.org/cgi/doi/10.1101/gr.948003.
References
- Bandelt, H.J., Forster, P., and Rohl, A. 1999. Median-joining networks for inferring intraspecific phylogenies. Mol. Biol. Evol. 16: 37–48. [DOI] [PubMed] [Google Scholar]
- Barbujani, G., Magagni, A., Minch, E., and Cavalli-Sforza, L.L. 1997. An apportionment of human DNA diversity. Proc. Natl. Acad. Sci. 94: 4516–4519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bell, G.I., Horita, S., and Karam, J.H. 1984. A polymorphic locus near the human insulin gene is associated with insulin-dependent diabetes mellitus. Diabetes 33: 176–183. [DOI] [PubMed] [Google Scholar]
- Bennett, S.T. and Todd, J.A. 1996. Human type 1 diabetes and the insulin gene: Principles of mapping polygenes. Annu. Rev. Genet. 30: 343–370. [DOI] [PubMed] [Google Scholar]
- Cavalli-Sforza, L., Menozzi, P., and Piazza, A. 1994. History and geography of human genes, chapter 2. Princeton University Press, Princeton, NJ.
- Chen, F.C. and Li, W.H. 2001. Genomic divergences between humans and other hominoids and the effective population size of the common ancestor of humans and chimpanzees. Am. J. Hum. Genet. 68: 444–456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark, A.G., Weiss, K.M., Nickerson, D.A., Taylor, S.L., Buchanan, A., Stengard, J., Salomaa, V., Vartiainen, E., Perola, M., Boerwinkle, E., et al. 1998. Haplotype structure and population genetic inferences from nucleotide-sequence variation in human lipoprotein lipase. Am. J. Hum. Genet. 63: 595–612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doria, A., Lee, J., Warram, J.H., and Krolewski, A.S. 1996. Diabetes susceptibility at IDDM2 cannot be positively mapped to the VNTR locus of the insulin gene. Diabetologia 39: 594–599. [DOI] [PubMed] [Google Scholar]
- Elbein, S.C., Corsetti, L., and Permutt, M.A. 1985. New polymorphisms at the insulin locus increase its usefulness as a genetic marker. Diabetes 34: 1139–1144. [DOI] [PubMed] [Google Scholar]
- Excoffier, L., Smouse, P.E., and Quattro, J.M. 1992. Analysis of molecular variance inferred from metric distances among DNA haplotypes: Application to human mitochondrial DNA restriction data. Genetics 131: 479–491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fullerton, S.M., Clark, A.G., Weiss, K.M., Nickerson, D.A., Taylor, S.L., Stengard, J.H., Salomaa, V., Vartiainen, E., Perola, M., Boerwinkle, E., et al. 2000. Apolipoprotein E variation at the sequence haplotype level: Implications for the origin and maintenance of a major human polymorphism. Am. J. Hum. Genet. 67: 881–900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gabriel, S.B., Schaffner, S.F., Nguyen, H., Moore, J.M., Roy, J., Blumenstiel, B., Higgins, J., DeFelice, M., Lochner, A., Faggart, M., et al. 2002. The structure of haplotype blocks in the human genome. Science 296: 2225–2229. [DOI] [PubMed] [Google Scholar]
- Hill, W. and Robertson, A. 1968. Linkage disequilibrium in finite populations. Theor. Appl. Genet. 38: 226–231. [DOI] [PubMed] [Google Scholar]
- Hudson, R.R., Kreitman, M., and Aguade, M. 1987. A test of neutral molecular evolution based on nucleotide data. Genetics 116: 153–159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeffreys, A.J., MacLeod, A., Tamaki, K., Neil, D.L., and Monckton, D.G. 1991. Minisatellite repeat coding as a digital approach to DNA typing. Nature 354: 204–209. [DOI] [PubMed] [Google Scholar]
- Jeffreys, A.J., Murray, J., and Neumann, R. 1998. High-resolution mapping of crossovers in human sperm defines a minisatellite-associated recombination hotspot. Mol. Cell 2: 267–273. [DOI] [PubMed] [Google Scholar]
- Jeffreys, A.J., Kauppi, L., and Neumann, R. 2001. Intensely punctate meiotic recombination in the class II region of the major histocompatibility complex. Nat. Genet. 29: 217–222. [DOI] [PubMed] [Google Scholar]
- Johnson, G.C., Esposito, L., Barratt, B.J., Smith, A.N., Heward, J., Di Genova, G., Ueda, H., Cordell, H.J., Eaves, I.A., Dudbridge, F., et al. 2001. Haplotype tagging for the identification of common disease genes. Nat. Genet. 29: 233–237. [DOI] [PubMed] [Google Scholar]
- Jorde, L.B., Bamshad, M., and Rogers, A.R. 1998. Using mitochondrial and nuclear DNA markers to reconstruct human evolution. Bioessays 20: 126–136. [DOI] [PubMed] [Google Scholar]
- Jorde, L.B., Watkins, W.S., and Bamshad, M.J. 2001. Population genomics: A bridge from evolutionary history to genetic medicine. Hum. Mol. Genet. 10: 2199–2207. [DOI] [PubMed] [Google Scholar]
- Jukes, T. and Cantor, C. 1969. Evolution of protein molecules. In Mammalian protein metabolism (ed. H. Munro), pp. 21–132. Academic Press, New York.
- Julier, C., Hyer, R.N., Davies, J., Merlin, F., Soularue, P., Briant, L., Cathelineau, G., Deschamps, I., Rotter, J.I., Froguel, P., et al. 1991. Insulin-IGF2 region on chromosome 11p encodes a gene implicated in HLA-DR4-dependent diabetes susceptibility. Nature 354: 155–159. [DOI] [PubMed] [Google Scholar]
- Kelsoe, J.R., Stubblefield, B.K., and Ginns, E.I. 1988. Human tyrosine hydroxylase (TH) genomic fragment (pHGTH4) identifies a PstI polymorphism. Nucleic Acids Res. 16: 7760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewontin, R. 1964. The interaction of selection and linkage. I. General considerations; heterotic models. Genetics 49: 49–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lucassen, A.M., Julier, C., Beressi, J.P., Boitard, C., Froguel, P., Lathrop, M., and Bell, J.I. 1993. Susceptibility to insulin dependent diabetes mellitus maps to a 4.1 kb segment of DNA spanning the insulin gene and associated VNTR. Nat. Genet. 4: 305–310. [DOI] [PubMed] [Google Scholar]
- Moore, G.E., Abu-Amero, S.N., Bell, G., Wakeling, E.L., Kingsnorth, A., Stanier, P., Jauniaux, E., and Bennett, S.T. 2001. Evidence that insulin is imprinted in the human yolk sac. Diabetes 50: 199–203. [DOI] [PubMed] [Google Scholar]
- Oda, N., Nakai, A., Fujiwara, K., Imamura, S., Fujita, T., Hamagishi, M., Kato, T., Kobayashi, T., Himeno, Y., Yamamoto, K., et al. 2001. Polymorphisms of the insulin gene among Japanese subjects. Metabolism 50: 631–634. [DOI] [PubMed] [Google Scholar]
- Owerbach, D. and Gabbay, K.H. 1993. Localization of a type I diabetes susceptibility locus to the variable tandem repeat region flanking the insulin gene. Diabetes 42: 1708–1714. [DOI] [PubMed] [Google Scholar]
- Relethford, J.H. and Harpending, H.C. 1994. Craniometric variation, genetic theory, and modern human origins. Am. J. Phys. Anthropol. 95: 249–270. [DOI] [PubMed] [Google Scholar]
- Relethford, J.H. and Jorde, L.B. 1999. Genetic evidence for larger African population size during recent human evolution. Am. J. Phys. Anthropol. 108: 251–260. [DOI] [PubMed] [Google Scholar]
- Rotwein, P., Yokoyama, S., Didier, D.K., and Chirgwin, J.M. 1986. Genetic analysis of the hypervariable region flanking the human insulin gene. Am. J. Hum. Genet. 39: 291–299. [PMC free article] [PubMed] [Google Scholar]
- Rozas, J. and Rozas, R. 1999. DnaSP version 3: An integrated program for molecular population genetics and molecular evolution analysis. Bioinformatics 15: 174–175. [DOI] [PubMed] [Google Scholar]
- Sabeti, P.C., Reich, D.E., Higgins, J.M., Levine, H.Z., Richter, D.J., Schaffner, S.F., Gabriel, S.B., Platko, J.V., Patterson, N.J., McDonald, G.J., et al. 2002. Detecting recent positive selection in the human genome from haplotype structure. Nature 419: 832–837. [DOI] [PubMed] [Google Scholar]
- Sachidanandam, R., Weissman, D., Schmidt, S.C., Kakol, J.M., Stein, L.D., Marth, G., Sherry, S., Mullikin, J.C., Mortimore, B.J., Willey, D.L., et al. 2001. A map of human genome sequence variation containing 1.42 million single nucleotide polymorphisms. Nature 409: 928–933. [DOI] [PubMed] [Google Scholar]
- Schneider, S., Roessli, D., and Excoffier, L. 2000. Arlequin. Genetics and Biometry Laboratory, University of Geneva, Geneva, Switzerland.
- Seino, S., Bell, G.I., and Li, W.H. 1992. Sequences of primate insulin genes support the hypothesis of a slower rate of molecular evolution in humans and apes than in monkeys. Mol. Biol. Evol. 9: 193–203. [DOI] [PubMed] [Google Scholar]
- Smith, N.G. and Lercher, M.J. 2002. Regional similarities in polymorphism in the human genome extend over many megabases. Trends Genet. 18: 281–283. [DOI] [PubMed] [Google Scholar]
- Stead, J.D. and Jeffreys, A.J. 2000. Allele diversity and germline mutation at the insulin minisatellite. Hum. Mol. Genet. 9: 713–723. [DOI] [PubMed] [Google Scholar]
- Stead, J.D. and Jeffreys, A.J. 2002. Structural analysis of insulin minisatellite alleles reveals unusually large differences in diversity between Africans and non-Africans. Am. J. Hum. Genet. 71: 1273–1284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stead, J.D., Buard, J., Todd, J.A., and Jeffreys, A.J. 2000. Influence of allele lineage on the role of the insulin minisatellite in susceptibility to type 1 diabetes. Hum. Mol. Genet. 9: 2929–2935. [DOI] [PubMed] [Google Scholar]
- Stephens, M., Smith, N.J., and Donnelly, P. 2001. A new statistical method for haplotype reconstruction from population data. Am. J. Hum. Genet. 68: 978–989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strimmer, K. and von Haeseler, A. 1996. Quartet puzzling: A quartet maximum likelihood method for reconstructing tree topologies. Mol. Biol. Evol. 13: 964–969. [Google Scholar]
- Sved, J.A. 1971. Linkage disequilibrium and homozygosity of chromosome segments in finite populations. Theor. Popul. Biol. 2: 125–141. [DOI] [PubMed] [Google Scholar]
- Tajima, F. 1989. Statistical method for testing the neutral mutation hypothesis by DNA polymorphism. Genetics 123: 585–595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahata, N., Lee, S.H., and Satta, Y. 2001. Testing multiregionality of modern human origins. Mol. Biol. Evol. 18: 172–183. [DOI] [PubMed] [Google Scholar]
- Templeton, A.R., Clark, A.G., Weiss, K.M., Nickerson, D.A., Boerwinkle, E., and Sing, C.F. 2000. Recombinational and mutational hotspots within the human lipoprotein lipase gene. Am. J. Hum. Genet. 66: 69–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tishkoff, S.A. and Williams, S.M. 2002. Genetic analysis of African populations: Human evolution and complex disease. Nat. Rev. Genet. 3: 611–621. [DOI] [PubMed] [Google Scholar]
- Wood, W.I., Gitschier, J., Lasky, L.A., and Lawn, R.M. 1985. Base composition-independent hybridization in tetramethylammonium chloride: A method for oligonucleotide screening of highly complex gene libraries. Proc. Natl. Acad. Sci. 82: 1585–1588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu, A., Zhao, C., Fan, Y., Jang, W., Mungall, A.J., Deloukas, P., Olsen, A., Doggett, N.A., Ghebranious, N., Broman, K.W., et al. 2001. Comparison of human genetic and sequence-based physical maps. Nature 409: 951–953. [DOI] [PubMed] [Google Scholar]
WEB SITE REFERENCES
- http://lgb.unige.ch/arlequin; ARLEQUIN: software for population genetic data analysis.
- http://www.ncbi.nlm.nih.gov/SNP; dbSNP: for details of polymorphisms described in this study.
- http://www.ub.es/dnasp/;DnaSP software.
- http://www.ncbi.nlm.nih.gov/GenBank; GenBank, accession numbers for the insulin region: human L15440, AY138589, AY138590; chimpanzee AY137496, AY137497; gorilla AY137498, AY137499, AY137500; orangutan AY137501, AY137502, AY137503.
- http://www.stats.ox.ac.uk/mathgen/software.html; Mathematical Genetics Group software (for PHASE software).
- http://www.fluxus-engineering.com/sharenet.htm; NETWORK 2.0 software.
- http://www.ncbi.nlm.nih.gov/Omim; Online Mendelian Inheritance in Man (OMIM); for INS VNTR [MIM 125852] and LPL [MIM 238600].
- http://www.leicester.ac.uk/genetics/ajj/insulin; Authors' INS VNTR MVR and SNP database.
- http://www.tree-puzzle.de/; TREE-PUZZLE software.