

Abstract
Protein farnesylation and geranylgeranylation, together referred to as prenylation, are lipid post-translational modifications that are required for the transforming activity of many oncogenic proteins, including some RAS family members. This observation prompted the development of inhibitors of farnesyltransferase (FT) and geranylgeranyltransferase 1 (GGT1) as potential anticancer drugs. In this Review, we discuss the mechanisms by which FT and GGT1 inhibitors (FTIs and GGTIs, respectively) affect signal transduction pathways, cell cycle progression, proliferation and cell survival. In contrast to their preclinical efficacy, only a small subset of patients responds to FTIs. Identifying tumours that depend on farnesylation for survival remains a challenge, and strategies to overcome this are discussed. One GGTI has recently entered the clinic, and the safety and efficacy of GGTIs await results from clinical trials.
Interest in developing inhibitors of farnesylation as anticancer drugs was prompted by the realization more than 20 years ago that a sizable proportion of some, but not all, human cancers harbour activating oncogenic mutations in the RAS genes (between 8% and 93%, depending on the tumour type)1, and that RAS GTPases require this lipid post-translational modification (PTM) for their malignant transforming activity2. Furthermore, many of the signal transduction pathways that are activated by RAS involve proteins that require farnesylation or geranylgeranylation (together referred to as prenylation) for their ability to mediate tumour cell survival, growth, proliferation, migration and metastasis (FIG. 1). This, coupled with the fact that it has notoriously been difficult to design small GTPase inhibitors per se3, prompted a global quest to develop farnesyltransferase (FT) inhibitors (FTIs) and geranylgeranyltransferase 1 (GGT1) inhibitors (GGTIs) (together referred to as prenyltransferase (PT) inhibitors (PTIs)) as potential anticancer drugs.
Figure 1. RAS signalling pathways in mammalian cells.
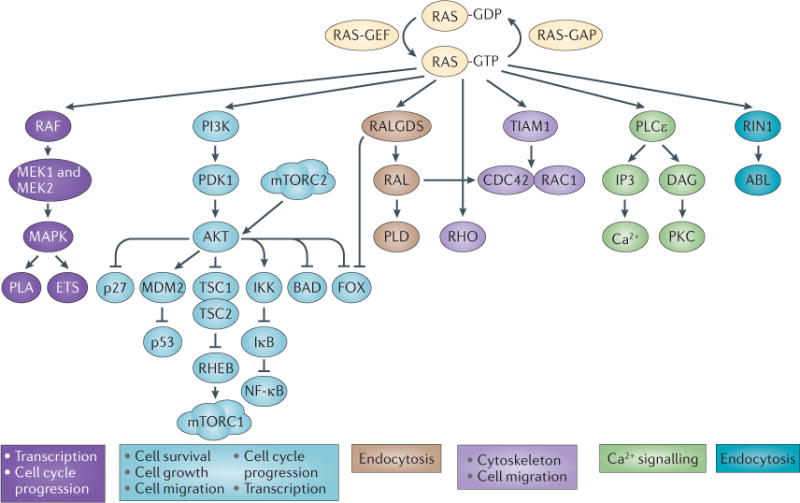
Active (farnesylated, membrane-bound and GTP-bound) RAS modulates a number of signalling pathways. Oncogenic RAS mutations tend to lock RAS in its GTP-bound state, resulting in constitutive RAS signalling. The major RAS effector pathways are shown. The two best-studied pathways that are activated by RAS are the RAF–MEK–MAPK signalling cascade and the PI3K–AKT pathway. The RAF–MEK–MAPK pathway ultimately activates the ETS family of transcription factors, which induce multiple genes that promote cell cycle progression and cell migration. Likewise, AKT phosphorylates multiple cellular proteins, leading to the inhibition of several tumour suppressors (such as p27, p53, tuberous sclerosis 1 (TSC1), TSC2 and BCL-2 antagonist of cell death (BAD)) or leading to the activation of several oncogene products. RAS also activates other small GTPases such as RALA and RALB, which have recently been shown to mediate RAS transformation in human pancreatic tumours, for example. Farnesyltransferase inhibitors (FTIs) were originally developed to block the function of RAS. However, as numerous studies in vitro and in vivo have shown, their antitumour activity is not correlated to the mutation status of KRAS isoforms. This suggests that the antitumour activity of FTIs relies on blocking the activity of other prenylated proteins. However, the inhibition of RAS protein function may still be important, particularly for tumours with mutant HRAS and tumours addicted to wild-type RAS. CDC42, cell division cycle 42; DAG, diacylglycerol; FOX, forkhead transcription factor; GAP, GTPase-activating protein; GEF, guanine nucleotide exchange factor; IKK, IκB kinase; IP3, inositol-1,4,5-trisphosphate; mTORC, mTOR complex; NF-κB, nuclear factor-κB; PDK1, phosphoinositide-dependent kinase 1; PKC, protein kinase C; PLA, phospholipase A; PLCε, phospholipase Cε; PLD, phospholipase D; RALGDS, RAL guanine nucleotide dissociation stimulator; RHEB, RAS homologue enriched in brain; RIN1, RAS and RAB interactor 1; TIAM1, T cell lymphoma invasion and metastasis 1.
Preclinical studies in the 1990s demonstrated that FTIs are highly successful in killing cancer cells in vitro and in animals with very little toxicity, thus generating much excitement and raising the hope that, finally, a RAS inhibitor may be developed as a novel anticancer drug. Contrary to expectations, however, responses to FTIs, whether in cells, animals or human patients, do not seem to depend on RAS mutations; and the inhibition of KRAS farnesylation leads to its geranylgeranylation (discussed below). Furthermore, in most clinical trials FTIs have not been as successful as expected, with no survival advantages, for example, to patients with advanced solid cancers4–6 or with acute myeloid leukaemia (AML)7. However, monotherapy with FTIs demonstrates antitumour activity in a subset of cancer patients, particularly those with haematological malignancies, whereas combinations of FTIs with cytotoxic agents improve the responses of patients with locally advanced breast cancer or other advanced solid tumours8–11. At present, we do not understand why some tumours are resistant while others are sensitive to FTIs. Clearly, the identification of the farnesylated proteins the inhibition of which is responsible for the antitumour effects of FTIs will lead to a better understanding of their mechanism of action and to the selection of patients whose tumours are sensitive to FTI treatment.
GGTIs as potential anticancer agents were developed for several reasons. First, KRAS (predominantly the alternatively spliced KRAS4B variant), which is the most frequently mutated isoform of RAS1, and NRAS become geranylgeranylated and remain fully functional when cells are treated with FTIs12–15. Second, in some human malignancies, such as pancreatic cancer with KRAS mutated in 90% of patients1, pathways that are mediated by geranylgeranylated proteins downstream of RAS, such as RALA and RALB, may be more relevant to oncogenesis than those mediated by MEK or AKT16,17. Third, the exclusively geranylgeranylated RHOC has an essential role in metastasis18,19. Fourth, the small GTPases cell division cycle 42 (CDC42) and RAC, which are exclusively geranylgeranylated, are crucial downstream targets for RAS-dependent transformation in rodent fibroblasts20,21. Furthermore, RAC1 is required to induce KRAS-driven lung cancer in mice22. Thus, GGTIs should be more efficient in cancer cells that are addicted to geranylgeranylated proteins, whereas FTI–GGTI combinations or dual prenylation inhibitors might be required to combat KRAS-dependent human tumours23. Similar to FTIs, GGTIs have shown promising results in vitro and in animal models, and one GGTI (GGTI-2418) has recently entered Phase I clinical trials24.
A better understanding of the aberrant signalling pathways that a given tumour is addicted to and the effects of PTIs on these pathways will lead to strategies that exploit the vulnerabilities of individual tumours and ultimately to predicting which patient populations are most likely to respond to PTIs, either alone or in combination. Work has begun in pursuing this goal with the identification of a two-gene expression ratio that potentially predicts the response of patients with AML to the FTI tipifarnib (also known as R115777)25,26 (discussed below).
General aspects of protein prenylation
The importance of FT and GGT1 for normal physiology and tumorigenesis: lessons from knockout mice
The biochemistry of prenylation and pioneering findings in the field of prenylation research are summarized in BOX 1 and the TIMELINE, respectively. Protein prenylation is required for the membrane localization of otherwise cytosolic proteins. Studies in yeast27 and mammalian cells28 suggest that protein prenylation is also required for the normal function of at least some proteins. Additionally, defective prenylation has been attributed to the pathogenesis of several diseases other than cancer (BOX 2) Contemporary reviews have estimated that several hundred proteins are subject to prenylation29,30. Therefore, the inhibition of FT activity probably prevents many proteins from functioning properly, and it is perhaps not surprising that genetic disruption of the catalytic FT β-subunit (Fntb) in mice causes embryos to die very early in development31 (TABLE 1). Whether the constitutive ablation of GGT1 activity in mammals is also embryonically lethal has not yet been determined. However, studies conducted in Saccharomyces cerevisiae32, Drosophila melanogaster33 and mouse embryonic fibroblasts34, as well as the fact that more proteins are geranylgeranylated than are farnesylated35, suggest that GGT1 function is essential for survival and development, and that the functions of FT and GGT1 are not redundant.
Box 1. Biochemistry of protein prenylation.
Prenylation is a universal lipid post-translational modification (PTM) of cysteine residues near the carboxyl terminus that facilitates membrane association174,175. Using farnesyl diphosphate (FPP) or geranylgeranyl diphosphate (GGPP) as the lipid donor, this enzymatically catalysed PTM transfers either a C15 farnesyl (F) or a C20 geranylgeranyl (GG) group to the sulphydryl group of the cysteine residue, thus forming covalent thioether bonds. In eukaryotic cells, prenylation is catalysed by three ’housekeeping‘ enzymes, farnesyltransferase (FT), gernaylgeranyltransferase 1 (GGT1) and GGT2. We focus on FT and GGT1. FT and GGT1 are cytosolic heterodimeric proteins that share a common α-subunit176, but that have homologous but distinct β-subunits177. The crystal structure of FT shows a crescent-shaped helical hairpin domain and an α–α barrel domain178. Both FT and GGT1 are metalloenzymes that require zinc for catalysing the covalent binding of the prenyl group carbon to the CaaX cysteine thiol179.
Enzyme kinetics and other biochemical studies indicated that FPP first binds to FT, which is followed by the binding of the protein substrate, with prenylation of the substrate occurring much faster than the release of the farnesylated protein product. Proteins modified by FT or GGT1 seem to share a conserved C-terminal CaaX recognition motif (in which C is cysteine, a is an aliphatic amino acid and X is variable). The nature of the C-terminal residue X specifies whether a protein is a substrate for FT or for GGT1: whereas FT prefers X to be methionine, serine, glutamine or cysteine, GGT1 prefers X to be leucine or isoleucine180. However, these rules are not absolute: for example, a CaaX protein with a C-terminal phenylalanine can be farnesylated or geranylgeranylated181. And although GGT1 clearly prefers X to be leucine182, some CaaL motifs can also be farnesylated by FT183. Furthermore, at least one protein, RHOB (CaaX box sequence: CKVL), is naturally both farnesylated and geranylgeranylated35. Proteins with a C-terminal CaaX box can undergo up to three additional PTMs (see part a of the figure). Some proteins such as KRAS-4B (CaaX box sequence: CVIM) and NRAS (CaaX box sequence: CVVM) are naturally only farnesylated, but can be geranylgeranylated and remain fully functional in the presence of an FTI (see part b of the figure). To block KRAS function would thus require the inhibition of both FT and GGT1 (REFS 12–15). It is not known how many other farnesylated proteins can be cross-prenylated by GGT1, but the presence of a C-terminal methionine seems to be important for the ability of proteins to undergo cross-prenylation.
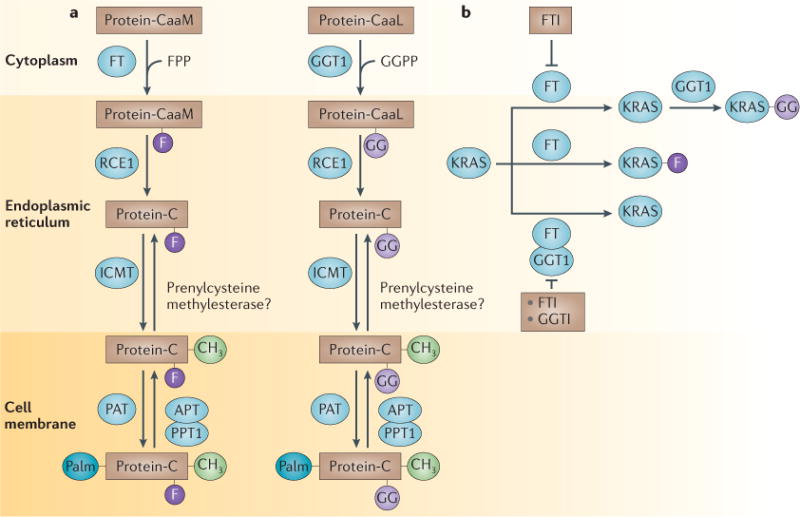
APT, acylprotein thioesterase; ICMT, isoprenylcysteine methyltransferase; PAT, protein acyltransferase; PPT1, palmitoylthioesterase 1; RCE1, RAS-converting enzyme 1.
Timeline.
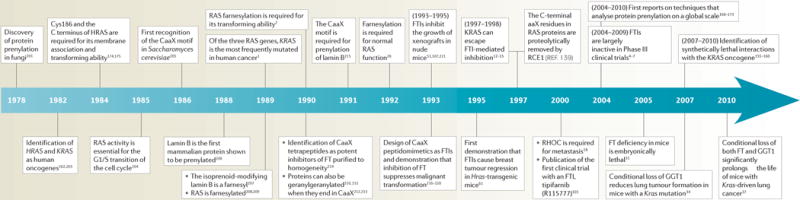
Protein prenylation and human cancer
FT, farnesyltransferase; FTI, FT inhibitor; GGT1, geranylgeranyl transferase 1; RCE1, RAS-converting enzyme 1.
Box 2. The use of PTIs in other diseases.
An unexpected benefit of farnesyltransferase (FT) inhibitors (FTIs) may be their potential to treat diseases other than cancer. Hutchinson–Gilford progeria syndrome (HGPS) is a genetic disease that is associated with premature ageing and death, normally from heart failure, at about 13 years of age. HGPS is linked to a mutation in LMNA that prevents prelamin A (a substrate for FT) from proper maturation into lamin A. Normal processing of prelamin A involves, among other steps, the removal of a carboxy-terminal peptide containing farnesylcysteine. Children with HGPS are unable to perform this step, which results in a mutant form of lamin A, termed progerin184, which is persistently farnesylated and non-functional185. In fibroblasts derived from patients with HGPS and mice, as well as HeLa cells, FTIs can prevent the aberrant nuclear morphology of cells that express progerin186–188. In mouse models of HGPS, FTIs greatly improve the phenotype of HGPS with respect to lifespan, body weight and bone integrity189. Furthermore, the crucial target for FTI treatment was confirmed to be progerin190. These results suggest that FTIs may be beneficial for children with HGPS, and lonafarnib is currently being tested in clinical trials.
A common problem in the treatment of cardiovascular diseases is the hyperproliferation of smooth muscle cells, as seen in restenosis (intimal hyperplasia) of coronary arteries following balloon angioplasty or bypass surgery. Local administration of FTIs or geranylgeranyl transferase inhibitors (GGTIs) can prevent restenosis by blocking neointima formation191. GGTIs may also assist in the therapy of cardiovascular diseases by increasing nitric oxide synthase expression192.
Parasitic diseases such as malaria, Chagas disease, African sleeping sickness, Toxoplasmosis and Leishmaniasis cause millions of deaths in tropical and subtropical regions, and the therapeutic potential of FTIs for these diseases has also been explored193. FTIs specifically designed to inhibit parasitic FT and not mammalian FT are significantly more toxic to parasitic protozoa194,195. Recently, a GGT1 from Trypanosoma cruzi, the parasite responsible for Chagas disease, was cloned196, and GGTIs may also be effective against these diseases.
FTIs also show strong antiviral activity in mice infected with the hepatitis δ virus197. Other diseases that may also be amenable to therapy with prenyltransferase (PT) inhibitors (PTIs) are multiple sclerosis198 and metabolic bone disorders199, as well as a wide variety of undesirable fibrotic reactions200.
Table 1.
Effects of PT-targeted gene deletions
| Affected gene product | Animal model | Type of gene deletion | Tissue | Phenotypes | Refs |
|---|---|---|---|---|---|
| GGT1 β-subunit | Saccharomyces cerevisiae | Constitutive | NA | Lethal | 32 |
|
| |||||
| GGT1 β-subunit | Drosophila melanogaster | Constitutive | NA | Lethal | 33 |
|
| |||||
| FT β-subunit | Mouse | Constitutive | NA | Embryonically lethal | 31 |
|
|
|||||
| Conditional | Lung | 31 | |||
|
|
|||||
| Conditional | Lung | Lack of HRAS association with cell membranes
|
37 | ||
| Delay of KRAS-induced lung tumours | |||||
| Conditional | Haematopoietic cells | Reduced severity of MPD | 38 | ||
|
| |||||
| GGT1 β-subunit | Mouse | Conditional | Lung |
|
34 |
|
| |||||
| FT β-subunit and GGT1 β-subunit | Mouse | Conditional | Lung |
|
37 |
FT, farnesyltransferase; GGT1, geranylgeranyltransferase 1; MPD, myeloproliferative disease; NA, not applicable; PT, prenyltransferase.
Owing to a ‘leaky’ null allele these results have been called into question36.
More interesting in this context are the results obtained with conditional PT deletions. The first such study by Barbacid and colleagues31 suggested that FT is not required for tumour initiation in mice, either in mice developing KRAS-G12V-induced lung adenocarcinoma or in mice subjected to carcinogen-induced skin carcinoma, but that it is required for tumour progression and maintenance31. The study also suggested that adult mice lacking Fntb show normal tissue homeostasis except for slight defects in wound healing or liver regeneration. Finally, in these conditional FT-knockout mice, the authors suggested that wild-type HRAS still associates with cellular membranes31. Recently, these unexpected results have been called into question by Yang et al.36 who re-analysed the conditional Fntb-null allele generated by the Barbacid laboratory: it produced a transcript that encoded a protein with a short in-frame deletion rather than, as expected, a transcript with a frameshift mutation that resulted in a true null allele. Thus, the results described above may have been due to a ‘leaky’ null allele that permitted the expression of partially active FT.
Two other recent studies by Bergo and colleagues have shown that conditional Fntb deficiency37 or conditional Pggt1b (which encodes the catalytic β-subunit of GGT1) deficiency34 reduces the formation of KRAS-G12D-induced lung cancer in mice. Furthermore, simultaneous knockout of both Fntb and Pggt1b has a far greater effect on KRAS-G12D-induced lung tumour onset and progression than either deletion alone37. Also, loss of both Fntb and Pggt1b significantly extends the lifespan of mice that express activated KRAS-G12D in their lungs, which validates FT and GGT1 as important targets for cancer therapy. In contrast to the earlier study by Barbacid et al.31, Bergo and colleagues demonstrated that Fntb transcripts were not detectable, and HRAS does not associate with cell membranes in the absence of FT37, confirming earlier cell-based studies.
Bergo and colleagues have also recently described the effect of GGT1 deficiency in the haematopoietic system38. Mice harbouring an inducible KrasG12D oncogene in haematopoietic cells develop a lethal myeloproliferative disease (MPD), and up to one-third also develop acute lymphoblastic leukaemia (ALL). Whereas the absence of GGT1 markedly reduced the severity of MPD, it had no effect on ALL38. As KRAS can be farnesylated in the absence of GGT1, the antitumour effects of GGT1 depletion with regards to MPD can be attributed to defective prenylation of other GGT1 targets38 that are downstream of KRAS, such as RALA and RALB.
Regulation of PTs
Given the importance of PTs, it is surprising that their regulation in response to external signals has not been investigated in great detail. The α-subunit of FT and GGT1, FNTα, has been shown to be phosphorylated in a transforming growth factor-β (TGFβ)-dependent manner, which either does not affect39 or decreases FT activity40. FNTβ was also shown to be phosphorylated41, raising the possibility that both of these events are necessary to modulate FT activity. Furthermore, insulin stimulates activating phosphorylation of FNTα by a member of the RAF1–MEK–MAPK pathway42.
FNTα is cleaved by caspase 3 during apoptosis43, suggesting that some signals may induce apoptosis by indirectly inhibiting the prenylation and the function of proteins that are involved in cell survival. Interestingly, dietary fish oil, which contains high levels of Ω3 polyunsaturated fatty acids, inhibits the expression of FT and colon tumorigenesis in rats44, which is consistent with the observation that FT activity is increased in human colon cancer45. These findings also raise the possibility that FTIs may function as chemopreventive agents, an idea that has received experimental support in mouse models of lung cancer46,47.
Mechanism of action of FTIs
The observation that FT and GGT1 may be dispensable for adult tissue homeostasis, but may be required for KRAS-driven tumorigenesis34,37, further validated the concept of developing PTIs as novel anticancer drugs. Several strategies, including structure-based drug design and high-throughput screens, were used to identify a variety of PTIs (see Supplementary information S1 (table) for structures and potencies of representative compounds).
Depending on the context, the treatment of cancer cells with FTIs results in the induction of apoptosis, cell cycle arrest and the inhibition of anchorage-dependent and anchorage-independent cell proliferation, cell migration and angiogenesis (see Supplementary information S2 (table)). The exact mechanisms by which FTIs induce the antitumour effects described above are unknown mainly because the identity of the crucial farnesylated proteins, the inhibition of which mediates these FTI effects, is unknown (discussed further below). However, several studies have shown that FTIs affect oncogenic and survival signal transduction pathways, which can explain some of their antitumour effects.
Cell proliferation
FTIs inhibit signalling pathways that are involved in anchorage-dependent and anchorage-independent proliferation. In well-defined systems in which mutant HRAS drives the transformation of NIH3T3 cells, FTIs inhibit the farnesylation of HRAS, prevent its association with the plasma membrane, inhibit downstream signal transduction pathways such as RAF–MEK–MAPK48 and inhibit tumour growth15. In human tumour cell lines with multiple genetic alterations, FTIs inhibit PI3K–AKT signalling, particularly in ovarian and pancreatic cancer cells that overexpress AKT2 (REF. 49), although this seems to be context-specific, as, in lung cancer cells with low or undetectable levels of phospho-AKT, lonafarnib (also known as SCH66336)-induced apoptosis does not rely on AKT inhibition50. In nude mice, many FTIs inhibit the growth of human tumours harbouring a variety of genetic alterations, including KRAS mutations, TP53 deletions and silenced cyclin-dependent kinase inhibitor 2A (CDKN2AINK4A)51. Similarly, in transgenic mice that express mutant KRAS or that overexpress wild-type NRAS, FTIs only inhibit tumour growth and do not induce tumour regression52,53. However, in mutant Hras-transgenic mice, FTIs cause tumour regression (see below) (see Supplementary information S3 (table)).
Cell cycle progression
FTIs primarily accumulate cells at prometaphase by preventing bipolar spindle formation and chromosome alignment54,55, which may rely on the inhibition of the farnesylation of the centromere-associated protein E (CENPE) and CENPF56,57, as well as phosphatase of regenerating liver (PRL) protein tyrosine phosphatases (PTPs)58. However, in some human cancer cell lines FTIs can induce G1 phase arrest. For example, L-744,832 induces p21 accumulation and inhibition of RB phosphorylation59. Similarly, in Rat1 fibroblasts transformed with HRASG12V, the FTI HR-12 causes the accumulation of p27, which results in the inhibition of cyclin-dependent kinase 2 (CDK2) and subsequent G1 arrest60. This compound inhibits both anchorage-dependent and anchorage-independent cell growth and blocks cell motility in wound healing assays. Rat1 cells transformed with myristoylated HRASG12V are resistant to HR-12 (REF. 60), indicating that the inhibition of HRAS farnesylation is responsible for HR-12 effects.
Apoptosis
FTIs cause breast tumour regression very effectively in mutant Hras-transgenic mice61 that express Myc or that lack Trp53, but not in Erbb2-transgenic mice62, suggesting that these drugs may induce apoptosis, which was confirmed by studies in cultured cells. For example, FTIs inhibit integrin-mediated and growth factor-mediated activation of the PI3K–AKT pathway, which results in the dephosphorylation of AKT substrates, including the pro-apoptotic BCL-2 family member BCL-2 antagonist of cell death (BAD), which leads to its activation. In this setting, overexpression of constitutively active AKT2 rescues FTI-induced apoptosis49. However, in other cells, FTIs induce apoptosis only when deprived of growth factors or of substratum attachment, suggesting that growth factors and integrins can rescue FTI-induced apoptosis63–65. FTIs can also induce apoptosis by enhancing death receptor signals66 or by inhibiting nuclear factor-κB (NF-κB)-dependent induction of cyclin D1, survivin, inhibitor of apoptosis proteins (IAPs) and BCL-2 (REF. 67). Another FTI that has been explored in preclinical and clinical studies, BMS-214662, has pro-apoptotic activity and induces tumour regression in nude mouse xenografts, but this seems to be related to the inhibition of GGT2 and not FT or GGT1 (REF. 68).
Angiogenesis
Lonafarnib inhibits angiogenesis in lung and head and neck tumour cells by decreasing hypoxia and insulin-like growth factor 1 (IGF1)-stimulated hypoxia inducible factor 1α (HIF1α) expression69. This study also showed that lonafarnib inhibits vascular endothelial growth factor A (VEGFA) production by inhibiting HIF1α binding to heat shock protein 90 (HSP90), which results in the degradation of HIF1α. Consistent with this, other FTIs, such as L-744,832 (REF. 70) and tipifarnib71, affect angiogenesis, possibly by inhibiting HIF1α expression and hypoxia. Finally, another FTI, LB42708, inhibits angiogenesis, possibly by inhibiting pathways that are mediated by MAPK and AKT72.
Combinations
In cell culture, structurally unrelated FTIs can enhance the growth inhibitory and apoptotic effects of radiation73, taxanes74,75, cisplatin76,77, 5-fluorouracil78, MEK inhibitors79, CDK inhibitors80 and the breakpoint cluster region (BCR)–ABL inhibitor imatinib (also known as STI-571)81. In nude mouse xenografts, beneficial combinations have been reported for lonafarnib and cytotoxic agents such as cyclophosphamide, 5-fluorouracil, vincristine82 and paclitaxel83, or for FTI-2148 and paclitaxel, cisplatin and gemcitabine84. We have recently found that a combination of tipifarnib and the inhibitor of AKT activation triciribine phosphate (TCNP)85 (but not the single agents alone) causes breast tumour regression in Erbb2-transgenic mice86.
Several studies by Giannakakou and colleagues have recently provided a mechanistic explanation for the commonly observed synergy between FTIs and taxanes. First, they showed that lonafarnib in combination with paclitaxel enhances tubulin acetylation more than the effect of either drug alone, and that this is due to the inhibition of histone deacetylase 6 (HDAC6)87, an enzyme that functions as a tubulin deacetylase and that is involved in stress response, microtubule stability and cell migration. FTIs increase the amount of microtubule-bound paclitaxel, even in cells that are resistant to paclitaxel alone, and this is dependent on functional HDAC6 (REF. 88). Finally, FT and HDAC6 physically associate with each other at microtubules, and FTIs induce the dissociation of FT from microtubules, resulting in the inhibition of HDAC6 activity, an effect that was duplicated by stable knockdown of FNTα using short hairpin RNA (shRNA)89. Most interestingly, HDAC6 does not contain a carboxy-terminal CaaX motif and is thus a very unlikely FT substrate. It is possible that other FT targets that are present in the microtubule–protein complex mediate FT regulation of HDAC6.
GGTI effects in cultured cells and in vivo
Similar to FTIs, GGTIs induce apoptosis and inhibit tumour cell growth, both in cultured cells and in animal models (see Supplementary information S3 (table)). In contrast to FTIs, GGTIs result in G1 arrest and not mitotic arrest90. Their ability to induce G1 arrest may be due to inducing the expression of the CDK inhibitors p21 and p27, inhibition of CDK2 and CDK4, and hypophosphorylation of RB91. GGTIs induce the accumulation of p27 in the nucleus through the inhibition of CDK2-mediated phosphorylation of Thr187 in p27, and this is important for their ability to induce tumour cell death92. Tamanoi and colleagues recently described P61-A6, a GGTI that causes G1 arrest, probably (at least in part) by inhibiting RHOA geranylgeranylation and inducing p21 expression (REFS 93,94). Furthermore, GGTI-induced apoptosis may also depend on their ability to reduce the levels of phosphorylated and thus activated AKT and survivin95. GGTIs can also induce apoptosis by increasing death receptor 5 (DR5; also known as TNFRSF10B) expression, decreasing cellular FLICE-like inhibitory protein (cFLIP; also known as CFLAR) expression and enhancing TNF-related apoptosis-inducing ligand (TRAIL)-induced apoptosis in human non-small-cell lung cancer (NSCLC) cells96. In addition, GGTIs inhibit platelet-derived growth factor (PDGF)-stimulation of PDGF receptor (PDGFR)-mediated tyrosine phosphorylation and MAPK signalling, suggesting that PDGFR phosphorylation is mediated by a GGT1 substrate97.
The ability of our lead GGTI, GGTI-2418 (currently in Phase I clinical trials), to inhibit anchorage-dependent and anchorage-independent growth may also depend on its ability to inhibit the geranylgeranylation of RALB or RALA, respectively98. In addition, GGTI-2418 effectively prevents xenograft tumour growth in nude mice and results in breast tumour regression in Erbb2-transgenic mice92. Casey and colleagues described GGTI-DU40 (REF. 99), which is a highly potent and selective GGT1 inhibitor that inhibits the prenylation of several cellular proteins, including RHO GTPases. In MDA-MB-231 breast cancer cells, GGTI-DU40, but not the FTI L-744,832, inhibits thrombin-induced cell rounding. GGTIs with novel scaffolds were recently identified by a virtual screen of 9.5 million compounds in conjunction with quantitative structure–activity relationship modelling100.
FTIs in the clinic
Starting in 2000 (REF. 101), four FTIs have been evaluated in at least 75 clinical trials: tipifarnib, lonafarnib, BMS-214662 and L-778123. In 64 of these studies (with 35 being Phase I trials), the clinical response has been determined (TABLES 2,3; see Supplementary information S4 and S5 (tables)). Sixteen years after curing transgenic mice that developed mouse mammary tumour virus (MMTV)–Hras-driven breast tumours with FTIs61, and after accumulating other very impressive preclinical data, it has become clear that in human clinical trials, monotherapy with FTIs shows limited antitumour activity in haematopoietic cancers, and generally no or very little activity in solid tumours. Thirty-eight of the 64 clinical trials (59%) concerned tipifarnib, either alone or in combination with other agents. Eighteen of the 64 trials (28%) — all but one of which was conducted in patients with solid tumours — reported no objective responses. Seventeen of the 18 trials (94%) with no objective responses were conducted with FTI monotherapy. Twenty-three of the 64 trials (36%) conducted in both solid and haematological malignancies showed very little antitumour activity (the rate of objective responses was <15%). The median objective response was 2.3% for monotherapy with FTIs and 11.4% for combinations with FTIs. Furthermore, in 28 of the 64 trials (44%), FT activity or the prenylation status of marker proteins (most commonly HDJ2 (also known as DNAJA1) or prelamin A) was assessed to determine whether the FTI treatment had affected its intended targets. There was no correlation between FT inhibition and clinical responses. Most importantly, two FTIs that have so far advanced to Phase III clinical trials are lonafarnib6 and tipifarnib4,5,7, and, unfortunately, these drugs were unable to improve the outcome for advanced pancreatic cancer4, advanced colon cancer5, advanced NSCLC6 or AML7, whether alone7,5 or in combination with carboplatin and paclitaxel6 or with gemcitabine4.
Table 2.
Clinical trials with FTIs*
| Disease | Phase | Patients (median age) | Clinical response and number of patients‡ | FT activity or prenylation | Response rate (%) | Other comments |
|---|---|---|---|---|---|---|
| Tipifarnib | ||||||
| Acute leukaemia | I | 34 (63) | 2 CR and 8 PR | FT ↓§ and HDJ2 ↓ | 29.4 | No NRAS mutations in patient tumours |
| Advanced bladder cancer | II | 34 (64) | 2 PR and 13 SD | ND | 5.9 | Response rate does not warrant further investigation |
| Advanced breast cancer | II | 76 (54) | 9 PR and 9 SD | ND | 11.8 | All responders had wild-type RAS genes |
| Advanced colon cancer | II | 55 (69) | 1 PR, 11 SD, and 31 PD and MD | ND | 1.8 | Tipifarnib is ineffective |
| Advanced NSCLC | II | 44 (71) | 7 SD | HDJ2 ↓ and prelamin A ↓. FT ↓ in 83% of patients | 0 | No objective response. Future studies should be done with combinations |
| Advanced solid tumours | I | 25 (58) | 0 CR, 0 PR, 8 SD, and 17 PD and MD | Data not shown | 0 | No objective response |
| Advanced solid tumours | I | 9 (53) | 1 SD, and 8 PD and MD | ND | 0 | No objective response |
| Advanced solid tumours | I | 28 (56) | 2 PR and 3 SD | ND | 7.1 | 5 of 15 patients had KRAS mutations |
| Advanced solid tumours | I | 21 | 6 SD | ND | 0 | No objective response. Phase II trial recommended |
| AML | II | 252 (62) | 11 CR and 8 PR | ND | 7.5 | MS for patients with CR: 369 days |
| AML | II | 145 (74) | 22 CR, 3 PR, 50 SD and 58 PD and MD | HDJ2 ↓ | 17.2 | Median duration of CR: 7.3 months |
| AML | III | 228 (76) | 18 CR, 20 PR, 105 SD, and 36 PD and MD | ND | 16.7 | MS: 107 days. 8% of patients had a CR with an MS of 666 days |
| 229‖ | 0 CR, 3 PR, 130 SD, and 46 PD and MD | 1.3 | MS: 109 days | |||
| Brain tumours | II | 81 (11) | 2 PR | ND | 2.5 | Very little activity |
| CML, myelofibrosis and multiple myeloma | 40 (57) | 7 CR and 7 PR | ND | 17.5 | Clinical activity in CML and myelofibrosis | |
| Metastatic pancreatic cancer | II | 20 (61) | 1 SD | FT ↓ by 50% and HDJ2 ↓ by 33% | 0 | No objective response. MS: 19.7 weeks |
| Multiple myeloma | II | 36 (62) | 0 CR, 0 PR, 23 SD, and 13 PD and MD | FT ↓ and HDJ2 ↓ | 0 | No objective response. No correlation between FT ↓ and disease stabilization |
| MDS | I | 20 (66) | 1 CR, 5 PR, and 1 PD and MD | FT ↓ and HDJ2 ↓ | 30 | No correlation between FT ↓ and response. No correlation between RAS mutation status and response |
| MDS | II | 27 (66) | 2 CR and 1 PR | ND | 11.1 | Modest antitumour activity |
| MDS | II | 82 (67) | 26 PR and 37 SD | ND | 31.7 | Median duration of CR: 11.5 months |
| MDS | I | 61 (68) | 3 CR and 13 PR | FT ↓ by 75% | 26.2 | Only one responder with a KRAS mutation. No correlation between FT ↓ and dose |
| Neuro-fibromatosis and neurofibromas | I | 40 (≤15) | FT ↓ by 43% and HDJ2 ↓ | 0 | No objective response | |
| Pancreatic cancer | II | 53 (65) | ND | 0 | No objective response. MS: 2.6 months | |
| Small-cell lung cancer | II | 20 (62) | 1 SD | ND | 0 | No objective response. MS: 6.8 months. Progression-free MS: 1.4 months |
| Advanced colon cancer | III | 235 (61) | 0 CR, 1 PR, 57 SD, and 155 PD and MD | ND | 0.4 | MS: 174 days. SD >3 months: 24% |
| 133¶ (62) | 0 CR, 0 PR, 17 SD, and 107 PD and MD | 0 | MS: 185 days. SD >3 months: 13% | |||
| Lonafarnib | ||||||
| Advanced solid tumours | I | 24 (57) | 0 CR, 0 PR and 2 SD | ND | 0 | No objective response |
| Advanced solid tumours | I | 12 (61) | 0 CR and 0 PR | Prelamin A ↓ | 0 | No objective response |
| Advanced solid tumours | I | 22 (54) | 1 CR and 1 PR | FT ↓ | 12.5# | Sponsor terminated study early owing to negative interim efficacy. FT ↓ not correlated with response |
| Advanced solid tumours | II | 15 (57) | 0 CR, 0 PR and 7 SD | ND | 0 | No objective response |
| CML | Pilot | 13 (62) | 0 CR and 2 PR | ND | 15.5 | |
| CNS tumours | I | 48 (12) | 0 CR, 1 PR and 9 SD | ND | 2.1 | |
| Metastatic colon cancer | II | 21 (64) | 0 CR, 0 PR and 3 SD | ND | 0 | No objective response |
| MDS or sAML | II | 16 (70) | 1 PR | ND | 6.7 | |
| NSCLC | II | 29 (58) | 3 PR, 11 SD, and 15 PD and MD | ND | 10.3 | MS: 39 months. Well-tolerated. Further clinical trials recommended |
| Refractory urothelial cancer (transitional cell carcinoma) | II | 10 (65) | 0 CR, 0 PR, 2 SD, and 8 PD and MD | Small HDJ2 ↓ | 0 | No objective response |
| Solid tumours | I | 20 (59) | 1 PR and 8 SD | Prelamin A ↓ | 5.0 | |
| BMS-214662 | ||||||
| Acute leukaemia | I | 30 (53) | 4 CR and 1 PR | Short-lived FT ↓ | 16.7 | |
| Advanced solid tumours | I | 44 (54) | 0 CR, 0 PR and 1 SD | Transient FT ↓ by 89.5% | 0 | No objective response. One patient with pancreatic cancer survived for >3.5 years |
| Advanced solid tumours | I | 68 (60) | 0 CR, 0 PR and 5 SD | Short-lived FT ↓ | 0 | No objective response |
| Advanced solid tumours | I | 19 (55) | 1 SD, and 18 PD and MD | ND | 0 | No objective response |
| Solid tumours | I | 25 (57) | 1 PR, 16 SR, and 8 PD and MD | Short-lived FT ↓ | 4.0 | Response was minor |
AML, acute myeloid leukaemia; CML, chronic myeloid leukaemia; CNS, central nervous system; CR, complete response; FNTB, farnesyltransferase β-subunit; FT, farnesyltransferase; FTIs, FT inhibitors; GGTI, geranylgeranyltransferase 1 inhibitor; HI, haematological improvement; MD, metastatic disease; MDS, myelodysplastic syndrome; MS, median survival; ND, not determined; NSCLC, non-small-cell lung cancer; OS, overall survival; PD, progressive disease; PR, partial response; sAML, secondary acute myeloid leukaemia; SD, stable disease.
See Supplementary information S4 (table) for a table with references. This table only considers FTIs because, to our knowledge, only one GGTI, GGTI-2418, is currently in clinical trials. Studies that did not evaluate tumour response are not included. Median ages are rounded to the closest integer. The number of evaluable patients is stated whenever possible. The response rate was calculated by dividing the sum of complete and partial responses by the number of evaluable patients.
PR includes haematological improvement.
Downward arrows indicate a reduction of enzyme activity (in the case of FT) or a reduction in farnesylation (in the case of HDJ2 or prelamin A).
This patient cohort received best supportive care.
In this study all patients received best supportive care, with 235 receiving tipifarnib and 133 receiving a placebo.
Number differs from the authors’ calculation as they included SD, resulting in a response rate of 37.5%.
Table 3.
Clinical trials with combinations including FTIs*
| Drugs | Disease | Phase | Patients (median age) | Clinical response and number of patients‡ | FT activity or prenylation | Response rate (%) | Other comments |
|---|---|---|---|---|---|---|---|
| Tipifarnib + capecitabine | Advanced solid tumours | I | 41 (57) | 5 PR and 11 SD | FT ↓§ and HDJ2 ↓ | 12.2 | No correlation between FT ↓ and response |
| Tipifarnib + doxorubicin + cyclophosphamide | Advanced breast cancer | I and II | 32 (51) | 7 CR | FT ↓ by 55–100% | 21.9 | |
| Stage IIB–IIIC breast cancer | II | 44 (51) | 11 CR | Median FT ↓ by 91% | 25 | ||
| Tipifarnib + etoposide | AML | I | 84 (77) | 20 CR | p-S6 ↓ | 23.4 | |
| Tipifarnib + gemcitabine | Advanced solid tumours | I | 19 (59) | 2 PR | HDJ2 ↓ | 10.5 | |
| Tipifarnib + gemcitabine | Advanced pancreatic cancer | III | 341 (61) | 6 CR, 6 PR, 53 SD, and 28 PD and MD | ND | 1.8 | MS: 193 days. 6-month survival: 53%. 1-year survival: 27% |
| 347‖ (62) | 8 CR, 8 PR, 52 SD, and 30 PD and MD | ND | 2.3 | MS: 182 days. 6-month survival: 49%. 1-year survival: 24% | |||
| Tipifarnib + gemcitabine + cisplatin | Advanced solid tumours | I | 27 (58) | 1 CR and 8 PR | Prelamin A ↓ | 33.3 | |
| Advanced solid tumours | I | 31 (58) | 8 PR and 12 SD | ND | 25.8 | Phase II trial recommended | |
| Tipifarnib + idarubicin + cytarabine | AML and MDS | I/II | 95 (50) | 61# CR and 9 PR | ND | 74 | MS: 17 months |
| 108¶ (52) | 65# | 70 | MS: 13 months | ||||
| Tipifarnib + imatinib | CML | I | 25 (62) | 17 PR | ND | 68 | 11 patients withdrew (lack of response) |
| Tipifarnib + irinotecan | Solid tumours | I | 35 (52) | 3 PR, 14 SD, and 13 PD and MD | ND | 8.6 | |
| Tipifarnib + letrozole | Advanced breast cancer | II | 74 (60) | 3 CR, 19 PR, 29 SD, and 23 PD and MD | ND | 29.7 | No improvement by tipifarnib |
| 39‖ (61) | 1 CR, 14 PR, 15 SD, and 9 PD and MD | ND | 38.5 | ||||
| Tipifarnib + sorafenib | Advanced solid tumours | I | 43 (56) | 3 PR, 15 SD, and 20 PD and MD | 25% of patients with >50% FT ↓ | 7.0 | |
| Tipifarnib + tamoxifen | Metastatic breast cancer | I | 12 (50) | 2 PR and 1 SD | FT ↓ 42–54% | 16.7 | |
| Lonafarnib + carboplatin + paclitaxel | Advanced NSCLC | III | 308 (unknown) | NA | NA | OS: 144 days. TTP: 137 days | |
| 308‖ (unknown) | NA | OS: 168 days. TTP: 152 days | |||||
| Lonafarnib + docetaxel | Advanced solid tumours | I | 29 | 1 CR and 6 SD | 3.4 | Response (CR + SD) correlates with low FNTB mRNA levels | |
| Lonafarnib + gemcitabine | Advanced bladder cancer | II | 31 (64) | 1 CR and 9 PR | ND | 32.3 | MS: 11.5 months. TTP: 7 months |
| Lonafarnib + imatinib | CML | I | 23 (55) | 6 CR and 2 PR | ND | 34.7 | |
| Lonafarnib + paclitaxel | Solid tumours | I | 21 (60) | 6 PR | ND | 28.6 | 6 patients previously treated |
| BMS-214662 + cisplatin | Advanced solid tumours | I | 23 (57) | 15 SD | Short-lived FT ↓ | 0 | No objective response |
| BMS-214662 + paclitaxel | Advanced solid tumours | I | 26 (60) | 2? PR | Short-lived FT ↓ | ≤7.7 | |
| BMS-214662 + paclitaxel + carboplatin | Advanced solid tumours | I | 30 (58) | 3 PR and 8 SD | Short-lived FT ↓ and HDJ2 ↓ | 10.0 | No correlation between dose and HDJ2 ↓ |
| L-778,123 + radiotherapy | Advanced solid tumours | I | 7 (59) | 5 CR, 1 PR, and 6 PD and MD | ND | 85.7 | No RAS mutations |
| Advanced pancreatic cancer | I | 10 (59) | 1 PR, 5 SD, and 4 PD and MD | HDJ2 ↓ | 10.0 | 3 out of 4 patients examined have KRAS mutations |
AML, acute myeloid leukaemia; CML, chronic myeloid leukaemia; CR, complete response; FNTB, farnesyltransferase β-subunit; FT, farnesyltransferase; FTIs, FT inhibitors; GGTI, geranylgeranyltransferase 1 inhibitor; HI, haematological improvement; MD, metastatic disease; MDS, myelodysplastic syndrome; MS, median survival; NA, not applicable; ND, not determined; NSCLC, non-small-cell lung cancer; OS, overall survival; p, phosphorylated; PD, progressive disease; PR, partial response; S6, ribosomal protein S6; SD, stable disease; TTP, median time to progression.
See Supplementary information S5 (table) for a table with references. This table only considers FTIs because, to our knowledge, only one GGTI, GGTI-2418, is currently in clinical trials. Studies that did not evaluate tumour response are not included. Median ages are rounded to the closest integer. The number of evaluable patients is stated whenever possible. The response rate was calculated by dividing the sum of complete and partial responses by the number of evaluable patients.
PR includes haematological improvement.
Downward arrows indicate a reduction of enzyme activity (in the case of FT) or a reduction in farnesylation (in the case of HDJ2 or prelamin A).
This patient cohort received a placebo instead of the FTI.
This patient cohort received idarubicin plus cytarabine (referred to by the authors as a historical control).
As the reference only provides percentage response rates, the patient numbers were calculated.
What accounts for this discrepancy between laboratory findings and clinical data? First, in humans, KRAS, and not HRAS, is most frequently mutated1. Second, unlike HRAS, which is exclusively farnesylated, KRAS (and possibly NRAS) can escape FTI-mediated inhibition as it can be alternatively prenylated by GGT1 and thus is fully functional12–15 (BOX 1). Third, the lack of antitumour activity may be due to the fact that most of the clinical trials enrolled patients with advanced and/or metastatic disease. It is also important to note that even though it was known preclinically that KRAS function is resistant to FTIs, Phase III clinical trials were carried out in patients whose tumours harboured mutant KRAS (that is, patients with pancreatic cancer). Perhaps this is because preclinical studies had shown that some cancer cells that harbour mutant KRAS are sensitive to FTIs, possibly owing to the inhibition of exclusively farnesylated proteins downstream of KRAS. In another clinical trial, attempts were made to directly inhibit KRAS function by using L-778123, which inhibits both FT (half-maximal inhibitory concentration (IC50) = 2 nM) and GGT1 (IC50 = 98 nM). Unfortunately, in peripheral blood mononuclear cells from patients treated with this drug, KRAS prenylation was not inhibited102.
When used in combination with other agents, FTIs have fared better. For example, Phase I studies based on a combination of tipifarnib with gemcitabine and cisplatin have shown some promise in advanced solid tumours (33.3% complete response rate or 26% partial response rate)8,9. Similarly, in Phase II neoadjuvant settings, tipifarnib increases the rate of pathological complete responses from the historical 10% to 25% when combined with chemotherapy (doxorubicin and cyclophosphamide) in patients with locally advanced breast cancer10,11. The fact that some patients respond to FTIs suggests that some human tumours depend on farnesylated proteins for survival. The identification of these proteins remains a challenge that must be overcome in order to select patients whose tumours are most likely to respond to FTIs. To this end, a strategy to predict clinical response to FTIs was recently developed by Raponi et al. Following the analysis of gene expression profiles from patients with untreated AML, these authors found that a high ratio of expression of two genes, RAS guanyl releasing protein 1 (RASGRP1), which encodes a RAS guanine nucleotide exchange factor (GEF) that activates RAS, and aprataxin (APTX), which encodes a protein involved in DNA excision repair, predicts a tipifarnib-positive response of patients with AML25,26. Moreover, in patients with advanced solid cancers, low mRNA levels of FNTB, but not FNTA, are associated with improved response to lonafarnib plus taxane and significantly better survival103. Similar studies are needed for other types of cancer.
Targets crucial for the antitumour activity of FTIs
As the data described above have illustrated, and as can be expected of drugs that affect a large number of PT substrates, PTIs trigger a plethora of molecular and cellular effects, whether in cell culture, animal models or in human cancer patients. A key question is, what are the crucial PT substrates that mediate these effects? The answer to this question may uncover why FTIs and GGTIs are only effective in a subset of cells and tumours.
RAS proteins as crucial targets for FTIs
FTIs were originally developed to inhibit RAS function. However, the ability of FTIs to inhibit tumour growth is not correlated with mutations in RAS, whether in cells104,105, animals105 or human patients106 (discussed above). KRAS, the most frequently mutated human oncoprotein, becomes geranylgeranylated and is fully functional in tumour cells treated with FTIs12–15. However, FTIs are effective at inhibiting the growth of mutant KRAS-harbouring tumours in nude mice107 and transgenic mouse models53, suggesting that the inhibition of KRAS farnesylation is not required for FTI antitumour activity, and that, in these models, tumours are addicted to farnesylated proteins other than KRAS. Similar considerations apply to NRAS, because it can also escape FTI-mediated inhibition. HRAS, conversely, is not alternatively geranylgeranylated in cells treated with FTIs. Therefore, the inhibition of HRAS farnesylation can still contribute to FTI antitumour activity in tumours that are addicted to mutant or wild-type HRAS for survival. Thus, it may be worthwhile to design clinical trials that involve FTIs for patients with HRAS-mutant bladder cancers, a tumour that has clearly been understudied so far (TABLES 2,3).
RHEB as a crucial target for FTIs
Like HRAS, RAS homologue enriched in brain (RHEB) is exclusively farnesylated75. The GTPase-activating protein (GAP) for RHEB is the tumour suppressor tuberous sclerosis complex TSC1–TSC2 (REFS 108–110). AKT phosphorylates and inactivates TSC1–TSC2, causing activation of RHEB111–113. RHEB stimulates the protein kinase mTOR, which results in activating phosphorylation of the mTOR substrates S6 kinase (S6K) and eukaryotic translation initiation factor 4E-binding protein 1 (4EBP1)114,115, which is consistent with RHEB being essential for cell growth and cell cycle progression in D. melanogaster116. RHEB may activate mTOR either by directly binding to it117 or by binding to the mTOR antagonist FKBP38 (also known as FKBP8)118. Most importantly, the ability of RHEB to stimulate mTOR depends on its farnesylation75,114. Therefore, cancer cells that are addicted to RHEB may be sensitive to FTIs. For example, this could include tumours that overexpress RHEB or that harbour persistently activated pathways that lead to constitutive RHEB activation (including, PTEN deficiency, PI3K and AKT mutations, AKT overexpression or TSC1–TSC2 deficiency). Consistent with this idea, RHEB is frequently upregulated in transformed cells and human cancer cells75,119, and, in several NSCLC cell lines, the ability of FTIs to inhibit proliferation or induce apoptosis depends on RHEB expression levels, which in turn are correlated to the degree of S6K phosphorylation by mTOR77. Similarly, RHEB is overexpressed in some lymphomas, which have increased mTOR activity and enhanced sensitivity to FTIs120. The fact that an exclusively geranylgeranylated mutant of RHEB (RHEB-GG) renders PTEN-deficient lymphoma cells resistant to FTIs further supports the idea that the inhibition of RHEB farnesylation contributes to the antitumour activity of FTIs120. Similar results were obtained when RHEB-GG was shown to rescue the ability of FTIs to synergize with paclitaxel75 and cisplatin77. As mTOR is an inhibitor of autophagy121, and RHEB activates mTOR, it may not be surprising that FTIs can induce autophagy, possibly by blocking RHEB function122. The induction of autophagy may be particularly important in cancer cells that are resistant to apoptosis. Collectively, these findings suggest that the inhibition of RHEB farnesylation contributes to the antitumour activity of FTIs, which should perhaps be evaluated in clinical trials for patients with tumours that express high levels of RHEB, as well as for patients with tuberous sclerosis, which is a syndrome caused by the loss of TSC1 or TSC2 function.
Inhibition of RHOB farnesylation is unlikely to contribute to the antitumour activity of FTIs
Under physiological conditions, RHOB is found both geranylgeranylated (RHOB-GG), which accounts for 70% of all RHOB, and farnesylated (RHOB-F), which accounts for the remaining 30%, in cells. It has been suggested that the inhibition of RHOB farnesylation accumulates RHOB-GG and contributes to FTI-induced apoptosis123,124. Consistent with this, in murine fibroblasts, RHOB-GG but not RHOB-F, suppresses RAS-induced transformation125. However, the fact that a large proportion of RHOB is already in the geranylgeranylated form in the absence of FTI treatment argues against a major contribution of inhibition of RHOB farnesylation to FTI antitumour activity. More importantly, in human cancer cells of epithelial origin, both RHOB-F and RHOB-GG have tumour suppressive activity, further arguing against the inhibition of RHOB farnesylation playing a part in FTI antitumour activity126. Consistent with RHOB functioning as a tumour suppressor, RHOB is downregulated in several human cancers127–129. Although RHOB was shown to be farnesylated in vitro by FT, and the treatment of cultured cells with the FTI L-739749 increases the levels of RHOB-GG130, the laboratory of Goldstein and Brown has shown using purified components that RHOB is farnesylated by GGT1 but not by FT131. This, of course, would disqualify RHOB as a target.
However, FTI treatment activates the RHOB promoter and accumulates large amounts of the RHOB protein132. Furthermore, mutant HRAS-transformed RHOB−/− cells are less sensitive to FTI-induced apoptosis and FTI inhibition of anchorage-dependent but not anchorage-independent tumour growth. Taken together, these results suggest that increased RHOB protein levels, not inhibition of RHOB farnesylation, may contribute to some of the effects of FTIs133.
Other possible targets
PTIs are bound to have effects not only on oncogenic RAS family members, but also on other prenylated proteins some of which are not yet known. Some candidate targets for prenylation include tumour suppressors, ARHI (also known as NOEY2 and DIRAS3)134, RAS-related and oestrogen-regulated growth inhibitor (RERG)135, deleted in breast cancer 2 (DBC2; also known as RHOBTB2)136, RAS-related inhibitor of cell growth (RIG)137, RAS-related protein on chromosome 22 (RRP22; also known as RASL10A)138 and maybe others. The non-discriminatory action of PTIs may abrogate crucially important tumour suppressor functions and may at least partially compromise their effects on oncogenic pathways. Therefore, it needs to be established whether the proteins mentioned above are exclusively farnesylated or geranylgeranylated, and whether their prenylation is crucial to their growth inhibitory function. For proteins that pass these tests it is recommended that their functional status is assessed in future clinical trials.
The inhibition of the farnesylation of several other proteins could contribute to the antitumour activity of FTIs. These include CENPE, CENPF, the PRL phosphatases PRL1 (also known as PTP4A1), PRL2 (also known as PTP4A2) and PRL3 (also known as PTP4A3), lamins A and B, HDJ2, RND3, peroxisomal biogenesis factor 19 (PEX19), RHOD, RHO6, RHO7 (also known as RHON), TC10 (also known as RHOQ) and prostacyclin receptor (PTGIR). Although some of these may be associated with known effects of FTIs such as the possible role that CENPE and CENPF and PRLs may have in FTI-induced mitotic arrest, the contributions of others require further investigations.
Alternative approaches
RCE1 and ICMT inhibitors
Interestingly, prenylation seems to be constitutive, but prenylated proteins can undergo up to three additional PTMs, the last two of which are reversible (BOX 1). First, the last three C-terminal amino acids, aaX, are proteolytically removed by RAS-converting enzyme 1 (RCE1)139. Second, the carboxyl group in the now C-terminal prenylated cysteine is methylated by isoprenylcysteine carboxyl methyltransferase (ICMT)140. Third, many prenylated proteins become palmitoylated on upstream cysteines by membrane-bound palmitoyl transferases141. The carboxymethylation, which neutralizes the carboxyl negative charge, coupled with the palmitoylation, further stabilizes membrane association and anchoring.
RCE1 and ICMT, which function downstream of PTs, have also attracted attention as potential targets for cancer therapy. Conditional lack of Rce1 expression in skin carcinoma cells that express activated HRAS has much less severe effects on cell proliferation than the effect of lonafarnib142. By contrast, conditional deletion of Icmt efficiently blocks transformation by either human KRAS-G12V or human BRAF-V599E oncogenic mutants140. Considering these results, ICMT inhibitors are more likely to be successful than RCE1 inhibitors. Indeed, pharmacological inhibition of ICMT inhibits the growth of HepG2 tumour xenografts in nude mice143. As postprenylation inhibitors will affect both farnesylated and geranylgeranylated proteins, these drugs may be more toxic than FTIs or GGTIs.
Prenylated proteins that are phosphorylated
Several small GTPases are subject to reversible phosphorylation. For example, protein kinase C (PKC)- or PKA-mediated phosphorylation of CDC42, KRAS, RAP1A and RHOA induces their removal from the cell membrane144–147. The question is whether this relocation translates into a loss or change of function. With regard to KRAS, this has recently been investigated. Interestingly, PKC-mediated phosphorylation of Ser181 in KRAS promotes its relocation to mitochondrial membranes where it associates with BCL-XL (also known as BCL2L1) and promotes apoptosis. Overexpression of KRAS-S181E is sufficient to induce apoptosis, an effect that is rescued by co-transfection with BCL-2 (REF. 145). This raises the intriguing possibility that PKC agonists such as bryostatin 1145 may be more efficient for killing and/or less toxic to KRAS-dependent tumours than PTIs. Phosphorylation of RAB6, RHOE and RALA activates their function148–151. Most interestingly, aurora kinase A enhances, and protein phosphatase 2A (PP2A) inhibits, the transforming activity of RALA150,151. Provided that activating phosphorylation can occur independently of prenylation, these findings may indicate that GGTI-mediated inhibition of RALA may not be sufficient to completely block its transforming ability. However, these findings also suggest that GGTIs that inhibit RALA might synergize with aurora kinase A inhibitors or with PP2A agonists. Phosphorylation of CDC42, RHOB and RHEB inhibits their function152–154, cautioning against combinations of PTIs and protein kinase inhibitors to target CDC42- or RHEB-dependent tumours.
Synthetic lethality
As is well documented, many human cancers depend on oncogenic KRAS for survival, and this dependency confers a vulnerability that is unique to these cancer cells. Indeed, recent unbiased RNA interference-based screens155–159 and other approaches160 identified six genes the knockdown of which kills only human tumours that depend on mutant KRAS. Such a synthetically lethal strategy would be of benefit if some of the identified targets are more druggable than KRAS (see Supplementary information S6 (figure)). Of the six studies cited above, the one identifying the lethal interaction between oncogenic KRAS and CDK4 is particularly interesting160. It has been previously reported that RAS-mediated transformation requires the expression of functional RB in mouse fibroblasts161. This is consistent with the fact that activated RAS induces cyclin D1 (REF. 162), which activates CDK4 and other kinases and ultimately leads to inhibitory phosphorylation of RB, thus permitting the G1/S transition163. These considerations might explain why human tumours very rarely display loss-of-function mutations in RB together with activating RAS mutations164. Furthermore, they provide a rationale for investigating whether PTIs and CDK inhibitors act in a synergistic fashion, both in animals and in clinical trials.
Considering the enormous number of proteins affected by the RAS signalling network, we predict that further synthetic lethal interactions will be identified. For example, small interfering RNA (siRNA) screens silencing protein kinases or protein phosphatases may reveal molecular targets the inhibition of which sensitizes cancer cells to PTIs. Conversely, siRNA screens silencing the prenylome may reveal crucial prenylated proteins the inhibition of which sensitizes cancer cells to drugs that target other signal transduction pathways; for example, PI3K–AKT, RAF–MEK–MAPK and CDKs.
Future directions and challenges
Despite the conceptual advances that have been made over the past decade, in our opinion, the major challenge in this field is the following question: which PT substrates are crucial for the proliferation or survival of different cancer types? Or, in other words, does the antitumour activity of PTIs in a given tumour depend on the inhibition of certain prenylated proteins? At present, the exact size of the prenylome is unknown165. Although more than 100 proteins have been experimentally confirmed to undergo prenylation166, a recent search of the UniProt database (release 20 April 2010; see Further information) returned 587 human genes that encode proteins bearing a C-terminal CXXX motif. Although not all of these proteins will qualify as PT substrates, this suggests that many prenylated proteins have not yet been identified.
Current standard methods to characterize protein prenylation are, for the most part, designed to follow individual proteins167 and are thus not practical to address the above questions. Therefore, this field should develop and streamline techniques that are capable of analysing prenylation on a global scale, in a manner that is reasonably rapid, feasible and convenient for many laboratories. As a step towards characterizing the entire prenylome, Maurer-Stroh et al.165 have recently developed a sequence-based software suite that is designed to predict whether proteins hitherto unknown to be prenylated are likely to be modified by FT, GGT1 and/or GGT2. Combining this with approaches that uncover actual prenylation patterns in various cancer cells, as well as changes in prenylation patterns in response to PTIs, will eventually reveal which prenylated proteins the inhibition of which is responsible for the antitumour effects of PTIs and which patients are most likely to respond to treatment with these inhibitors. Several techniques pursuing this goal have recently been described168–173. For example, labelling cells with modified tractable prenyl donors in lieu of the natural farnesyl diphosphate (FPP) or geranylgeranyl diphosphate (GGPP) is a step in that direction. This can involve the labelling of cells with azido-farnesyl, followed by the affinity purification of farnesylated proteins with a biotinylated phosphine capture reagent168. Similarly, labelling cells with azido-GG analogues, followed by the selective labelling of the resulting azido-GG proteins with a modified rhodamine, can be used to detect geranylgeranylated proteins by fluorescent imaging172.
It is our belief that proteome-wide or prenylome-wide approaches, such as those discussed above, are urgently needed to identify the subsets of prenylated proteins that are affected by FTIs and/or GGTIs, which in turn should help to link the physiological effects of various PTIs to their molecular targets, and thus will help to design improved clinical trials.
Supplementary Material
At a glance.
Post-translational modifications with the lipids farnesyl or geranylgeranyl (together referred to as prenyl) are catalysed by farnesyltransferase (FT) or geranylgeranyltransferase 1 (GGT1) and are required for the cellular localization, function and cancer-causing activities of some proteins. Among the hundreds of proteins that are estimated to be prenylated most are either exclusively farnesylated (for example, HRAS and RAS homologue enriched in brain (RHEB)) or geranylgeranylated (for example, RHOA, RHOC, RALA and RALB); some are both farnesylated and geranylgeranylated (RHOB), and others are naturally farnesylated but become geranylgeranylated when FT is inhibited (for example, KRAS and NRAS).
These and other important observations prompted the design and development of inhibitors of FT (FTIs) and GGT1 (GGTIs) as potential anticancer drugs. Several FTIs have been tested clinically but only one GGTI has recently entered clinical trials.
Further validation of FT and GGT1 as anticancer drug targets was recently provided by genetic mouse models: conditional loss of FT and/or GGT1 hampers mutant KRAS-induced tumorigenesis and extends the lifespan of mice.
FTI treatment results in the reversal of several hallmarks of cancer, including mitotic arrest at prometaphase, induction of apoptosis, inhibition of anchorage-dependent and anchorage-independent growth, invasion, angiogenesis and tumour growth, as well as induction of tumour regression in animal models. These effects seem to be mediated by interference with aberrant signal transduction pathways such as RAF–MEK–ERK, PI3K–AKT, and other oncogenic and survival pathways.
GGTI treatment also results in the reversal of the cancer hallmarks mentioned above except that they block cells in the G1 phase of the cell cycle, and this seems to be owing to their ability to induce the accumulation of the cyclin-dependent kinase (CDK) inhibitors p21 and p27 and to inhibit CDKs and induce hypophosphorylation of RB. GGTI treatment also decreases the levels of phospho-AKT and survivin, and this seems to mediate their ability to induce apoptosis.
Although in preclinical models FTIs are highly effective as antitumour agents, in clinical trials limited efficacy was observed. This is primarily due to poor patient selection. This in turn is due to our lack of understanding of the mechanism of action of FTIs. In the future, a major effort must be dedicated to identifying the prenylated proteins the inhibition of which is responsible for the antitumour effects of PTIs. This will be of great value not only for enhancing our understanding of the mechanism of action of FTIs and GGTIs, but also for selecting patients whose tumours are addicted to specific prenylated proteins and who are more likely to respond to these agents. Recent advances in techniques to characterize the human prenylome are likely to accelerate achieving these crucial goals in the prenylation field.
Acknowledgments
This work was partially supported by US National Institutes of Health grants CA067771 and CA098473 to S.M.S.
Glossary
- Farnesylation
One of two types of prenylation. This involves the transfer of a farnesyl moiety to the cysteine of the C-terminal CaaX box of the target protein. Catalysed by farnesyltransferase
- Geranylgeranylation
This prenylation is catalysed by geranylgeranyltransferase 1 (GGT1) or GGT2. GGT1 transfers a geranylgeranyl moiety to the cysteine of the C-terminal CaaX box, and GGT2 acts on the cysteines of C-terminal CXC or CC motifs
- Prenylation
Also known as isoprenylation. An irreversible post-translational modification of proteins consisting of the covalent attachment of an isoprenyl lipid to a cysteine within four residues of the C terminus
- Myristoylated
A universal and irreversible co-translational modification of proteins involving the covalent attachment of a myristoyl group to an N-terminal amino acid of a nascent polypeptide. It is important for membrane targeting of the modified protein
- CaaX motif
This refers to the last four C-terminal amino acids that serve as a recognition motif for farnesyltransferase or geranylgeranyltransferase 1. C (cysteine) is the amino acid being modified, a is an aliphatic residue and X is any residue
- Intimal hyperplasia
The thickening of the innermost layer of a blood vessel as a complication of a reconstruction procedure or endarterectomy. It is the universal response of a vessel to injury and is an important reason for late bypass graft failure, particularly in vein and synthetic vascular grafts
- Neointima
A new or thickened layer of arterial intima (innermost layer of an artery or a vein) formed especially on a prosthesis or in atherosclerosis by migration and proliferation of cells from the media
- Palmitoylated
A post-translational modification, consisting of the covalent attachment of fatty acids to cysteine residues of membrane proteins, thought to further enhance membrane anchoring of previously prenylated proteins. In contrast to prenylation and myristoylation, it is reversible
- Prenylome
The subset of proteins in a cell or organism that is modified by prenylation
Footnotes
Competing interests statement
The authors declare competing financial interests. See Web version for details.
DATABASES
ClinicalTrials.gov: http://clinicaltrials.gov/)
National Cancer Institute Drug Dictionary: http://www.cancer.gov/drugdictionary 5-fluorouracil | BMS-214662 | bryostatin 1 | carboplatin | cisplatin | cyclophosphamide | doxorubicin | gemcitabine | imatinib | lonafarnib | paclitaxel | tipifarnib | triciribine phosphate | vincristine
UniProt: http://www.uniprot.org/
FURTHER INFORMATION
Saïd M. Sebti’s homepage:
Catalogue of Somatic Mutations in Cancer (COSMIC): http://www.sanger.ac.uk/cosmic
PrePS – Prenylation Prediction Suite: http://mendel.imp.ac.at/sat/PrePS/
SUPPLEMENTARY INFORMATION
See online article: S1 (table) | S2 (table) | S3 (table) | S4 (table) | S5 (table) | S6 (figure)
References
- 1.Bos JL. ras oncogenes in human cancer: a review. Cancer Res. 1989;49:4682–4689. [PubMed] [Google Scholar]
- 2.Jackson JH, et al. Farnesol modification of Kirstenras exon 4B protein is essential for transformation. Proc Natl Acad Sci USA. 1990;87:3042–3046. doi: 10.1073/pnas.87.8.3042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bommi-Reddy A, Kaelin WG. Slaying RAS with a synthetic lethal weapon. Cell Res. 2010;20:119–121. doi: 10.1038/cr.2010.16. [DOI] [PubMed] [Google Scholar]
- 4.Van Cutsem E, et al. Phase III trial of gemcitabine plus tipifarnib compared with gemcitabine plus placebo in advanced pancreatic cancer. J Clin Oncol. 2004;22:1430–1438. doi: 10.1200/JCO.2004.10.112. [DOI] [PubMed] [Google Scholar]
- 5.Rao S, et al. Phase III double-blind placebo-controlled study of farnesyl transferase inhibitor R115777 in patients with refractory advanced colorectal cancer. J Clin Oncol. 2004;22:3950–3957. doi: 10.1200/JCO.2004.10.037. [DOI] [PubMed] [Google Scholar]
- 6.Blumenschein G, et al. A randomized phase III trial comparing lonafarnib/carboplatin/paclitaxel versus carboplatin/paclitaxel (CP) in chemotherapy-naïve patients with advanced or metastatic non-small cell lung cancer. Lung Cancer. 2005;49:S30. [Google Scholar]
- 7.Harousseau JL, et al. A randomized phase 3 study of tipifarnib compared with best supportive care, including hydroxyurea, in the treatment of newly diagnosed acute myeloid leukemia in patients 70 years or older. Blood. 2009;114:1166–1173. doi: 10.1182/blood-2009-01-198093. References 4–7 describe the results of Phase III clinical trials with lonafarnib or tipifarnib. Whether alone or in combination the FTIs failed to even slightly improve the outcome for patients with advanced NSCLC, advanced pancreatic cancer, advanced colon cancer or AML. [DOI] [PubMed] [Google Scholar]
- 8.Adjei AA, et al. A Phase I trial of the farnesyl protein transferase inhibitor R115777 in combination with gemcitabine and cisplatin in patients with advanced cancer. Clin Cancer Res. 2003;9:2520–2526. [PubMed] [Google Scholar]
- 9.Siegel-Lakhai WS, et al. Phase I and pharmacological study of the farnesyltransferase inhibitor tipifarnib (Zarnestra, R115777) in combination with gemcitabine and cisplatin in patients with advanced solid tumours. Br J Cancer. 2005;93:1222–1229. doi: 10.1038/sj.bjc.6602850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sparano JA, et al. Targeted inhibition of farnesyltransferase in locally advanced breast cancer: a phase I and II trial of tipifarnib plus dose-dense doxorubicin and cyclophosphamide. J Clin Oncol. 2006;24:3013–3018. doi: 10.1200/JCO.2005.04.9114. [DOI] [PubMed] [Google Scholar]
- 11.Sparano JA, et al. Phase II trial of tipifarnib plus neoadjuvant doxorubicin-cyclophosphamide in patients with clinical stage IIB-IIIC breast cancer. Clin Cancer Res. 2009;15:2942–2948. doi: 10.1158/1078-0432.CCR-08-2658. References 8–11 stand out from the bulk of clinical trials with FTIs in that they demonstrate that a combination of tipifarnib with chemotherapy can make a difference, even in solid advanced tumours. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Rowell CA, Kowalczyk JJ, Lewis MD, Garcia AM. Direct demonstration of geranylgeranylation and farnesylation of Ki-Ras in vivo. J Biol Chem. 1997;272:14093–14097. doi: 10.1074/jbc.272.22.14093. [DOI] [PubMed] [Google Scholar]
- 13.Whyte DB, et al. K- and N-Ras are geranylgeranylated in cells treated with farnesyl protein transferase inhibitors. J Biol Chem. 1997;272:14459–14464. doi: 10.1074/jbc.272.22.14459. [DOI] [PubMed] [Google Scholar]
- 14.Lerner EC, et al. Inhibition of the prenylation of K-Ras, but not H- or N-Ras, is highly resistant to CAAX peptidomimetics and requires both a farnesyltransferase and a geranylgeranyltransferase I inhibitor in human tumor cell lines. Oncogene. 1997;15:1283–1288. doi: 10.1038/sj.onc.1201296. [DOI] [PubMed] [Google Scholar]
- 15.Sun J, Qian Y, Hamilton AD, Sebti SM. Both farnesyltransferase and geranylgeranyltransferase I inhibitors are required for inhibition of oncogenic K-Ras prenylation but each alone is sufficient to suppress human tumor growth in nude mouse xenografts. Oncogene. 1998;16:1467–1473. doi: 10.1038/sj.onc.1201656. References 12–15 by three independent groups show that KRAS can escape FTI-mediated inhibition and remain fully functional through undergoing cross-prenylation by GGT1. As KRAS is the most frequently mutated human oncogene, this finding was disappointing as it meant that KRAS function could not be inhibited with FTIs. [DOI] [PubMed] [Google Scholar]
- 16.Hamad NM, et al. Distinct requirements for Ras oncogenesis in human versus mouse cells. Genes Dev. 2002;16:2045–2057. doi: 10.1101/gad.993902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lim KH, et al. Activation of RalA is critical for Ras-induced tumorigenesis of human cells. Cancer Cell. 2005;7:533–545. doi: 10.1016/j.ccr.2005.04.030. References 16 and 17 show that exclusively geranylgeranylated RALA and RALB, which are downstream of RAS, may be more important for some human cancers than the RAF–MEK–ERK or PI3K–AKT pathways. [DOI] [PubMed] [Google Scholar]
- 18.Clark EA, Golub TR, Lander ES, Hynes RO. Genomic analysis of metastasis reveals an essential role for RhoC. Nature. 2000;406:532–535. doi: 10.1038/35020106. [DOI] [PubMed] [Google Scholar]
- 19.Hakem A, et al. RhoC is dispensable for embryogenesis and tumor initiation but essential for metastasis. Genes Dev. 2005;19:1974–1979. doi: 10.1101/gad.1310805. References 18 and 19 provide evidence that the exclusively geranylgeranylated RHOC is not necessary for embryonic development but is essential for metastasis. These results, together with those of references 16 and 17, can be regarded as a major incentive for developing GGTIs to treat advanced cancers. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Qiu RG, Abo A, McCormick F, Symons M. Cdc42 regulates anchorage-independent growth and is necessary for Ras transformation. Mol Cell Biol. 1997;17:3449–3458. doi: 10.1128/mcb.17.6.3449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Joyce PL, Cox AD. Rac1 and Rac3 are targets for geranylgeranyltransferase I inhibitor-mediated inhibition of signaling, transformation, and membrane ruffling. Cancer Res. 2003;63:7959–7967. [PubMed] [Google Scholar]
- 22.Kissil JL, et al. Requirement for Rac1 in a K-ras induced lung cancer in the mouse. Cancer Res. 2007;67:8089–8094. doi: 10.1158/0008-5472.CAN-07-2300. [DOI] [PubMed] [Google Scholar]
- 23.Lobell RB, et al. Evaluation of farnesyl:protein transferase and geranylgeranyl:protein transferase inhibitor combinations in preclinical models. Cancer Res. 2001;61:8758–8768. [PubMed] [Google Scholar]
- 24.O’Dwyer PJ, Gallagher M, Nguyen B, Waddell MJ, Chiorean EG. Phase I accelerated dose-escalating safety and pharmacokinetic (PK) study of GGTI-2418, a novel geranylgeranyltransferase I inhibitor in patients with refractory solid tumors. Ann Oncol. 2010;21:ii42. [Google Scholar]
- 25.Raponi M, et al. Identification of molecular predictors of response in a study of tipifarnib treatment in relapsed and refractory acute myelogenous leukemia. Clin Cancer Res. 2007;13:2254–2260. doi: 10.1158/1078-0432.CCR-06-2609. [DOI] [PubMed] [Google Scholar]
- 26.Raponi M, et al. A 2-gene classifier for predicting response to the farnesyltransferase inhibitor tipifarnib in acute myeloid leukemia. Blood. 2008;111:2589–2596. doi: 10.1182/blood-2007-09-112730. These two studies have advanced approaches to correctly predict clinical outcome following FTI therapy. The authors have identified a signature two-gene expression ratio (RASGRPS1/APTX) as a predictor for the response to tipifarnib in patients with AML. [DOI] [PubMed] [Google Scholar]
- 27.Yang W, Urano J, Tamanoi F. Protein farnesylation is critical for maintaining normal cell morphology and canavanine resistance in Schizosaccharomyces pombe. J Biol Chem. 2000;275:429–438. doi: 10.1074/jbc.275.1.429. [DOI] [PubMed] [Google Scholar]
- 28.Cox AD, Hisaka MM, Buss JE, Der CJ. Specific isoprenoid modification is required for function of normal, but not oncogenic, Ras protein. Mol Cell Biol. 1992;12:2606–2615. doi: 10.1128/mcb.12.6.2606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Perez-Sala D. Protein isoprenylation in biology and disease: general overview and perspectives from studies with genetically engineered animals. Front Biosci. 2007;12:4456–4472. doi: 10.2741/2401. [DOI] [PubMed] [Google Scholar]
- 30.Sebti SM. Protein farnesylation: implications for normal physiology, malignant transformation, and cancer therapy. Cancer Cell. 2005;7:297–300. doi: 10.1016/j.ccr.2005.04.005. [DOI] [PubMed] [Google Scholar]
- 31.Mijimolle N, et al. Protein farnesyltransferase in embryogenesis, adult homeostasis, and tumor development. Cancer Cell. 2005;7:313–324. doi: 10.1016/j.ccr.2005.03.004. [DOI] [PubMed] [Google Scholar]
- 32.Ohya Y, et al. Yeast CAL1 is a structural and functional homologue to the DPR1 (RAM) gene involved in ras processing. J Biol Chem. 1991;266:12356–12360. [PubMed] [Google Scholar]
- 33.Therrien M, et al. KSR, a novel protein kinase required for RAS signal transduction. Cell. 1995;83:879–888. doi: 10.1016/0092-8674(95)90204-x. [DOI] [PubMed] [Google Scholar]
- 34.Sjogren AK, et al. GGTase-I deficiency reduces tumor formation and improves survival in mice with K-RAS-induced lung cancer. J Clin Invest. 2007;117:1294–1304. doi: 10.1172/JCI30868. This article shows that targeted deletion of Ggt1 in the lung reduces Kras-driven tumour formation and increases the lifespan of mice with Kras-induced lung cancer. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Reid TS, Terry KL, Casey PJ, Beese LS. Crystallographic analysis of CaaX prenyltransferases complexed with substrates defines rules of protein substrate selectivity. J Mol Biol. 2004;343:417–433. doi: 10.1016/j.jmb.2004.08.056. [DOI] [PubMed] [Google Scholar]
- 36.Yang SH, et al. Caution! Analyze transcripts from conditional knockout alleles. Transgenic Res. 2009;18:483–489. doi: 10.1007/s11248-008-9237-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Liu M, et al. Targeting the protein prenyltransferases efficiently reduces tumor development in mice with K-RAS-induced lung cancer. Proc Natl Acad Sci USA. 2010;107:6471–6476. doi: 10.1073/pnas.0908396107. This article demonstrates that concomitant conditional loss of both FT and GGT1 in mice effectively reduces Kras-induced lung carcinogenesis, and extends the lifespan of these mice considerably more than FT or GGT1 deficiency alone, suggesting that the simultaneous inhibition of FT and GGT1 may be therapeutically beneficial in cancer patients. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Sjogren AK, et al. Inactivating GGTase-I reduces disease phenotypes in a mouse model of K-RAS-induced myeloproliferative disease. Leukemia. 2011;25:186–189. doi: 10.1038/leu.2010.242. [DOI] [PubMed] [Google Scholar]
- 39.Wang T, et al. The p21(RAS) farnesyltransferase α subunit in TGF-β and activin signaling. Science. 1996;271:1120–1122. doi: 10.1126/science.271.5252.1120. [DOI] [PubMed] [Google Scholar]
- 40.Kumar A, Beresini MH, Dhawan P, Mehta KD. α-subunit of farnesyltransferase is phosphorylated in vivo: effect of protein phosphatase-1 on enzymatic activity. Biochem Biophys Res Commun. 1996;222:445–452. doi: 10.1006/bbrc.1996.0764. [DOI] [PubMed] [Google Scholar]
- 41.Kumar A, Mehta K. D p21ras farnesyltransferase α- and β ubunits are phosphorylated in PC-12 cells: TGF-β signaling pathway independent phosphorylation. Neurosci Lett. 1997;231:143–146. doi: 10.1016/s0304-3940(97)00549-1. [DOI] [PubMed] [Google Scholar]
- 42.Goalstone M, Carel K, Leitner JW, Draznin B. Insulin stimulates the phosphorylation and activity of farnesyltransferase via the Ras-mitogen-activated protein kinase pathway. Endocrinology. 1997;138:5119–5124. doi: 10.1210/endo.138.12.5621. [DOI] [PubMed] [Google Scholar]
- 43.Kim KW, et al. Inactivation of farnesyltransferase and geranylgeranyltransferase I by caspase-3: cleavage of the common α subunit during apoptosis. Oncogene. 2001;20:358–366. doi: 10.1038/sj.onc.1204099. [DOI] [PubMed] [Google Scholar]
- 44.Singh J, Hamid R, Reddy BS. Dietary fish oil inhibits the expression of farnesyl protein transferase and colon tumor development in rodents. Carcinogenesis. 1998;19:985–989. doi: 10.1093/carcin/19.6.985. [DOI] [PubMed] [Google Scholar]
- 45.Caruso MG, et al. Increased farnesyltransferase activity in human colorectal cancer: relationship with clinicopathological features and K-ras mutation. Scand J Gastroenterol. 2003;38:80–85. doi: 10.1080/00365520310000483. [DOI] [PubMed] [Google Scholar]
- 46.Lantry LE, et al. Chemopreventive efficacy of promising farnesyltransferase inhibitors. Exp Lung Res. 2000;26:773–790. doi: 10.1080/01902140150216819. [DOI] [PubMed] [Google Scholar]
- 47.Zhang Z, et al. Farnesyltransferase inhibitors are potent lung cancer chemopreventive agents in A/J. mice with a dominant-negative p53 and/or heterozygous deletion of Ink4a/Arf. Oncogene. 2003;22:6257–6265. doi: 10.1038/sj.onc.1206630. [DOI] [PubMed] [Google Scholar]
- 48.Lerner EC, et al. Ras CAAX peptidomimetic FTI-277 selectively blocks oncogenic Ras signaling by inducing cytoplasmic accumulation of inactive Ras-Raf complexes. J Biol Chem. 1995;270:26802–26806. doi: 10.1074/jbc.270.45.26802. [DOI] [PubMed] [Google Scholar]
- 49.Jiang K, et al. The phosphoinositide 3-OH kinase/AKT2 pathway as a critical target for farnesyltransferase inhibitor-induced apoptosis. Mol Cell Biol. 2000;20:139–148. doi: 10.1128/mcb.20.1.139-148.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Sun SY, Zhou Z, Wang R, Fu H, Khuri FR. The farnesyltransferase inhibitor Lonafarnib induces growth arrest or apoptosis of human lung cancer cells without downregulation of Akt. Cancer Biol Ther. 2004;3:1092–1098. doi: 10.4161/cbt.3.11.1176. discussion 1099–1101. [DOI] [PubMed] [Google Scholar]
- 51.Sun J, Qian Y, Hamilton AD, Sebti SM. Ras CAAX peptidomimetic FTI 276 selectively blocks tumor growth in nude mice of a human lung carcinoma with K-Ras mutation and p53 deletion. Cancer Res. 1995;55:4243–4247. [PubMed] [Google Scholar]
- 52.Mangues R, et al. Antitumor effect of a farnesyl protein transferase inhibitor in mammary and lymphoid tumors overexpressing N-ras in transgenic mice. Cancer Res. 1998;58:1253–1259. [PubMed] [Google Scholar]
- 53.Omer CA, et al. Mouse mammary tumor virus-Ki-rasB transgenic mice develop mammary carcinomas that can be growth-inhibited by a farnesyl:protein transferase inhibitor. Cancer Res. 2000;60:2680–2688. [PubMed] [Google Scholar]
- 54.Crespo NC, Ohkanda J, Yen TJ, Hamilton AD, Sebti SM. The farnesyltransferase inhibitor, FTI-2153, blocks bipolar spindle formation and chromosome alignment and causes prometaphase accumulation during mitosis of human lung cancer cells. J Biol Chem. 2001;276:16161–16167. doi: 10.1074/jbc.M006213200. [DOI] [PubMed] [Google Scholar]
- 55.Crespo NC, et al. The farnesyltransferase inhibitor, FTI-2153, inhibits bipolar spindle formation during mitosis independently of transformation and Ras and p53 mutation status. Cell Death Differ. 2002;9:702–709. doi: 10.1038/sj.cdd.4401023. [DOI] [PubMed] [Google Scholar]
- 56.Ashar HR, et al. Farnesyl transferase inhibitors block the farnesylation of CENP-E and CENP-F and alter the association of CENP-E with the microtubules. J Biol Chem. 2000;275:30451–30457. doi: 10.1074/jbc.M003469200. [DOI] [PubMed] [Google Scholar]
- 57.Hussein D, Taylor SS. Farnesylation of Cenp-F is required for G2/M progression and degradation after mitosis. J Cell Sci. 2002;115:3403–3414. doi: 10.1242/jcs.115.17.3403. [DOI] [PubMed] [Google Scholar]
- 58.Wang J, Kirby CE, Herbst R. The tyrosine phosphatase PRL-1 localizes to the endoplasmic reticulum and the mitotic spindle and is required for normal mitosis. J Biol Chem. 2002;277:46659–46668. doi: 10.1074/jbc.M206407200. [DOI] [PubMed] [Google Scholar]
- 59.Sepp-Lorenzino L, Rosen N. A farnesyl-protein transferase inhibitor induces p21 expression and G1 block in p53 wild type tumor cells. J Biol Chem. 1998;273:20243–20251. doi: 10.1074/jbc.273.32.20243. [DOI] [PubMed] [Google Scholar]
- 60.Reuveni H, Klein S, Levitzki A. The inhibition of Ras farnesylation leads to an increase in p27Kip1 and G1 cell cycle arrest. Eur J Biochem. 2003;270:2759–2772. doi: 10.1046/j.1432-1033.2003.03647.x. [DOI] [PubMed] [Google Scholar]
- 61.Kohl NE, et al. Inhibition of farnesyltransferase induces regression of mammary and salivary carcinomas in ras transgenic mice. Nature Med. 1995;1:792–797. doi: 10.1038/nm0895-792. Using transgenic mice expressing mutant HRAS in the mammary glands, this is the first demonstration that FTIs cause persistent tumour regression in vivo. [DOI] [PubMed] [Google Scholar]
- 62.Barrington RE, et al. A farnesyltransferase inhibitor induces tumor regression in transgenic mice harboring multiple oncogenic mutations by mediating alterations in both cell cycle control and apoptosis. Mol Cell Biol. 1998;18:85–92. doi: 10.1128/mcb.18.1.85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Lebowitz PF, Sakamuro D, Prendergast GC. Farnesyl transferase inhibitors induce apoptosis of Ras-transformed cells denied substratum attachment. Cancer Res. 1997;57:708–713. [PubMed] [Google Scholar]
- 64.Suzuki N, Urano J, Tamanoi F. Farnesyltransferase inhibitors induce cytochrome c release and caspase 3 activation preferentially in transformed cells. Proc Natl Acad Sci USA. 1998;95:15356–15361. doi: 10.1073/pnas.95.26.15356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Oh SH, Jin Q, Kim ES, Khuri FR, Lee HY. Insulin-like growth factor-I receptor signaling pathway induces resistance to the apoptotic activities of SCH66336 (lonafarnib) through Akt/mammalian target of rapamycin-mediated increases in survivin expression. Clin Cancer Res. 2008;14:1581–1589. doi: 10.1158/1078-0432.CCR-07-0952. [DOI] [PubMed] [Google Scholar]
- 66.Zhang B, Prendergast GC, Fenton RG. Farnesyltransferase inhibitors reverse Ras-mediated inhibition of Fas gene expression. Cancer Res. 2002;62:450–458. [PubMed] [Google Scholar]
- 67.Takada Y, Khuri FR, Aggarwal BB. Protein farnesyltransferase inhibitor (SCH 66336) abolishes NF-κB activation induced by various carcinogens and inflammatory stimuli leading to suppression of NF-κB-regulated gene expression and up-regulation of apoptosis. J Biol Chem. 2004;279:26287–26299. doi: 10.1074/jbc.M400963200. [DOI] [PubMed] [Google Scholar]
- 68.Lackner MR, et al. Chemical genetics identifies Rab geranylgeranyl transferase as an apoptotic target of farnesyl transferase inhibitors. Cancer Cell. 2005;7:325–336. doi: 10.1016/j.ccr.2005.03.024. [DOI] [PubMed] [Google Scholar]
- 69.Han JY, et al. Hypoxia-inducible factor 1α and antiangiogenic activity of farnesyltransferase inhibitor SCH66336 in human aerodigestive tract cancer. J Natl Cancer Inst. 2005;97:1272–1286. doi: 10.1093/jnci/dji251. [DOI] [PubMed] [Google Scholar]
- 70.Cohen-Jonathan E, et al. The farnesyltransferase inhibitor L744, 832 reduces hypoxia in tumors expressing activated H-ras. Cancer Res. 2001;61:2289–2293. [PubMed] [Google Scholar]
- 71.Delmas C, et al. The farnesyltransferase inhibitor R115777 reduces hypoxia and matrix metalloproteinase 2 expression in human glioma xenograft. Clin Cancer Res. 2003;9:6062–6068. [PubMed] [Google Scholar]
- 72.Kim CK, et al. The farnesyltransferase inhibitor LB42708 suppresses vascular endothelial growth factor-induced angiogenesis by inhibiting ras-dependent mitogen-activated protein kinase and phosphatidylinositol 3-kinase/Akt signal pathways. Mol Pharmacol. 2010;78:142–150. doi: 10.1124/mol.110.063586. [DOI] [PubMed] [Google Scholar]
- 73.Bernhard EJ, et al. The farnesyltransferase inhibitor FTI-277 radiosensitizes H-ras-transformed rat embryo fibroblasts. Cancer Res. 1996;56:1727–1730. [PubMed] [Google Scholar]
- 74.Moasser MM, et al. Farnesyl transferase inhibitors cause enhanced mitotic sensitivity to taxol and epothilones. Proc Natl Acad Sci USA. 1998;95:1369–1374. doi: 10.1073/pnas.95.4.1369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Basso AD, et al. The farnesyl transferase inhibitor (FTI) SCH66336 (lonafarnib) inhibits Rheb farnesylation and mTOR signaling. Role in FTI enhancement of taxane and tamoxifen anti-tumor activity. J Biol Chem. 2005;280:31101–31108. doi: 10.1074/jbc.M503763200. [DOI] [PubMed] [Google Scholar]
- 76.Adjei AA, Davis JN, Bruzek LM, Erlichman C, Kaufmann SH. Synergy of the protein farnesyltransferase inhibitor SCH66336 and cisplatin in human cancer cell lines. Clin Cancer Res. 2001;7:1438–1445. [PubMed] [Google Scholar]
- 77.Zheng H, et al. Ras homologue enriched in brain is a critical target of farnesyltransferase inhibitors in non-small cell lung cancer cells. Cancer Lett. 2010;297:117–125. doi: 10.1016/j.canlet.2010.05.004. In this paper, the response to FTIs was correlated to the expression levels of RHEB in human lung tumour tissue and cell lines. [DOI] [PubMed] [Google Scholar]
- 78.Russo P, Malacarne D, Falugi C, Trombino S, O’Connor PM. RPR-115135, a farnesyltransferase inhibitor, increases 5-FU- cytotoxicity in ten human colon cancer cell lines: role of p53. Int J Cancer. 2002;100:266–275. doi: 10.1002/ijc.10461. [DOI] [PubMed] [Google Scholar]
- 79.Brassard DL, et al. Inhibitors of farnesyl protein transferase and MEK1, 2 induce apoptosis in fibroblasts transformed with farnesylated but not geranylgeranylated H-Ras. Exp Cell Res. 2002;273:138–146. doi: 10.1006/excr.2001.5440. [DOI] [PubMed] [Google Scholar]
- 80.Edamatsu H, Gau CL, Nemoto T, Guo L, Tamanoi F. Cdk inhibitors, roscovitine and olomoucine, synergize with farnesyltransferase inhibitor (FTI) to induce efficient apoptosis of human cancer cell lines. Oncogene. 2000;19:3059–3068. doi: 10.1038/sj.onc.1203625. [DOI] [PubMed] [Google Scholar]
- 81.Hoover RR, Mahon FX, Melo JV, Daley GQ. Overcoming STI571 resistance with the farnesyl transferase inhibitor SCH66336. Blood. 2002;100:1068–1071. doi: 10.1182/blood.v100.3.1068. [DOI] [PubMed] [Google Scholar]
- 82.Liu M, et al. Antitumor activity of SCH 66336, an orally bioavailable tricyclic inhibitor of farnesyl protein transferase, in human tumor xenograft models and wap-ras transgenic mice. Cancer Res. 1998;58:4947–4956. [PubMed] [Google Scholar]
- 83.Shi B, et al. The farnesyl protein transferase inhibitor SCH66336 synergizes with taxanes in vitro and enhances their antitumor activity in vivo. Cancer Chemother Pharmacol. 2000;46:387–393. doi: 10.1007/s002800000170. [DOI] [PubMed] [Google Scholar]
- 84.Sun J, et al. Antitumor efficacy of a novel class of non-thiol-containing peptidomimetic inhibitors of farnesyltransferase and geranylgeranyltransferase I: combination therapy with the cytotoxic agents cisplatin, Taxol, and gemcitabine. Cancer Res. 1999;59:4919–4926. [PubMed] [Google Scholar]
- 85.Berndt N, et al. The Akt activation inhibitor TCN-P inhibits Akt phosphorylation by binding to the PH domain of Akt and blocking its recruitment to the plasma membrane. Cell Death Differ. 2010;17:1795–1804. doi: 10.1038/cdd.2010.63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Balasis ME, et al. Combination of farnesyltransferase and Akt inhibitors is synergistic in breast cancer cells and causes significant tumor regression in ErbB2 transgenic mice. Clin Cancer Res. 2011;17:2852–2862. doi: 10.1158/1078-0432.CCR-10-2544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Marcus AI, et al. The synergistic combination of the farnesyl transferase inhibitor lonafarnib and paclitaxel enhances tubulin acetylation and requires a functional tubulin deacetylase. Cancer Res. 2005;65:3883–3893. doi: 10.1158/0008-5472.CAN-04-3757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Marcus AI, et al. Farnesyltransferase inhibitors reverse taxane resistance. Cancer Res. 2006;66:8838–8846. doi: 10.1158/0008-5472.CAN-06-0699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Zhou J, et al. The protein farnesyltransferase regulates HDAC6 activity in a microtubule-dependent manner. J Biol Chem. 2009;284:9648–9655. doi: 10.1074/jbc.M808708200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Vogt A, Sun J, Qian Y, Hamilton AD, Sebti SM. The geranylgeranyltransferase-I inhibitor GGTI-298 arrests human tumor cells in G0/G1 and induces p21(WAF1/CIP1/SDI1) in a p53-independent manner. J Biol Chem. 1997;272:27224–27229. doi: 10.1074/jbc.272.43.27224. [DOI] [PubMed] [Google Scholar]
- 91.Sun J, et al. The geranylgeranyltransferase I inhibitor GGTI-298 induces hypophosphorylation of retinoblastoma and partner switching of cyclin-dependent kinase inhibitors. A potential mechanism for GGTI-298 antitumor activity. J Biol Chem. 1999;274:6930–6934. doi: 10.1074/jbc.274.11.6930. [DOI] [PubMed] [Google Scholar]
- 92.Kazi A, et al. Blockade of protein geranylgeranylation inhibits Cdk2-dependent p27Kip1 phosphorylation on Thr187 and accumulates p27Kip1 in the nucleus: implications for breast cancer therapy. Mol Cell Biol. 2009;29:2254–2263. doi: 10.1128/MCB.01029-08. This paper demonstrates that the antitumour activity of GGTI-2417 and GGTI-2418 depends on stabilizing p27 via blocking its phosphorylation at Thr187. In vivo, GGTI-2418 prevents human breast tumour growth in nude mice and causes breast tumour regression in MMTV-Erbb2 transgenic mice. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Watanabe M, et al. Inhibitors of protein geranylgeranyltransferase I and Rab geranylgeranyltransferase identified from a library of allenoate-derived compounds. J Biol Chem. 2008;283:9571–9579. doi: 10.1074/jbc.M706229200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Lu J, et al. In vivo antitumor effect of a novel inhibitor of protein geranylgeranyltransferase-I. Mol Cancer Ther. 2009;8:1218–1226. doi: 10.1158/1535-7163.MCT-08-1122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Dan HC, et al. Phosphatidylinositol-3-OH kinase/AKT and survivin pathways as critical targets for geranylgeranyltransferase I inhibitor-induced apoptosis. Oncogene. 2004;23:706–715. doi: 10.1038/sj.onc.1207171. [DOI] [PubMed] [Google Scholar]
- 96.Chen S, et al. Dissecting the roles of DR4, DR5 and c-FLIP in the regulation of geranylgeranyltransferase I inhibition-mediated augmentation of TRAIL-induced apoptosis. Mol Cancer. 2010;9:23. doi: 10.1186/1476-4598-9-23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.McGuire TF, Qian Y, Vogt A, Hamilton AD, Sebti SM. Platelet-derived growth factor receptor tyrosine phosphorylation requires protein geranylgeranylation but not farnesylation. J Biol Chem. 1996;271:27402–27407. doi: 10.1074/jbc.271.44.27402. [DOI] [PubMed] [Google Scholar]
- 98.Falsetti SC, et al. Geranylgeranyltransferase I inhibitors target RalB to inhibit anchorage-dependent growth and induce apoptosis and RalA to inhibit anchorage-independent growth. Mol Cell Biol. 2007;27:8003–8014. doi: 10.1128/MCB.00057-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Peterson YK, Kelly P, Weinbaum CA, Casey PJ. A novel protein geranylgeranyltransferase-I inhibitor with high potency, selectivity, and cellular activity. J Biol Chem. 2006;281:12445–12450. doi: 10.1074/jbc.M600168200. [DOI] [PubMed] [Google Scholar]
- 100.Peterson YK, Wang XS, Casey PJ, Tropsha A. Discovery of geranylgeranyltransferase-I inhibitors with novel scaffolds by the means of quantitative structure-activity relationship modeling, virtual screening, and experimental validation. J Med Chem. 2009;52:4210–4220. doi: 10.1021/jm8013772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Zujewski J, et al. Phase I and pharmacokinetic study of farnesyl protein transferase inhibitor R115777 in advanced cancer. J Clin Oncol. 2000;18:927–941. doi: 10.1200/JCO.2000.18.4.927. This is, to our knowledge, the first published report of a clinical trial with an FTI (tipifarnib). [DOI] [PubMed] [Google Scholar]
- 102.Lobell RB, et al. Preclinical and clinical pharmacodynamic assessment of L-778, 123, a dual inhibitor of farnesyl:protein transferase and geranylgeranyl:protein transferase type-I. Mol Cancer Ther. 2002;1:747–758. [PubMed] [Google Scholar]
- 103.Kauh J, et al. Farnesyl transferase expression determines clinical response to the docetaxel-lonafarnib combination in patients with advanced malignancies. Cancer. 2011;117:4049–4059. doi: 10.1002/cncr.26004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Sepp-Lorenzino L, et al. A peptidomimetic inhibitor of farnesyl:protein transferase blocks the anchorage-dependent and -independent growth of human tumor cell lines. Cancer Res. 1995;55:5302–5309. [PubMed] [Google Scholar]
- 105.End DW, et al. Characterization of the antitumor effects of the selective farnesyl protein transferase inhibitor R115777 in vivo and in vitro. Cancer Res. 2001;61:131–137. [PubMed] [Google Scholar]
- 106.Kurzrock R, et al. Farnesyltransferase inhibitor R115777 in myelodysplastic syndrome: clinical and biologic activities in the phase 1 setting. Blood. 2003;102:4527–4534. doi: 10.1182/blood-2002-11-3359. [DOI] [PubMed] [Google Scholar]
- 107.Kohl NE, et al. Protein farnesyltransferase inhibitors block the growth of ras-dependent tumors in nude mice. Proc Natl Acad Sci USA. 1994;91:9141–9145. doi: 10.1073/pnas.91.19.9141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Tee AR, Manning BD, Roux PP, Cantley IC, Blenis J. Turberous sclerosis complex gene products, tuberin and hamartin, control mTOR signaling by acting as a GTPase-activating protein complex toward Rheb. Curr Biol. 2003;13:1259–1268. doi: 10.1016/s0960-9822(03)00506-2. [DOI] [PubMed] [Google Scholar]
- 109.Garami A, et al. Insulin activation of Rheb, a mediator of mTOR/S6K/4E-BP signaling, is inhibited by TSC1 and 2. Mol Cell. 2003;11:1457–1466. doi: 10.1016/s1097-2765(03)00220-x. [DOI] [PubMed] [Google Scholar]
- 110.Zhang Y, et al. Rheb is a direct target of the tuberous sclerosis tumor suppressor proteins. Nature Cell Biol. 2003;5:578–581. doi: 10.1038/ncb999. [DOI] [PubMed] [Google Scholar]
- 111.Inoki K, Li Y, Zhu T, Wu J, Guan KL. TSC2 is phosphorylated and inhibited by Akt and suppresses mTOR signalling. Nature Cell Biol. 2002;4:648–657. doi: 10.1038/ncb839. [DOI] [PubMed] [Google Scholar]
- 112.Potter CJ, Pedraza LG, Xu T. Akt regulates growth by directly phosphorylating Tsc2. Nature Cell Biol. 2002;4:658–665. doi: 10.1038/ncb840. [DOI] [PubMed] [Google Scholar]
- 113.Manning BD, Tee AR, Logsdon MN, Blenis J, Cantley LC. Identification of the tuberous sclerosis complex-2 tumor suppressor gene product tuberin as a target of the phosphoinositide 3-kinase/akt pathway. Mol Cell. 2002;10:151–162. doi: 10.1016/s1097-2765(02)00568-3. [DOI] [PubMed] [Google Scholar]
- 114.Castro AF, Rebhun JF, Clark GJ, Quilliam LA. Rheb binds tuberous sclerosis complex 2 (TSC2) and promotes S6 kinase activation in a rapamycin- and farnesylation-dependent manner. J Biol Chem. 2003;278:32493–32496. doi: 10.1074/jbc.C300226200. [DOI] [PubMed] [Google Scholar]
- 115.Tabancay AP, et al. Identification of dominant negative mutants of Rheb GTPase and their use to implicate the involvement of human Rheb in the activation of p70S76K. J Biol Chem. 2003;278:39921–39930. doi: 10.1074/jbc.M306553200. [DOI] [PubMed] [Google Scholar]
- 116.Patel PH, et al. Drosophila Rheb GTPase is required for cell cycle progression and cell growth. J Cell Sci. 2003;116:3601–3610. doi: 10.1242/jcs.00661. This paper shows that RHEB is required for cell growth (and cell cycle progression). [DOI] [PubMed] [Google Scholar]
- 117.Long X, Lin Y, Ortiz-Vega S, Yonezawa K, Avruch J. Rheb binds and regulates the mTOR kinase. Curr Biol. 2005;15:702–713. doi: 10.1016/j.cub.2005.02.053. [DOI] [PubMed] [Google Scholar]
- 118.Bai X, et al. Rheb activates mTOR by antagonizing its endogenous inhibitor, FKBP38. Science. 2007;318:977–980. doi: 10.1126/science.1147379. [DOI] [PubMed] [Google Scholar]
- 119.Gromov PS, Madsen P, Tomerup N, Celis JE. A novel approach for expression cloning of small GTPases: identification, tissue distribution and chromosome mapping of the human homolog of rheb. FEBS Lett. 1995;377:221–226. doi: 10.1016/0014-5793(95)01349-0. [DOI] [PubMed] [Google Scholar]
- 120.Mavrakis KJ, et al. Tumorigenic activity and therapeutic inhibition of Rheb GTPase. Genes Dev. 2008;22:2178–2188. doi: 10.1101/gad.1690808. In this paper, RHEB was identified as a factor capable of enhancing lymphomagenesis in the Eμ-Myc transgenic mouse. Importantly, inhibition of RHEB farnesylation by FTI-277 was identified as a major factor contributing to the antitumour effect of the FTI, suggesting that in lymphomas, and perhaps other haematological malignancies, RHEB is an important target for FTIs. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Ravikumar B, et al. Inhibition of mTOR induces autophagy and reduces toxicity of polyglutamine expansions in fly and mouse models of Huntington disease. Nature Genet. 2004;36:585–595. doi: 10.1038/ng1362. [DOI] [PubMed] [Google Scholar]
- 122.Pan J, et al. Autophagy induced by farnesyltransferase inhibitors in cancer cells. Cancer Biol Ther. 2008;7:1679–1684. doi: 10.4161/cbt.7.10.6661. [DOI] [PubMed] [Google Scholar]
- 123.Du W, Lebowitz PF, Prendergast GC. Cell growth inhibition by farnesyltransferase inhibitors is mediated by gain of geranylgeranylated RhoB. Mol Cell Biol. 1999;19:1831–1840. doi: 10.1128/mcb.19.3.1831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Du W, Prendergast GC. Geranylgeranylated RhoB mediates suppression of human tumor cell growth by farnesyltransferase inhibitors. Cancer Res. 1999;59:5492–5496. [PubMed] [Google Scholar]
- 125.Mazières J, et al. Geranylgeranylated, but not farnesylated, RhoB suppresses Ras transformation of NIH-3T3 cells. Exp Cell Res. 2005;304:354–364. doi: 10.1016/j.yexcr.2004.10.019. [DOI] [PubMed] [Google Scholar]
- 126.Chen Z, et al. Both farnesylated and geranylgeranylated RhoB inhibit malignant transformation and suppress human tumor growth in nude mice. J Biol Chem. 2000;275:17974–17978. doi: 10.1074/jbc.C000145200. [DOI] [PubMed] [Google Scholar]
- 127.Adnane J, Muro-Cacho C, Mathews L, Sebti SM, Munoz-Antonia T. Suppression of rho B expression in invasive carcinoma from head and neck cancer patients. Clin Cancer Res. 2002;8:2225–2232. [PubMed] [Google Scholar]
- 128.Forget MA, et al. The expression of rho proteins decreases with human brain tumor progression: potential tumor markers. Clin Exp Metastasis. 2002;19:9–15. doi: 10.1023/a:1013884426692. [DOI] [PubMed] [Google Scholar]
- 129.Mazières J, et al. Loss of RhoB expression in human lung cancer progression. Clin Cancer Res. 2004;10:2742–2750. doi: 10.1158/1078-0432.ccr-03-0149. [DOI] [PubMed] [Google Scholar]
- 130.Lebowitz PF, Casey PJ, Prendergast GC, Thissen JA. Farnesyltransferase inhibitors alter the prenylation and growth-stimulating function of RhoB. J Biol Chem. 1997;272:15591–15594. doi: 10.1074/jbc.272.25.15591. [DOI] [PubMed] [Google Scholar]
- 131.Armstrong SA, Hannah VC, Goldstein JL, Brown MS. CAAX geranylgeranyl transferase transfers farnesyl as efficiently as geranylgeranyl to RhoB. J Biol Chem. 1995;270:7864–7868. doi: 10.1074/jbc.270.14.7864. [DOI] [PubMed] [Google Scholar]
- 132.Delarue FL, et al. Farnesyltransferase and geranylgeranyltransferase I inhibitors upregulate RhoB expression by HDAC1 dissociation, HAT association and histone acetylation of the RhoB promoter. Oncogene. 2007;26:633–640. doi: 10.1038/sj.onc.1209819. [DOI] [PubMed] [Google Scholar]
- 133.Liu A, Du W, Liu JP, Jessell TM, Prendergast GC. RhoB alteration is necessary for apoptotic and antineoplastic responses to farnesyltransferase inhibitors. Mol Cell Biol. 2000;20:6105–6113. doi: 10.1128/mcb.20.16.6105-6113.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Xu F, et al. The human ARHI tumor suppressor gene inhibits lactation and growth in transgenic mice. Cancer Res. 2000;60:4913–4920. [PubMed] [Google Scholar]
- 135.Finlin BS, et al. RERG is a novel ras-related, estrogen-regulated and growth-inhibitory gene in breast cancer. J Biol Chem. 2001;276:42259–42267. doi: 10.1074/jbc.M105888200. [DOI] [PubMed] [Google Scholar]
- 136.Hamaguchi M, et al. DBC2, a candidate for a tumor suppressor gene involved in breast cancer. Proc Natl Acad Sci USA. 2002;99:13647–13652. doi: 10.1073/pnas.212516099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Ellis CA, et al. Rig is a novel Ras-related protein and potential neural tumor suppressor. Proc Natl Acad Sci USA. 2002;99:9876–9881. doi: 10.1073/pnas.142193799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Elam C, et al. RRP22 is a farnesylated, nucleolar, ras-related protein with tumor suppressor potential. Cancer Res. 2005;65:3117–3125. doi: 10.1158/0008-5472.CAN-04-0749. [DOI] [PubMed] [Google Scholar]
- 139.Boyartchuk VL, Ashby MN, Rine J. Modulation of Ras and α-factor function by carboxyl-terminal proteolysis. Science. 1997;275:1796–1800. doi: 10.1126/science.275.5307.1796. [DOI] [PubMed] [Google Scholar]
- 140.Bergo MO, et al. Inactivation of Icmt inhibits transformation by oncogenic K-Ras and B-Raf. J Clin Invest. 2004;113:539–550. doi: 10.1172/JCI18829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Linder ME, Deschenes RJ. Palmitoylation: policing protein stability and traffic. Nature Rev Mol Cell Biol. 2007;8:74–84. doi: 10.1038/nrm2084. [DOI] [PubMed] [Google Scholar]
- 142.Bergo MO, et al. Absence of the CAAX endoprotease Rce1: effects on cell growth and transformation. Mol Cell Biol. 2002;22:171–181. doi: 10.1128/MCB.22.1.171-181.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Wang M, et al. Inhibition of isoprenylcysteine carboxylmethyltransferase induces autophagic-dependent apoptosis and impairs tumor growth. Oncogene. 2010;29:4959–4970. doi: 10.1038/onc.2010.247. [DOI] [PubMed] [Google Scholar]
- 144.Forget MA, Desrosiers RR, Gingras D, Beliveau R. Phosphorylation states of Cdc42 and RhoA regulate their interactions with Rho GDP dissociation inhibitor and their extraction from biological membranes. Biochem J. 2002;361:243–254. doi: 10.1042/0264-6021:3610243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Bivona TG, et al. PKC regulates a farnesyl-electrostatic switch on K-Ras that promotes its association with Bcl-XL on mitochondria and induces apoptosis. Mol Cell. 2006;21:481–493. doi: 10.1016/j.molcel.2006.01.012. This article highlights a long neglected aspect of KRAS regulation, namely phosphorylation of KRAS at Ser181 and its functional consequences: PKC-mediated phosphorylation at this site, which is located within the polybasic region of the KRAS C terminus, converts KRAS from an oncogenic protein into a pro-apoptotic protein. [DOI] [PubMed] [Google Scholar]
- 146.Rundell CJ, Repellin CE, Yarwood SJ. Protease inhibitors prevent the protein kinase A-dependent loss of Rap1 GTPase from the particulate fraction of COS1 cells. Biochem Biophys Res Commun. 2004;315:1077–1081. doi: 10.1016/j.bbrc.2004.01.161. [DOI] [PubMed] [Google Scholar]
- 147.Lang P, et al. Protein kinase A phosphorylation of RhoA mediates the morphological and functional effects of cyclic AMP in cytotoxic lymphocytes. EMBO J. 1996;15:510–519. [PMC free article] [PubMed] [Google Scholar]
- 148.Fitzgerald ML, Reed GL. Rab6 is phosphorylated in thrombin-activated platelets by a protein kinase C-dependent mechanism: effects on GTP/GDP binding and cellular distribution. Biochem J. 1999;342:353–360. [PMC free article] [PubMed] [Google Scholar]
- 149.Riento K, et al. RhoE function is regulated by ROCK I-mediated phosphorylation. EMBO J. 2005;24:1170–1180. doi: 10.1038/sj.emboj.7600612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Sablina AA, et al. The tumor suppressor PP2A Aβ regulates the RalA GTPase. Cell. 2007;129:969–982. doi: 10.1016/j.cell.2007.03.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Lim KH, et al. Aurora-A phosphorylates, activates, and relocalizes the small GTPase RalA. Mol Cell Biol. 2010;30:508–523. doi: 10.1128/MCB.00916-08. References 150 and 151 demonstrate that the transforming activity of RALA not only depends on prenylation, but also on phosphorylation at Ser194, which is controlled by aurora kinase A and PP2A. These findings may pave the way for new therapeutic opportunities, such as combining GGTIs with aurora kinase inhibitors. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Kwon T, Kwon DY, Chun J, Kim JH, Kang SS. Akt protein kinase inhibits Rac1-GTP binding through phosphorylation at serine 71 of Rac1. J Biol Chem. 2000;275:423–428. doi: 10.1074/jbc.275.1.423. [DOI] [PubMed] [Google Scholar]
- 153.Tillement V, et al. Phosphorylation of RhoB by CK1 impedes actin stress fiber organization and epidermal growth factor receptor stabilization. Exp Cell Res. 2008;314:2811–2821. doi: 10.1016/j.yexcr.2008.06.011. [DOI] [PubMed] [Google Scholar]
- 154.Zheng M, et al. Inactivation of Rheb by PRAK-mediated phosphorylation is essential for energy-depletion-induced suppression of mTORC1. Nature Cell Biol. 2011;13:263–272. doi: 10.1038/ncb2168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Sarthy AV, et al. Survivin depletion preferentially reduces the survival of activated K-Ras-transformed cells. Mol Cancer Ther. 2007;6:269–276. doi: 10.1158/1535-7163.MCT-06-0560. [DOI] [PubMed] [Google Scholar]
- 156.Barbie DA, et al. Systematic RNA interference reveals that oncogenic KRAS-driven cancers require TBK1. Nature. 2009;462:108–112. doi: 10.1038/nature08460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Scholl C, et al. Synthetic lethal interaction between oncogenic KRAS dependency and STK33 suppression in human cancer cells. Cell. 2009;137:821–834. doi: 10.1016/j.cell.2009.03.017. [DOI] [PubMed] [Google Scholar]
- 158.Luo J, et al. A genome-wide RNAi screen identifies multiple synthetic lethal interactions with the Ras oncogene. Cell. 2009;137:835–848. doi: 10.1016/j.cell.2009.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Singh A, et al. A gene expression signature associated with “K-Ras addiction” reveals regulators of EMT and tumor cell survival. Cancer Cell. 2009;15:489–500. doi: 10.1016/j.ccr.2009.03.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Puyol M, et al. A synthetic lethal interaction between K-Ras oncogenes and Cdk4 unveils a therapeutic strategy for non-small cell lung carcinoma. Cancer Cell. 2010;18:63–73. doi: 10.1016/j.ccr.2010.05.025. References 155–160 are particularly important as they identify novel promising targets for cancer patients whose tumours depend on KRAS for survival. Importantly, the four protein kinases identified are not essential for the survival of normal cells, but their loss of function in combination with activated KRAS is synthetically lethal. Reference 159 also emphasizes the point that not all KRAS-mutant cell lines are addicted to KRAS, suggesting that in a subset of tumours, KRAS may not be a suitable target. [DOI] [PubMed] [Google Scholar]
- 161.Williams JP, et al. The retinoblastoma protein is required for Ras-induced oncogenic transformation. Mol Cell Biol. 2006;26:1170–1182. doi: 10.1128/MCB.26.4.1170-1182.2006. This paper provides evidence that RAS-dependent transformation requires the presence of functional RB, a major tumour suppressor. Although this study uses murine fibroblasts, its findings may explain the fact that activating mutations of RAS and inactivating mutations of RB rarely occur together in human cancers. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Filmus J, et al. Induction of cyclin D1 overexpression by activated ras. Oncogene. 1994;9:3627–3633. [PubMed] [Google Scholar]
- 163.Weinberg RA. The retinoblastoma protein and cell cycle control. Cell. 1995;81:323–330. doi: 10.1016/0092-8674(95)90385-2. [DOI] [PubMed] [Google Scholar]
- 164.Kaye FJ. RB and cyclin dependent kinase pathways: defining a distinction between RB and p16 loss in lung cancer. Oncogene. 2002;21:6908–6914. doi: 10.1038/sj.onc.1205834. [DOI] [PubMed] [Google Scholar]
- 165.Maurer-Stroh S, et al. Towards complete sets of farnesylated and geranylgeranylated proteins. PLoS Comp Biol. 2007;3:e66. doi: 10.1371/journal.pcbi.0030066. More than 100 proteins have been experimentally confirmed to undergo prenylation, but the exact size of the prenylome is unknown. This paper describes methods to better predict whether a potentially prenylatable protein is a substrate for FT, GGT1 or GGT2. These take into account the requirement for specific residues within the CaaX box, evolutionary conservation of the prenylation motif across phyla and physicochemical constraints extending up to 11 residues upstream of the prenylatable Cys. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Lane KT, Beese LS. Thematic review series: lipid posttranslational modifications. Structural biology of protein farnesyltransferase and geranylgeranyltransferase type I. J Lipid Res. 2006;47:681–699. doi: 10.1194/jlr.R600002-JLR200. [DOI] [PubMed] [Google Scholar]
- 167.Berndt N, Sebti SM. Measuring protein farnesylation and geranylgeranylation and using prenyltransferase inhibitors as chemical probes and anticancer agents. Nature Protoc. (in the press) [Google Scholar]
- 168.Kho Y, et al. A tagging-via-substrate technology for detection and proteomics of farnesylated proteins. Proc Natl Acad Sci USA. 2004;101:12479–12484. doi: 10.1073/pnas.0403413101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Troutman JM, Roberts MJ, Andres DA, Spielmann HP. Tools to analyze protein farnesylation in cells. Bioconjug Chem. 2005;16:1209–1217. doi: 10.1021/bc050068+. [DOI] [PubMed] [Google Scholar]
- 170.Nguyen UT, et al. Analysis of the eukaryotic prenylome by isoprenoid affinity tagging. Nature Chem Biol. 2009;5:227–235. doi: 10.1038/nchembio.149. [DOI] [PubMed] [Google Scholar]
- 171.Onono FO, et al. A tagging-via-substrate approach to detect the farnesylated proteome using two-dimensional electrophoresis coupled with Western blotting. Mol Cell Proteomics. 2010;9:742–751. doi: 10.1074/mcp.M900597-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Chan LN, et al. A novel approach to tag and identify geranylgeranylated proteins. Electrophoresis. 2009;30:3598–3606. doi: 10.1002/elps.200900259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Degraw AJ, et al. Evaluation of alkyne-modified isoprenoids as chemical reporters of protein prenylation. Chem Biol Drug Des. 2010;76:460–471. doi: 10.1111/j.1747-0285.2010.01037.x. References 168–173 considerably advance our abilities to describe the effects of PTIs on the entire prenylome rather than just individual proteins. This is needed to identify prenylated proteins the inhibition of which contributes to the antitumour effects of PTIs. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Willumsen BM, Christensen A, Hubbert NL, Papageorge AG, Lowy DR. The p21 ras C-terminus is required for transformation and membrane association. Nature. 1984;310:583–586. doi: 10.1038/310583a0. [DOI] [PubMed] [Google Scholar]
- 175.Willumsen BM, Norris K, Papageorge AG, Hubbert NL, Lowy DR. Harvey murine sarcoma virus p21 ras protein: biological and biochemical significance of the cysteine nearest the carboxy terminus. EMBO J. 1984;3:2581–2585. doi: 10.1002/j.1460-2075.1984.tb02177.x. Although when references 174 and 175 were published it was unknown that RAS is farnesylated, these studies are important as they demonstrated that the C terminus of RAS is essential for both its membrane binding and its transforming activity. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Seabra MC, Reiss Y, Casey PJ, Brown MS, Goldstein JL. Protein farnesyltransferase and geranylgeranyltransferase share a common α subunit. Cell. 1991;65:429–434. doi: 10.1016/0092-8674(91)90460-g. [DOI] [PubMed] [Google Scholar]
- 177.Zhang FL, et al. cDNA cloning and expression of rat and human protein geranylgeranyltransferase type-I. J Biol Chem. 1994;269:3175–3180. [PubMed] [Google Scholar]
- 178.Park HW, Boduluri SR, Moomaw JF, Casey PJ, Beese LS. Crystal structure of protein farnesyltransferase at 2.25 angstrom resolution. Science. 1997;275:1800–1804. doi: 10.1126/science.275.5307.1800. [DOI] [PubMed] [Google Scholar]
- 179.Huang CC, Casey PJ, Fierke CA. Evidence for a catalytic role of zinc in protein farnesyltransferase. Spectroscopy of Co2+-farnesyltransferase indicates metal coordination of the substrate thiolate. J Biol Chem. 1997;272:20–23. doi: 10.1074/jbc.272.1.20. [DOI] [PubMed] [Google Scholar]
- 180.Zhang FL, Casey PJ. Protein prenylation: molecular mechanisms and functional consequences. Annu Rev Biochem. 1996;65:241–269. doi: 10.1146/annurev.bi.65.070196.001325. [DOI] [PubMed] [Google Scholar]
- 181.Carboni JM, et al. Farnesyltransferase inhibitors are inhibitors of Ras but not R-Ras2/TC21, transformation. Oncogene. 1995;10:1905–1913. [PubMed] [Google Scholar]
- 182.Moores SL, et al. Sequence dependence of protein isoprenylation. J Biol Chem. 1991;266:14603–14610. [PubMed] [Google Scholar]
- 183.Boutin JA, et al. Chromatographic assay and peptide substrate characterization of partially purified farnesyl- and geranylgeranyltransferases from rat brain cytosol. Arch Biochem Biophys. 1998;354:83–94. doi: 10.1006/abbi.1998.0678. [DOI] [PubMed] [Google Scholar]
- 184.Eriksson M, et al. Recurrent de novo point mutations in lamin A cause Hutchinson-Gilford progeria syndrome. Nature. 2003;423:293–298. doi: 10.1038/nature01629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Young SG, Fong LG, Michaelis S. Prelamin A, Zmpste24, misshapen cell nuclei, and progeria-new evidence suggesting that protein farnesylation could be important for disease pathogenesis. J Lipid Res. 2005;46:2531–2558. doi: 10.1194/jlr.R500011-JLR200. [DOI] [PubMed] [Google Scholar]
- 186.Mallampalli MP, Huyer G, Bendale P, Gelb MH, Michaelis S. Inhibiting farnesylation reverses the nuclear morphology defect in a HeLa cell model for Hutchinson-Gilford progeria syndrome. Proc Natl Acad Sci USA. 2005;102:14416–14421. doi: 10.1073/pnas.0503712102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Toth JI, et al. Blocking protein farnesyltransferase improves nuclear shape in fibroblasts from humans with progeroid syndromes. Proc Natl Acad Sci USA. 2005;102:12873–12878. doi: 10.1073/pnas.0505767102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Yang SH, et al. Blocking protein farnesyltransferase improves nuclear blebbing in mouse fibroblasts with a targeted Hutchinson-Gilford progeria syndrome mutation. Proc Natl Acad Sci USA. 2005;102:10291–10296. doi: 10.1073/pnas.0504641102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Fong LG, et al. A protein farnesyltransferase inhibitor ameliorates disease in a mouse model of progeria. Science. 2006;311:1621–1623. doi: 10.1126/science.1124875. [DOI] [PubMed] [Google Scholar]
- 190.Yang SH, et al. Assessing the efficacy of protein farnesyltransferase inhibitors in mouse models of progeria. J Lipid Res. 2010;51:400–405. doi: 10.1194/jlr.M002808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Work LM, et al. Short-term local delivery of an inhibitor of Ras farnesyltransferase prevents neointima formation in vivo after porcine coronary balloon angioplasty. Circulation. 2001;104:1538–1543. doi: 10.1161/hc3801.095661. [DOI] [PubMed] [Google Scholar]
- 192.Finder JD, et al. Inhibition of protein geranylgeranylation causes a superinduction of nitric-oxide synthase-2 by interleukin-1β in vascular smooth muscle cells. J Biol Chem. 1997;272:13484–13488. doi: 10.1074/jbc.272.21.13484. [DOI] [PubMed] [Google Scholar]
- 193.Eastman RT, Buckner FS, Yokoyama K, Gelb MH, Van Voorhis WC. Thematic review series: lipid posttranslational modifications. Fighting parasitic disease by blocking protein farnesylation. J Lipid Res. 2006;47:233–240. doi: 10.1194/jlr.R500016-JLR200. [DOI] [PubMed] [Google Scholar]
- 194.Carrico D, et al. In vitro and in vivo antimalarial activity of peptidomimetic protein farnesyltransferase inhibitors with improved membrane permeability. Bioorg Med Chem. 2004;12:6517–6526. doi: 10.1016/j.bmc.2004.09.020. [DOI] [PubMed] [Google Scholar]
- 195.Nallan L, et al. Protein farnesyltransferase inhibitors exhibit potent antimalarial activity. J Med Chem. 2005;48:3704–3713. doi: 10.1021/jm0491039. [DOI] [PubMed] [Google Scholar]
- 196.Yokoyama K, Gillespie JR, Van Voorhis WC, Buckner FS, Gelb MH. Protein geranylgeranyltransferase-I of Trypanosoma cruzi. Mol Biochem Parasitol. 2008;157:32–43. doi: 10.1016/j.molbiopara.2007.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Bordier BB, et al. In vivo antiviral efficacy of prenylation inhibitors against hepatitis delta virus. J Clin Invest. 2003;112:407–414. doi: 10.1172/JCI17704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Walters, et al. Inhibition of Rho GTPases with protein prenyltransferase inhibitors prevents leukocyte recruitment to the central nervous system and attenuates clinical signs of disease in an animal model of multiple sclerosis. J Immunol. 2002;168:4087–4094. doi: 10.4049/jimmunol.168.8.4087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Coxon FP, et al. Protein geranylgeranylation is required for osteoclast formation, function, and survival: inhibition by bisphosphonates and GGTI-298. J Bone Miner Res. 2000;15:1467–1476. doi: 10.1359/jbmr.2000.15.8.1467. [DOI] [PubMed] [Google Scholar]
- 200.Kucich U, et al. Requirement for geranylgeranyl transferase I and acyl transferase in the TGF-β-stimulated pathway leading to elastin mRNA stabilization. Biochem Biophys Res Commun. 1998;252:111–116. doi: 10.1006/bbrc.1998.9544. [DOI] [PubMed] [Google Scholar]
- 201.Kamiya Y, Sakurai A, Tamura S, Takahashi N. Structure of rhodotorucine A, a novel lipopeptide, inducing mating tube formation in Rhodosporidium toruloides. Biochem Biophys Res Commun. 1978;83:1077–1083. doi: 10.1016/0006-291x(78)91505-x. [DOI] [PubMed] [Google Scholar]
- 202.Der CJ, Krontiris TG, Cooper GM. Transforming genes of human bladder and lung carcinoma cell lines are homologous to the ras genes or Harvey and Kirsten sarcoma viruses. Proc Natl Acad Sci USA. 1982;79:3637–3640. doi: 10.1073/pnas.79.11.3637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203.Parada LF, Tabin CJ, Shih C, Weinberg RA. Human EJ bladder carcinoma oncogene is homologue of Harvey sarcoma virus ras gene. Nature. 1982;297:474–478. doi: 10.1038/297474a0. [DOI] [PubMed] [Google Scholar]
- 204.Mulcahy LS, Smith MR, Stacey DW. Requirement for ras proto-oncogene function during serum-stimulated growth of NIH 3T3 cells. Nature. 1985;313:241–243. doi: 10.1038/313241a0. [DOI] [PubMed] [Google Scholar]
- 205.Powers S, et al. RAM, a gene of yeast required for a functional modification of RAS proteins and for production of mating pheromone a-factor. Cell. 1986;47:413–422. doi: 10.1016/0092-8674(86)90598-2. [DOI] [PubMed] [Google Scholar]
- 206.Wolda SL, Glomset JA. Evidence for modification of lamin B by a product of mevalonic acid. J Biol Chem. 1988;263:5997–6000. [PubMed] [Google Scholar]
- 207.Farnsworth CC, Wolda SL, Gelb MH, Glomset JA. Human lamin B contains a farnesylated cysteine residue. J Biol Chem. 1989;264:20422–20429. [PMC free article] [PubMed] [Google Scholar]
- 208.Casey PJ, Solski PA, Der CJ, Buss J. E p21ras is modified by a farnesyl isoprenoid. Proc Natl Acad Sci USA. 1989;86:8323–8327. doi: 10.1073/pnas.86.21.8323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209.Hancock JF, Magee AI, Childs JE, Marshall CJ. All ras proteins are polyisoprenylated but only some are palmitoylated. Cell. 1989;57:1167–1177. doi: 10.1016/0092-8674(89)90054-8. [DOI] [PubMed] [Google Scholar]
- 210.Rilling HC, Breunger E, Epstein WW, Crain PF. Prenylated proteins: the structure of the isoprenoid group. Science. 1990;247:318–320. doi: 10.1126/science.2296720. [DOI] [PubMed] [Google Scholar]
- 211.Farnsworth CC, Gelb MH, Glomset JA. Identification of geranylgeranyl-modified proteins in HeLa cells. Science. 1990;247:320–322. doi: 10.1126/science.2296721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212.Yamane HK, et al. Brain G protein γ subunits contain an all-trans-geranylgeranylcysteine methyl ester at their carboxyl termini. Proc Natl Acad Sci USA. 1990;87:5868–5872. doi: 10.1073/pnas.87.15.5868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213.Mumby SM, Casey PJ, Gilman AG, Gutowski S, Sternweis PC. G protein γ subunits contain a 20-carbon isoprenoid. Proc Natl Acad Sci USA. 1990;87:5873–5877. doi: 10.1073/pnas.87.15.5873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214.Reiss Y, Goldstein JL, Seabra MC, Casey PJ, Brown MS. Inhibition of purified p21ras farnesyl:protein transferase by Cys-AAX tetrapeptides. Cell. 1990;62:81–88. doi: 10.1016/0092-8674(90)90242-7. [DOI] [PubMed] [Google Scholar]
- 215.Kitten GT, Nigg EA. The CaaX motif is required for isoprenylation, carboxyl methylation, and nuclear membrane association of lamin B2. J Cell Biol. 1991;113:13–23. doi: 10.1083/jcb.113.1.13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 216.Kohl NE, et al. Selective inhibition of ras-dependent transformation by a farnesyltransferase inhibitor. Science. 1993;260:1934–1937. doi: 10.1126/science.8316833. [DOI] [PubMed] [Google Scholar]
- 217.Garcia AM, Rowell C, Ackermann K, Kowalczyk JJ, Lewis MD. Peptidomimetic inhibitors of Ras farnesylation and function in whole cells. J Biol Chem. 1993;268:18415–18418. [PubMed] [Google Scholar]
- 218.Nigam M, Seong CM, Qian Y, Hamilton AD, Sebti SM. Potent inhibition of human tumor p21ras farnesyltransferase by A1A2-lacking p21ras CA1A2X peptidomimetics. J Biol Chem. 1993;268:20695–20698. [PubMed] [Google Scholar]
- 219.James GL, et al. Benzodiazepine peptidomimetics: potent inhibitors of Ras farnesylation in animal cells. Science. 1993;260:1937–1942. doi: 10.1126/science.8316834. [DOI] [PubMed] [Google Scholar]
- 220.Gibbs JB, et al. Selective inhibition of farnesyl-protein transferase blocks ras processing in vivo. J Biol Chem. 1993;268:7617–7620. [PubMed] [Google Scholar]
- 221.Hara M, et al. Identification of Ras farnesyltransferase inhibitors by microbial screening. Proc Natl Acad Sci USA. 1993;90:2281–2285. doi: 10.1073/pnas.90.6.2281. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.