

Abstract
The prevalence of diabetes is increasing rapidly world-wide. A cardinal feature of most forms of diabetes is the lack of insulin-producing capability, due to the loss of insulin-producing β-cells, impaired glucose-sensitive insulin secretion from the β-cell, or a combination thereof, the reasons for which largely remain elusive. Reversible phosphorylation is an important and versatile mechanism for regulating the biological activity of many intracellular proteins, which, in turn, controls a variety of cellular functions. For instance, significant changes in protein kinase activities and in protein phosphorylation patterns occur subsequent to stimulation of insulin release by glucose. Therefore, the molecular mechanisms regulating phosphorylation of proteins involved in the insulin secretory process by the β-cell have been extensively investigated. However, far less is known about the role and regulation of protein dephosphorylation by various protein phosphatases. Herein we review extant data implicating serine/threonine and tyrosine phosphatases in various aspects of healthy and diabetic islet biology, ranging from control of hormonal stimulus-secretion coupling to mitogenesis and apoptosis.
Keywords: Diabetes, Islet, Protein phosphatase, High-fat diet, Knockout mouse, Obesity, Insulin
Type 2 diabetes: a growing epidemic
Type 2 diabetes (T2D) is a syndrome characterized by disordered metabolism, resulting in hyperglycemia. The most common and dreaded long-term complication of diabetes is cardiovascular disease, which accounts for 75-80 % of all diabetes related deaths (Meetoo, et al. 2007). Diabetes is widespread and it is the fourth leading cause of death in the U.S. (Meetoo et al. 2007). The expenses by diabetes have been shown to be a major drain on health- and productivity-related resources for healthcare systems and governments. In the U.S. alone, the annual cost for diabetes amounts to the considerable sum of $ 245 billion, of which ~ 97 % is targeted to T2D (American Diabetes 2013). Improved glycemia is a main focus of T2D therapy and glycated hemoglobin (HbA1c) levels of 5-6 % (DCCT standard; corresponding to 31-42 mmol/mol by IFCC standard) are recommended treatment goals. However, more than 50 % of patients with T2D have a HbA1c level of >7 % (53 mmol/mol by IFCC standard) and are thus inadequately controlled (Koro, et al. 2004).
Loss of glucose-sensitive insulin secretion of the pancreatic β-cell is an early pathogenic event and contributes significantly to the development of the diabetic state (Bell and Polonsky 2001; Grimsby, et al. 2003; Ward, et al. 1984). The changes in β-cell function in diabetes include decline in glucose-sensitive insulin secretory output (Ward et al. 1984), disturbances in pulsatile insulin release (Tengholm and Gylfe 2009) and impaired insulin synthesis (Kahn and Halban 1997). Thus, improvement of β-cell function is a major goal in the clinical management of the disease.
Inadequacy of the pancreatic β-cell also results from a combination of impaired secretory function and insufficient β-cell mass. The ability of the β-cell to expand its proliferative capacity in response to an increased insulin demand may be of critical regulatory significance for the development of diabetes (Lee and Nielsen 2009; Sjöholm 1996). T2D patients exhibit a reduced β-cell mass, possibly due to increased rates of apoptosis (Butler, et al. 2003). Maintaining islet β-cell mass and adequate insulin secretion to meet metabolic demands is crucial to avoid glucose intolerance and the development of T2D.
There is a progressive and relentless deterioration in β-cell function over time in T2D, regardless of therapy allocation, such as insulin, glibenclamide, or metformin treatment (1998a, b), eventually leaving many patients reliant on exogenous insulin replacement therapy.
The role of declining β-cell mass and function in the development of T2D has drawn attention to the need for agents that can halt this process. Moreover, in individuals with established T2D, inhibition of the increased apoptosis may lead to restoration of β-cell mass and it may also prevent pre-diabetic subjects to progress into overt T2D.
Regulation of insulin secretion
Pancreatic β-cells are equipped to rapidly sense ambient glycemia. In order for the cells to respond appropriately with insulin secretion, glucose must be metabolized within the β-cells (Ashcroft and Rorsman 2012; Hedeskov 1980). Glucose rapidly enters the cells via the efficient glucose transporter 2 (GLUT2 [GLUT1 in human islets]) that enables a balance between the extracellular and intracellular concentration of glucose (Meglasson and Matschinsky 1986; Newgard and McGarry 1995). Following entry, glucose is phosphorylated by glucokinase, which acts as a glucose sensor by controlling the amount of glucose that traverses through the glycolytic pathway (Matschinsky, et al. 1998). Glucose metabolism results, among other things, in increased production of adenosine triphosphate (ATP), leading to an increased ATP/adenosine diphosphate (ADP) ratio (Detimary, et al. 1995), which (like sulfonylurea drugs) closes the ATP-sensitive K+ (KATP) channels (Ashcroft, et al. 1984; Cook and Hales 1984). This causes depolarization of the plasma membrane, opening of voltage-dependent Ca2+ channels (VDCC) and influx of extracellular Ca2+. Elevation of cytosolic Ca2+ is the main trigger of granule translocation and insulin exocytosis (Jonas, et al. 1998). However, experiments indicate that glucose retains an excellent ability to secrete insulin even in the presence of maximally effective concentrations of K+ and diazoxide, which acts by opening K+ channels (Gembal, et al. 1992; Komatsu, et al. 1997). Thus, although signaling molecules other than ATP and Ca2+ must be involved in glucose sensing in the β-cell, the precise nature by which these complementary signals promote secretion and the KATP-independent signaling pathways activated by glucose have remained elusive. Insulin secretion is a complex process, tuned by many mechanisms, and has been the topic of excellent reviews (Ashcroft and Rorsman 2012; Rorsman and Braun 2013).
Introduction to reversible protein phosphorylation and protein phosphatases
In 1992, the Nobel Prize in Physiology or Medicine was awarded jointly to Edmond H. Fischer and Edwin G. Krebs, for their earlier discoveries revealing that the reversible covalent attachment of phosphate to a protein functions as a mechanism to regulate biological activity. The protein that was reversibly phosphorylated was glycogen phosphorylase, and the proteins that catalyzed phosphorylation and dephosphorylation were termed phosphorylase kinase and phosphorylase phosphatase, respectively (Fischer, et al. 1959; Krebs, et al. 1959; Sutherland and Wosilait 1955). Today, this simple reaction, in which a kinase catalyzes the transfer of phosphate from the gamma position of a high energy phosphonucleotide (usually ATP) to the side chain hydroxyl of a protein (usually serine, threonine or tyrosine) and a phosphatase catalyzes phosphate hydrolysis, is established as a fundamental, if not paramount, mechanism by which eukaryotic cells regulate virtually all aspects of cell biology. Accordingly, there has been an intensive global effort to identify and characterize the biological roles of these important regulatory enzymes.
Sequence data from the human genome indicate humans express ~520 protein kinases, with ~90 acting as tyr-kinases and 428 acting as ser/thr kinases (Johnson and Hunter 2005). Many kinases are highly conserved in nature. However, the tyrosine kinases appear to have evolved more recently, with the evolution of multicellular eukaryote organisms (Alonso, et al. 2004; Johnson and Hunter 2005). To counter these kinases, humans have a nearly equal number (~107) of phospho-tyr-phosphatases, suggesting each tyr-kinase is countered by a single tyrphosphatase (Alonso et al. 2004). In contrast, the number of genes encoding proteins capable of catalyzing the hydrolysis of phospho-ser/thr residues is more limited (~40 genes). Estimates of total protein phosphate from studies using [32P] labeling (Hunter and Sefton 1980; Hunter, et al. 1980) or proteomic analysis of phosphorylation sites of human proteins (Olsen, et al. 2006) are in agreement, indicating that ~98% of the phosphate in proteins is attached to ser/thr residues, with <2% attached to tyrosine. Thus, the tyrosine kinases/phosphatases may be viewed as “thoroughbreds” acting extensively only in the restricted arena of multicellular eukaryotes. Ser/thr kinases/phosphatases are more primitive and are more likely to act as the “work horses” of reversible phosphorylation in both single and multicellular eukaryotes. Clearly, both families are important and several excellent reviews focus on ser/thr and tyrosine kinases (Endicott, et al. 2012; Manning, et al. 2002; Nolen, et al. 2004; Taylor and Kornev 2011).
When compared to their kinase counter parts, less is known about the biological roles played by protein phosphatases. This is due, in part, to technical difficulties associated with accurately measuring protein dephosphorylation, and to early and lingering misconceptions that phosphatases act as simple “housekeeping” or pleiotropic enzymes. Today we know that phosphatases are not simple housekeeping enzymes, rather they play specific, active and sometimes even dominant roles in controlling both the levels of phosphorylation in cells and the regulation of physiological processes (Alonso et al. 2004). In this review we will focus on protein phosphatases, emphasizing their roles in pancreatic islets.
PTPs (phosphotyrosine phosphatases) have received the most recognition for playing precise and regulated roles in cell signaling, and the 107 genes encoding PTPs can be divided into four families (Alonso et al. 2004) (Figure 1). The largest family (class I Cys-base PTPs) contains 99 genes. Class I PTPs share a common catalytic mechanism (Figure 2). In this reaction, during the cleavage of the scissile P-O bond a covalent phospho-cysteine intermediate is produced at the catalytic site. Hydrolysis of the cysteinyl-phosphate intermediate is then facilitated by protonation of phenolic oxygen by a conserved aspartic acid and the positioning of an activated water by a conserved active site glutamine and serine. Mutation of the conserved aspartic acid to alanine can aid the identification of substrates, by producing substrate-trapping mutants that retain the covalent attachment at the catalytic cysteine (Blanchetot, et al. 2005; Flint, et al. 1997).
Figure 1. Family tree of PTPs.

Figure 2. Comparison of PTPase and PPP- catalytic mechanism.

A) Schematic representation of PTP-Cys-mediated hydrolysis of substrate derived from the crystal structure of PTP1B (Barford, et al. 1994). Schematic representation of metal ion-mediated hydrolysis of substrate derived from the crystal structure of PP5C (Swingle et al. 2004). The attacking hydroxide W1 is shown in blue, and the leaving group of the substrate is in green. The substrate, the planar PO3 moiety of the transition state, and the phosphate product are all shown in red. Solid lines to the metal ions denote metal-ligand bonds, and solid or dashed wedges indicate metal-ligand bonds directed above or below the plane of the page, respectively. Wavy lines to the metal ions indicate strained contacts with poor coordination geometry. Dotted lines indicate hydrogen bonds, and the nearly dissociated axial bonds in the transition state are shown by half-dotted double lines.
The class I PTPs can be further divided into four subgroups, with the most studied subgroup referred to as the classical PTPs (38 genes). Classical PTPs are strictly tyrosine specific and come in two forms, transmembrane PTPases (21 genes) and non-receptor PTPase (17 genes). The transmembrane PTPs have external “ligand-binding” domains, a membrane-spanning domain, and a cytosolic catalytic domain(s). In many ways these PTPs mimic the general characteristics of receptor-tyrosine kinases, the enzymes that they commonly counter in a cell. The non-receptor PTPs lack the extracellular and transmembrane domains. They are generally cytosolic proteins, some of which are anchored to membranes by prenylation.
The largest and most diverse family (in terms of substrate specificity) of class-I cys-based PTPs are called dual-specificity phosphatases (DSPs or DUSPs; 69 genes). DSPs share the cys-based catalytic mechanism, and as their name implies can act on phosphotyrosine and phosphothreonine residues. Eleven have MAP kinase targeting motifs and may act exclusively at specific phosphotyrosine or phosphothreonine sites on MAP kinases. Nineteen are considered atypical DSPs and they represent a poorly characterized family of enzymes that lack MAP kinase targeting motifs (Alonso et al. 2004). PTENS (5 genes) and myotubularins (16 genes) are contained in the DSP family, but they appear to have evolved to specifically dephosphorylate the D3-phosphate of inositol phospholipids (Wishart and Dixon 2002).
The human genome has only one gene encoding a class II cysteine based PTPs (ACP1) which encodes a low (18 kDa) molecular weight protein. Humans express three Class III cysteine based PTPs, which encode CDC25A, CDC25B and CDC25C. The CDC25 PTPs are well-characterized phosphatases that function to dephosphorylate cyclin dependent kinases (Cdks) at their inhibitory dually phosphorylated thr/tyr motifs. For further details on the PTPs, see the excellent review by Alonso et al. (Alonso et al. 2004).
Phospho-ser/thr-phosphatases (PSPs) are divided into three major families based on different catalytic mechanisms (PPPs, phosphoprotein phosphatases; PPMs, metal-dependent protein phosphatases; and FCP/SCP, aspartate-based phosphatases (Shi 2009) (Figure 3). Although the nomenclature may suggest otherwise, the catalytic mechanism employed by both PPPs and PPMs require two metal ions (Figure 2B). All PPP-family members share a common catalytic domain, with 10 absolutely conserved amino acids at the active site (Swingle, et al. 2004). Six act to coordinate two metal ions (Fe/Zn) needed for the activation of a water molecule, which functions as the critical nucleophile during catalysis. The others position the incoming substrate for near perfect inline nucleophilic attack by the activated water (Swingle et al. 2004). PPMs are Mn2+/Mg2+-dependent phosphatases. PPMs evolved a different folding strategy to produce a similar catalytic mechanism that also utilizes metal ions in the activation of a water molecule for the dephosphorylation reaction (Shi 2009). Unlike PTPs, a covalent intermediate is not produced during the reaction. The aspartate-based catalysis mechanism utilized by FCP/SCP is different and may be limited to a limited number of substrates that contain random repeats of SYPTSPS (for review see: (Shi 2009)).
Figure 3. Family tree of PSPs.
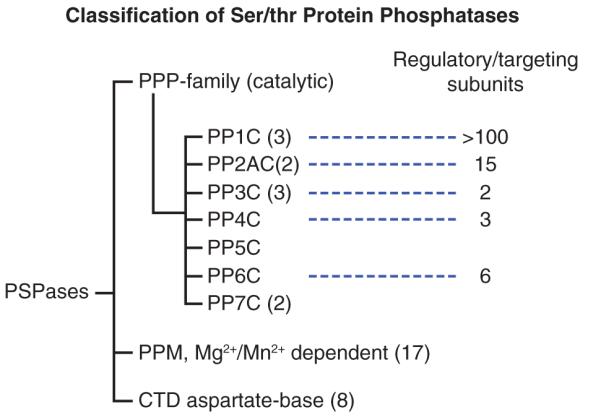
Based on the number of genes encoding proteins with phosphatase catalytic activity, PPMs represent the largest family of human PSPs. The PPM family included pyruvate dehydrogenase phosphatase, and ~16 genes encoding >20 isoforms of the PP2C (Lammers and Lavi 2007). These enzymes are insensitive to natural inhibitors (i.e. okadaic acid, microcystin, cantharidin and calyculin A), and the actions of most PPMs are poorly understood. However, due to their unique expression and subcellular localization patterns, most are predicted to act on a single or limited substrates (Lammers and Lavi 2007).
The PPP-family contains 7 subfamilies (PP1-PP7) (Figure 3), which are encoded by only 13 human genes yet together catalyze over 90 % of all protein dephosphorylation occurring in eukaryotic cells (Moorhead, et al. 2007; Virshup and Shenolikar 2009). Humans contain 3 genes encoding four isoforms of PP1, (PP1cα, PP1cβ, PP1cγ1, and PP1cγ2, with the PP1cγ2 isoform produced by alternate splicing of the PP1γ1gene). Two human genes encode for nearly identical (98%) isoforms of PP2A (PP2Acα, PP2Acβ). PP4 and PP6 share 65% identity with PP2Ac, but are encoded by distinct genes (Honkanen and Golden 2002). Humans express three highly homologous isoforms of PP2B/calcineurin (PP2Bα, PP2Bβ and PP2Bγ) and two genes encode isoforms of PP7 (also called PPEF). PP5 is unique in the respect that humans only express a single isoform of PP5. All PPP-family members are highly conserved in nature (e.g. the ortholog of PP2Aα in Neurospora crassa [bread mold] shares 87 % amino acid identity with human PP2Aα). See Figure 4 for a structural comparison of PP1-MYTP1, PP2Ac-A-B and PP5.
Figure 4. Structural comparison of PP1-MYTP1, PP2Ac-A-B and PP.
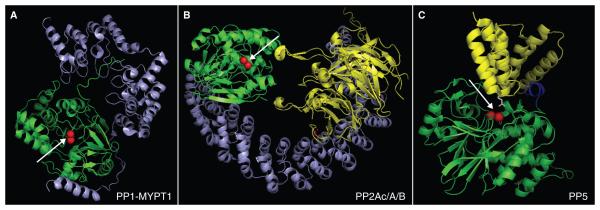
A) PP1 (green) in complex with myosin phosphatase targeting subunit MYPT1 (blue). B) PP2A holoenzyme: PP2A catalytic subunit (green) in complex with the PP2A scaffold A (blue) and a B55-regulatory targeting subunit (yellow). C). PP5 in an inactive conformation. The catalytic domain is shown ingreen, N-terminal inhibitory/TPR-targeting domain in yellow, and a unique C-terminal inhibitory domain in blue. The images were generated using PyMol based on protein data bank accession number 1S70 (Terrak, et al. 2004) (PP1-MYTP1), 3DW8 (Xu, et al. 2008) (PP2Ac/A/B) and 1WA0 (Yang et al. 2005) (PP5). Arrows indicate the catalytic site with metal ions shown as red spheres.
The ability of <15 gene products to counter ~90% of all cellular protein phosphorylation produced the lingering misconception that PPP family enzymes act as pleiotropic or simple housekeeping enzymes. More recently this popular, yet erroneous, belief has given way to overwhelming data that indicate the actions of most PPPs are dynamic and highly regulated. What the early studies failed to reveal was that, although PPPs share a structurally related catalytic core and identical catalytic mechanisms, they function in the cell as multi-subunit protein complexes. In cells each PPP family member can achieve many specific functions because the protein encoded by a PPP gene represents a catalytic subunit that can interact with a distinct set of substrates and interaction proteins. PP1 and PP2A are the most studied, and to date nearly 200 PP1-interacting proteins have been validated (Heroes, et al.). These PP1-interacting proteins share little or no structural similarity beyond their PP1-interacting domains and many are only expressed in differentiated or highly specialized cells (Heroes et al. ; Virshup and Shenolikar 2009). Therefore, PP1 actually represents a vast array of PP1c-containing holoenzymes, in which the structurally unrelated binding partners control the subcellular localization, activity and substrate specificity of PP1 (Bollen, et al. ; Heroes et al.).
PP2A, PP4 and PP6 also gain regulation and substrate specificity by assembling into a number of different multi-subunit holoenzymes that share a common catalytic subunit. For this family PP2A is best studied. PP2A commonly functions as a three-protein holoenzyme. Most human cells express both the catalytic subunit (PP2Ac) and an A-subunit that functions as a scaffold to tether PP2Ac to a number of different regulatory/targeting B-subunit. In humans there are 15 genes encoding four families of B-subunits that produce >21 B-isoforms, many of which are expressed only in certain types of cells or during different stages of development (Shi 2009; Virshup and Shenolikar 2009). The final composition of the PP2A-holoenzyme is then derived from the combinatorial assembly of one of the two isoforms of PP2Ac, one of the two isoforms of PP2A-A, and one of the >20 B-subunits (Sents, et al. ; Virshup and Shenolikar 2009). Therefore, substrate specificity, subcellular targeting and control of PP2A holoenzyme activity is usually regulated by assembly and mainly determined by the regulatory B-subunits (Lambrecht, et al. ; Sents et al. ; Virshup and Shenolikar 2009). Similar regulatory, targeting and control mechanisms are starting to emerge from studies of PP4 and PP6, which have their own scaffold and regulatory proteins (Chen, et al. 2008; Couzens, et al.). In addition, there are a few examples (i.e. alpha 4) in which interaction of certain B-type regulatory proteins are shared by PP2A, PP4 and PP6 (Breitkreutz, et al. ; Chen, et al. 1998; Kloeker, et al. 2003).
PP2B, more commonly called calcineurin, is the target of cyclosporin A, which is useful in a clinical setting as a strong immunosuppressive agent. Both calcineurin and PP7 are insensitive to okadaic acid and microcystin (Honkanen and Golden 2002; Huang and Honkanen 1998), and both calcineurin and PP7 are regulated by calcium. For calcineurin, the catalytic-A subunit is maintained in an inactive/inhibited state by the binding of an inhibitory protein, commonly call calcineurin B. Calcineurin only becomes active upon the calcium-mediated association with Ca2+-bound calmodulin (Shi 2009). PP7 is also activated by calcium; however, the C-terminal domain of PP7 contains calmodulin-like EF-had like domains that appear to directly bind Ca2+ (Honkanen and Golden 2002; Huang and Honkanen 1998; Huang, et al. 2000). Calcineurin expression is high in brain, while the expression of PP7 is limited, mostly to retina (Huang and Honkanen 1998; Shi 2009).
PP5 is unique in the respect that the catalytic, regulatory and substrate targeting domains of PP5 are encoded by a single gene and expressed as a single polypeptide (Golden, et al. 2008; Honkanen and Golden 2002). The catalytic core of PP5 is similar in structure to the catalytic subunit of PP1, PP2A, PP4 and PP6, and like these PSPs, PP5 is sensitive to inhibition by okadaic acid, calyculin A, cantharidin and microcystins (Honkanen and Golden 2002; Swingle, et al. 2007; Swingle et al. 2004; Virshup and Shenolikar 2009). Indeed the vast majority of the studies that use these natural inhibitors to study PPP-actions in cells draw conclusions that implicate PP1 and PP2A in the process that are being studied, failing to acknowledge that PP4, PP5 and PP6 are also widely expressed in human tissues and potently inhibited by these natural compounds (Swingle et al. 2007; Swingle et al. 2004; Virshup and Shenolikar 2009). Unlike PP1 and PP2A, the catalytic activity of PP5 is minimal when PP5 is not associated in a complex with other proteins (Golden et al. 2008). This is because when PP5 is alone, the N-terminal domain of PP5 folds back over the catalytic site producing an auto-inhibitory complex that blocks substrate access to the catalytic site (Golden et al. 2008). The N-terminal region of PP5 also contains three tetratricopeptide repeat (TPR)-domains, which mediate the binding of PP5 to proteins that contain TPR-docking sites. The best-studied interaction for PP5 is the interaction with heat shock protein 90 (HSP-90) (Skarra, et al.). PP5 binds HSP-90 via interactions between the N-terminal TPR domains of PP5 and a C-terminal TPR-docking domain in HSP-90 (Ramsey, et al. 2000; Russell, et al. 1999; Silverstein, et al. 1997; Skarra et al.). Upon binding, PP5 appears to undergo a conformational change allowing substrate access to the active site (Yang, et al. 2005). To date ~110 proteins have been identified with TPR-docking domains, suggesting that PP5 could also play many unique cellular roles. Currently, PP5 is known to affect stress, hormone (i.e. GR) and metabolic mediated signaling cascades (Amable, et al. ; Grankvist, et al. ; Grankvist, et al. ; Skarra et al.). However, the mechanism controlling PP5 interactions and activity remain largely unexplained.
Protein phosphatases in β-cell proliferation and apoptosis
Protein tyrosine phosphatases
Protein tyrosine phosphatases (PTPs) are a superfamily of enzymes which oppose the roles of their protein tyrosine kinase counter parts (Andersen, et al. 2001). In relation to β-cell apoptosis, PTPN2 (also known as TC-PTP or PTP-S2; a member of the first nontransmembrane (NT1) subfamily of PTPs), has attracted interest. PTPN2 was identified a candidate gene for type 1 diabetes that is expressed in pancreatic β-cells (Todd, et al. 2007; Ylipaasto, et al. 2005). Furthermore, PTPN2 expression was regulated by cytokines (Cardozo, et al. 2001; Moore, et al. 2009). Transfection with PTPN2 siRNAs inhibited basal- and cytokine-induced PTPN2 expression in rat β-cells and dispersed human islets cells. Decreased PTPN2 expression exacerbated interleukin (IL)-β + interferon (IFN)-γ-induced β-cell apoptosis and turned IFN-γ alone into a proapoptotic signal (Moore et al. 2009). Inhibition of PTPN2 amplified IFN-γ-induced STAT1 phosphorylation, whereas double knockdown of both PTPN2 and STAT1 protected β-cells against cytokine-induced apoptosis, suggesting that STAT1 hyperactivation is responsible for the aggravation of cytokine-induced β-cell death in PTPN2-deficient cells (Moore et al. 2009). Further studies have shown that PTPN2 modulates pancreatic β-cell apoptosis via regulation of the BH3-only protein Bim (Santin, et al. 2011). PTPN2 knockdown exacerbated type I IFN-induced apoptosis in INS-1E, primary rat, and human β-cells. PTPN2 silencing and exposure to type I and II IFNs induced BAX translocation to the mitochondria, cytochrome c release, and caspase 3 activation. There was also an increase in Bim phosphorylation that was at least in part regulated by JNK1. From these data, it can be concluded that PTPN2 confers cytoprotective effects to pancreatic β-cells. However, such an anti-apoptotic role of PTPN2 cannot be generalized to other PTPs.
In contrast to the anti-apoptotic role played by PTPN2, ablation of protein tyrosine phosphatase 1B (PTP1B) increases β-cell proliferation in vivo {Fernandez-Ruiz, 2014 #249}. Morphometric analysis of pancreatic islets from Ptp1b−/− mice showed a higher β-cell area, concomitantly with higher β-cell proliferation and a lower β-cell apoptosis when compared to islets from their respective wild-type cognates {Fernandez-Ruiz, 2014 #249}. At a functional level, isolated islets from Ptp1b−/− mice exhibited enhanced glucose-stimulated insulin secretion. Moreover, Ptp1b−/−mice were able to partially reverse streptozotocin-induced β-cell loss, all indicating that inhibition of PTP1B activity in islet cells may be a therapeutic avenue to promote islet function.
PTP-BL is a non-receptor protein tyrosine phosphatase that is expressed in β-cells under the control of the MODY5 gene product, HNF-1β (Lee, et al. 1999; Thomas, et al. 2004). In mature β-cells HNF-1β expression is low, but forced induction of HNF-1β leads to enhanced rates of apoptosis, altered regulation of the cell cycle and inhibition of stimulated insulin secretion in β-cells, suggesting that control of HNF-1β expression may be important for the regulation of β-cell viability and function (Welters, et al. 2006). Stably transfected insulin-producing cells, expressing the wild-type form of PTP-BL, display compromised cell proliferation but no change in the rate of cell apoptosis (Welters, et al. 2008). Furthermore, cells over-expressing PTP-BL were less responsive towards mitogenic stimulation by Wnt3a. Although only performed in a β-cell line, these data suggest that PTP-BL may play a role in the regulation of cell cycle progression in β-cells, and that it interacts functionally with components of the Wnt signaling pathway. Future studies are needed to determine if PTP-BL plays a regulatory role during β-cell proliferation in vivo. For example, it will be interesting to determine if forced expression of PTPBL inhibits adaptive β-cell proliferation in response to reduced insulin sensitivity.
Ser/thr protein phosphatase 1 (PP1)
PP1 is regulated by its interaction with a variety of protein subunits that target the catalytic subunit (PP1C) to specific subcellular compartments and determines its localization, activity and substrate selectivity (Cohen 2002). In the field of β-cell research, the PP1 regulatory subunit Ppp1R15A has attracted special interest. Ppp1R15A targets PP1 to the endoplasmic reticulum (ER) and is induced under conditions of ER stress (Rutkowski, et al. 2006). The physiological response to ER stress is a collection of cellular events aiming to alleviate ER stress by decreasing overall protein synthesis through phosphorylation of the eukaryotic initiation factor 2α (eIF2α), through enhanced protein folding capacity by increased expression of chaperones and through activation of mechanisms for protein degradation (Ortsäter and Sjöholm 2007). The function of Ppp1R15A is to serve as the regulatory subunit of the PP1A catalytic domain. Through their interaction, Ppp1R15A and PP1C dephosphorylate eIF2α and thus exert a regulatory feed-back that can allow for re-initiation of protein synthesis and thereby allows for expression of stress-induced genes (Novoa, et al. 2003). Inhibition of PP1-mediated dephosphorylation of eIF2α by the compound salubrinal was found to be cytotoxic by itself in β-cells and in isolated islets of Langerhans. In addition, salubrinal potentiated fatty acid-induced ER stress and apoptosis (Cnop, et al. 2007; Ladriere, et al. 2010). Besides being a part of ER stress signaling, PP1 plays a pivotal role in glucose-induced stimulation of overall translation in β-cells, which depends on a PP1-mediated decrease in Ser51 phosphorylation of eIF2α (Vander Mierde, et al. 2007). Thus, the steady-state level of eIF2α phosphorylation in β-cells is the result of a balance between folding-load-induced phosphorylation and PP1-dependent dephosphorylation. Because defects in the pancreatic ER kinase-eIF2α signaling system lead to β-cell failure and diabetes, deregulation of the PP1 system could likewise lead to cellular dysfunction and disease.
Ser/thr protein phosphatase 2A (PP2A)
The PP2A family of enzymes is a major class of ser/thr protein phosphatases. They are also one of the most abundant cellular proteins, accounting for ~ 1% of total cellular protein and some 80 % of all cellular ser/thr protein phosphatase activity (Janssens and Goris 2001; Shi 2009). Evidence suggests that PP2A activation can be linked to apoptosis, e.g. activation of caspase-3 causes cleavage of the regulatory A subunit of PP2A, which in turn increases PP2A activity (Santoro, et al. 1998). As discussed below, PP2A may have a critical role in β-cell survival and demise. Exposure (for at least 24 h hours) of insulin-secreting cells to the phosphatase inhibitor, okadaic acid, at concentrations inhibiting PP1, PP2A,PP4, PP5 and PP6 reduces cell proliferation and insulin secretion. The reduced proliferation was found to be related to the induction of apoptosis as evident by morphological criteria and the occurrence of DNA fragmentation (Krautheim, et al. 1999). Of particular interest is that PP2A is hyper-activated by chronic exposure to high glucose (Arora, et al. 2013) and ceramide (Kowluru and Metz 1997), which are both well-known inducers of β-cell apoptosis. In the case of glucose, it was found that small interfering RNA-mediated knockdown of the catalytic subunit of PP2A (PP2Ac) markedly attenuates glucose-induced activation of PP2A (Arora et al. 2013). Moreover, metabolizable --but not non-metabolizable -- glucose derivatives induce Leu309 methylation of the catalytic subunit of PP2A. As a consequence, knockdown of the cytosolic leucine carboxymethyl transferase 1 (LCMT1), which carboxymethylates PP2Ac, significantly attenuates PP2A activation induced by high glucose. It was also found that glucose exposure induced LCMT1 expression, as well as the PP2A regulatory subunit B55α. Taken together, the data indicate that high glucose exposure hyperactivates PP2A via the induction of the methylating enzyme LCMT1 and the regulatory subunit B55α. Recent experiments have established a link between glucose-induced activation of PP2A and nuclear import of forkhead box O1 (FOXO1) in β-cells (Yan, et al. 2012). Under conditions of oxidative stress evoked by high glucose stimulation, FOXO1 associates with the PP2A holoenzyme composed of the catalytic C, structural A and B55α regulatory subunits. Knockdown of B55α in INS-1 cells reduced FOXO1 dephosphorylation, inhibited FOXO1 nuclear translocation and attenuated oxidative stress-induced cell death (Yan et al. 2012). This mechanism may be relevant also in vivo since both B55α and nuclear FOXO1 levels were increased under hyperglycemic conditions in db/db mouse islets, an animal model of T2D (Yan et al. 2012). Taken together, these data tell us that PP2A may play a role for glucotoxicity in β-cells via dephosphorylation of FOXO1 and that prevention of PP2A hyperactivation may confer protection against glucotoxicity.
Ser/thr protein phosphatase 2B/calcineurin (PP2B)
PP2B or calcineurin is a two-subunit enzyme, with a 58 to 64-kDa catalytic and calmodulin-binding subunit -- calcineurin A -- that is tightly bound to a regulatory 19-kDa calcium-binding regulatory subunit -- calcineurin B (Klee, et al. 1988).
Calcineurin is a Ca2+-activated cytosolic phosphatase that is critical for antigen-stimulated T lymphocyte activation (Crabtree and Olson 2002). Therefore, pharmacologic calcineurin inhibition is highly effective in preventing allograft rejection. However, calcineurin is also expressed in β-cells (Ebihara, et al. 1996; Redmon, et al. 1996; Tamura, et al. 1995), where it has two well described molecular targets, the nuclear factor of activated T cell (NFAT)2 family of transcription factors (Lawrence, et al. 2001), and the cAMP-responsive element-binding protein (CREB) transcriptional co-activator, transducer of regulated CREB activity-2 (TORC2) (Screaton, et al. 2004). Through dephosphorylation-mediated nuclear localization of these targets, calcineurin integrates Ca2+ and cAMP signals generated by physiologic stimuli, such as hyperglycemia and incretin receptor activation, to alter gene expression (Lawrence, et al. 2002; Lawrence et al. 2001; Screaton et al. 2004). CREB is a cAMP- and Ca2+-responsive transcriptional activator that is required for β-cell proliferation and survival (Hussain, et al. 2006; Inada, et al. 2004; Jhala, et al. 2003). Glucose and incretin hormones promote synergistic CREB activity by inducing the nuclear re-localization of TORC2, a co-activator for CREB (Koo, et al. 2005; Screaton et al. 2004; Shaw, et al. 2005). In islet cells under basal conditions, when CREB activity is low, TORC2 is phosphorylated and sequestered in the cytoplasm by 14-3-3 proteins (Screaton et al. 2004). In response to feeding stimuli, TORC2 is dephosphorylated, enters the nucleus, and binds to CREB located at target gene promoters (Bittinger, et al. 2004; Koo et al. 2005; Screaton et al. 2004). Ser275 of TORC2 is a 14-3-3 binding site that is phosphorylated under low glucose conditions and which becomes dephosphorylated by calcineurin in response to glucose influx (Jansson, et al. 2008). Dephosphorylation of Ser275 is essential for both glucoseand cAMP-mediated activation of CREB in β-cells and islets, demonstrating the essential role of calcineurin activity in β-cell physiology.
Given this role of calcineurin in β-cell biology, it is not surprising that pharmacologic calcineurin inhibition -- necessary to prevent rejection in the setting of islet transplantation -- is associated with post-transplant β-cell failure. New-onset diabetes mellitus after transplantation is a frequent complication after kidney transplantation, with an incidence of 15% to 30% (Cosio, et al. 2002; Kasiske, et al. 2003).
Several studies show that calcineurin inhibitors can target β-cells directly. Tacrolimus (FK506), a calcineurin inhibitor used in clinical practice to suppress islet graft rejection, induces β-cell apoptosis as evident by TUNEL staining in cultured human islets within 48 hours of exposure (Soleimanpour, et al. 2010). This study identified insulin receptor substrate-2 (Irs2), a known cAMP-responsive element-binding protein target and upstream regulator of the PI3K/Akt pathway, as a calcineurin target in β-cells. It was found that tacrolimus decreased Akt phosphorylation, suggesting that calcineurin could regulate replication and survival via the PI3K/Akt pathway (Soleimanpour et al. 2010). Similarly, rapamycin and cyclosporin A (also calcineurin inhibitors) decrease cell viability in human and rat pancreatic islets (Barlow, et al. 2012; Ozbay, et al. 2011) and in clonal insulin producing cells (Plaumann, et al. 2008). Mechanistically, calcineurin inhibition activates the dual leucine-zipper-bearing kinase (DLK), which in turn activates apoptotic MAPK signaling (Merritt, et al. 1999; Plaumann et al. 2008). Human β-cell proliferation decreases exponentially with increasing age (Meier, et al. 2008).
Thus, studies of human β-cells, which are often performed on islets from elderly donors, often fail to detect β-cell proliferation. Therefore, studies of β-cell proliferation are often performed on β-cells obtained from rodent donors. In such studies, tacrolimus decreased β-cell proliferation by 72% in C57Bl/6 mice compared with vehicle-treated controls (Goodyer, et al. 2012). These results lend support to experiments performed in mice lacking calcineurin in β-cells (Heit, et al. 2006). Mice with a β-cell-specific deletion of the calcineurin phosphatase regulatory subunit b1 develop age-dependent diabetes characterized by decreased β-cell proliferation and mass, reduced pancreatic insulin content and hypoinsulinemia. Moreover, β-cells lacking calcineurin activity have a reduced expression of established regulators of β-cell proliferation, like MafA, Beta2 and Pdx1 (Heit et al. 2006). The impact of calcineurin on β-cell function is complex since, transgenic overexpression of active calcineurin in β-cells phenocopied mice with a β-cell-specific deletion of the calcineurin and resulted in decreased β-cell mass and hyperglycemia (Bernal-Mizrachi, et al. 2010). These mice, which express a constitutively active form of calcineurin under the insulin gene promoter, exhibit glucose intolerance (Bernal-Mizrachi et al. 2010). In vitro studies of islets isolated from suchmice demonstrated decreased β-cell mass that was accompanied by decreased proliferation and enhanced apoptosis (Bernal-Mizrachi et al. 2010). Taken together, these results demonstrate that pharmacological inhibition of calcineurin and genetic calcineurin deletion markedly inhibit rodent β-cell proliferation and promote β-cell apoptosis, which should be taken into account when treating patients in the need of immunosuppression. This may be especially important with patients displaying insulin resistance as the diabetogenic effect of tacrolimus and cyclosporin A is more pronounced in insulin resistant obese rats (Rodriguez-Rodriguez, et al. 2013).
While the vast majority of data suggest that calcineurin inhibition reduces β-cell viability and may cause a diabetes phenotype, the situation is different when it comes to β-cell death induced by either proinflammatory cytokines (Grunnet, et al. 2009) or glucocorticoids (Ranta, et al. 2006; Ranta, et al. 2008).
Treatment of isolated rats or human islets with cytokines promotes β-cell apoptosis by the intrinsic apoptotic pathway along with dephosphorylation of the proapoptotic protein Bad at ser136 (Grunnet et al. 2009). This particular serine residue is a target for calcineurin (Wang, et al. 1999). In concordance, supplementation of tacrolimus to the cytokine-containing cell media prevented Bad dephosphorylation and cytokine-induced cytotoxicity (Grunnet et al. 2009), showing that -- under these circumstances -- calcineurin inhibition is favorable for β-cell viability.
The situation is similar under conditions of glucocorticoid exposure in culture. Although glucocorticoid-induced β-cell apoptosis has not been demonstrated in vivo, it is clear that these steroid hormones are cytotoxic to β-cells during ex vivo culture conditions (Avram, et al. 2008; Fransson, et al. 2013; Ranta et al. 2006; Ranta et al. 2008; Reich, et al. 2012). Glucocorticoids activate calcineurin, which in turn dephosphorylates the apoptotic protein Bad (Tumlin, et al. 1997). Such a mechanism has also been demonstrated in insulin-producing cells (Ranta et al. 2006). Inhibition of calcineurin activity by tacrolimus and deltamethrin in insulin-secreting INS-1 cells reduced apoptosis provoked by the synthetic glucocorticoid analogue dexamethasone (Ranta et al. 2008). Thus, direct inhibition of calcineurin activity in β-cells decreases cell viability and reduces β-cell function. Of note, the situation is different for β-cell death induced by cytokines and glucocorticoids. In such cases, inhibition of calcineurin counteracts the cytotoxic effect of cytokines and glucocorticoids. Pharmacological inhibitors are never 100 % specific so these seemingly contradictory findings may be, at least partly, explained by effects that are independent of calcineurin. For example, tacrolimus can inhibit NF-κ-B activity, leading to inhibition of NO formation (Tunon, et al. 2003).
Ser/thr protein phosphatases 2C (PP2C)
Identification of PP2C isoforms traces back to the 1980s (Hiraga, et al. 1981; Pato and Adelstein 1983). PP2C enzymes act on a variety of substrate classes, e.g. kinases, receptors, channels and transcription factors, thereby affecting quite diverse physiological effects, e.g. stress response, metabolism and cell cycle (Klumpp, et al. 2006). In contrast to most other ser/thr protein phosphatases, inhibitors like okadaic acid, microcystin, tautomycin or inhibitor proteins I1 and I2 have no effect on PP2C isoenzymes. Hitherto PP2C isoforms have not been implicated in β-cell apoptosis. However, they are sensitive to stimulation by unsaturated fatty acids (Krieglstein, et al. 2008). In this aspect PP2C isoforms have been linked to fatty acid-induced apoptosis in neural and endothelial cells (Schwarz, et al. 2006) and similar mechanisms may be operative in pancreatic β-cells.
Ser/thr protein phosphatases 4, 6 and 7
The ser/thr protein phosphatases 4, 6 and 7 have not been studied with regards to their possible implications in β-cell apoptosis. The catalytic subunit of PP4 is expressed in islets of Langerhans as evident by immunohistochemistry (http://www.proteinatlas.org/), where it is located in the nucleus (Veluthakal, et al. 2006). Neither PP6 nor PP7 catalytic subunit expression has been documented in β-cells.
Ser/thr protein phosphatase 5 (PP5)
PP5 is another member if the PPP family (Andreeva and Kutuzov 1999; Swingle et al. 2004) that is highly conserved among species and expressed in most, if not all, mammalian cells. However, the roles of PP5 in biology and disease are only beginning to emerge (Amable et al. 2011; Hinds, et al. 2011; Yong, et al. 2007), and the influence of PP5 on β-cell function is still unknown. The human gene encoding PP5 (PPP5C) is localized on chromosome 19 (Xu, et al. 1996). PP5 has been reported to be present both in the nucleus and cytosol (Chinkers 2001). It has proven difficult to studying the biological role of PP5, partly because until recently, only a few physiological substrates have been identified. The polyunsaturated fatty acid, arachidonic acid, and the structural component of caveolae, caveolin-1, have both been shown to activate PP5 (Ramsey and Chinkers 2002; Taira and Higashimoto 2013). A high-throughput screening effort identified chaulmoogric acid as a compound which can activate PP5 at fairly high concentrations (Cher, et al. 2009). Suramin was identified as a novel PP5 activator by its competitively binding of a domain of PP5 and thereby causing its activation (Yamaguchi, et al. 2013). During standard conditions, PP5 is predominately in an inactive state (Sinclair, et al. 1999), causing a very low basal activity that represent less than 1 % of the total measurable phosphatase activity. A unique characteristic of PP5 is that it is expressed as a single polypeptide, which consists of a phosphatase catalytic domain near its C-terminus and a regulatory domain at the N-terminus (Becker, et al. 1994; Golden and Honkanen 2003). An additional feature, unique for PP5 among its family members, is the extended N-terminal region containing multiple tetratricopeptide repeat (TPR) domains, by which PP5 mediates protein-protein interactions (Das, et al. 1998). PP5 is associated with numerous proteins involved in diverse signaling networks, including heat shock protein 90 (Hsp90) in complex with the glucocorticoid receptor (GR) (Chen, et al. 1996; Silverstein et al. 1997), the cell division cycle (CDC16/CDC27/CDC37) subunits of the anaphase-promoting complex (Ollendorff and Donoghue 1997; Vaughan, et al. 2008), cryptochrome 2 (Zhao and Sancar 1997), ataxia-telangiectasia and Rad3-related (Zhang, et al. 2005) ataxia-telangiectasia and Rad3 mutated (Ali, et al. 2004) DNA-dependent protein kinase catalytic subunit (Wechsler, et al. 2004), apoptosis signal regulating kinase 1 (ASK1) (Morita, et al. 2001), Hsp90-dependent heme-regulated eIF2α kinase (Shao, et al. 2002), Rac GTP-binding protein (Gentile, et al. 2006), the A-regulatory subunit of PP2A (Lubert, et al. 2001), Raf proto-oncogene ser/thr protein kinase (von Kriegsheim, et al. 2006), stress-induced phosphoprotein 1 (Skarra et al. 2011), and the Gα12/Gα13 subunits of heterotrimeric GTP-binding proteins (Yamaguchi, et al. 2002).
PP5 was recently shown for the first time to play a role in the β-cells (Grankvist et al. 2012). During the progression towards T2D, β-cells are often exposed to a combination of high levels of glucose and fatty acids, producing so-called glucolipotoxicity, which is associated with increased production of reactive oxygen species (ROS) (Oprescu, et al. 2007). In turn, increased levels of ROS cause initiation of apoptosis, resulting in a reduced β-cell mass (Butler et al. 2003). Several studies have indicated that PP5 is acting in the regulation of signaling cascades activated by oxidative stress.
Elevated levels of ROS can induce the association of PP5 with ASK1, leading to reduced ASK1 phosphorylation at Thr845, and thereby causing ASK1 inactivation (Huang, et al. 2004; Kutuzov, et al. 2005; Morita et al. 2001; Zhou, et al. 2004). This suggests that PP5 can suppress the oxidative stress-induced apoptosis by averting sustained activation of ASK1 and its downstream target c-Jun N-terminal kinase (JNK). PP5 may accordingly act as a negative regulator of the ASK1/JNK signaling pathway and in so doing protecting cells from apoptosis (Kutuzov et al. 2005; Mkaddem, et al. 2009; Morita et al. 2001). This concept was supported by the recent publication (Grankvist et al. 2012) showing that islets from mice lacking PP5 were more susceptible towards stress-induced apoptosis than wild-type cognates. Additionally, PP5-deficient mice had lower fasting glycemia and improved glucose tolerance compared to the wild-type mice, suggesting a novel role for PP5 in the regulation of glucose homeostasis. These findings cannot be explained by a difference in islet mass between the PP5-deficient and wild-type mice, since no difference was observed (Grankvist et al. 2012). Furthermore, a high-fat diet treatment for 10 weeks revealed that the mice lacking PP5 gained markedly less weight, did not accumulate visceral fat and displayed enhanced insulin sensitivity compared to the wild-type littermates (Grankvist et al. 2013). Another group (Hinds et al. 2011) also recently published studies showing embryonic fibroblasts from PP5 knockout mice didn’t accumulate lipids after adipogenic stimuli. Together, these studies suggest that PP5 may play a previously unrecognized role in both glucose and lipid metabolism. Nevertheless, additional studies are necessary to further address the role of PP5 in glucose homeostasis and β-cell function. See Table 1 for a summarized view on the role of different PSPs in β-cell biology.
Table 1.
Summary of the protein phosphatases’ effects on pancreatic β-cells
| Protein name | Intervention | Biological material |
Effect on β-cells | Reference |
|---|---|---|---|---|
| Protein tyrosine phosphatases |
||||
| PTPN2 | Downregulation with siRNA |
INS-1E cells, rat and human islets |
Promotes cytokine- induced apoptosis |
(Moore et al. 2009; Santin et al. 2011) |
| PTP1B | Global genetic deletion |
Mice | Promotion of β-cell proliferation and reduced islet cell apoptosis |
{Fernandez-Ruiz, 2014 #249} |
| PTP-BL | Stable over expression |
INS-1 cells | Compromised cell proliferation but no change in the rate of cell apoptosis |
(Welters et al. 2008) |
| Se/thr protein phosphatases |
||||
| Ppp1R15A | Pharmacological inhibition by salubrinal |
INS-1E cells, rat and human islets |
Induce apoptosis and augment fatty acid- induced apoptosis and controls glucose- mediated translation. |
(Cnop et al. 2007; Ladriere et al. 2010; Vander Mierde et al. 2007) |
| PP2A | High glucose or ceramide exposure |
INS-1 832/13 cells and rat islets |
Enhanced PP2A activity and loss of GSIS |
(Arora et al. 2013; Kowluru and Metz 1997) |
| PP2A | High glucose exposure |
INS-1 and βTC- 3 cells |
FOXO1 activation | (Yan et al. 2012) |
| PP2B (calcineurin) |
Pharmacological inhibition by tacrolimus, rapamycin and cyclosporin A |
Human and rat islets, MIN6 cells |
Increased apoptosis | (Barlow et al. 2012; Ozbay et al. 2011; Plaumann et al. 2008; Soleimanpour et al. 2010) |
| PP2B (calcineurin) |
Pharmacological inhibition by tacrolimus |
C57Bl/6j mice in vivo |
Inhibition of β–cell proliferation |
(Goodyer et al. 2012) |
| PP2B (calcineurin) |
B-cell specific genetic deletion |
Mice | Develops age-dependent diabetes alongside loss of β–cell mass |
(Heit et al. 2006) |
| PP2B (calcineurin) |
B-cell specific transgenic overexpression |
Mice | Glucose intolerance and loss of β–cell mass |
(Bernal-Mizrachi et al. 2010) |
| PP2B (calcineurin) |
Pharmacological inhibition by tacrolimus or deltamethrin |
Human and rat islets |
Attenuation of cytokine- and glucocorticoid- induced apoptosis |
(Grunnet et al. 2009; Ranta et al. 2008) |
| PP5 | Downregulation with siRNA or use of islets isolated from Ppp5c−/− mice |
MIN6 cells and mouse islets |
Promotes glucocorticoid- and palmitate-induced apoptosis |
(Fransson et al. 2013; Grankvist et al. 2012) |
| PP5 | Global genetic deletion |
MIN6 cells and mice |
Improves glucose tolerance |
(Grankvist et al. 2012; Grankvist et al. 2013) |
Protein phosphatases and islet hormone secretion
Significant changes in protein kinase activities and in protein phosphorylation patterns occur subsequent to stimulation of β-cell insulin release by nutrients (Jones and Persaud 1998; Newgard and McGarry 1995; Sjöholm 1998). Therefore, the molecular mechanisms regulating phosphorylation by protein kinases of proteins involved in the insulin secretory process by the β-cell have been extensively investigated. However, far less is known about the role and regulation of protein dephosphorylation by various protein phosphatases.
While early investigators reported the presence of phosphatase activity in pancreatic islets (Colca, et al. 1984; Lernmark, et al. 1980; Lipson, et al. 1979; Taljedal 1967), the identity of these enzymes was unknown at the time. More contemporary studies have established that i) the β-cell contains ser/thr and tyrosine PP activity (Chen and Ostenson 2005; Gagliardino, et al. 1991; Sjöholm, et al. 1993b); ii) stimulation of protein phosphorylation by direct activation of PKA and PKC with forskolin or phorbol ester, respectively, results in a stimulated insulin secretion (Arkhammar, et al. 1994; Hisatomi, et al. 1996; Sjöholm 1991); iii) physiological stimuli of insulin secretion increase β-cell phosphorylation state (Jones and Persaud 1998); iv) short-term treatment of β-cells or permeabilized rat pancreatic islets with the specific PP inhibitor okadaic acid promotes Ca2+ entry and insulin exocytosis (Haby, et al. 1994; Hisatomi et al. 1996; Larsson, et al. 1997; Ämmälä, et al. 1994). These combined findings point to an important functional role for protein (de)phosphorylation in regulation of the stimulus-secretion coupling in the β-cell. The role of PPs in β-cell function and insulin secretion is nonetheless poorly understood.
Ser/Thr-PPases
Identification and characterization
PPP types 1 and 2A were identified in crude RINm5F β-cell homogenates by both enzymatic assay and Western blot analysis (Sjöholm et al. 1993b). They were also characterized in terms of their sensitivity to the inhibitory actions of several compounds isolated from cyanobacteria, marine dinoflagellates and marine sponges, (viz. okadaic acid, microcystin-LR, calyculin-A and nodularin). It was found that okadaic acid was the least potent inhibitor (IC approximately 10−9 50 M, IC approximately 10−6 100 M), while the other compounds exhibited IC50 values of approximately 5 × 10−10 M and IC100 approximately 5 × 10−9 M (Sjöholm et al. 1993b).
Role in insulin stimulus-secretion coupling
The mechanisms that regulate insulin secretion were electrophysiologically investigated in single β-cells (Haby et al. 1994). The secretory responses were substantially increased by conditions that promote protein phosphorylation, such as activation of protein kinases A and C or inhibition of PPP-family members (PP1, PP2A, PP4-PP6) by okadaic acid. These results suggest that, although Ca2+ is required for the initiation of exocytosis, modulation of exocytosis by protein kinases and phosphatases is of much greater quantitative importance. Similar findings were reported by other groups (Mayer, et al. 1994). It should be noted, however, that not all investigators have arrived at this conclusion (Ammon, et al. 1996; Krautheim et al. 1999; Murphy and Jones 1996; Sato, et al. 1998; Tamagawa, et al. 1992). In some of these studies, PP inhibitors (e.g. okadaic acid) were added to intact cells, sometimes for long periods of time. It is important to keep in mind that inhibitors like okadaic acid may exert non-specific effects and toxicity when added to intact cells. The agent is known to interfere with membrane integrity by non-specific mechanisms. Loss of membrane integrity will disrupt for instance the Ca2+ gradient over the membrane, causing massive uncontrolled Ca2+ influx that may cause apoptosis. A more physiologic way of studying roles of PP by using inhibitors is to apply these acutely to permeabilized cells or in patch clamp settings, in which Ca2+ gradients are not operative. Indeed, the inhibitory effect of okadaic acid on GSIS in intact cells was mimicked by the inactive analogue 1-nor-okadaone; in contrast, okadaic acid stimulated insulin secretion from permeabilized cells (Ratcliff and Jones 1993).
In one study, it was shown that the inhibitory effect of leptin on insulin secretion in rat and human islets is associated with decreased expression and activation of a PP1-like enzyme (Kuehnen, et al. 2011).
In another study (Sjöholm, et al. 1995), the effects of known insulin secretagogues and intracellular second messengers on the activities of cat-ion independent ser/thr PPs in insulin-secreting RINm5F β-cells were investigated. The stimulation of intact RINm5F cells with the insulin secretagogues L-arginine, L-glutamine, K+, or extracellular ATP elicited time-dependent changes in PPP- activities with an early decrease in type 1-like and/or type 2A-like PPP-activity that gradually returned to normal levels. Addition of cAMP, cGMP, or prostaglandins E2 and F1α at widely different concentrations to RINm5F cell homogenates failed to affect PPP activities. In contrast, addition of physiological concentrations of adenine nucleotides, known to increase upon nutrient stimulation, to RINm5F β-cell homogenates inhibited PP2A-like and, to a lesser extent, PP1-like, PPP activity. It was concluded that insulin secretagogues cause time- and concentration-dependent inhibitory effects on RINm5F β-cell PPP activities, which may contribute to the increase in the phosphorylation state that occurs after stimulation of insulin release (Sjöholm et al. 1995). Thus, inhibition of protein dephosphorylation may be a regulatory mechanism controlling the stimulus-secretion coupling in insulin-producing cells. However, there are also contradictory findings: Murphy et al reported that PP1/2A was stimulated by glucose and required for GSIS in rat islets (Murphy and Jones 1996). The reasons for these discrepancies remain unknown at this time, but may relate to different models used.
In another study (Sjöholm, et al. 2002), it was demonstrated that glycolytic and Krebs cycle intermediates, whose concentrations increase upon glucose stimulation, not only dose dependently inhibited ser/thr PP enzymatic activities, but also directly promote insulin exocytosis from permeabilized β-cells. Thus, fructose-1,6-bisphosphate, phosphoenolpyruvate, 3-phosphoglycerate, citrate, and oxaloacetate inhibited PPPs and significantly enhanced insulin exocytosis, non-additive to that of okadaic acid, at micromolar Ca2+ concentrations. In contrast, the effect of GTP was potentiated by okadaic acid, suggesting that the action of GTP does not require PPP inactivation. It was concluded that specific glucose metabolites and GTP inhibit β-cell PP activities and directly stimulate Ca2+-independent insulin exocytosis. The glucose metabolites, but not GTP, seem to require PP inactivation for their stimulatory effect on exocytosis. Thus, an increase in phosphorylation state, through inhibition of protein dephosphorylation by metabolic intermediates, may link glucose sensing to insulin exocytosis in the β-cell.
Although disputed (MacDonald and Fahien 2000), a messenger role has been postulated for L-glutamate in linking glucose stimulation to sustained insulin exocytosis in the β-cell (Maechler and Wollheim 1999), but the precise nature by which L-glutamate controls insulin secretion remains elusive. Effects of L-glutamate on the activities of PPPs and Ca2+-regulated insulin exocytosis in INS-1E β-cells were investigated (Lehtihet, et al. 2005). Glucose was found to increase L-glutamate contents and promote insulin secretion from INS-1E cells. L-glutamate also dose-dependently inhibited PP enzyme activities mimicking the specific PPP inhibitor, okadaic acid. L-glutamate and okadaic acid directly and non-additively promoted insulin exocytosis from permeabilized INS-1E cells in a Ca2+-independent manner. Thus, an inhibition of protein dephosphorylation by glucose-derived L-glutamate may link glucose sensing to sustained insulin exocytosis.
It was additionally demonstrated that inositol hexakisphosphate (InsP6), whose concentration in β-cells transiently increases upon glucose stimulation (Larsson et al. 1997), dose-dependently and differentially inhibits enzyme activities of ser/thr PPPs in physiologically relevant concentrations (Lehtihet, et al. 2004). However, and in contrast to previous findings in rat islets (Gagliardino, et al. 1997), none of the hypoglycemic sulfonylureas tested (glipizide, glibenclamide, tolbutamide) affected PP1 or -2A activity at clinically relevant concentrations in RINm5F cells. The reasons for this discrepancy remain elusive at this time; however, in part they may be due to different cell subclones, experimental conditions, and phosphoprotein substrates used.
The insulin secretagogue L-arginine, an immediate metabolic precursor to polyamines, was reported to cause a rapid and transient decrease in PP1 activity in RINm5F β-cells (Sjöholm et al. 1995). It was previously reported that polyamines dose-dependently suppress PP1-like activity when added to RINm5F cell homogenates at physiologic concentrations, while having minor and inconsistent effects on PP2A-like activity (Sjöholm and Honkanen 2000). The IC50 value for spermine on PP1-like activity was approximately 4 mM. The inhibitory effect was reproduced and of comparable magnitude on purified PPs types 1 and 2A. On the other hand, when endogenous polyamine pools were exhausted by 4 days of exposure to the specific L-ornithine decarboxylase inhibitor DL-α-difluoromethylornithine (Sjöholm, et al. 1993a), there was an increase in PP2A-like activity. Quantitative Western analysis revealed that the amount of PP2A protein did not change after this treatment. It was concluded that polyamines cause time-and concentration-dependent inhibitory effects on RINm5F β-cell PPP activities, which may contribute to the increase in phosphorylation state that occurs after secretory stimulation. Please see Figure 5 for a proposed model of PPP regulation of insulin stimulus-secretion coupling. Elegant work by the Kowluru laboratory has elucidated in great detail how PP2A is regulated (Kowluru 2005). The catalytic subunit of PP2A is subject to several means of post-translational modification: 1) Reversible carboxylmethylation at Leu309, catalyzed by a PP methyltransferase, results in activation of the enzyme, holoenzyme assembly and substrate association (Kowluru, et al. 1996). As ebelactone, an inhibitor of methyl esterases, not only delayed demethylation of PP2A, but also decreased GSIS, a negative role for PP2A in normal rat islet GSIS was suggested (Kowluru et al. 1996). On the other hand, genetic silencing of the catalytic subunit of PP2A in INS-1 832/13 insulinoma cells by siRNA was found to abrogate GSIS (Jangati, et al. 2007). Carboxylmethylation of the catalytic subunit of PP2A was inhibited by certain glucose metabolites and by increased cytosolic Ca2+ (Palanivel, et al. 2004), leading to inactivation of the enzyme. It was suggested that this mechanism facilitates hyperphosphorylation of exocytotic proteins, thereby augmenting insulin secretion. 2) Phosphorylation at Tyr307 has been shown to inhibit PP2A catalytic activity, whereas nitration of Tyr307 alleviates the enzyme from inactivation by phosphorylation (Kowluru and Matti 2012). 3) Phosphorylation at Thr304 results in inactivation of PP2A (Kowluru and Matti 2012).
Figure 5. Regulation of β-cell PP activities and their effects on the insulin stimulus-secretion coupling.

Glucose, the β-cell’s main stimulus, is taken up across the plasma membrane by the facilitative glucose transporter GLUT-2. The sugar is further metabolized in the glycolytic pathway and TCA cycle to yield coupling factors suppressing PP activity, thereby activating influx of Ca2+ that sets in motion the exocytotic release of insulin. The ATP generated during glucose catabolism also serves to close K+ channels, causing depolarization, and as a substrate for cAMP formation. Receptor-operated, G protein-coupled signaling pathways through phospholipase C-protein kinase C and adenylyl cyclase are also depicted. See text for details. AC, adenylyl cyclase; ATP, adenosine trisphosphate; cAMP, cyclic AMP; DAG, diacylglycerol; ER, endoplasmic reticulum; G, GTP-binding protein; Gln, glutamine; Glu, glutamate; GLUT-2, glucose transporter 2; GTP, guanosine trisphosphate; InsP3, inositol trisphosphate; InsP6, inositol hexakisphosphate; KATP, ATP-dependent K+ channel; OA, okadaic acid; PIP2, phosphatidylinositol bisphosphate; PKC, protein kinase C; PLC, phospholipase C; PP, protein phosphatase; R, receptor; TPA, 12-O-tetradecanoyl phorbolacetate; VGCC, voltage-gated Ca2+ channel.
Not only secretion, but also protein synthesis may also be translationally regulated by reversible phosphorylation. It was suggested that glucose-stimulated translation in the β-cell requires a PP-1-mediated decrease in Ser51 phosphorylation of eukaryotic translation initiation factor 2α, an important factor controlling translational fidelity (Vander Mierde et al. 2007). Additionally, in INS-1 832/13 cells, glucose dephosphorylates elongation factor 2, probably through activation of PP2A (Yan, et al. 2003). This suggests that INS-1 832/13 cell protein translation rates are controlled by glucose-induced reversible phosphorylation of elongation factor 2.
Control of transcription factors may also be regulated by reversible phosphorylation: In INS-1E cells, high glucose down-regulates the expression of PPAR-α, leading to decreased fatty acid oxidation, through activation of PP2A and inactivation of AMPK, a cellular energy gauge (Ravnskjaer, et al. 2006). Also other important enzymes of critical regulatory role in GSIS appear regulated by phosphorylation. For instance, acetyl-CoA-carboxylase -- which catalyzes malonyl-CoA formation -- was found to be activated by magnesium and glutamate probably through an okadaic acid-sensitive PP2A-like enzyme (Kowluru, et al. 2001), an effect that may stimulate β-cell anaplerosis through provision of long-chain fatty acids.
PP2B (calcineurin) has also been implicated in control of islet hormone secretion: Renström et al showed that the inhibitory effect of several neurotransmittors known to inhibit insulin secretion (viz. somatostatin, galanin and epinephrine) was associated with an activation of PP2B (Renstrom, et al. 1996). Conversely, this inhibition of secretion was prevented by PP2B inhibitors. Since PP2B inhibitors (e.g. cyclosporine-A) are used in islet transplantation for immunosuppressive purposes, the physiological role of the enzyme in islets is clinically very relevant. While short term PP2B inhibition stimulates insulin secretion (Ebihara et al. 1996), long-term inhibition of the enzyme -- or overexpression of its inhibitory regulators (Peiris, et al. 2012) -- may cause β-cell functional suppression and demise (Sjöholm 1994). PP2B also appears required for proper cAMP-stimulated gene transcription in HIT β-cells (Schwaninger, et al. 1995).
PTPs
Vanadate inhibits most PTPs and has been shown to exert direct glucose-dependent insulinotropic effects in isolated rodent islets by mechanisms involving phosphoinositide hydrolysis and Ca2+ handling (Fagin, et al. 1987; Zhang, et al. 1991). Interestingly, vanadium salts have also been found to exert anti-diabetic and islet-protective effects in various and widely different diabetic animal models, such as streptozotocin (Pederson, et al. 1989), 90 % pancreatectomy (Nakamura, et al. 1995) and Zucker diabetic fatty rats (Winter, et al. 2005), adding further credence to PTPs as inhibitors of insulin secretion also in vivo.
The PTPs IA-2 (ICA-512) and IA-2β (phogrin) are major autoantigens in T1D (Torii 2009), located in secretory granules, but developmentally differentially regulated. Whereas expression of phogrin appears insensitive to factors that influence β-cell function, IA-2 expression seems regulated by glucose, cAMP and autocrine insulin (Lobner, et al. 2002). In vivo, genetic disruption of IA-2 or phogrin results in glucose intolerance due to impaired insulin secretion (Henquin, et al. 2008). However, it is likely both enzymes are regulating the stability and/or loading of secretory granules, rather than influencing the exocytotic process per se. Thus, the main effects of PTPs on insulin secretion seem inhibitory.
Protein phosphatases and diabetes
Surprisingly little is known regarding the role of different PP in islets in diabetic states, in spite of the fact that PTPs IA-2 and phogrin are important autoantigens in T1D. Nonetheless, work from the Östenson laboratory (Östenson, et al. 2002) reported 60 % overexpression of PTP σ in islets of Goto-Kakizaki rats, a lean genetic model of T2D. Importantly, down-regulation of PTP σled to increased GSIS in these normally “glucose-blind” islets. The authors concluded that increased expression of PTP σ may be of pathogenetic significance for the defective insulin secretion in GK rat islets. Interestingly, the same group reported that genetic variation in receptor PTP σ is associated with T2D in Swedish Caucasians (Langberg, et al. 2007).
Another connection to both T1D and T2D may be ceramide, which is formed during sphingomyelin breakdown by proinflammatory cytokines like interleukin-1 (IL-1) (Mullen, et al. 2012). Ceramide may inhibit β-cell mitogenesis and insulin production (Sjöholm 1995), possibly through activation of JNK and the transcription factor ATF2 (Welsh 1996). Many effects of ceramide are believed to be mediated by a ceramide-activated PPP (CAPP), a PP2A-like enzyme expressed in islets (Kowluru and Metz 1997). The genetic silencing of the α isoform of the PP2A catalytic subunit, achieved through siRNA knockdown, was found to significantly reduce CAPP enzymatic activity in INS 832/13 cells (Jangati, et al. 2006).
The Kowluru lab also reported that the catalytic subunit of PP4, present in INS-1 cell nuclei, can be regulated by IL-1: Exposure of the INS-1 cells to IL-1 led to the expected increase in NO formation, but also reduced the expression, carboxylmethylation and enzymatic activity of PP4 (Veluthakal et al. 2006). PP4 catalytic subunit was found to form complex with nuclear lamin-B, which regulates nuclear envelope assembly. The authors proposed that IL-1/NO-induced inhibition of PP4 expression and enzymatic activity may aid keeping lamin-B phosphorylated and thereby make it amenable for pro-apoptotic caspases that may lead to β-cell death (Veluthakal et al. 2006).
This effector system may also be relevant in T2D, as studies have shown that cytokines like IL-1 are produced by islet cells and increased by glucotoxicity (Maedler, et al. 2002).
Another connection between T2D and ceramide is the lipotoxicity prevailing in T2D. While IL-1-induced ceramide is formed from sphingolipids, islet ceramide accumulating under conditions of hyperlipidemia appears to be derived from de novo synthesis from free fatty acids (FFAs) (Shimabukuro, et al. 1998). Thus, islet ceramide and CAPP may be increased by two different mechanisms in T2D: A glucotoxic pathway involving paracrine/autocrine IL-1 promoting sphingomyelin breakdown, and a lipotoxic pathway in which ceramide is generated from FFAs. These two pathways, which normally potentiate each other’s toxicity, may thus additively or synergistically activate islet CAPP.
In islets of type 2 diabetic Goto-Kakizaki rats, the magnesium and glutamate-sensitive PP2A-like enzyme mentioned above (Kowluru et al. 2001) appears to be dysregulated in that the activation of ACC by magnesium and glutamate seems to be markedly reduced (Palanivel, et al. 2005). The pathophysiological relevance of this derangement is unclear at this time, but could conceivably result in reduced formation of long-chain fatty acids and contribute to the loss of GSIS in this widely used animal model.
Elegant studies from the Kowluru laboratory have provided in-depth mechanistic insights into the role of PP2A in islets under diabetes-like glucotoxic conditions (Arora et al. 2013). During chronic hyperglycemia, mimicked by high glucose in vitro, PP2A becomes hyperactivated - an effect coupled to loss of GSIS. Knock-down by siRNA of the PP2A catalytic subunit prevented this hyperactivation. Also, glucose, but not non-metabolizable sugars, augmented the carboxylmethylation of Leu307 of the catalytic subunit (Arora et al. 2013). High glucose increased the expression of a regulatory subunit of PP2A, which has been implicated in islet dysfunction during glucotoxicity. No clear role for ER stress in glucose-induced activation of PP2A could be found. Authors proposed that exposure of the β-cell to high glucose results in exaggerated PP2A activity and subsequent loss of GSIS.
Future prospects
To understand how protein dephosphorylation is regulated within the islet, and how this controls hormone secretion, apoptosis and proliferation, thorough and deep mechanistic studies are clearly needed. With the exception of PP5, which is the only member of the PPP family with the catalytic and regulatory subunit encoded by one gene, knock-down strategies targeting the catalytic subunits will not likely be a fruitful avenue to follow in order to explore the function of ser/thr protein phosphatases. Elimination of the regulatory subunits is likely to be more exact in targeting specific cellular functions. Knock-down experiments should be followed by an investigation of changes in the phoshoproteome to further pinpoint which proteins that are targeted. Furthermore, characterization of the different PSPs/PTPs expressed in the various islet cell types may prove important not only from a diabetes pathogenic perspective but may also offer clues to development of novel antidiabetic drugs.
Acknowledgments
Funding Work from the authors’ laboratories cited in this paper was supported by grants from the Swedish Diabetes Foundation (Diabetesfonden), Folksam Research Foundation, the Diabetes Research Wellness Foundation, the Tore Nilsson Foundation, the Lars Hierta’s memorial foundation, Tornspiran Foundation, Berth von Kantzow’s Foundation, Golje’s Memorial Foundation, Eva and Oscar Ahrén’s Foundation, and NIH CA 60750.
References
- Ali A, Zhang J, Bao S, Liu I, Otterness D, Dean NM, Abraham RT, Wang XF. Requirement of protein phosphatase 5 in DNA-damage-induced ATM activation. Genes Dev. 2004;18:249–254. doi: 10.1101/gad.1176004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alonso A, Sasin J, Bottini N, Friedberg I, Friedberg I, Osterman A, Godzik A, Hunter T, Dixon J, Mustelin T. Protein tyrosine phosphatases in the human genome. Cell. 2004;117:699–711. doi: 10.1016/j.cell.2004.05.018. [DOI] [PubMed] [Google Scholar]
- Amable L, Grankvist N, Largen JW, Ortsater H, Sjoholm A, Honkanen RE. Disruption of serine/threonine protein phosphatase 5 (PP5:PPP5c) in mice reveals a novel role for PP5 in the regulation of ultraviolet light-induced phosphorylation of serine/threonine protein kinase Chk1 (CHEK1) J Biol Chem. 2011;286:40413–40422. doi: 10.1074/jbc.M111.244053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- American Diabetes A. Economic costs of diabetes in the U.S. in 2012. Diabetes Care. 2013;36:1033–1046. doi: 10.2337/dc12-2625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ammon HP, Heurich RO, Kolb HA, Lang F, Schaich R, Drews G, Leiers T. The phosphatase inhibitor okadaic acid blocks KCl-depolarization-induced rise of cytosolic calcium of rat insulinoma cells (RINm5F) Naunyn Schmiedebergs Arch Pharmacol. 1996;354:95–101. doi: 10.1007/BF00178708. [DOI] [PubMed] [Google Scholar]
- Andersen JN, Mortensen OH, Peters GH, Drake PG, Iversen LF, Olsen OH, Jansen PG, Andersen HS, Tonks NK, Moller NP. Structural and evolutionary relationships among protein tyrosine phosphatase domains. Mol Cell Biol. 2001;21:7117–7136. doi: 10.1128/MCB.21.21.7117-7136.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andreeva AV, Kutuzov MA. RdgC/PP5-related phosphatases: novel components in signal transduction. Cell Signal. 1999;11:555–562. doi: 10.1016/s0898-6568(99)00032-7. [DOI] [PubMed] [Google Scholar]
- Arkhammar P, Juntti-Berggren L, Larsson O, Welsh M, Nanberg E, Sjoholm A, Kohler M, Berggren PO. Protein kinase C modulates the insulin secretory process by maintaining a proper function of the beta-cell voltage-activated Ca2+ channels. J Biol Chem. 1994;269:2743–2749. [PubMed] [Google Scholar]
- Arora DK, Machhadieh B, Matti A, Wadzinski BE, Ramanadham S, Kowluru A. High Glucose Exposure Promotes Activation of Protein Phosphatase 2A in Rodent Islets and INS-1 832/13 beta-Cells by Increasing the Posttranslational Carboxylmethylation of Its Catalytic Subunit. Endocrinology. 2013;155:380–391. doi: 10.1210/en.2013-1773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ashcroft FM, Harrison DE, Ashcroft SJ. Glucose induces closure of single potassium channels in isolated rat pancreatic β-cells. Nature. 1984;312:446–448. doi: 10.1038/312446a0. [DOI] [PubMed] [Google Scholar]
- Ashcroft FM, Rorsman P. Diabetes mellitus and the beta cell: the last ten years. Cell. 2012;148:1160–1171. doi: 10.1016/j.cell.2012.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Avram D, Ranta F, Hennige AM, Berchtold S, Hopp S, Haring HU, Lang F, Ullrich S. IGF-1 protects against dexamethasone-induced cell death in insulin secreting INS-1 cells independent of AKT/PKB phosphorylation. Cell Physiol Biochem. 2008;21:455–462. doi: 10.1159/000129638. [DOI] [PubMed] [Google Scholar]
- Barford D, Flint AJ, Tonks NK. Crystal structure of human protein tyrosine phosphatase 1B. Science. 1994;263:1397–1404. [PubMed] [Google Scholar]
- Barlow AD, Xie J, Moore CE, Campbell SC, Shaw JA, Nicholson ML, Herbert TP. Rapamycin toxicity in MIN6 cells and rat and human islets is mediated by the inhibition of mTOR complex 2 (mTORC2) Diabetologia. 2012;55:1355–1365. doi: 10.1007/s00125-012-2475-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Becker W, Kentrup H, Klumpp S, Schultz JE, Joost HG. Molecular cloning of a protein serine/threonine phosphatase containing a putative regulatory tetratricopeptide repeat domain. J Biol Chem. 1994;269:22586–22592. [PubMed] [Google Scholar]
- Bell GI, Polonsky KS. Diabetes mellitus and genetically programmed defects in beta-cell function. Nature. 2001;414:788–791. doi: 10.1038/414788a. [DOI] [PubMed] [Google Scholar]
- Bernal-Mizrachi E, Cras-Meneur C, Ye BR, Johnson JD, Permutt MA. Transgenic overexpression of active calcineurin in beta-cells results in decreased beta-cell mass and hyperglycemia. PLoS One. 2010;5:e11969. doi: 10.1371/journal.pone.0011969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bittinger MA, McWhinnie E, Meltzer J, Iourgenko V, Latario B, Liu X, Chen CH, Song C, Garza D, Labow M. Activation of cAMP response element-mediated gene expression by regulated nuclear transport of TORC proteins. Curr Biol. 2004;14:2156–2161. doi: 10.1016/j.cub.2004.11.002. [DOI] [PubMed] [Google Scholar]
- Blanchetot C, Chagnon M, Dube N, Halle M, Tremblay ML. Substrate-trapping techniques in the identification of cellular PTP targets. Methods. 2005;35:44–53. doi: 10.1016/j.ymeth.2004.07.007. [DOI] [PubMed] [Google Scholar]
- Bollen M, Peti W, Ragusa MJ, Beullens M. The extended PP1 toolkit: designed to create specificity. Trends Biochem Sci. 2010;35:450–458. doi: 10.1016/j.tibs.2010.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Breitkreutz A, Choi H, Sharom JR, Boucher L, Neduva V, Larsen B, Lin ZY, Breitkreutz BJ, Stark C, Liu G, et al. A global protein kinase and phosphatase interaction network in yeast. Science. 2010;328:1043–1046. doi: 10.1126/science.1176495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butler AE, Janson J, Bonner-Weir S, Ritzel R, Rizza RA, Butler PC. Beta-cell deficit and increased beta-cell apoptosis in humans with type 2 diabetes. Diabetes. 2003;52:102–110. doi: 10.2337/diabetes.52.1.102. [DOI] [PubMed] [Google Scholar]
- Cardozo AK, Kruhoffer M, Leeman R, Orntoft T, Eizirik DL. Identification of novel cytokine-induced genes in pancreatic beta-cells by high-density oligonucleotide arrays. Diabetes. 2001;50:909–920. doi: 10.2337/diabetes.50.5.909. [DOI] [PubMed] [Google Scholar]
- Chen GI, Tisayakorn S, Jorgensen C, D’Ambrosio LM, Goudreault M, Gingras AC. PP4R4/KIAA1622 forms a novel stable cytosolic complex with phosphoprotein phosphatase 4. J Biol Chem. 2008;283:29273–29284. doi: 10.1074/jbc.M803443200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen J, Ostenson CG. Inhibition of protein-tyrosine phosphatases stimulates insulin secretion in pancreatic islets of diabetic Goto-Kakizaki rats. Pancreas. 2005;30:314–317. doi: 10.1097/01.mpa.0000161887.25115.6c. [DOI] [PubMed] [Google Scholar]
- Chen J, Peterson RT, Schreiber SL. Alpha 4 associates with protein phosphatases 2A, 4, and 6. Biochem Biophys Res Commun. 1998;247:827–832. doi: 10.1006/bbrc.1998.8792. [DOI] [PubMed] [Google Scholar]
- Chen MS, Silverstein AM, Pratt WB, Chinkers M. The tetratricopeptide repeat domain of protein phosphatase 5 mediates binding to glucocorticoid receptor heterocomplexes and acts as a dominant negative mutant. J Biol Chem. 1996;271:32315–32320. doi: 10.1074/jbc.271.50.32315. [DOI] [PubMed] [Google Scholar]
- Cher C, Tremblay MH, Barber JR, Chung Ng S, Zhang B. Identification of Chaulmoogric Acid as a Small Molecule Activator of Protein Phosphatase 5. Appl Biochem Biotechnol. 2009 doi: 10.1007/s12010-009-8647-3. [DOI] [PubMed] [Google Scholar]
- Chinkers M. Protein phosphatase 5 in signal transduction. Trends Endocrinol Metab. 2001;12:28–32. doi: 10.1016/s1043-2760(00)00335-0. [DOI] [PubMed] [Google Scholar]
- Cnop M, Ladriere L, Hekerman P, Ortis F, Cardozo AK, Dogusan Z, Flamez D, Boyce M, Yuan J, Eizirik DL. Selective Inhibition of Eukaryotic Translation Initiation Factor 2α Dephosphorylation Potentiates Fatty Acid-induced Endoplasmic Reticulum Stress and Causes Pancreatic β-Cell Dysfunction and Apoptosis. J Biol Chem. 2007;282:3989–3997. doi: 10.1074/jbc.M607627200. [DOI] [PubMed] [Google Scholar]
- Cohen PT. Protein phosphatase 1--targeted in many directions. J Cell Sci. 2002;115:241–256. doi: 10.1242/jcs.115.2.241. [DOI] [PubMed] [Google Scholar]
- Colca JR, Kotagal N, Lacy PE, Brooks CL, Norling L, Landt M, McDaniel ML. Glucose-stimulated protein phosphorylation in the pancreatic islet. Biochem J. 1984;220:529–537. doi: 10.1042/bj2200529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cook DL, Hales CN. Intracellular ATP directly blocks K+ channels in pancreatic B-cells. Nature. 1984;311:271–273. doi: 10.1038/311271a0. [DOI] [PubMed] [Google Scholar]
- Cosio FG, Pesavento TE, Kim S, Osei K, Henry M, Ferguson RM. Patient survival after renal transplantation: IV. Impact of post-transplant diabetes. Kidney Int. 2002;62:1440–1446. doi: 10.1111/j.1523-1755.2002.kid582.x. [DOI] [PubMed] [Google Scholar]
- Couzens AL, Knight JD, Kean MJ, Teo G, Weiss A, Dunham WH, Lin ZY, Bagshaw RD, Sicheri F, Pawson T, et al. Protein interaction network of the Mammalian hippo pathway reveals mechanisms of kinase-phosphatase interactions. Sci Signal. 2013;6:rs15. doi: 10.1126/scisignal.2004712. [DOI] [PubMed] [Google Scholar]
- Crabtree GR, Olson EN. NFAT signaling: choreographing the social lives of cells. Cell. 2002;109(Suppl):S67–79. doi: 10.1016/s0092-8674(02)00699-2. [DOI] [PubMed] [Google Scholar]
- Das AK, Cohen PW, Barford D. The structure of the tetratricopeptide repeats of protein phosphatase 5: implications for TPR-mediated protein-protein interactions. Embo J. 1998;17:1192–1199. doi: 10.1093/emboj/17.5.1192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Detimary P, Jonas JC, Henquin JC. Possible links between glucose-induced changes in the energy state of pancreatic B cells and insulin release. Unmasking by decreasing a stable pool of adenine nucleotides in mouse islets. J Clin Invest. 1995;96:1738–1745. doi: 10.1172/JCI118219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ebihara K, Fukunaga K, Matsumoto K, Shichiri M, Miyamoto E. Cyclosporin A stimulation of glucose-induced insulin secretion in MIN6 cells. Endocrinology. 1996;137:5255–5263. doi: 10.1210/endo.137.12.8940343. [DOI] [PubMed] [Google Scholar]
- Endicott JA, Noble ME, Johnson LN. The structural basis for control of eukaryotic protein kinases. Annu Rev Biochem. 2012;81:587–613. doi: 10.1146/annurev-biochem-052410-090317. [DOI] [PubMed] [Google Scholar]
- Fagin JA, Ikejiri K, Levin SR. Insulinotropic effects of vanadate. Diabetes. 1987;36:1448–1452. doi: 10.2337/diab.36.12.1448. [DOI] [PubMed] [Google Scholar]
- Fischer EH, Graves DJ, Crittenden ER, Krebs EG. Structure of the site phosphorylated in the phosphorylase b to a reaction. J Biol Chem. 1959;234:1698–1704. [PubMed] [Google Scholar]
- Flint AJ, Tiganis T, Barford D, Tonks NK. Development of “substrate-trapping” mutants to identify physiological substrates of protein tyrosine phosphatases. Proc Natl Acad Sci U S A. 1997;94:1680–1685. doi: 10.1073/pnas.94.5.1680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fransson L, Rosengren V, Saha TK, Grankvist N, Islam T, Honkanen RE, Sjöholm Å , Ortsäter H. Mitogen-activated protein kinases and protein phosphatase 5 mediate glucocorticoid-induced cytotoxicity in pancreatic islets and beta-cells. Mol Cell Endocrinol. 2013;383:126–136. doi: 10.1016/j.mce.2013.12.010. [DOI] [PubMed] [Google Scholar]
- Gagliardino JJ, Krinks MH, Gagliardino EE. Identification of the calmodulin-regulated protein phosphatase, calcineurin, in rat pancreatic islets. Biochim Biophys Acta. 1991;1091:370–373. doi: 10.1016/0167-4889(91)90202-9. [DOI] [PubMed] [Google Scholar]
- Gagliardino JJ, Rossi PF, Garcia ME. Inhibitory effect of sulfonylureas on protein phosphatase activity in rat pancreatic islets. Acta Diabetol. 1997;34:6–9. doi: 10.1007/s005920050057. [DOI] [PubMed] [Google Scholar]
- Gembal M, Gilon P, Henquin JC. Evidence that glucose can control insulin release independently from its action on ATP-sensitive K+ channels in mouse B cells. J Clin Invest. 1992;89:1288–1295. doi: 10.1172/JCI115714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gentile S, Darden T, Erxleben C, Romeo C, Russo A, Martin N, Rossie S, Armstrong DL. Rac GTPase signaling through the PP5 protein phosphatase. Proc Natl Acad Sci U S A. 2006;103:5202–5206. doi: 10.1073/pnas.0600080103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Golden T, Swingle M, Honkanen RE. The role of serine/threonine protein phosphatase type 5 (PP5) in the regulation of stress-induced signaling networks and cancer. Cancer Metastasis Rev. 2008;27:169–178. doi: 10.1007/s10555-008-9125-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Golden TA, Honkanen RE. Regulating the expression of protein phosphatase type 5. Methods Enzymol. 2003;366:372–390. doi: 10.1016/s0076-6879(03)66028-3. [DOI] [PubMed] [Google Scholar]
- Goodyer WR, Gu X, Liu Y, Bottino R, Crabtree GR, Kim SK. Neonatal beta cell development in mice and humans is regulated by calcineurin/NFAT. Dev Cell. 2012;23:21–34. doi: 10.1016/j.devcel.2012.05.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grankvist N, Amable L, Honkanen RE, Sjöholm Å , Ortsäter H. Serine/threonine protein phosphatase 5 regulates glucose homeostasis in vivo and apoptosis signalling in mouse pancreatic islets and clonal MIN6 cells. Diabetologia. 2012;55:2005–2015. doi: 10.1007/s00125-012-2541-1. [DOI] [PubMed] [Google Scholar]
- Grankvist N, Honkanen RE, Sjöholm Å , Ortsäter H. Genetic disruption of protein phosphatase 5 in mice prevents high-fat diet feeding-induced weight gain. FEBS Lett. 2013;587:3869–3874. doi: 10.1016/j.febslet.2013.10.022. [DOI] [PubMed] [Google Scholar]
- Grimsby J, Sarabu R, Corbett WL, Haynes NE, Bizzarro FT, Coffey JW, Guertin KR, Hilliard DW, Kester RF, Mahaney PE, et al. Allosteric activators of glucokinase: potential role in diabetes therapy. Science. 2003;301:370–373. doi: 10.1126/science.1084073. [DOI] [PubMed] [Google Scholar]
- Group US Effect of intensive blood-glucose control with metformin on complications in overweight patients with type 2 diabetes (UKPDS 34). UK Prospective Diabetes Study (UKPDS) Group. Lancet. 1998a;352:854–865. [PubMed] [Google Scholar]
- Group US Intensive blood-glucose control with sulphonylureas or insulin compared with conventional treatment and risk of complications in patients with type 2 diabetes (UKPDS 33). UK Prospective Diabetes Study (UKPDS) Group. Lancet. 1998b;352:837–853. [PubMed] [Google Scholar]
- Grunnet LG, Aikin R, Tonnesen MF, Paraskevas S, Blaabjerg L, Storling J, Rosenberg L, Billestrup N, Maysinger D, Mandrup-Poulsen T. Proinflammatory cytokines activate the intrinsic apoptotic pathway in beta-cells. Diabetes. 2009;58:1807–1815. doi: 10.2337/db08-0178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haby C, Larsson O, Islam MS, Aunis D, Berggren PO, Zwiller J. Inhibition of serine/threonine protein phosphatases promotes opening of voltage-activated L-type Ca2+ channels in insulin-secreting cells. Biochem J. 1994;298(Pt 2):341–346. doi: 10.1042/bj2980341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hedeskov CJ. Mechanism of glucose-induced insulin secretion. Physiol Rev. 1980;60:442–509. doi: 10.1152/physrev.1980.60.2.442. [DOI] [PubMed] [Google Scholar]
- Heit JJ, Apelqvist AA, Gu X, Winslow MM, Neilson JR, Crabtree GR, Kim SK. Calcineurin/NFAT signalling regulates pancreatic beta-cell growth and function. Nature. 2006;443:345–349. doi: 10.1038/nature05097. [DOI] [PubMed] [Google Scholar]
- Henquin JC, Nenquin M, Szollosi A, Kubosaki A, Notkins AL. Insulin secretion in islets from mice with a double knockout for the dense core vesicle proteins islet antigen-2 (IA-2) and IA-2beta. J Endocrinol. 2008;196:573–581. doi: 10.1677/JOE-07-0496. [DOI] [PubMed] [Google Scholar]
- Heroes E, Lesage B, Gornemann J, Beullens M, Van Meervelt L, Bollen M. The PP1 binding code: a molecular-lego strategy that governs specificity. Febs J. 2013;280:584–595. doi: 10.1111/j.1742-4658.2012.08547.x. [DOI] [PubMed] [Google Scholar]
- Hinds TD, Jr., Stechschulte LA, Cash HA, Whisler D, Banerjee A, Yong W, Khuder SS, Kaw MK, Shou W, Najjar SM, et al. Protein phosphatase 5 mediates lipid metabolism through reciprocal control of glucocorticoid receptor and peroxisome proliferator-activated receptor-gamma (PPARgamma) J Biol Chem. 2011;286:42911–42922. doi: 10.1074/jbc.M111.311662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hiraga A, Kikuchi K, Tamura S, Tsuiki S. Purification and characterization of Mg2+- dependent glycogen synthase phosphatase (phosphoprotein phosphatase IA) from rat liver. Eur J Biochem. 1981;119:503–510. doi: 10.1111/j.1432-1033.1981.tb05636.x. [DOI] [PubMed] [Google Scholar]
- Hisatomi M, Hidaka H, Niki I. Ca2+/calmodulin and cyclic 3,5′ adenosine monophosphate control movement of secretory granules through protein phosphorylation/dephosphorylation in the pancreatic beta-cell. Endocrinology. 1996;137:4644–4649. doi: 10.1210/endo.137.11.8895328. [DOI] [PubMed] [Google Scholar]
- Honkanen RE, Golden T. Regulators of serine/threonine protein phosphatases at the dawn of a clinical era? Curr Med Chem. 2002;9:2055–2075. doi: 10.2174/0929867023368836. [DOI] [PubMed] [Google Scholar]
- Huang S, Shu L, Easton J, Harwood FC, Germain GS, Ichijo H, Houghton PJ. Inhibition of mammalian target of rapamycin activates apoptosis signal-regulating kinase 1 signaling by suppressing protein phosphatase 5 activity. J Biol Chem. 2004;279:36490–36496. doi: 10.1074/jbc.M401208200. [DOI] [PubMed] [Google Scholar]
- Huang X, Honkanen RE. Molecular cloning, expression, and characterization of a novel human serine/threonine protein phosphatase, PP7, that is homologous to Drosophila retinal degeneration C gene product (rdgC) J Biol Chem. 1998;273:1462–1468. doi: 10.1074/jbc.273.3.1462. [DOI] [PubMed] [Google Scholar]
- Huang X, Swingle MR, Honkanen RE. Photoreceptor serine/threonine protein phosphatase type 7: cloning, expression, and functional analysis. Methods Enzymol. 2000;315:579–593. doi: 10.1016/s0076-6879(00)15869-0. [DOI] [PubMed] [Google Scholar]
- Hunter T, Sefton BM. Transforming gene product of Rous sarcoma virus phosphorylates tyrosine. Proc Natl Acad Sci U S A. 1980;77:1311–1315. doi: 10.1073/pnas.77.3.1311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hunter T, Sefton BM, Beemon K. Studies on the structure and function of the avian sarcoma virus transforming-gene product. Cold Spring Harb Symp Quant Biol. 1980;44(Pt 2):931–941. doi: 10.1101/sqb.1980.044.01.100. [DOI] [PubMed] [Google Scholar]
- Hussain MA, Porras DL, Rowe MH, West JR, Song WJ, Schreiber WE, Wondisford FE. Increased pancreatic beta-cell proliferation mediated by CREB binding protein gene activation. Mol Cell Biol. 2006;26:7747–7759. doi: 10.1128/MCB.02353-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inada A, Hamamoto Y, Tsuura Y, Miyazaki J, Toyokuni S, Ihara Y, Nagai K, Yamada Y, Bonner-Weir S, Seino Y. Overexpression of inducible cyclic AMP early repressor inhibits transactivation of genes and cell proliferation in pancreatic beta cells. Mol Cell Biol. 2004;24:2831–2841. doi: 10.1128/MCB.24.7.2831-2841.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jangati GR, Veluthakal R, Kowluru A. siRNA-mediated depletion of endogenous protein phosphatase 2Acalpha markedly attenuates ceramide-activated protein phosphatase activity in insulin-secreting INS-832/13 cells. Biochem Biophys Res Commun. 2006;348:649–652. doi: 10.1016/j.bbrc.2006.07.100. [DOI] [PubMed] [Google Scholar]
- Jangati GR, Veluthakal R, Susick L, Gruber SA, Kowluru A. Depletion of the catalytic subunit of protein phosphatase-2A (PP2Ac) markedly attenuates glucose-stimulated insulin secretion in pancreatic beta-cells. Endocrine. 2007;31:248–253. doi: 10.1007/s12020-007-0046-3. [DOI] [PubMed] [Google Scholar]
- Janssens V, Goris J. Protein phosphatase 2A: a highly regulated family of serine/threonine phosphatases implicated in cell growth and signalling. Biochem J. 2001;353:417–439. doi: 10.1042/0264-6021:3530417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jansson D, Ng AC, Fu A, Depatie C, Al Azzabi M, Screaton RA. Glucose controls CREB activity in islet cells via regulated phosphorylation of TORC2. Proc Natl Acad Sci U S A. 2008;105:10161–10166. doi: 10.1073/pnas.0800796105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jhala US, Canettieri G, Screaton RA, Kulkarni RN, Krajewski S, Reed J, Walker J, Lin X, White M, Montminy M. cAMP promotes pancreatic beta-cell survival via CREB-mediated induction of IRS2. Genes Dev. 2003;17:1575–1580. doi: 10.1101/gad.1097103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson SA, Hunter T. Kinomics: methods for deciphering the kinome. Nat Methods. 2005;2:17–25. doi: 10.1038/nmeth731. [DOI] [PubMed] [Google Scholar]
- Jonas JC, Gilon P, Henquin JC. Temporal and quantitative correlations between insulin secretion and stably elevated or oscillatory cytoplasmic Ca2+ in mouse pancreatic β-cells. Diabetes. 1998;47:1266–1273. doi: 10.2337/diab.47.8.1266. [DOI] [PubMed] [Google Scholar]
- Jones PM, Persaud SJ. Protein kinases, protein phosphorylation, and the regulation of insulin secretion from pancreatic beta-cells. Endocr Rev. 1998;19:429–461. doi: 10.1210/edrv.19.4.0339. [DOI] [PubMed] [Google Scholar]
- Kahn SE, Halban PA. Release of incompletely processed proinsulin is the cause of the disproportionate proinsulinemia of NIDDM. Diabetes. 1997;46:1725–1732. doi: 10.2337/diab.46.11.1725. [DOI] [PubMed] [Google Scholar]
- Kasiske BL, Snyder JJ, Gilbertson D, Matas AJ. Diabetes mellitus after kidney transplantation in the United States. Am J Transplant. 2003;3:178–185. doi: 10.1034/j.1600-6143.2003.00010.x. [DOI] [PubMed] [Google Scholar]
- Klee CB, Draetta GF, Hubbard MJ. Calcineurin. Adv Enzymol Relat Areas Mol Biol. 1988;61:149–200. doi: 10.1002/9780470123072.ch4. [DOI] [PubMed] [Google Scholar]
- Kloeker S, Reed R, McConnell JL, Chang D, Tran K, Westphal RS, Law BK, Colbran RJ, Kamoun M, Campbell KS, et al. Parallel purification of three catalytic subunits of the protein serine/threonine phosphatase 2A family (PP2A(C), PP4(C), and PP6(C)) and analysis of the interaction of PP2A(C) with alpha4 protein. Protein Expr Purif. 2003;31:19–33. doi: 10.1016/s1046-5928(03)00141-4. [DOI] [PubMed] [Google Scholar]
- Klumpp S, Thissen MC, Krieglstein J. Protein phosphatases types 2Calpha and 2Cbeta in apoptosis. Biochem Soc Trans. 2006;34:1370–1375. doi: 10.1042/BST0341370. [DOI] [PubMed] [Google Scholar]
- Komatsu M, Schermerhorn T, Noda M, Straub SG, Aizawa T, Sharp GW. Augmentation of insulin release by glucose in the absence of extracellular Ca2+: new insights into stimulus-secretion coupling. Diabetes. 1997;46:1928–1938. doi: 10.2337/diab.46.12.1928. [DOI] [PubMed] [Google Scholar]
- Koo SH, Flechner L, Qi L, Zhang X, Screaton RA, Jeffries S, Hedrick S, Xu W, Boussouar F, Brindle P, et al. The CREB coactivator TORC2 is a key regulator of fasting glucose metabolism. Nature. 2005;437:1109–1111. doi: 10.1038/nature03967. [DOI] [PubMed] [Google Scholar]
- Koro CE, Bowlin SJ, Bourgeois N, Fedder DO. Glycemic control from 1988 to 2000 among U.S. adults diagnosed with type 2 diabetes: a preliminary report. Diabetes Care. 2004;27:17–20. doi: 10.2337/diacare.27.1.17. [DOI] [PubMed] [Google Scholar]
- Kowluru A. Novel regulatory roles for protein phosphatase-2A in the islet beta cell. Biochem Pharmacol. 2005;69:1681–1691. doi: 10.1016/j.bcp.2005.03.018. [DOI] [PubMed] [Google Scholar]
- Kowluru A, Chen HQ, Modrick LM, Stefanelli C. Activation of acetyl-CoA carboxylase by a glutamate- and magnesium-sensitive protein phosphatase in the islet beta-cell. Diabetes. 2001;50:1580–1587. doi: 10.2337/diabetes.50.7.1580. [DOI] [PubMed] [Google Scholar]
- Kowluru A, Matti A. Hyperactivation of protein phosphatase 2A in models of glucolipotoxicity and diabetes: potential mechanisms and functional consequences. Biochem Pharmacol. 2012;84:591–597. doi: 10.1016/j.bcp.2012.05.003. [DOI] [PubMed] [Google Scholar]
- Kowluru A, Metz SA. Ceramide-activated protein phosphatase-2A activity in insulin-secreting cells. FEBS Lett. 1997;418:179–182. doi: 10.1016/s0014-5793(97)01379-3. [DOI] [PubMed] [Google Scholar]
- Kowluru A, Seavey SE, Rabaglia ME, Nesher R, Metz SA. Carboxylmethylation of the catalytic subunit of protein phosphatase 2A in insulin-secreting cells: evidence for functional consequences on enzyme activity and insulin secretion. Endocrinology. 1996;137:2315–2323. doi: 10.1210/endo.137.6.8641181. [DOI] [PubMed] [Google Scholar]
- Krautheim A, Rustenbeck I, Steinfelder HJ. Phosphatase inhibitors induce defective hormone secretion in insulin-secreting cells and entry into apoptosis. Exp Clin Endocrinol Diabetes. 1999;107:29–34. doi: 10.1055/s-0029-1212069. [DOI] [PubMed] [Google Scholar]
- Krebs EG, Graves DJ, Fischer EH. Factors affecting the activity of muscle phosphorylase b kinase. J Biol Chem. 1959;234:2867–2873. [PubMed] [Google Scholar]
- Krieglstein J, Hufnagel B, Dworak M, Schwarz S, Kewitz T, Reinbold M, Klumpp S. Influence of various fatty acids on the activity of protein phosphatase type 2C and apoptosis of endothelial cells and macrophages. Eur J Pharm Sci. 2008;35:397–403. doi: 10.1016/j.ejps.2008.08.007. [DOI] [PubMed] [Google Scholar]
- Kuehnen P, Laubner K, Raile K, Schofl C, Jakob F, Pilz I, Path G, Seufert J. Protein phosphatase 1 (PP-1)-dependent inhibition of insulin secretion by leptin in INS-1 pancreatic beta-cells and human pancreatic islets. Endocrinology. 2011;152:1800–1808. doi: 10.1210/en.2010-1094. [DOI] [PubMed] [Google Scholar]
- Kutuzov MA, Andreeva AV, Voyno-Yasenetskaya TA. Regulation of apoptosis signal-regulating kinase 1 (ASK1) by polyamine levels via protein phosphatase 5. J Biol Chem. 2005;280:25388–25395. doi: 10.1074/jbc.M413202200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ladriere L, Igoillo-Esteve M, Cunha DA, Brion JP, Bugliani M, Marchetti P, Eizirik DL, Cnop M. Enhanced signaling downstream of ribonucleic Acid-activated protein kinase-like endoplasmic reticulum kinase potentiates lipotoxic endoplasmic reticulum stress in human islets. J Clin Endocrinol Metab. 2010;95:1442–1449. doi: 10.1210/jc.2009-2322. [DOI] [PubMed] [Google Scholar]
- Lambrecht C, Haesen D, Sents W, Ivanova E, Janssens V. Structure, regulation, and pharmacological modulation of PP2A phosphatases. Methods Mol Biol. 2013;1053:283–305. doi: 10.1007/978-1-62703-562-0_17. [DOI] [PubMed] [Google Scholar]
- Lammers T, Lavi S. Role of type 2C protein phosphatases in growth regulation and in cellular stress signaling. Crit Rev Biochem Mol Biol. 2007;42:437–461. doi: 10.1080/10409230701693342. [DOI] [PubMed] [Google Scholar]
- Langberg EC, Gu HF, Nordman S, Efendic S, Ostenson CG. Genetic variation in receptor protein tyrosine phosphatase sigma is associated with type 2 diabetes in Swedish Caucasians. Eur J Endocrinol. 2007;157:459–464. doi: 10.1530/EJE-07-0114. [DOI] [PubMed] [Google Scholar]
- Larsson O, Barker CJ, Sjoholm A, Carlqvist H, Michell RH, Bertorello A, Nilsson T, Honkanen RE, Mayr GW, Zwiller J, et al. Inhibition of phosphatases and increased Ca2+ channel activity by inositol hexakisphosphate. Science. 1997;278:471–474. doi: 10.1126/science.278.5337.471. [DOI] [PubMed] [Google Scholar]
- Lawrence MC, Bhatt HS, Easom RA. NFAT regulates insulin gene promoter activity in response to synergistic pathways induced by glucose and glucagon-like peptide-1. Diabetes. 2002;51:691–698. doi: 10.2337/diabetes.51.3.691. [DOI] [PubMed] [Google Scholar]
- Lawrence MC, Bhatt HS, Watterson JM, Easom RA. Regulation of insulin gene transcription by a Ca(2+)-responsive pathway involving calcineurin and nuclear factor of activated T cells. Mol Endocrinol. 2001;15:1758–1767. doi: 10.1210/mend.15.10.0702. [DOI] [PubMed] [Google Scholar]
- Lee SH, Shin MS, Park WS, Kim SY, Kim HS, Lee JH, Han SY, Lee HK, Park JY, Oh RR, et al. Immunohistochemical localization of FAP-1, an inhibitor of Fas-mediated apoptosis, in normal and neoplastic human tissues. APMIS. 1999;107:1101–1108. doi: 10.1111/j.1699-0463.1999.tb01515.x. [DOI] [PubMed] [Google Scholar]
- Lee YC, Nielsen JH. Regulation of beta cell replication. Mol Cell Endocrinol. 2009;297:18–27. doi: 10.1016/j.mce.2008.08.033. [DOI] [PubMed] [Google Scholar]
- Lehtihet M, Honkanen RE, Sjoholm A. Inositol hexakisphosphate and sulfonylureas regulate beta-cell protein phosphatases. Biochem Biophys Res Commun. 2004;316:893–897. doi: 10.1016/j.bbrc.2004.02.144. [DOI] [PubMed] [Google Scholar]
- Lehtihet M, Webb DL, Honkanen RE, Sjoholm A. Glutamate inhibits protein phosphatases and promotes insulin exocytosis in pancreatic beta-cells. Biochem Biophys Res Commun. 2005;328:601–607. doi: 10.1016/j.bbrc.2005.01.024. [DOI] [PubMed] [Google Scholar]
- Lernmark A, Nielsen DA, Parman AU, Sehlin J, Steiner DF, Taljedal IB. Cation-activated phosphatase activities in islet cell plasma membrane preparations. Horm Metab Res Suppl. 1980;10(Suppl):55–61. [PubMed] [Google Scholar]
- Lipson LG, Siegel E, Wollheim CB, Sharp GW. Insulin release during fasting: studies on adenylate cyclase, phosphodiesterase, protein kinase, and phosphoprotein phosphatase in isolated islets of langerhans of the rat. Endocrinology. 1979;105:702–707. doi: 10.1210/endo-105-3-702. [DOI] [PubMed] [Google Scholar]
- Lobner K, Steinbrenner H, Roberts GA, Ling Z, Huang GC, Piquer S, Pipeleers DG, Seissler J, Christie MR. Different regulated expression of the tyrosine phosphatase-like proteins IA-2 and phogrin by glucose and insulin in pancreatic islets: relationship to development of insulin secretory responses in early life. Diabetes. 2002;51:2982–2988. doi: 10.2337/diabetes.51.10.2982. [DOI] [PubMed] [Google Scholar]
- Lubert EJ, Hong Y, Sarge KD. Interaction between protein phosphatase 5 and the A subunit of protein phosphatase 2A: evidence for a heterotrimeric form of protein phosphatase 5. J Biol Chem. 2001;276:38582–38587. doi: 10.1074/jbc.M106906200. [DOI] [PubMed] [Google Scholar]
- MacDonald MJ, Fahien LA. Glutamate is not a messenger in insulin secretion. J Biol Chem. 2000;275:34025–34027. doi: 10.1074/jbc.C000411200. [DOI] [PubMed] [Google Scholar]
- Maechler P, Wollheim CB. Mitochondrial glutamate acts as a messenger in glucose-induced insulin exocytosis. Nature. 1999;402:685–689. doi: 10.1038/45280. [DOI] [PubMed] [Google Scholar]
- Maedler K, Sergeev P, Ris F, Oberholzer J, Joller-Jemelka HI, Spinas GA, Kaiser N, Halban PA, Donath MY. Glucose-induced beta cell production of IL-1beta contributes to glucotoxicity in human pancreatic islets. J Clin Invest. 2002;110:851–860. doi: 10.1172/JCI15318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manning G, Whyte DB, Martinez R, Hunter T, Sudarsanam S. The protein kinase complement of the human genome. Science. 2002;298:1912–1934. doi: 10.1126/science.1075762. [DOI] [PubMed] [Google Scholar]
- Matschinsky FM, Glaser B, Magnuson MA. Pancreatic β-cell glucokinase: closing the gap between theoretical concepts and experimental realities. Diabetes. 1998;47:307–315. doi: 10.2337/diabetes.47.3.307. [DOI] [PubMed] [Google Scholar]
- Mayer P, Jochum C, Schatz H, Pfeiffer A. Okadaic acid indicates a major function for protein phosphatases in stimulus-response coupling of RINm5F rat insulinoma cells. Exp Clin Endocrinol. 1994;102:313–319. doi: 10.1055/s-0029-1211297. [DOI] [PubMed] [Google Scholar]
- Meetoo D, McGovern P, Safadi R. An epidemiological overview of diabetes across the world. Br J Nurs. 2007;16:1002–1007. doi: 10.12968/bjon.2007.16.16.27079. [DOI] [PubMed] [Google Scholar]
- Meglasson MD, Matschinsky FM. Pancreatic islet glucose metabolism and regulation of insulin secretion. Diabetes Metab Rev. 1986;2:163–214. doi: 10.1002/dmr.5610020301. [DOI] [PubMed] [Google Scholar]
- Meier JJ, Butler AE, Saisho Y, Monchamp T, Galasso R, Bhushan A, Rizza RA, Butler PC. Beta-cell replication is the primary mechanism subserving the postnatal expansion of beta-cell mass in humans. Diabetes. 2008;57:1584–1594. doi: 10.2337/db07-1369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merritt SE, Mata M, Nihalani D, Zhu C, Hu X, Holzman LB. The mixed lineage kinase DLK utilizes MKK7 and not MKK4 as substrate. J Biol Chem. 1999;274:10195–10202. doi: 10.1074/jbc.274.15.10195. [DOI] [PubMed] [Google Scholar]
- Mkaddem SB, Werts C, Goujon JM, Bens M, Pedruzzi E, Ogier-Denis E, Vandewalle A. Heat shock protein gp96 interacts with protein phosphatase 5 and controls toll-like receptor 2 (TLR2)-mediated activation of extracellular signal-regulated kinase (ERK) 1/2 in post-hypoxic kidney cells. J Biol Chem. 2009;284:12541–12549. doi: 10.1074/jbc.M808376200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moore F, Colli ML, Cnop M, Esteve MI, Cardozo AK, Cunha DA, Bugliani M, Marchetti P, Eizirik DL. PTPN2, a candidate gene for type 1 diabetes, modulates interferon-gamma-induced pancreatic beta-cell apoptosis. Diabetes. 2009;58:1283–1291. doi: 10.2337/db08-1510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moorhead GB, Trinkle-Mulcahy L, Ulke-Lemee A. Emerging roles of nuclear protein phosphatases. Nat Rev Mol Cell Biol. 2007;8:234–244. doi: 10.1038/nrm2126. [DOI] [PubMed] [Google Scholar]
- Morita K, Saitoh M, Tobiume K, Matsuura H, Enomoto S, Nishitoh H, Ichijo H. Negative feedback regulation of ASK1 by protein phosphatase 5 (PP5) in response to oxidative stress. Embo J. 2001;20:6028–6036. doi: 10.1093/emboj/20.21.6028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mullen TD, Hannun YA, Obeid LM. Ceramide synthases at the centre of sphingolipid metabolism and biology. Biochem J. 2012;441:789–802. doi: 10.1042/BJ20111626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murphy LI, Jones PM. Phospho-serine/threonine phosphatases in rat islets of Langerhans: identification and effect on insulin secretion. Mol Cell Endocrinol. 1996;117:195–202. doi: 10.1016/0303-7207(95)03747-0. [DOI] [PubMed] [Google Scholar]
- Nakamura S, Tanigawa K, Kawaguchi M, Inoue Y, Xu G, Nagami H, Teramoto M, Kato Y, Tamura K. Effect of chronic vanadate administration in partially depancreatized rats. Diabetes Res Clin Pract. 1995;27:51–59. doi: 10.1016/0168-8227(94)01012-o. [DOI] [PubMed] [Google Scholar]
- Newgard CB, McGarry JD. Metabolic coupling factors in pancreatic β-cell signal transduction. Annu Rev Biochem. 1995;64:689–719. doi: 10.1146/annurev.bi.64.070195.003353. [DOI] [PubMed] [Google Scholar]
- Nolen B, Taylor S, Ghosh G. Regulation of protein kinases; controlling activity through activation segment conformation. Mol Cell. 2004;15:661–675. doi: 10.1016/j.molcel.2004.08.024. [DOI] [PubMed] [Google Scholar]
- Novoa I, Zhang Y, Zeng H, Jungreis R, Harding HP, Ron D. Stress-induced gene expression requires programmed recovery from translational repression. Embo J. 2003;22:1180–1187. doi: 10.1093/emboj/cdg112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ollendorff V, Donoghue DJ. The serine/threonine phosphatase PP5 interacts with CDC16 and CDC27, two tetratricopeptide repeat-containing subunits of the anaphase-promoting complex. J Biol Chem. 1997;272:32011–32018. doi: 10.1074/jbc.272.51.32011. [DOI] [PubMed] [Google Scholar]
- Olsen JV, Blagoev B, Gnad F, Macek B, Kumar C, Mortensen P, Mann M. Global, in vivo, and site-specific phosphorylation dynamics in signaling networks. Cell. 2006;127:635–648. doi: 10.1016/j.cell.2006.09.026. [DOI] [PubMed] [Google Scholar]
- Oprescu AI, Bikopoulos G, Naassan A, Allister EM, Tang C, Park E, Uchino H, Lewis GF, Fantus IG, Rozakis-Adcock M, et al. Free fatty acid-induced reduction in glucose-stimulated insulin secretion: evidence for a role of oxidative stress in vitro and in vivo. Diabetes. 2007;56:2927–2937. doi: 10.2337/db07-0075. [DOI] [PubMed] [Google Scholar]
- Ortsäter H, Sjöholm A. A busy cell--endoplasmic reticulum stress in the pancreatic β-cell. Mol Cell Endocrinol. 2007;277:1–5. doi: 10.1016/j.mce.2007.06.006. [DOI] [PubMed] [Google Scholar]
- Ozbay LA, Smidt K, Mortensen DM, Carstens J, Jorgensen KA, Rungby J. Cyclosporin and tacrolimus impair insulin secretion and transcriptional regulation in INS-1E beta-cells. Br J Pharmacol. 2011;162:136–146. doi: 10.1111/j.1476-5381.2010.01018.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palanivel R, Veluthakal R, Kowluru A. Regulation by glucose and calcium of the carboxylmethylation of the catalytic subunit of protein phosphatase 2A in insulin-secreting INS-1 cells. Am J Physiol Endocrinol Metab. 2004;286:E1032–1041. doi: 10.1152/ajpendo.00587.2003. [DOI] [PubMed] [Google Scholar]
- Palanivel R, Veluthakal R, McDonald P, Kowluru A. Further evidence for the regulation of acetyl-CoA carboxylase activity by a glutamate- and magnesium-activated protein phosphatase in the pancreatic beta cell: defective regulation in the diabetic GK rat islet. Endocrine. 2005;26:71–77. doi: 10.1385/ENDO:26:1:071. [DOI] [PubMed] [Google Scholar]
- Pato MD, Adelstein RS. Characterization of a Mg2+-dependent phosphatase from turkey gizzard smooth muscle. J Biol Chem. 1983;258:7055–7058. [PubMed] [Google Scholar]
- Pederson RA, Ramanadham S, Buchan AM, McNeill JH. Long-term effects of vanadyl treatment on streptozocin-induced diabetes in rats. Diabetes. 1989;38:1390–1395. doi: 10.2337/diab.38.11.1390. [DOI] [PubMed] [Google Scholar]
- Peiris H, Raghupathi R, Jessup CF, Zanin MP, Mohanasundaram D, Mackenzie KD, Chataway T, Clarke JN, Brealey J, Coates PT, et al. Increased expression of the glucose-responsive gene, RCAN1, causes hypoinsulinemia, beta-cell dysfunction, and diabetes. Endocrinology. 2012;153:5212–5221. doi: 10.1210/en.2011-2149. [DOI] [PubMed] [Google Scholar]
- Plaumann S, Blume R, Borchers S, Steinfelder HJ, Knepel W, Oetjen E. Activation of the dual-leucine-zipper-bearing kinase and induction of beta-cell apoptosis by the immunosuppressive drug cyclosporin A. Mol Pharmacol. 2008;73:652–659. doi: 10.1124/mol.107.040782. [DOI] [PubMed] [Google Scholar]
- Ramsey AJ, Chinkers M. Identification of potential physiological activators of protein phosphatase 5. Biochemistry. 2002;41:5625–5632. doi: 10.1021/bi016090h. [DOI] [PubMed] [Google Scholar]
- Ramsey AJ, Russell LC, Whitt SR, Chinkers M. Overlapping sites of tetratricopeptide repeat protein binding and chaperone activity in heat shock protein 90. J Biol Chem. 2000;275:17857–17862. doi: 10.1074/jbc.M001625200. [DOI] [PubMed] [Google Scholar]
- Ranta F, Avram D, Berchtold S, Dufer M, Drews G, Lang F, Ullrich S. Dexamethasone induces cell death in insulin-secreting cells, an effect reversed by exendin-4. Diabetes. 2006;55:1380–1390. doi: 10.2337/db05-1220. [DOI] [PubMed] [Google Scholar]
- Ranta F, Dufer M, Stork B, Wesselborg S, Drews G, Haring HU, Lang F, Ullrich S. Regulation of calcineurin activity in insulin-secreting cells: stimulation by Hsp90 during glucocorticoid-induced apoptosis. Cell Signal. 2008;20:1780–1786. doi: 10.1016/j.cellsig.2008.06.003. [DOI] [PubMed] [Google Scholar]
- Ratcliff H, Jones PM. Effects of okadaic acid on insulin secretion from rat islets of Langerhans. Biochim Biophys Acta. 1993;1175:188–191. doi: 10.1016/0167-4889(93)90022-h. [DOI] [PubMed] [Google Scholar]
- Ravnskjaer K, Boergesen M, Dalgaard LT, Mandrup S. Glucose-induced repression of PPARalpha gene expression in pancreatic beta-cells involves PP2A activation and AMPK inactivation. J Mol Endocrinol. 2006;36:289–299. doi: 10.1677/jme.1.01965. [DOI] [PubMed] [Google Scholar]
- Redmon JB, Olson LK, Armstrong MB, Greene MJ, Robertson RP. Effects of tacrolimus (FK506) on human insulin gene expression, insulin mRNA levels, and insulin secretion in HIT T15 cells. J Clin Invest. 1996;98:2786–2793. doi: 10.1172/JCI119105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reich E, Tamary A, Sionov RV, Melloul D. Involvement of thioredoxin-interacting protein (TXNIP) in glucocorticoid-mediated beta cell death. Diabetologia. 2012;55:1048–1057. doi: 10.1007/s00125-011-2422-z. [DOI] [PubMed] [Google Scholar]
- Renstrom E, Ding WG, Bokvist K, Rorsman P. Neurotransmitter-induced inhibition of exocytosis in insulin-secreting beta cells by activation of calcineurin. Neuron. 1996;17:513–522. doi: 10.1016/s0896-6273(00)80183-x. [DOI] [PubMed] [Google Scholar]
- Rodriguez-Rodriguez AE, Trinanes J, Velazquez-Garcia S, Porrini E, Vega Prieto MJ, Diez Fuentes ML, Arevalo M, Salido Ruiz E, Torres A. The higher diabetogenic risk of tacrolimus depends on pre-existing insulin resistance. A study in obese and lean Zucker rats. Am J Transplant. 2013;13:1665–1675. doi: 10.1111/ajt.12236. [DOI] [PubMed] [Google Scholar]
- Rorsman P, Braun M. Regulation of insulin secretion in human pancreatic islets. Annu Rev Physiol. 2013;75:155–179. doi: 10.1146/annurev-physiol-030212-183754. [DOI] [PubMed] [Google Scholar]
- Russell LC, Whitt SR, Chen MS, Chinkers M. Identification of conserved residues required for the binding of a tetratricopeptide repeat domain to heat shock protein 90. J Biol Chem. 1999;274:20060–20063. doi: 10.1074/jbc.274.29.20060. [DOI] [PubMed] [Google Scholar]
- Rutkowski DT, Arnold SM, Miller CN, Wu J, Li J, Gunnison KM, Mori K, Sadighi Akha AA, Raden D, Kaufman RJ. Adaptation to ER stress is mediated by differential stabilities of pro-survival and pro-apoptotic mRNAs and proteins. PLoS Biol. 2006;4:e374. doi: 10.1371/journal.pbio.0040374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santin I, Moore F, Colli ML, Gurzov EN, Marselli L, Marchetti P, Eizirik DL. PTPN2, a candidate gene for type 1 diabetes, modulates pancreatic beta-cell apoptosis via regulation of the BH3-only protein Bim. Diabetes. 2011;60:3279–3288. doi: 10.2337/db11-0758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santoro MF, Annand RR, Robertson MM, Peng YW, Brady MJ, Mankovich JA, Hackett MC, Ghayur T, Walter G, Wong WW, et al. Regulation of protein phosphatase 2A activity by caspase-3 during apoptosis. J Biol Chem. 1998;273:13119–13128. doi: 10.1074/jbc.273.21.13119. [DOI] [PubMed] [Google Scholar]
- Sato Y, Mariot P, Detimary P, Gilon P, Henquin JC. Okadaic acid-induced decrease in the magnitude and efficacy of the Ca2+ signal in pancreatic beta cells and inhibition of insulin secretion. Br J Pharmacol. 1998;123:97–105. doi: 10.1038/sj.bjp.0701578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwaninger M, Blume R, Kruger M, Lux G, Oetjen E, Knepel W. Involvement of the Ca(2+)-dependent phosphatase calcineurin in gene transcription that is stimulated by cAMP through cAMP response elements. J Biol Chem. 1995;270:8860–8866. doi: 10.1074/jbc.270.15.8860. [DOI] [PubMed] [Google Scholar]
- Schwarz S, Hufnagel B, Dworak M, Klumpp S, Krieglstein J. Protein phosphatase type 2Calpha and 2Cbeta are involved in fatty acid-induced apoptosis of neuronal and endothelial cells. Apoptosis. 2006;11:1111–1119. doi: 10.1007/s10495-006-6982-1. [DOI] [PubMed] [Google Scholar]
- Screaton RA, Conkright MD, Katoh Y, Best JL, Canettieri G, Jeffries S, Guzman E, Niessen S, Yates JR, 3rd, Takemori H, et al. The CREB coactivator TORC2 functions as a calcium-and cAMP-sensitive coincidence detector. Cell. 2004;119:61–74. doi: 10.1016/j.cell.2004.09.015. [DOI] [PubMed] [Google Scholar]
- Sents W, Ivanova E, Lambrecht C, Haesen D, Janssens V. The biogenesis of active protein phosphatase 2A holoenzymes: a tightly regulated process creating phosphatase specificity. Febs J. 2013;280:644–661. doi: 10.1111/j.1742-4658.2012.08579.x. [DOI] [PubMed] [Google Scholar]
- Shao J, Hartson SD, Matts RL. Evidence that protein phosphatase 5 functions to negatively modulate the maturation of the Hsp90-dependent heme-regulated eIF2alpha kinase. Biochemistry. 2002;41:6770–6779. doi: 10.1021/bi025737a. [DOI] [PubMed] [Google Scholar]
- Shaw RJ, Lamia KA, Vasquez D, Koo SH, Bardeesy N, Depinho RA, Montminy M, Cantley LC. The kinase LKB1 mediates glucose homeostasis in liver and therapeutic effects of metformin. Science. 2005;310:1642–1646. doi: 10.1126/science.1120781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi Y. Serine/threonine phosphatases: mechanism through structure. Cell. 2009;139:468–484. doi: 10.1016/j.cell.2009.10.006. [DOI] [PubMed] [Google Scholar]
- Shimabukuro M, Higa M, Zhou YT, Wang MY, Newgard CB, Unger RH. Lipoapoptosis in beta-cells of obese prediabetic fa/fa rats. Role of serine palmitoyltransferase overexpression. J Biol Chem. 1998;273:32487–32490. doi: 10.1074/jbc.273.49.32487. [DOI] [PubMed] [Google Scholar]
- Silverstein AM, Galigniana MD, Chen MS, Owens-Grillo JK, Chinkers M, Pratt WB. Protein phosphatase 5 is a major component of glucocorticoid receptor.hsp90 complexes with properties of an FK506-binding immunophilin. J Biol Chem. 1997;272:16224–16230. doi: 10.1074/jbc.272.26.16224. [DOI] [PubMed] [Google Scholar]
- Sinclair C, Borchers C, Parker C, Tomer K, Charbonneau H, Rossie S. The tetratricopeptide repeat domain and a C-terminal region control the activity of Ser/Thr protein phosphatase 5. J Biol Chem. 1999;274:23666–23672. doi: 10.1074/jbc.274.33.23666. [DOI] [PubMed] [Google Scholar]
- Sjöholm Å . Phorbol ester stimulation of pancreatic beta-cell replication, polyamine content and insulin secretion. FEBS Lett. 1991;294:257–260. doi: 10.1016/0014-5793(91)81442-b. [DOI] [PubMed] [Google Scholar]
- Sjöholm Å . Inhibitory effects of cyclosporin A on rat insulinoma cell proliferation, polyamine content and insulin secretion. Mol Cell Endocrinol. 1994;99:21–24. doi: 10.1016/0303-7207(94)90141-4. [DOI] [PubMed] [Google Scholar]
- Sjöholm Å . Ceramide inhibits pancreatic beta-cell insulin production and mitogenesis and mimics the actions of interleukin-1 beta. FEBS Lett. 1995;367:283–286. doi: 10.1016/0014-5793(95)00470-t. [DOI] [PubMed] [Google Scholar]
- Sjöholm Å . Diabetes mellitus and impaired pancreatic beta-cell proliferation. J Intern Med. 1996;239:211–220. doi: 10.1046/j.1365-2796.1996.377740000.x. [DOI] [PubMed] [Google Scholar]
- Sjöholm Å . Aspects of novel sites of regulation of the insulin stimulus-secretion coupling in normal and diabetic pancreatic islets. Endocrine. 1998;9:1–13. doi: 10.1385/ENDO:9:1:1. [DOI] [PubMed] [Google Scholar]
- Sjöholm Å , Arkhammar P, Welsh N, Bokvist K, Rorsman P, Hallberg A, Nilsson T, Welsh M, Berggren PO. Enhanced stimulus-secretion coupling in polyamine-depleted rat insulinoma cells. An effect involving increased cytoplasmic Ca2+, inositol phosphate generation, and phorbol ester sensitivity. J Clin Invest. 1993a;92:1910–1917. doi: 10.1172/JCI116784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sjöholm Å , Honkanen RE. Polyamines regulate serine/threonine protein phosphatases in insulin-secreting cells. Pancreas. 2000;20:32–37. doi: 10.1097/00006676-200001000-00005. [DOI] [PubMed] [Google Scholar]
- Sjöholm Å , Honkanen RE, Berggren PO. Characterization of serine/threonine protein phosphatases in RINm5F insulinoma cells. Biosci Rep. 1993b;13:349–358. doi: 10.1007/BF01150479. [DOI] [PubMed] [Google Scholar]
- Sjöholm Å , Honkanen RE, Berggren PO. Inhibition of serine/threonine protein phosphatases by secretagogues in insulin-secreting cells. Endocrinology. 1995;136:3391–3397. doi: 10.1210/endo.136.8.7628374. [DOI] [PubMed] [Google Scholar]
- Sjöholm Å , Lehtihet M, Efanov AM, Zaitsev SV, Berggren PO, Honkanen RE. Glucose metabolites inhibit protein phosphatases and directly promote insulin exocytosis in pancreatic beta-cells. Endocrinology. 2002;143:4592–4598. doi: 10.1210/en.2002-220672. [DOI] [PubMed] [Google Scholar]
- Skarra DV, Goudreault M, Choi H, Mullin M, Nesvizhskii AI, Gingras AC, Honkanen RE. Label-free quantitative proteomics and SAINT analysis enable interactome mapping for the human Ser/Thr protein phosphatase 5. Proteomics. 2011;11:1508–1516. doi: 10.1002/pmic.201000770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soleimanpour SA, Crutchlow MF, Ferrari AM, Raum JC, Groff DN, Rankin MM, Liu C, De Leon DD, Naji A, Kushner JA, et al. Calcineurin signaling regulates human islet {beta}-cell survival. J Biol Chem. 2010;285:40050–40059. doi: 10.1074/jbc.M110.154955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sutherland EW, Jr., Wosilait WD. Inactivation and activation of liver phosphorylase. Nature. 1955;175:169–170. doi: 10.1038/175169a0. [DOI] [PubMed] [Google Scholar]
- Swingle M, Ni L, Honkanen RE. Small-molecule inhibitors of ser/thr protein phosphatases: specificity, use and common forms of abuse. Methods Mol Biol. 2007;365:23–38. doi: 10.1385/1-59745-267-X:23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swingle MR, Honkanen RE, Ciszak EM. Structural basis for the catalytic activity of human serine/threonine protein phosphatase-5. J Biol Chem. 2004;279:33992–33999. doi: 10.1074/jbc.M402855200. [DOI] [PubMed] [Google Scholar]
- Taira J, Higashimoto Y. Caveolin-1 interacts with protein phosphatase 5 and modulates its activity in prostate cancer cells. Biochem Biophys Res Commun. 2013;431:724–728. doi: 10.1016/j.bbrc.2013.01.051. [DOI] [PubMed] [Google Scholar]
- Taljedal IB. Electrophoretic studies on phosphatases from the pancreatic islets of obese-hyperglycaemic mice. Acta Endocrinol (Copenh) 1967;55:153–162. [PubMed] [Google Scholar]
- Tamagawa T, Iguchi A, Uemura K, Miura H, Nonogaki K, Ishiguro T, Sakamoto N. Effects of the protein phosphatase inhibitors okadaic acid and calyculin A on insulin release from rat pancreatic islets. Endocrinol Jpn. 1992;39:325–329. doi: 10.1507/endocrj1954.39.325. [DOI] [PubMed] [Google Scholar]
- Tamura K, Fujimura T, Tsutsumi T, Nakamura K, Ogawa T, Atumaru C, Hirano Y, Ohara K, Ohtsuka K, Shimomura K, et al. Transcriptional inhibition of insulin by FK506 and possible involvement of FK506 binding protein-12 in pancreatic beta-cell. Transplantation. 1995;59:1606–1613. [PubMed] [Google Scholar]
- Taylor SS, Kornev AP. Protein kinases: evolution of dynamic regulatory proteins. Trends Biochem Sci. 2011;36:65–77. doi: 10.1016/j.tibs.2010.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tengholm A, Gylfe E. Oscillatory control of insulin secretion. Mol Cell Endocrinol. 2009;297:58–72. doi: 10.1016/j.mce.2008.07.009. [DOI] [PubMed] [Google Scholar]
- Terrak M, Kerff F, Langsetmo K, Tao T, Dominguez R. Structural basis of protein phosphatase 1 regulation. Nature. 2004;429:780–784. doi: 10.1038/nature02582. [DOI] [PubMed] [Google Scholar]
- Thomas H, Senkel S, Erdmann S, Arndt T, Turan G, Klein-Hitpass L, Ryffel GU. Pattern of genes influenced by conditional expression of the transcription factors HNF6, HNF4alpha and HNF1beta in a pancreatic beta-cell line. Nucleic Acids Res. 2004;32:e150. doi: 10.1093/nar/gnh144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Todd JA, Walker NM, Cooper JD, Smyth DJ, Downes K, Plagnol V, Bailey R, Nejentsev S, Field SF, Payne F, et al. Robust associations of four new chromosome regions from genome-wide analyses of type 1 diabetes. Nat Genet. 2007;39:857–864. doi: 10.1038/ng2068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Torii S. Expression and function of IA-2 family proteins, unique neuroendocrine-specific protein-tyrosine phosphatases. Endocr J. 2009;56:639–648. doi: 10.1507/endocrj.k09e-157. [DOI] [PubMed] [Google Scholar]
- Tumlin JA, Lea JP, Swanson CE, Smith CL, Edge SS, Someren JS. Aldosterone and dexamethasone stimulate calcineurin activity through a transcription-independent mechanism involving steroid receptor-associated heat shock proteins. J Clin Invest. 1997;99:1217–1223. doi: 10.1172/JCI119278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tunon MJ, Sanchez-Campos S, Gutierrez B, Culebras JM, Gonzalez-Gallego J. Effects of FK506 and rapamycin on generation of reactive oxygen species, nitric oxide production and nuclear factor kappa B activation in rat hepatocytes. Biochem Pharmacol. 2003;66:439–445. doi: 10.1016/s0006-2952(03)00288-0. [DOI] [PubMed] [Google Scholar]
- Vander Mierde, Scheuner D, Quintens R, Patel R, Song B, Tsukamoto K, Beullens M, Kaufman RJ, Bollen M, Schuit FC. Glucose activates a protein phosphatase-1-mediated signaling pathway to enhance overall translation in pancreatic beta-cells. Endocrinology. 2007;148:609–617. doi: 10.1210/en.2006-1012. [DOI] [PubMed] [Google Scholar]
- Wang HG, Pathan N, Ethell IM, Krajewski S, Yamaguchi Y, Shibasaki F, McKeon F, Bobo T, Franke TF, Reed JC. Ca2+-induced apoptosis through calcineurin dephosphorylation of BAD. Science. 1999;284:339–343. doi: 10.1126/science.284.5412.339. [DOI] [PubMed] [Google Scholar]
- Ward WK, Bolgiano DC, McKnight B, Halter JB, Porte D., Jr. Diminished B cell secretory capacity in patients with noninsulin-dependent diabetes mellitus. J Clin Invest. 1984;74:1318–1328. doi: 10.1172/JCI111542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaughan CK, Mollapour M, Smith JR, Truman A, Hu B, Good VM, Panaretou B, Neckers L, Clarke PA, Workman P, et al. Hsp90-dependent activation of protein kinases is regulated by chaperone-targeted dephosphorylation of Cdc37. Mol Cell. 2008;31:886–895. doi: 10.1016/j.molcel.2008.07.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wechsler T, Chen BP, Harper R, Morotomi-Yano K, Huang BC, Meek K, Cleaver JE, Chen DJ, Wabl M. DNA-PKcs function regulated specifically by protein phosphatase 5. Proc Natl Acad Sci U S A. 2004;101:1247–1252. doi: 10.1073/pnas.0307765100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Welsh N. Interleukin-1 beta-induced ceramide and diacylglycerol generation may lead to activation of the c-Jun NH2-terminal kinase and the transcription factor ATF2 in the insulin-producing cell line RINm5F. J Biol Chem. 1996;271:8307–8312. doi: 10.1074/jbc.271.14.8307. [DOI] [PubMed] [Google Scholar]
- Welters HJ, Oknianska A, Erdmann KS, Ryffel GU, Morgan NG. The protein tyrosine phosphatase-BL, modulates pancreatic beta-cell proliferation by interaction with the Wnt signalling pathway. J Endocrinol. 2008;197:543–552. doi: 10.1677/JOE-07-0262. [DOI] [PubMed] [Google Scholar]
- Welters HJ, Senkel S, Klein-Hitpass L, Erdmann S, Thomas H, Harries LW, Pearson ER, Bingham C, Hattersley AT, Ryffel GU, et al. Conditional expression of hepatocyte nuclear factor-1beta, the maturity-onset diabetes of the young-5 gene product, influences the viability and functional competence of pancreatic beta-cells. J Endocrinol. 2006;190:171–181. doi: 10.1677/joe.1.06768. [DOI] [PubMed] [Google Scholar]
- Veluthakal R, Wadzinski BE, Kowluru A. Localization of a nuclear serine/threonine protein phosphatase in insulin-secreting INS-1 cells: potential regulation by IL-1beta. Apoptosis. 2006;11:1401–1411. doi: 10.1007/s10495-006-8371-1. [DOI] [PubMed] [Google Scholar]
- Winter CL, Lange JS, Davis MG, Gerwe GS, Downs TR, Peters KG, Kasibhatla B. A nonspecific phosphotyrosine phosphatase inhibitor, bis(maltolato)oxovanadium(IV), improves glucose tolerance and prevents diabetes in Zucker diabetic fatty rats. Exp Biol Med (Maywood) 2005;230:207–216. doi: 10.1177/153537020523000307. [DOI] [PubMed] [Google Scholar]
- Virshup DM, Shenolikar S. From promiscuity to precision: protein phosphatases get a makeover. Mol Cell. 2009;33:537–545. doi: 10.1016/j.molcel.2009.02.015. [DOI] [PubMed] [Google Scholar]
- Wishart MJ, Dixon JE. PTEN and myotubularin phosphatases: from 3-phosphoinositide dephosphorylation to disease. Trends Cell Biol. 2002;12:579–585. doi: 10.1016/s0962-8924(02)02412-1. [DOI] [PubMed] [Google Scholar]
- von Kriegsheim A, Pitt A, Grindlay GJ, Kolch W, Dhillon AS. Regulation of the Raf-MEK-ERK pathway by protein phosphatase 5. Nat Cell Biol. 2006;8:1011–1016. doi: 10.1038/ncb1465. [DOI] [PubMed] [Google Scholar]
- Xu X, Lagercrantz J, Zickert P, Bajalica-Lagercrantz S, Zetterberg A. Chromosomal localization and 5′ sequence of the human protein serine/threonine phosphatase 5′ gene. Biochem Biophys Res Commun. 1996;218:514–517. doi: 10.1006/bbrc.1996.0092. [DOI] [PubMed] [Google Scholar]
- Xu Y, Chen Y, Zhang P, Jeffrey PD, Shi Y. Structure of a protein phosphatase 2A holoenzyme: insights into B55-mediated Tau dephosphorylation. Mol Cell. 2008;31:873–885. doi: 10.1016/j.molcel.2008.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamaguchi F, Yamamura S, Shimamoto S, Tokumitsu H, Tokuda M, Kobayashi R. Suramin is a Novel Activator of PP5 and Biphasically Modulates S100-Activated PP5 Activity. Appl Biochem Biotechnol. 2013 doi: 10.1007/s12010-013-0522-6. [DOI] [PubMed] [Google Scholar]
- Yamaguchi Y, Katoh H, Mori K, Negishi M. Galpha(12) and Galpha(13) interact with Ser/Thr protein phosphatase type 5 and stimulate its phosphatase activity. Curr Biol. 2002;12:1353–1358. doi: 10.1016/s0960-9822(02)01034-5. [DOI] [PubMed] [Google Scholar]
- Yan L, Guo S, Brault M, Harmon J, Robertson RP, Hamid R, Stein R, Yang E. The B55alpha-containing PP2A holoenzyme dephosphorylates FOXO1 in islet beta-cells under oxidative stress. Biochem J. 2012;444:239–247. doi: 10.1042/BJ20111606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan L, Nairn AC, Palfrey HC, Brady MJ. Glucose regulates EF-2 phosphorylation and protein translation by a protein phosphatase-2A-dependent mechanism in INS-1-derived 832/13 cells. J Biol Chem. 2003;278:18177–18183. doi: 10.1074/jbc.M301116200. [DOI] [PubMed] [Google Scholar]
- Yang J, Roe SM, Cliff MJ, Williams MA, Ladbury JE, Cohen PT, Barford D. Molecular basis for TPR domain-mediated regulation of protein phosphatase 5. Embo J. 2005;24:1–10. doi: 10.1038/sj.emboj.7600496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ylipaasto P, Kutlu B, Rasilainen S, Rasschaert J, Salmela K, Teerijoki H, Korsgren O, Lahesmaa R, Hovi T, Eizirik DL, et al. Global profiling of coxsackievirus- and cytokine-induced gene expression in human pancreatic islets. Diabetologia. 2005;48:1510–1522. doi: 10.1007/s00125-005-1839-7. [DOI] [PubMed] [Google Scholar]
- Yong W, Bao S, Chen H, Li D, Sanchez ER, Shou W. Mice Lacking Protein Phosphatase 5 Are Defective in Ataxia Telangiectasia Mutated (ATM)-mediated Cell Cycle Arrest. J Biol Chem. 2007;282:14690–14694. doi: 10.1074/jbc.C700019200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang AQ, Gao ZY, Gilon P, Nenquin M, Drews G, Henquin JC. Vanadate stimulation of insulin release in normal mouse islets. J Biol Chem. 1991;266:21649–21656. [PubMed] [Google Scholar]
- Zhang J, Bao S, Furumai R, Kucera KS, Ali A, Dean NM, Wang XF. Protein phosphatase 5 is required for ATR-mediated checkpoint activation. Mol Cell Biol. 2005;25:9910–9919. doi: 10.1128/MCB.25.22.9910-9919.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao S, Sancar A. Human blue-light photoreceptor hCRY2 specifically interacts with protein serine/threonine phosphatase 5 and modulates its activity. Photochem Photobiol. 1997;66:727–731. doi: 10.1111/j.1751-1097.1997.tb03214.x. [DOI] [PubMed] [Google Scholar]
- Zhou G, Golden T, Aragon IV, Honkanen RE. Ser/Thr protein phosphatase 5 inactivates hypoxia-induced activation of an apoptosis signal-regulating kinase 1/MKK-4/JNK signaling cascade. J Biol Chem. 2004;279:46595–46605. doi: 10.1074/jbc.M408320200. [DOI] [PubMed] [Google Scholar]
- Ämmälä C, Eliasson L, Bokvist K, Berggren PO, Honkanen RE, Sjöholm A, Rorsman P. Activation of protein kinases and inhibition of protein phosphatases play a central role in the regulation of exocytosis in mouse pancreatic beta cells. Proc Natl Acad Sci U S A. 1994;91:4343–4347. doi: 10.1073/pnas.91.10.4343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Östenson CG, Sandberg-Nordqvist AC, Chen J, Hallbrink M, Rotin D, Langel U, Efendic S. Overexpression of protein-tyrosine phosphatase PTP sigma is linked to impaired glucose-induced insulin secretion in hereditary diabetic Goto-Kakizaki rats. Biochem Biophys Res Commun. 2002;291:945–950. doi: 10.1006/bbrc.2002.6536. [DOI] [PubMed] [Google Scholar]