

Abstract
Vibrio cholerae has been shown to produce a cyclic dipeptide, cyclo(phenylalanine–proline) (cFP), that functions to repress virulence factor production. The objective of this study was to determine if heterologous cyclic dipeptides could repress V. cholerae virulence factor production. To that end, three synthetic cyclic dipeptides that differed in their side chains from cFP were assayed for virulence inhibitory activity in V. cholerae. The results revealed that cyclo(valine–valine) (cVV) inhibited virulence factor production by a ToxR-dependent process that resulted in the repression of the virulence regulator aphA. cVV-dependent repression of aphA was found to be independent of known aphA regulatory genes. The results demonstrated that V. cholerae was able to respond to exogenous cyclic dipeptides and implicated the hydrophobic amino acid side chains on both arms of the cyclo dipeptide scaffold as structural requirements for inhibitory activity. The results further suggest that cyclic dipeptides have potential as therapeutics for cholera treatment.
Introduction
Vibrio cholerae is a significant human health threat, particularly in the developing world, where it is estimated to cause 3–5 million cases of the severe acute diarrhoeal disease cholera each year. V. cholerae is a Gram-negative bacterium that inhabits aquatic ecosystems in many regions of the world. Humans get cholera from these aquatic reservoirs through the consumption of water and food contaminated with V. cholerae (Bennish, 1994; Nelson et al., 2009). Following ingestion, V. cholerae colonizes the small intestines where unknown environmental signals induce the expression of virulence genes that are essential for intestinal colonization and disease development (Bennish, 1994). The two most important virulence factors produced by V. cholerae are cholera toxin (CT) and the toxin coregulated pilus (TCP) (Kaper et al., 1995). CT is an A1B5 enterotoxin that is responsible for the voluminous secretory diarrhoea associated with cholera (Holmgren, 1981). The TCP is a type IV pilus that is essential for intestinal colonization (Herrington et al., 1988; Taylor et al., 1987).
The production of CT and TCP is tightly controlled by a hierarchical regulatory system called the ToxR regulon (Childers & Klose, 2007). The ToxR regulon coordinately activates the expression of virulence genes following entry into the host in response to largely unknown in vivo signals. Induction of the ToxR regulon begins with AphA and AphB (Kovacikova & Skorupski, 1999; Skorupski & Taylor, 1999), two cytoplasmic DNA-binding proteins that function together to activate tcpP expression. TcpP is a membrane-localized DNA-binding protein that is structurally similar to the virulence regulator ToxR (Häse & Mekalanos, 1998). TcpP and ToxR are thought to regulate the expression of their respective target genes in response to environmental cues (reviewed by Childers & Klose, 2007). When appropriately stimulated, TcpP and ToxR bind together at the toxT promoter and activate ToxT production. ToxT then directly activates the expression of the genes that encode CT and TCP production along with other virulence factors (Higgins & DiRita, 1994). Loss of function of any of the genes that encode the primary ToxR regulon regulatory proteins renders V. cholerae avirulent. This latter fact provides the rationale for the development of antivirulence therapeutics that target the ToxR regulon.
Cyclic dipeptides (CDPs) are bioactive molecules that are abundant in nature. They belong to the family of diketopiperazine secondary metabolites and are produced by both prokaryotes and eukaryotes (Borthwick, 2012). Several CDPs have been shown to exhibit biological activity, but their native biological functions in most micro-organisms remain unknown (Borthwick, 2012; Prasad, 1995). Previous studies have shown that the endogenously produced cyclic dipeptide cyclo(phenylalanine–proline) (cFP) accumulated in V. cholerae culture supernatant in a growth-dependent manner (Park et al., 2006). Subsequent studies in our laboratory showed that cFP activated a novel ToxR-dependent signal-transduction cascade that resulted in the downregulation of virulence factor production (Bina & Bina, 2010; Bina et al., 2013). Exposure of V. cholerae to cFP resulted in the ToxR-dependent activation of leuO expression. LeuO production then led to aphA repression, downregulation of the ToxR regulon, and the resultant attenuation of CT and TCP production. These results, combined with published data, suggested that cFP may function as a concentration-dependent negative effector of CT and TCP production in V. cholerae (Bina & Bina, 2010; Park et al., 2006).
The unravelling of the V. cholerae cFP signalling pathway illuminated a potential therapeutic approach for cholera in which cFP, or other CDPs, could be introduced into the gastrointestinal tract of cholera patients, or people at risk for cholera, to attenuate virulence factor production in the gut. This would have the effect of either blocking infection in at-risk populations or mitigating disease in cholera patients. As exemplified by the ongoing cholera outbreak in Haiti, alternative therapeutic interventions for cholera are needed to combat the rapid evolution of antibiotic resistance (Kitaoka et al., 2011) and to supplement current cholera control strategies. In the present study we tested the hypothesis that CDP antivirulence activity was not limited to cFP by assaying three CDPs that were not native to V. cholerae. Among the CDPs we tested, we identified cyclo(valine–valine) (cVV), which exhibited more potent antivirulence activity in V. cholerae than cFP. Characterization of cVV’s mechanism of action revealed that cVV inhibited virulence factor production by a ToxR-dependent process that resulted in repression of aphA transcription. However, the signal transduction pathway that led to aphA repression was independent of known aphA regulators, indicating that cVV functioned by a novel mechanism.
Methods
Bacterial strains, culture conditions and chemicals.
Bacterial strains and plasmids used in this study are listed in Table 1. Escherichia coli strain EC100Dpir+ was used for all cloning experiments. E. coli strain SM10λpir (Klose & Mekalanos, 1998) was used for conjugation of plasmids into V. cholerae. All V. cholerae strains used in this study were derivatives of O1 El Tor strain N16961 (Heidelberg et al., 2000). V. cholerae strain JB58 (N16961 ΔlacZ SmR) was used as the wild-type (WT) control strain in all experiments. All bacterial strains were grown in Luria–Bertani (LB) broth or on LB agar at 37 °C. V. cholerae strains were grown under the growth conditions for AKI medium to induce the ToxR regulon (Iwanaga et al., 1986). Bacterial stocks were maintained at −80 °C in LB broth containing 25 % glycerol. Carbenicillin (Cb), and streptomycin (Sm) were added to the growth medium at 100 µg ml−1 when required. Chloramphenicol was used at 2 µg ml−1 for selection in V. cholerae and 25 µg ml−1 in E. coli. Chemicals were purchased from ThermoFisher unless otherwise indicated. CDPs were purchased from Bachem and stock solutions were prepared in DMSO.
Table 1. Bacterial strains, plasmids and oligonucleotides used in this study.
| Strain/plasmid | Genotype/sequence | Source |
| Strain | ||
| Vibrio cholerae | ||
| JB58 | N16961 SmR ΔlacZ | Bina et al. (2006) |
| XBV222 | JB58 ΔleuO | Bina et al. (2013) |
| DT733 | N16961 SmR ΔlacZ ΔtoxRS | Bina et al. (2013) |
| VA127 | JB58 Δlrp : : Cm | This study |
| VA101 | JB58 ΔvpsR | This study |
| Escherichia coli | ||
| SM10λpir | thi-1 thr leu tonA lacY supE recA : : RP4-2-Tc : : Mu Kmr (λ pirR6K) | Miller & Mekalanos (1988) |
| EC100Dpir+ | F- mcrA D (mrr-hsdRMS-mcrBC) W80dlacZDM15 DlacX74 recA1 endA1 araD139 D (ara, leu)7697 galU galK l- rpsL (StrR) nupG pir+ | Epicentre |
| Plasmid | ||
| pWM91 | Suicide plasmid vector used for allelic exchange | Metcalf et al. (1996) |
| pVA42 | pWM91-ΔlrpR; lrp deletion construct | This study |
| pVA43 | pWM91-ΔvpsR; vpsR deletion construct | This study |
| pVA60 | pWM91-Δlrp : : Cm; lrp : : Cm marker exchange vector | This study |
| pXB208 | pBAD18 km : : aphA | This study |
| pBAD18 km | Arabinose inducible expression vector | Guzman et al. (1995) |
| Oligonucleotide primers (5′→3′) | ||
| qRT-PCR | ||
| leuO-F | GACCACTTCGCCACAAATCACCA | |
| leuO-R | CGTTGGATGGCGGAAAATGCG | |
| hfq-F | CGGCATTAAACTGCAAGGTCA | |
| hfq-R | CTGTGGTGGCTAACTGGACG | |
| qrr2-F | GGTGACCCTTGTTAAGCCGA | |
| qrr2-R | CTATTCACTAACAACGTCAGTTGGC | |
| qrr-4-F | TGACCCTTCTAAGCCGAGGG | |
| qrr-4-R | GAACAATGGTGTTCACTAACAACG | |
| aphA-F | GCAGAACCTTACCGTCTGCAA | |
| aphA-R | GCGTAATAAGCGGCTTCGATT | |
| aphB-F | ATCGGTGAAGTGAAAGACATTTTG | |
| aphB-R | GATGTTGATGCAACTCTTCAGCAT | |
| toxR-F | GTCAAAACGGTTCCGAAACG | |
| toxR-R | TGTCATGAGCAGCTTCGCTTT | |
| tcpP-F | GGTGGAGTTATGGCCAATGG | |
| tcpP-R | GTTATCCCCGGTAACCTTGCT | |
| toxT-F | CTGATGATCTTGATGCTATGGAGAAA | |
| toxT-R | TCATCCGATTCGTTCTTAATTCAC | |
| 16s RNA-F | CTTTTTGAATCGCAGCAGGT | |
| 16s RNA-R | GGTGGTCGAATCATGAGCTT | |
| gyrA-F | CAATGCCGGTACACTGGTACG | |
| gyrA-R | AAGTACGGATCAGGGTCACG | |
| vpsR-F | CCCTGGCTGGCTGTGTTGGA | |
| vpsR-R | TGAACGCCAGCCAACGGACT | |
| lrp-F | TCAACAAACACCAACAGTGAGGCG | |
| lrp-r | ACCCTGTCTTGAGCGTGTTCGTC | |
| leuO-F | GACCACTTCGCCACAAATCACCA | |
| leuO-R | CGTTGGATGGCGGAAAATGCG | |
| Cloning | ||
| aphA-F-EcoRI | GCGAATTCACCATGTCATTACCACACG | |
| aphA-R-XbaI | ACTCTAGAGGCTCTCTCTATCTCTGCTC | |
| vpsR-F1 | ACGAGCTCCACGTCACAACCCAGAATTCACC | |
| vpsR-R1 | GCCCCGGGTTAATCAGCGCCCATTTGTGGCCTCG | |
| vpsR-F2 | TCAGTACCTGGCCACAACTTGATCACCGATGAAAACTTC | |
| vpsR-R2 | GATCAAGTTGTGGCCAGGTACTGAATCCATACGGAATTGAGTGC | |
| lrp-F1 | GGGAGCTCTGCGTGAGTGGTAGGGTG | |
| lrp-R1 | AACCCGGGAGCAGGTTCGCGCTGCCTTGG | |
| lrp-F2 | GCCGTCTAAGGACCAAACCAACCAATTAGTGATTAAAACTCGC | |
| lrp-R2 | ATTGGTTGGTTTGGTCCTTAGACGGCTTCTTATAACTATCCAC | |
Construction of mutants.
Oligonucleotide PCR primers used for cloning are listed in Table 1. Deletion of vpsR (VC0665) in strain JB58 was constructed by homologous recombination using the allelic exchange plasmid pWM91 as previously described (Bina et al., 2006, 2008). Briefly, 1 kb regions flanking vpsR were amplified by PCR using vpsR-F1/R2 and vpsR-F2/R1 primer pairs. The resulting PCR amplicons were mixed together and used as a template for PCR using the vpsR-F1/R1 primer pair to generate the deletion construct. The resulting fragment was then restricted with SacI and XmaI restriction enzymes before being ligated into similarly digested pWM91 to generate pVA43. The integrity of the deletion construct was verified by DNA sequencing. pVA43 was then transformed into E. coli Sm10λpir before being mobilized into JB58. V. cholerae cointegrants were then selected on Cb and Sm. Several Cb/Sm-resistant colonies were then plated onto LB-no NaCl-5 % sucrose agar plates. Finally, sucrose-resistant colonies were screened replica plated onto LB/Cb and LB/sucrose. Cb-sensitive and sucrose-resistant colonies were then screened by PCR using flanking PCR primers to confirm the deletion. The lrp deletion was constructed by marker exchange mutagenesis in a similar fashion. Briefly, lrp-F1/R2 and lrp-F2/R1 primer pairs were used to amplify 1 kb regions that flanked lrp. The resulting PCR amplicons were then used as a template for PCR using the lrp-F1/R1 primer pair, and the resulting 2 kb fragment, containing the lrp deletion, was cloned into pWM91 to generate pVA42. A chloramphenicol resistance gene was then extracted from plasmid pTOPO-Cm by SacI/EcoRV digestion and blunt end cloned into the SpeI site of pVA42. The results of this cloning reaction replaced lrp with the chloramphenicol resistance gene to generate pVA60. The lrp mutant was then constructed by mobilizing the pVA60 into strain JB58 and selecting for chloramphenicol resistance followed by sucrose counter selection as described above. pXB208 was constructed by amplifying the aphA gene from N16961 using primers aphA-F-EcoRI and aphA-R-XbaI. The resulting PCR amplicon was digested with XbaI and EcoRI restriction enzymes before being ligated to similarly digested pBAD18km to generate pXB208.
Quantification of CT production.
CT production was quantified by GM1 ELISA in strains cultured under virulence gene-inducing conditions (i.e. AKI growth conditions) as described by Taylor et al. (2012) with slight modification. Briefly, fresh overnight LB broth cultures of the respective strains were diluted 10 000-fold in AKI broth. The inoculated broth was then dispensed in 2.5 ml aliquots into 13×100 mm test tubes. The tested cyclic dipeptides or an equivalent amount of DMSO carrier were then added to the cultures to a final concentration of 0.5 mM. The test tubes were then statically incubated for 4 h at 37 °C. One millilitre from each culture was then aseptically transferred to a 15×150 mm test tube and incubated with shaking at 37 °C for 18 h before CT production was determined by the GM1 ELISA as described previously (Bina et al., 2008). Purified CT (Sigma-Aldrich) was used as a standard to quantify CT production.
TcpA Western immunoblotting.
Western blots for TcpA production were performed on V. cholerae strains following growth under AKI conditions in the presence or absence of 0.5 mM cVV. Aliquots of the AKI-grown cultures were adjusted to an OD600 of 1.0 and the cells collected by centrifugation. The cell pellets were resuspended in 3× solubilization buffer and heated at 100 °C for 10 min. Equal volumes of each resulting cell lysate was then resolved by SDS 10 %-PAGE (SDS-PAGE) before being transferred to a PVDF membrane (GE Healthcare Life Sciences). The membrane was incubated with rabbit polyclonal antisera (10−4 dilution) against TcpA (kind gift of Dr Jun Zhu, University of Pennsylvania) followed by incubation with horseradish peroxidase-conjugated goat anti-rabbit IgG (Biomeda). Immunoreactive proteins were visualized using the SuperSignal West Pico Chemiluminescent detection kit (Pierce Biotechnology).
Quantitative real-time PCR (qRT-PCR).
V. cholerae cultures were grown under AKI conditions and culture aliquots were collected at the indicated time points. The cells were pelleted from the culture aliquots by centrifugation at 5000 g for 10 min at 4 °C. RNA was then extracted from the cell pellets using TRIZOL reagent according to the manufacturer’s instruction (Invitrogen). First-strand cDNA synthesis was carried out using SuperScript III reverse transcriptase (Life Technologies) and random primers at 50 °C for 1 h in the presence of 5 mM DTT. The reaction products were then purified using a Wizard SV gel and PCR clean-up system (Promega). The expression levels of specific genes were quantified by amplifying 25 ng cDNA with 10 pmol of gene-specific primers (Table 1) using the SYBR Green PCR mix (Life Technologies) on a StepOnePlus Real-time PCR System (Life Technologies). 16S RNA was used as the internal control for all experiments. Relative gene expression levels were calculated by the 2−ΔΔCt method (Livak & Schmittgen, 2001) and presented as mean±sd of a minimum of three biological replicates.
Results
cVV is a concentration-dependent inhibitor of CT production
Recent work in our laboratory showed that cFP, a CDP produced by V. cholerae, functioned as a negative effector of CT and TCP production (Bina & Bina, 2010). This suggested the possibility that cFP, or other CDPs, could be used as potential therapeutics for cholera treatment. We therefore sought to test the hypothesis that heterologous CDPs could inhibit virulence factor production in V. cholerae.
To test this hypothesis and gain insights into the structural feature(s) of CDPs that modulate V. cholerae virulence factor production we tested three CDPs, cyclo(glycine–tryptophan) (cGW), cyclo(leucine–tryptophan) (cLW) and cVV, for inhibitory activity against CT production in El Tor V. cholerae strain N16961 virulence (Fig. 1). These three CDPs were selected because they were not naturally produced by V. cholerae but shared partial structural features with cFP, namely a cyclic dipeptide scaffold with an aromatic and a hydrophobic side chain. In addition, these three CDPs were commercially available at a reasonable cost.
Fig. 1.
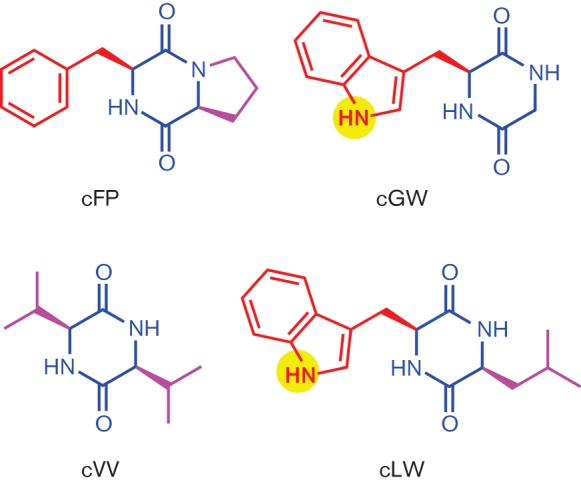
Structure of CDPs used in this study. The core CDP is shown in blue, the aromatic side chain is shown in red, hydrophobic non-aromatic side chains are shown in magenta and side chain polar residues are highlighted in yellow.
The CDPs were added to AKI broth at a final concentration of 0.5 mM with control cultures receiving an equal volume of DMSO. The AKI broth was then inoculated at a 1 : 10 000 ratio with an overnight LB broth culture of V. cholerae strain JB58. The culture was then incubated overnight under AKI conditions and CT production was quantified. The results of the analysis showed that 0.5 mM cFP inhibited CT production by approximately 50 % (Fig. 2a), consistent with previously published results (Bina & Bina, 2010). By contrast, 0.5 mM cVV exhibited greater inhibitory activity than cFP and resulted in an ~85 % reduction in CT production (Fig. 2a). cGW and cLW did not have a significant effect on CT production. The effect of cVV on V. cholerae growth was examined to rule out the possibility that the cVV antivirulence activity resulted from non-specific growth inhibition. In these experiments we grew V. cholerae under AKI conditions in the presence of 0.5 mM cVV or an equivalent amount of DMSO and monitored OD600 over time as an indicator of growth. The results showed that 0.5 mM cVV did not impact V. cholerae growth (data not shown). This indicated that the cVV inhibitory activity was specific for CT production.
Fig. 2.
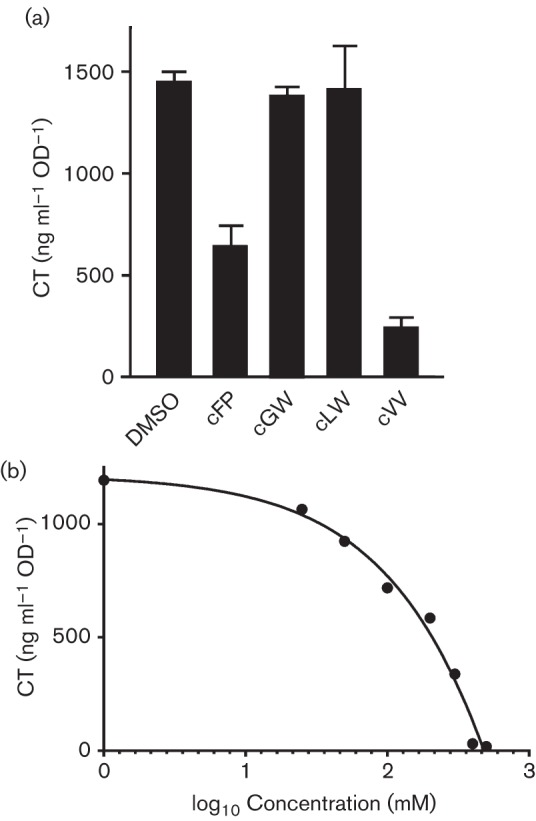
Effect of CDPs on CT production. (a) V. cholerae was cultured under virulence gene-inducing conditions in the presence of 0.5 mM of the indicated CDP or an equivalent amount of DMSO for 20 h before CT production was quantified by GM1 ELISA. The results represent the mean of three biological replicates±sd. (b) The effect of varying concentrations of cVV on CT production. V. cholerae was cultured overnight under virulence gene-inducing conditions in the presence of the indicated concentrations of cVV before quantification of CT.
We hypothesized that if cVV was specific for CT production, then cVV would exhibit concentration-dependent CT inhibition. To test this we generated a cVV dose–response curve for CT production. This was done by growing V. cholerae under AKI conditions in the absence of cVV and in the presence of 500, 400, 300, 200, 100, 50 and 25 µM cVV and then quantifying CT production by GM1 ELISA. CT production at each tested cVV concentration was then plotted against the log10 concentration of cVV (Fig. 2b). The results of this experiment confirmed that cVV was a concentration-dependent CT inhibitor. From these data we were also able to calculate the cVV IC50 for CT by fitting the line to a four-parameter sigmoid equation using SigmaPlot (Version 12.3; Systat Software). The resulting analysis showed that cVV exhibited an IC50 of ~166 µM, which confirmed that cVV exhibited more potent antivirulence activity relative to cFP, which exhibited an IC50 of ~300 µM (Bina & Bina, 2010; Bina et al., 2013).
cVV signals through the ToxR virulence regulon
CT production in V. cholerae is controlled by a hierarchical regulatory cascade called the ToxR regulon. We therefore designed experiments to determine if cVV affected expression of the ToxR regulon. We cultured V. cholerae under AKI growth conditions in the presence of 0.5 mM cVV with control cultures receiving an equivalent volume of the carrier DMSO. RNA was then collected at 4 h post-inoculation and used for qRT-PCR to quantify the effect of cVV on expression of the primary regulators in the ToxR regulon (i.e. aphA, aphB, tcpP, toxR and toxT).
The qRT-PCR results showed that cVV inhibited aphA expression by nearly fourfold, but did not significantly affect the expression of toxR or aphB (Fig. 3). As induction of the ToxR regulon is initiated by AphA- and AphB-dependent activation of tcpP transcription, reduced aphA expression suggested a potential mechanism by which cVV could inhibit virulence factor production. cVV also inhibited the expression of genes that were downstream of aphA in the ToxR regulon. For example, there was a 2.4-fold decrease in tcpP expression and a 3.9-fold decrease in toxT expression. As ToxT is the direct activator of the genes encoding CT production, this result was consistent with the decrease in CT production observed in Fig. 2. Taken together, these results suggested that cVV inhibited CT production by repression of aphA transcription.
Fig. 3.
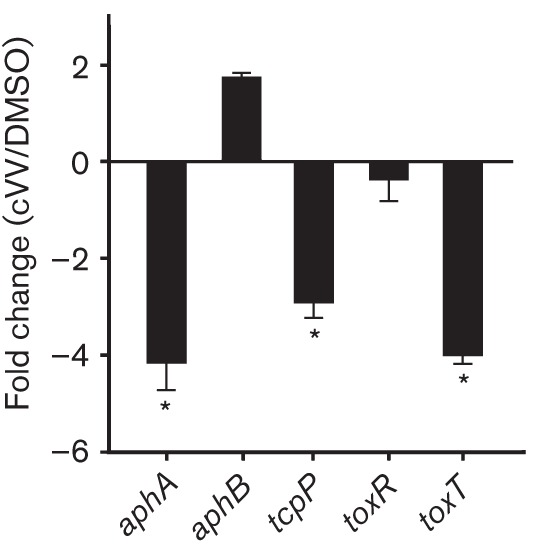
Effect of cVV on expression of the ToxR regulon. V. cholerae was grown under virulence gene-inducing conditions in the presence of 0.5 mM cVV or an equivalent volume of DMSO. Total RNA was isolated at 4 h and used for qRT-PCR. The expression ratios of the indicated genes in the cVV-treated versus DMSO control cultures were normalized to 16S RNA and are presented as the mean and sem from three biological replicates. *P<0.01 from a hypothetical value of 1.0.
Overexpression of aphA complements for CT production during growth in the presence of cVV
If cVV inhibited CT production by repressing aphA, then ectopic expression of aphA should restore virulence factor production in the presence of cVV. To test this, V. cholerae containing pXB208 (pBAD18km : : aphA) or the empty vector (pBAD18km) were cultured under AKI growth conditions in the presence or absence of 0.5 mM cVV plus 0.2 % arabinose (to induce aphA expression from the arabinose-inducible promoter in pBAD18km). Following overnight growth the cultures were analysed for CT production by GM1 ELISA and TCP production by a TcpA Western blot.
The results showed a significant reduction in CT production in the pBAD18km control strain when grown in the presence of cVV (Fig. 4a). By contrast, expression of aphA from the arabinose-inducible promoter in pXB208 alleviated the inhibitory effect of cVV on CT production. Ectopic expression of aphA also abolished the inhibitory effects of cVV on TcpA production (Fig. 4b). As the genes that encode TcpA and CT are coordinately regulated, these results supported the hypothesis that cVV inhibits virulence factor production by repression of aphA.
Fig. 4.
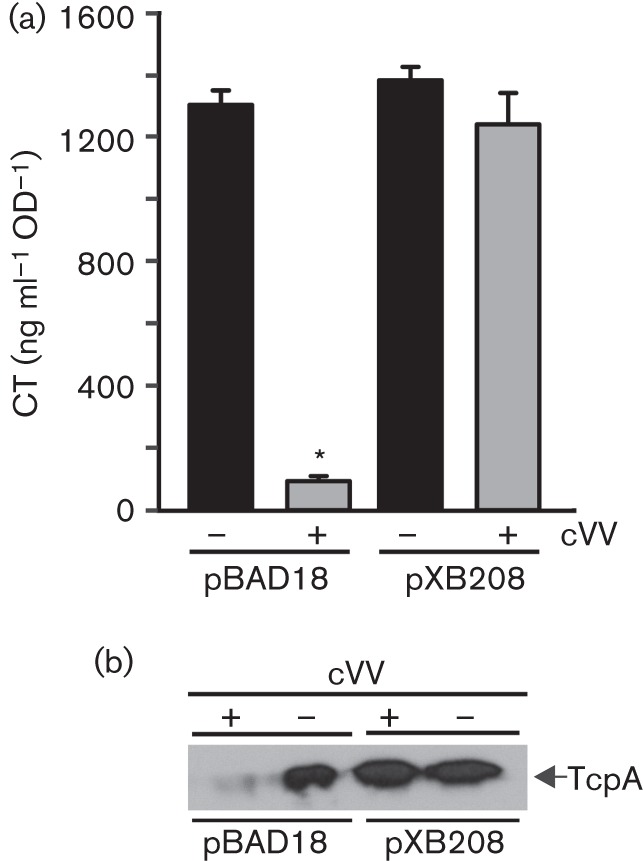
Overexpression of aphA attenuates cVV inhibitory activity. V. cholerae containing pBAD18km or pBAD18km–aphA (pXB208) was grown overnight under virulence gene-inducing conditions in the presence or absence of 0.5 mM cVV plus 0.2 % arabinose. (a) CT production. (b) TCP production. Analyses of CT and TcpA production were performed as described in Methods. The CT results represent the mean of three biological replicates±sd. The TcpA Western blot is representative of three independent experiments.
Effect of cVV on genes that are upstream of aphA in the ToxR regulon
Lrp, VpsR and LeuO have been shown to regulate aphA expression (Bina et al., 2013; Lin et al., 2007). We therefore examined whether cVV activity was dependent on any of these genes. To test this, V. cholerae was grown under AKI conditions in the presence or absence of cVV for 4 h, after which RNA was extracted and used for qRT-PCR using leuO-, lrp-, vpsR- and gyrA-specific primers (Table 1); gyrA encodes the A subunit of DNA gyrase and was included as a non-specific internal control. The results showed that cVV did not affect the expression of leuO, lrp or vpsR (Fig. 5a), suggesting that cVV activity was independent of these regulatory genes. cVV also did not affect gyrA expression, which suggested that it did not cause non-specific effects on the cell. The observation that cVV did not affect leuO expression was in contrast to the reported results for cFP, which was shown to activate leuO expression (Bina et al., 2013).
Fig. 5.

Effect of cVV on the expression of genes that regulate aphA expression. The indicated strains were grown under AKI conditions in the presence or absence of 0.5 mM cVV for 4 h before the relative expression of the indicated gene targets was determined by qRT-PCR. The fold change of the indicated genes was then calculated as the ratio of the indicated gene in the cVV-treated culture versus the DMSO control culture. The gyrA gene was used as a non-specific internal control. (a) Effect of cVV on expression of the indicated regulatory genes in wild-type strain JB58. (b) Effect of cVV on aphA expression in ΔleuO, ΔvpsR, Δlrp and ΔtoxRS mutant strains. (c) Effect of cVV on expression of hfq, luxO, qrr2, qrr4 and aphA in wild-type strain JB58. The data presented represent the sample mean±sem of three independent experiments that were performed in triplicate. *P<0.05.
To further confirm that cVV was not signalling though leuO, lrp or vpsR, we tested if cVV affected aphA expression in leuO, lrp and vpsR mutant backgrounds. We postulated that if cVV activity was dependent on any of these three genes, their deletion should abrogate cVV-dependent inhibition of aphA expression. We therefore cultured the respective mutants (Δlrp : : Cm, ΔvpsR, ΔtoxRS and ΔleuO) and JB58 under AKI conditions in the presence and absence of 0.5 mM cVV and quantified aphA expression by qRT-PCR. The toxRS mutant was included to test whether cVV activity was dependent on ToxR as was observed with cFP (Bina et al., 2013). The results showed that cVV exhibited inhibitory activity in the lrp, vpsR and leuO deletion backgrounds, as indicated by the cVV-dependent inhibition of aphA expression in each strain (Fig. 5b). This result provided further evidence to suggest that none of these three genes functioned in the cVV-dependent repression of aphA. By contrast, deletion of toxRS abolished cVV inhibitory activity, indicating that cVV activity was dependent upon ToxR. As the expression level of toxRS was unaffected by cVV (Fig. 2), we speculated that cVV was functioning to stimulate ToxR activity.
The expression of aphA is also under the influence of the V. cholerae quorum sensing (QS) systems. QS-dependent regulation of aphA is mediated through the master QS regulator HapR, which directly represses aphA expression (Kovacikova & Skorupski, 2002). We deemed it unlikely that HapR contributed to the cVV-dependent repression of aphA as N16961 is QS-insensitive due to a frame-shift mutation in hapR (Heidelberg et al., 2000). Although N16961 was hapR-negative, it was possible that genes upstream of hapR in the QS system could affect aphA expression in response to cVV (reviewed by Ng & Bassler, 2009). LuxO has been shown to activate expression of the QS regulated small (s)RNAs (Qrrs), which function with the Hfq chaperone to positively regulate AphA production at low cell density (Rutherford et al., 2011; Shao & Bassler, 2012). Therefore, one possible mechanism by which cVV could affect aphA expression is downregulation of luxO, Qrr’s or hfq. We therefore examined the effect of cVV on expression of luxO, qrr2, qrr4 and hfq. We did not examine qrr1 or qrr3 as we could not design unique primers for these sRNAs. The results showed that cVV did not have a significant effect on expression of luxO or hfq (Fig. 5c). By contrast, cVV appeared to activate the expression of qrr2 and qrr4 sRNAs while repressing aphA expression. It is likely that the upregulation of the qrr sRNAs resulted from reduced aphA expression as recent studies have shown that AphA functions to repress the expression of the sRNAs (Rutherford et al., 2011). Based on these results we concluded that cVV activity was independent of the QS systems.
Discussion
The present work was carried out to determine whether non-endogenous CDPs could be used to inhibit virulence factor production in V. cholerae. The results provided conclusive evidence that V. cholerae can respond to CDPs other than cFP. cVV exhibited more potent antivirulence activity than cFP (Fig. 2a). The finding that cVV, but not cGW or cLW, inhibited virulence factor production suggests that V. cholerae exhibits selectivity in CDP sensing and implicates hydrophobic side chains on both arms of the CDPs as structural requirements for the observed antivirulence activity. In light of the current studies, our results suggest that V. cholerae is able to respond to a broader range of diketopiperazines than just cFP. It remains to be determined if V. cholerae can also respond to diketopiperazines produced by resident microbial communities and enterocytes (Prasad, 1995). Environmental cues such as these could play a role in the spatial and temporal expression of the ToxR regulon that occurs during intestinal colonization (Lee et al., 1999). We are currently investigating whether such molecules affect V. cholerae virulence factor production.
cVV inhibitory activity was found to be dependent upon ToxR (Fig. 5b). This finding is similar to what was observed for cFP-dependent inhibition of virulence factor production. The observation that cVV activity was dependent upon ToxR is consistent with the presumed role of ToxR in environmental sensing. This finding also suggests that ToxR plays a critical role in sensing and responding to environmental CDPs. How this occurs in not clear. ToxR is a membrane-bound protein that contains a periplasmic sensing domain that is linked to a cytoplasmic DNA-binding domain by a transmembrane spanning domain. The topology of ToxR in the cytoplasmic membrane is thought to facilitate its role in sensing and transducing extracellular stimuli across the cytoplasmic membrane to modulate the expression of ToxR-dependent genes. At present it is unclear how ToxR senses cVV. It is tempting to speculate that cFP and cVV interact with ToxR by a similar mechanism. However, the fact that cVV and cFP appeared to activate different signalling cascades to repress aphA transcription (see below) suggests mechanistic differences in their respective effects on ToxR activity. At present we are unable to discriminate whether cVV interacts directly with ToxR or if cVV functions indirectly through another protein or via its interaction with the cytoplasmic membrane.
The mechanism by which cVV inhibited virulence factor production was linked to aphA repression. AphA is a virulence regulator that is essential for V. cholerae pathogenesis due to its requirement for CT and TCP production (Kovacikova & Skorupski, 1999). However, the mechanism by which cVV inhibited aphA expression remains unknown. Mutation of the known aphA regulators had no effect on cVV activity (Fig. 5). This suggests that cVV functioned by a novel mechanism and has implications for V. cholerae pathogenesis as it suggests that there is at least one additional unknown ToxR-dependent regulatory circuit that contributes to virulence gene expression.
In conclusion, we have shown that V. cholerae can sense and respond to the heterologous CDP cVV. This may have implications for V. cholerae pathogenesis in the gut where endogenously produced CDPs, from the host or resident microbial community, could impact virulence factor production. The collective results of this study also support the conclusion that cVV, and probably other CDPs, represent potential antivirulence therapeutics for cholera treatment. The antivirulence activity of cVV and cFP, in contrast to the ineffective nature of cLW and cGW, implicates the importance of two hydrophobic amino acid side chains on the CDP scaffold. This structure–activity relationship insight can be used to facilitate further efforts to design new molecules that could be used as antivirulence inhibitors for V. cholerae.
Acknowledgements
The research reported in this publication was supported by the National Institute of Allergy and Infectious Diseases of the National Institutes of Health under award numbers R21AI092007 and R01AI091845 and startup funds from the University of Pittsburgh, Department of Chemistry (X. L.). The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Abbreviations:
- Cb
carbenicillin
- CDP
cyclic dipeptide
- cFP
cyclo(phenylalanine–proline)
- cGW
cyclo(glycine–tryptophan)
- cLW
cyclo(leucine–tryptophan)
- CT
cholera toxin
- cVV
cyclo(valine–valine)
- Qrr
QS regulated small RNA
- qRT-PCR
quantitative real-time PCR
- QS
quorum sensing
- Sm
streptomycin
- sRNA
small RNA
- TCP
toxin coregulated pilus
References
- Bennish M. L. (1994). Cholera: pathophysiology, clinical features, and treatment. In Vibrio cholerae and Cholera: Molecular to Global Perspectives, pp. 229–255. Edited by Wachsmuth P. B. I. K., Olsvik O. Washington, DC: American Society for Microbiology. [Google Scholar]
- Bina X. R., Bina J. E. (2010). The cyclic dipeptide cyclo(Phe-Pro) inhibits cholera toxin and toxin-coregulated pilus production in O1 El Tor Vibrio cholerae. J Bacteriol 192, 3829–3832. 10.1128/JB.00191-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bina J. E., Provenzano D., Wang C., Bina X. R., Mekalanos J. J. (2006). Characterization of the Vibrio cholerae vexAB and vexCD efflux systems. Arch Microbiol 186, 171–181. 10.1007/s00203-006-0133-5 [DOI] [PubMed] [Google Scholar]
- Bina X. R., Provenzano D., Nguyen N., Bina J. E. (2008). Vibrio cholerae RND family efflux systems are required for antimicrobial resistance, optimal virulence factor production, and colonization of the infant mouse small intestine. Infect Immun 76, 3595–3605. 10.1128/IAI.01620-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bina X. R., Taylor D. L., Vikram A., Ante V. M., Bina J. E. (2013). Vibrio cholerae ToxR downregulates virulence factor production in response to cyclo(Phe-Pro). MBio 4, e00366-13. 10.1128/mBio.00366-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borthwick A. D. (2012). 2,5-Diketopiperazines: synthesis, reactions, medicinal chemistry, and bioactive natural products. Chem Rev 112, 3641–3716. 10.1021/cr200398y [DOI] [PubMed] [Google Scholar]
- Childers B. M., Klose K. E. (2007). Regulation of virulence in Vibrio cholerae: the ToxR regulon. Future Microbiol 2, 335–344. 10.2217/17460913.2.3.335 [DOI] [PubMed] [Google Scholar]
- Guzman L. M., Belin D., Carson M. J., Beckwith J. (1995). Tight regulation, modulation, and high-level expression by vectors containing the arabinose PBAD promoter. J Bacteriol 177, 4121–4130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Häse C. C., Mekalanos J. J. (1998). TcpP protein is a positive regulator of virulence gene expression in Vibrio cholerae. Proc Natl Acad Sci U S A 95, 730–734. 10.1073/pnas.95.2.730 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heidelberg J. F., Eisen J. A., Nelson W. C., Clayton R. A., Gwinn M. L., Dodson R. J., Haft D. H., Hickey E. K., Peterson J. D. & other authors (2000). DNA sequence of both chromosomes of the cholera pathogen Vibrio cholerae. Nature 406, 477–483. 10.1038/35020000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herrington D. A., Hall R. H., Losonsky G., Mekalanos J. J., Taylor R. K., Levine M. M. (1988). Toxin, toxin-coregulated pili, and the toxR regulon are essential for Vibrio cholerae pathogenesis in humans. J Exp Med 168, 1487–1492. 10.1084/jem.168.4.1487 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Higgins D. E., DiRita V. J. (1994). Transcriptional control of toxT, a regulatory gene in the ToxR regulon of Vibrio cholerae. Mol Microbiol 14, 17–29. 10.1111/j.1365-2958.1994.tb01263.x [DOI] [PubMed] [Google Scholar]
- Holmgren J. (1981). Actions of cholera toxin and the prevention and treatment of cholera. Nature 292, 413–417. 10.1038/292413a0 [DOI] [PubMed] [Google Scholar]
- Iwanaga M., Yamamoto K., Higa N., Ichinose Y., Nakasone N., Tanabe M. (1986). Culture conditions for stimulating cholera toxin production by Vibrio cholerae O1 El Tor. Microbiol Immunol 30, 1075–1083. 10.1111/j.1348-0421.1986.tb03037.x [DOI] [PubMed] [Google Scholar]
- Kaper J. B., Morris J. G., Jr, Levine M. M. (1995). Cholera. Clin Microbiol Rev 8, 48–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kitaoka M., Miyata S. T., Unterweger D., Pukatzki S. (2011). Antibiotic resistance mechanisms of Vibrio cholerae. J Med Microbiol 60, 397–407. 10.1099/jmm.0.023051-0 [DOI] [PubMed] [Google Scholar]
- Klose K. E., Mekalanos J. J. (1998). Differential regulation of multiple flagellins in Vibrio cholerae. J Bacteriol 180, 303–316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kovacikova G., Skorupski K. (1999). A Vibrio cholerae LysR homolog, AphB, cooperates with AphA at the tcpPH promoter to activate expression of the ToxR virulence cascade. J Bacteriol 181, 4250–4256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kovacikova G., Skorupski K. (2002). Regulation of virulence gene expression in Vibrio cholerae by quorum sensing: HapR functions at the aphA promoter. Mol Microbiol 46, 1135–1147. 10.1046/j.1365-2958.2002.03229.x [DOI] [PubMed] [Google Scholar]
- Lee S. H., Hava D. L., Waldor M. K., Camilli A. (1999). Regulation and temporal expression patterns of Vibrio cholerae virulence genes during infection. Cell 99, 625–634. 10.1016/S0092-8674(00)81551-2 [DOI] [PubMed] [Google Scholar]
- Lin W., Kovacikova G., Skorupski K. (2007). The quorum sensing regulator HapR downregulates the expression of the virulence gene transcription factor AphA in Vibrio cholerae by antagonizing Lrp- and VpsR-mediated activation. Mol Microbiol 64, 953–967. 10.1111/j.1365-2958.2007.05693.x [DOI] [PubMed] [Google Scholar]
- Livak K. J., Schmittgen T. D. (2001). Analysis of relative gene expression data using real-time quantitative PCR and the 2−ΔΔCtmethod. Methods 25, 402–408. 10.1006/meth.2001.1262 [DOI] [PubMed] [Google Scholar]
- Metcalf W. W., Jiang W., Daniels L. L., Kim S.-K., Haldimann A., Wanner B. L. (1996). Conditionally replicative and conjugative plasmids carrying lacZ α for cloning, mutagenesis, and allele replacement in bacteria. Plasmid 35, 1–13. 10.1006/plas.1996.0001 [DOI] [PubMed] [Google Scholar]
- Miller V. L., Mekalanos J. J. (1988). A novel suicide vector and its use in construction of insertion mutations: osmoregulation of outer membrane proteins and virulence determinants in Vibrio cholerae requires toxR. J Bacteriol 170, 2575–2583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelson E. J., Harris J. B., Morris J. G., Jr, Calderwood S. B., Camilli A. (2009). Cholera transmission: the host, pathogen and bacteriophage dynamic. Nat Rev Microbiol 7, 693–702. 10.1038/nrmicro2204 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ng W. L., Bassler B. L. (2009). Bacterial quorum-sensing network architectures. Annu Rev Genet 43, 197–222. 10.1146/annurev-genet-102108-134304 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park D.-K., Lee K.-E., Baek C.-H., Kim I. H., Kwon J. H., Lee W. K., Lee K. H., Kim B. S., Choi S. H., Kim K. S. (2006). Cyclo(Phe-Pro) modulates the expression of ompU in Vibrio spp. J Bacteriol 188, 2214–2221. 10.1128/JB.188.6.2214-2221.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prasad C. (1995). Bioactive cyclic dipeptides. Peptides 16, 151–164. 10.1016/0196-9781(94)00017-Z [DOI] [PubMed] [Google Scholar]
- Rutherford S. T., van Kessel J. C., Shao Y., Bassler B. L. (2011). AphA and LuxR/HapR reciprocally control quorum sensing in vibrios. Genes Dev 25, 397–408. 10.1101/gad.2015011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shao Y., Bassler B. L. (2012). Quorum-sensing non-coding small RNAs use unique pairing regions to differentially control mRNA targets. Mol Microbiol 83, 599–611. 10.1111/j.1365-2958.2011.07959.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skorupski K., Taylor R. K. (1999). A new level in the Vibrio cholerae ToxR virulence cascade: AphA is required for transcriptional activation of the tcpPH operon. Mol Microbiol 31, 763–771. 10.1046/j.1365-2958.1999.01215.x [DOI] [PubMed] [Google Scholar]
- Taylor R. K., Miller V. L., Furlong D. B., Mekalanos J. J. (1987). Use of phoA gene fusions to identify a pilus colonization factor coordinately regulated with cholera toxin. Proc Natl Acad Sci U S A 84, 2833–2837. 10.1073/pnas.84.9.2833 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor D. L., Bina X. R., Bina J. E. (2012). Vibrio cholerae VexH encodes a multiple drug efflux pump that contributes to the production of cholera toxin and the toxin co-regulated pilus. PLoS ONE 7, e38208. 10.1371/journal.pone.0038208 [DOI] [PMC free article] [PubMed] [Google Scholar]