

Abstract
While platelet activation is essential to maintain blood vessel patency and minimize loss of blood upon injury, untimely or excessive activity can lead to unwanted platelet activation and aggregation. Resultant thrombosis has the potential to block blood vessels, causing myocardial infarction or stroke. To tackle this major cause of mortality, clinical therapies that target platelet responsiveness (antiplatelet therapy) can successfully reduce cardiovascular events, especially in people at higher risk; however, all current antiplatelet therapies carry an increased probability of bleeding. This review will evaluate new and emerging targets for antithrombotics, focusing particularly on platelet glycoprotein VI, as blockade or depletion of this platelet-specific receptor conveys benefits in experimental models of thrombosis and thromboinflammation without causing major bleeding complications.
Keywords: platelet, bleeding, antithrombotic, hemostasis, glycoprotein, vessel, thrombosis
Introduction
The platelet response to vessel injury or infection is essential for normal hemostasis. Platelets also have multifaceted roles in inflammation and immunity.1 In response to vascular injury, circulating platelets rapidly adhere to exposed subendothelial matrix proteins, such as von Willebrand factor (VWF) and collagen.2,3 Adherent platelets become activated, spread, and release the contents of storage organelles. These are dense bodies that contain prothrombotic substances, including serotonin and adenosine diphosphate (ADP), membrane glycoproteins P-selectin and CD40 ligand, coagulation proteins, fibrinolytic proteins, growth factors, cytokines, and chemokines.4 The platelet response is enhanced by signaling pathways initiated through the thromboxane A2 receptor and P2Y1 and P2Y12 receptors for ADP, ultimately leading to the activation of the platelet-specific integrin αIIbβ3 and platelet aggregation, thereby maintaining blood vessel patency and minimizing the loss of blood upon injury.
Myocardial infarction or stroke can result from untimely or excessive platelet activity leading to unwanted platelet activation and aggregation, causing thrombosis. Antiplatelet therapies target the ADP receptor P2Y12 (thienopyridine-based inhibitors clopidogrel and prasugrel) or thromboxane generation (cyclooxygenase inhibitor, acetylsalicylic acid) and aim to modulate platelet responsiveness in people at risk of thrombosis.5 When given together (dual antiplatelet therapy), these treatments significantly reduce the risk of vascular events in high-risk patients.6,7 Critically, however, the risk of bleeding in patients also increases with the use of antiplatelet therapies.8,9
There is a clear need to develop new antiplatelet reagents ideally with high antithrombotic properties but negligible effects on normal hemostasis. Platelet glycoprotein (GP)VI represents an attractive new target as an antiplatelet reagent, because it is only expressed on platelets and platelet precursor cells in bone marrow (megakaryocytes), and GPVI blockade has demonstrated efficient antithrombotic potential in the experimental models of thrombosis without enhancing pathological bleeding. Targeting GPVI in the setting of myocardial infarction makes good sense as platelet levels of GPVI are elevated in people who have coronary syndromes.10–12 More recent data indicate that the blockade of GPVI may also be advantageous for therapies that target inflammatory processes involving platelet function. As several excellent reviews on this topic have been published elsewhere recently,13–16 we will briefly cover the background rationale for targeting GPVI before highlighting the existing and novel approaches to modulate GPVI function.
GPVI structural and functional features
GPVI is found exclusively on platelets and megakaryocytes and is the predominant platelet receptor for collagen.17 GPVI has also been shown to bind laminin, an additional extracellular matrix protein.18 Human GPVI is an approximately 64 kDa type I transmembrane glycoprotein of the immunoreceptor family, with two extracellular immunoglobulin (Ig)-like domains, an extracellular mucin-like domain, followed by a 19-amino acid transmembrane domain, and a cytoplasmic tail of approximately 50 residues that is important for the transmission of ligand-regulated signals.19 Platelets from healthy donors contain approximately 10,000 copies per platelet of GPVI.20 Critical residues within the transmembrane domain of GPVI enable this receptor to link with the fragment crystallizable (Fc) γ chain and form a cooperative signaling complex, which utilizes two immunoreceptor tyrosine-based activation motifs (ITAMs) within the cytoplasmic tail portions of dimerized Fcγ chain.21,22 The cytoplasmic tail of GPVI also contains a proline-rich region that can recruit sarcoma (Src) family kinase members23 and a calmodulin-binding sequence in the juxtamembrane region.24 The Ig domains of GPVI25 are able to bind specific glycine–proline–hydroxyproline motifs within collagen26 and, in doing so, cluster GPVI on the platelet membrane to enhance the signaling response triggered by binding collagen.17
Like many other Ig-like receptor family members,27–29 GPVI can exist as a dimer on the platelet surface.30,31 Dimerization is enhanced by ligand binding and by platelet activation.31,32 In the case of GPVI, dimers can be stabilized via the formation of a disulfide bond between cysteinyl thiol groups on the penultimate residues within the cytoplasmic tails of the adjacent GPVI molecules.27 Information gleaned from the crystal structure of the ligand-binding Ig-like domains of GPVI also hints that an interaction between adjacent GPVI ectodomains is likely.33 Such a receptor dimerization step is likely to enhance ligand-induced signaling and to strengthen collagen-binding, as the affinity of dimerized GPVI ectodomains for collagen is significantly greater than for the monomer.30,34
An additional feature of GPVI is that the receptor can be downregulated at the platelet membrane.35 This modulation of receptor levels can be achieved by the cleavage of the ectodomain upon the ligand engagement of GPVI36 or FcγRIIa37 by activation of the coagulation cascade38 or exposure to elevated levels of shear stress.39 Under certain experimental conditions, GPVI may also be internalized and degraded.40,41
Throughout biology, the shedding of the receptor ligand-binding ectodomains is a consistent recurring mechanism that is used to regulate the function of adhesion and signaling receptors.42–44 This irreversible process occurs within seconds to minutes of the exposure of the platelets to collagen or to pathophysiological levels of shear and may be a mechanism by which ligand- or shear-exposed platelets can reduce levels of functional GPVI at the membrane surface and downregulate activation signals. This process is mediated by members of the a disintegrin and metalloproteinase (ADAM) family of membrane-bound metalloproteinases, predominantly ADAM10 in the human system.45 In nucleated cells, ADAMs are synthesized and stored as proenzymes within the cytoplasmic vesicles. Upon appropriate stimulation, they are then enzymatically processed to remove the prodomain and brought to the surface of the cell as an active metalloproteinase.46 This activation process can take 4–16 hours. In platelets, the ADAM proteolytic processing of substrates can be detected within seconds to minutes of platelet activation. There is no evidence that platelets store zymogen forms of ADAMs, implying that the ADAMs proteins are present on the surface of nonactivated platelets (Qiao, Andrews, Arthur, Gardiner; unpublished data, 2012). A peptide with sequence matching residues 228–248 of the extracellular juxtamembrane sequence within GPVI could be proteolyzed by recombinant ADAM10 at position R242–Q243.45 This site is also presumably present and accessible for enzymatic cleavage on the circulating platelet surface. Cleavage within this region of GPVI results in the liberation of an approximately 55 kDa ectodomain fragment of GPVI and production of an approximately 10 kDa remnant portion that remains associated with the platelet surface. However, unlike other platelet receptors that are constitutively shed from the platelet surface, the majority of GPVI remains intact on platelets under resting conditions. The proteolytic cleavage of GPVI only occurs upon specific activation of platelets.
Platelet processes mediating thrombosis
Platelet adhesion and activation are mediated by one or more adhesion-signaling receptors on the platelet membrane (Figure 1). Principal players in these adhesive events are the receptors GPIb-IX-V that binds VWF as well as other important vascular proteins, (P-selectin, leukocyte integrin αMβ2, thrombin, high molecular weight kininogen, thrombospondin-1, factor XI, factor XII) and GPVI which binds collagen and also laminin,18 both of which are exposed within damaged vascular walls.
Figure 1.
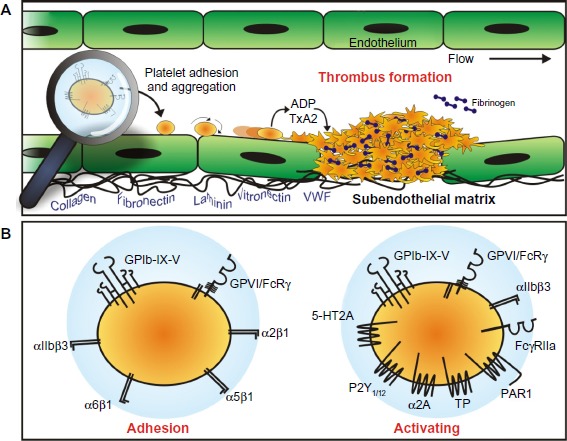
Platelet adhesion and aggregation.
Notes: (A) Platelets normally circulate through the vasculature in a nonadhesive state. Upon the detection of an exposed subendothelial matrix, platelets are induced to come into close contact with the vessel wall and roll, then arrest, at the site of vessel injury. The process of adhesion is orchestrated by the platelet adhesion receptors GPVI and GPIb-IX-V. The release of soluble agonists, such as ADP and thromboxane A2 (TxA2) amplify platelet activation. Platelet adhesion and activation, results in the formation of a platelet plug (thrombus). (B) Platelet engagement with the blood vessel wall is predominantly mediated by GPVI and GPIb-IX-V; however, the platelet surface possesses receptors that can engage matrix proteins. Additional involvement of these other adhesion proteins, including integrins α2β1, α5β1, and α6β1, which bind collagen, fibronectin, and laminin, respectively, and αIIbβ3 that binds VWF and fibrinogen, among others, help to stabilize the initial attachment and facilitate platelet recruitment and thrombus growth. Platelet activation occurs following agonist binding to GPIb-IX-V and GPVI, integrin αIIbβ3, FcγRIIa, and the G protein-coupled receptors for serotonin (5-HT2A), ADP (P2Y1/12), epinephrine (α2A adrenergic receptor), TxA2 (TP), and thrombin (PAR1).
Abbreviations: ADP, adenosine diphosphate; VWF, von Willebrand factor; GP, glycoprotein.
Importantly, GPIb-IX-V and GPVI engage their respective ligands differentially, according to local blood rheological conditions, with GPVI binding collagen exposed within the blood vessel walls at relatively low physiological shear rates17 and GPIb-IX-V binding VWF after exposure to high shear rates as found in arterioles and stenotic arteries.48
Engagement of either of these receptors leads to the activation of intracellular signaling pathways leading to the upregulation of platelet-specific αIIbβ3 integrin and resulting in enhanced ability of αIIbβ3 to bind the plasma protein fibrinogen.49,50 Triggering of these signaling pathways also initiates or enhances metalloproteolytic shedding of the extracellular ligand-binding portions of GPVI,36 and GPIbα and GPV.45,51
Interestingly, GPIbα and GPVI are directly and functionally linked on the platelet surface52 and activate common signaling pathways,53 further underscoring the extent of cooperation between these adhesion molecules. This differential involvement of GPIb-IX-V and GPVI in the initiation of platelet responses implies a coordinated response by platelets determined at least in part by specific vascular conditions and may provide an opportunity to selectively target a single prothrombotic process from one adhesion receptor while maintaining an adequate response from the other adhesion receptor.
Intracellular molecular signals that result from the engagement of either GPIb-IX-V or GPVI by their ligands may also represent attractive targets for antiplatelet therapeutics (Figure 2). The signaling cascades involve similar events where receptor binding results in the activation of the Src family kinases followed sequentially by spleen tyrosine kinase (Syk), lymphocyte cytosolic protein 2 (also know as SLP-76), phosphatidylinositide (PI) 3-kinase, Bruton’s tyrosine kinase (Btk), phospholipase (PL) Cγ2, and protein kinase (PK) C, ultimately resulting in the elevation of the cytosolic Ca2+ and αIIbβ3 activation.54,55 Small differences exist regarding the order of activation of the signaling molecules and some signaling/adaptor molecules. For example, Src and Lyn appear to be recruited to the GPIb-IX-V complex upon platelet activation, whereas Fyn and Lyn are constitutively associated with GPVI.
Figure 2.
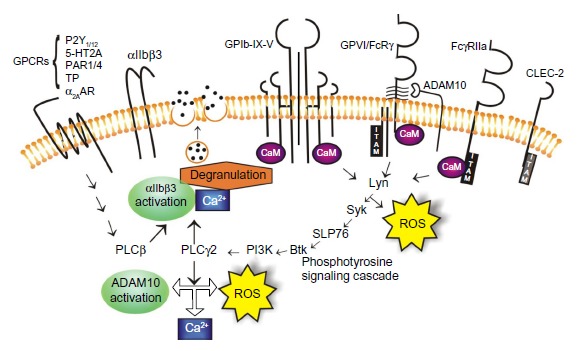
Signaling pathways orchestrate platelet activation and aggregation.
Notes: Engagement of platelet adhesion receptors triggers a phosphotyrosine-signaling cascade that leads to αIIbβ3 activation, ADAM10 activation, ROS production, calcium flux, and degranulation.
Abbreviations: GPCR, G protein-coupled receptor; GP, glycoprotein; ADAM, a disintegrin and metalloproteinase; ROS, reactive oxygen species; Syk, spleen tyrosine kinase; Btk, Bruton’s tyrosine kinase; CLEC-2, C-type lectin receptor-2; PLC, phospholipase C; PI3K, phosphatidylinositide 3-kinase; CaM, calmodulin; ITAM, immunoreceptor tyrosine-based activation motif.
Additionally, it is reported that GPVI activation induces significantly more inositol phosphate production and PLCγ2 activity relative to GPIb-IX-V activation.55 Differences regarding the tyrosine phosphorylation patterns downstream of GPIbα and GPVI, where the GPVI agonists induce more rapid tyrosine phosphorylation of platelet proteins relative to GPIbα engagement, are also evident.56 The engagement of GPVI also produces significant amounts of intracellular reactive oxygen species (ROS), possibly as part of the signaling process57 and the blockade of the ROS-generating nicotinamide adenine dinucleotide phosphate (NADP+)-oxidase downstream of GPVI engagement, reduces platelet activation and aggregation, and results in the formation of smaller thrombi,58,59 possibly via the ablation of thromboxane production.59 ROS-generating machinery is also associated with the cytoplasmic tails of the GPIb-IX-V complex;60 however, evidence that the engagement of GPIb-IX-V also leads to the generation of ROS has not yet emerged. These small differences in signaling responses between GPVI and GPIb-IX-V, once engaged by their respective ligands, may provide some biological leads for the design of agents that would selectively target one receptor over the other and so provide tools to mediate platelet activation under the differential shear conditions found in specific vascular beds.
GPVI involvement in thrombosis
In vascular lesions, where the endothelium has been denuded/disrupted or there exists plaques, collagen as well as VWF are exposed and form a prothrombotic surface, a potent trigger for the platelet adhesion and aggregation.2 Platelet engagement with the blood vessel wall is predominantly mediated by GPVI and GPIb-IX-V, which cooperate under a large range of blood shear conditions. Additional involvement of other adhesion proteins – including integrins α2β161,62 and α6β163 – helps to stabilize the initial attachment and facilitate platelet recruitment and thrombus growth.
However, the engagement of GPIb-IX-V and GPVI generates a cascade of signaling events that lead to Ca2+ mobilization, the rapid release of a battery of soluble agonists that includes ADP and thromboxane, inorganic polyphosphates,64 microparticles, and thiol oxidoreductase ERp57,65 and produces a negatively charged phosphatidylserine-expressing platelet surface66,67 to aid and enhance the generation of active tissue factor.
Consequences of reduction of GPVI function
Table 1 provides a summary of the known data collected from both mouse and human systems evaluating the contribution of GPVI to normal hemostasis and to thrombosis and other responses involving platelets. Platelets with reduced/absent GPVI, as found in people with platelet-targeting autoantibodies68,69 or genetic mutation,70–72 display a mild-to-more-severe bleeding diathesis that can include ecchymosis, epistaxis, easy bruising, and prolonged bleeding from mucosal membranes and gums. Experimentally, these platelets display reduced response to collagen and other GPVI agonists by aggregation or flow cytometry-based assays69,73 and a reduction in thrombus size when exposed to collagen in ex vivo blood flow-based assays.74
Table 1.
Extent of involvement of GPVI in onset of thrombotic and inflammatory disorders
| Type of injury | GPVI involvement | |
|---|---|---|
| Hemostasis | Minor vascular injury72,94 | Minor |
| Venous thrombosis | Low shear, blood stasis, coagulopathy80,83 | Minor |
| Arterial thrombosis | High shear, vascular damage77,114 | Major |
| Atherothrombosis | Plaque rupture, exposure of thrombogenic material, occlusion115 | Major |
| Ischemic stroke | Thromboembolic occlusion116,117 | Major |
| Inflammation | Collagen fragments, immune complexes ischemia/reperfusion injury95–97,118 | Major |
Abbreviation: GP, glycoprotein.
Similarly, platelets from mice genetically deficient for the GPVI/Fcγ chain75,76 or treated with anti-GPVI antibodies that cause the removal of GPVI from platelets76,77 do not respond efficiently or effectively to collagen in assays in vitro. Significantly, however, mice that are genetically deficient in GPVI do not display a prolonged bleeding time, but they do demonstrate a moderate-to-strong protection from thrombosis, depending on the injury model employed.75,78–81 Clearly, in animal models of thrombosis, the initiating event and the vascular bed being studied in part determine the extent of the contribution and the relative importance of GPVI to platelet activation and thrombus formation (Table 1).
Under normal or healthy blood rheological conditions, the contribution of GPVI is only minor, probably because other adhesion receptors (GPIb-IX-V and collagen-binding integrin α2β1, for example) deliver necessary platelet adhesion and thrombus stability properties, and the vascular conditions permit an accentuated contribution to platelet activation from thrombin and other soluble agonists.79,82,83 Keeping this in mind, the findings imply overall that GPVI may be a reasonable target for antithrombotic therapies as its nonessential role in hemostasis but important role in thrombosis means that the therapeutic targeting is unlikely to lead to unacceptable bleeding.
Existing clinical strategies to target platelet adhesion/activation
Numerous clinical therapeutics exist that target platelet activation and platelet adhesiveness (Table 2).84,85 The majority of the therapeutics aims to block the receptors involved in the initial and amplification stages of platelet activation; several reagents are often used in combination to achieve a potent antiplatelet effect. Dual and triple antiplatelet therapies have been shown to prevent ischemic events in high-risk patients with coronary artery disease or during percutaneous coronary interventions, but they can cause bleeding complications.8 Further, the incidence of recurrence of adverse vascular events remains of concern. The range of currently approved reagents includes:
Table 2.
Examples of existing and novel therapeutics targeting platelet receptors
| Platelet receptor | GPIbα | GPVI | PAR-1 | P2Y12 | Thromboxane receptor | αIIbβ3 |
|---|---|---|---|---|---|---|
| Copy number/platelet | 18,000–25,000 | 6,000–10,000 | 500–2,000 | Undefined | Undefined | 60,000–80,000 |
| Endogenous ligand | Shear-exposed VWF, P-selectin, αMβ2 | Subendothelial collagen, collagen fragments | Thrombin | ADP | Thromboxane | Fibrinogen, VWF, collagen |
| Phase of hemostasis | Primary | Primary | Primary | Amplification | Amplification | Stabilization |
| Existing therapeutics that directly target this receptor | Clopidogrel, Prasugrel, ticagrelor | Abciximab, tirofiban, eptifibatide | ||||
| Investigational therapeutics | 6B4-Fab | Revacept, kistomin, glaucocalyxin | Vorapaxar, atopaxar | Cangrelor, cilostazol | Terutroban |
Abbreviations: GP, glycoprotein; PAR-1, proteinase-activated receptor 1; VWF, von Willebrand factor; ADP, adenosine diphosphate; Fab, fragment antigen-binding.
Anticoagulants, such as lepirudin, warfarin, unfractionated heparin, low molecular weight heparin, bivalirudin, edoxaban, rivaroxaban, argatroban, and dabigatran86,87 that are direct or indirect inhibitors of thrombin and so block coagulation and interfere with the thrombin engagement of protease-activated receptors (PARs) on platelets
Clopidogrel, prasugrel, and ticagrelor,88 which serve to block platelet ADP receptor P2Y12
Aspirin, which blocks thromboxane generation (a strong platelet agonist) via inhibition of cyclooxygenases
Abciximab, eptifibatide, and tirofiban,89 which target fibrinogen interactions with the platelet-specific fibrinogen receptor αIIbβ3.
The main issue with these reagents is the elevated risk of adverse bleeding, due to the therapeutics targeting molecular pathways that are important for both thrombotic processes and normal hemostasis.90 This issue is compounded because the assessment of bleeding risk in patients receiving one or more of these therapies is complicated,91,92 generally requiring specialized platelet function analysis where the relationship between platelet function testing and bleeding in different patient groups on combinational therapy is not clear, due to limited data.
Targeting GPVI therapeutically
GPVI has emerged as a potential target for antithrombotic therapy for a number of reasons. First, GPVI is only expressed on platelets and megakaryocyte (platelet precursor cells in bone marrow) populations17 in relatively low abundance,93 thus permitting high specificity while minimizing potential side effects of a therapeutic agent. Second, GPVI appears to play only a supporting role in normal hemostasis,72,94 implying that targeting GPVI would not increase bleeding risk to unacceptable levels. Third, in animal models of thrombosis as well as in studies of inflammation95 and reperfusion injury following ischemia,96,97 a significant contribution of GPVI to tissue injury – together with experiments demonstrating the benefits of blockade of GPVI to reduce the extent of injury16 – have been well-documented.
Several options exist to modulate or inhibit GPVI-mediated platelet activation. GPVI–collagen interaction can be disturbed by collagen-binding molecules (GPVI mimics), by GPVI-function blocking reagents (aptamers, small molecules, and antibodies), or by GPVI depletion.
GPVI mimetics
Taking advantage of the stronger collagen-binding affinity of dimeric GPVI, a recombinant fusion protein was formed between the extracellular collagen-binding domain of GPVI and the C-terminal of human immunoglobulin Fc domain to form a soluble dimeric GPVI (GPVI-Fc).98 This reagent specifically bound to collagen with high affinity and attenuated platelet adhesion to immobilized collagen in vitro and to sites of vascular injury in vivo.98
Importantly, at doses sufficient to reduce platelet adhesion, the soluble form of GPVI only moderately prolonged tail bleeding times.99,100 Such a reagent holds several advantages over other types of GPVI inhibitors. The GPVI-Fc predominantly targets the exposed subendothelium at a site of vascular injury, suggesting that collagen exposed within a damaged vascular site is the primary site of binding. By directly targeting the site of interaction, there is no requirement for prolonged systemic inhibition of platelet function.99 Further, there have been no reports of aberrant platelet activation, loss of GPVI on circulating platelets, or thrombocytopenia associated with the use of this reagent in animal models. By the addition of an appropriate molecular tag to GPVI-Fc, it is possible that this reagent may also be developed as a bioimaging tool as it selectively binds to presumably prothrombotic regions of a vascular bed that is enriched for collagen.16,98,101
Injection of GPVI-Fc (Revacept; Janssen-Cilag GmbH, Neuss, Germany) has improved endothelial dysfunction and vascular morphology in atherosclerotic rabbits100 and reduced the cerebral infarct size and edema after ischemic stroke, with improved functional and prognostic outcome without intracranial bleeding.102 In a Phase I study, Revacept efficiently inhibited collagen-induced platelet aggregation ex vivo, with no alteration of primary hemostasis in 30 healthy donors;103 however, GPVI-Fc had only limited antithrombotic effects in animal models where the direct blockade of GPVI function was effective in preventing occlusive thrombus formation.104
While the experimental and early phase trial results with this fusion protein are encouraging, the precise clinical setting and the appropriate dosing and treatment regimens where this reagent may be useful still remain to be defined. Also, similar to other immunoglobulin fusion proteins and antibody therapies, the possibility of immunogenicity with repeated injections of Revacept remains a potential hazard.
GPVI-function blocking reagents
Anti-GPVI antibodies are of great interest as candidate antithrombotic reagents as they may be able to accomplish the dual purpose of interfering with collagen-GPVI interactions as well as triggering GPVI shedding and/or internalization. Numerous antibodies raised against human GPVI now exist,10,35 most of which activate platelets either directly through GPVI engagement or indirectly via the interaction with FcγRIIa when the antibody is intact. The fragment antigen-binding (Fab) fragments of most of these antibodies, including 9O12, 5C4, 1G5, and OM2 have strong-to-very-strong affinity for GPVI and inhibit GPVI-induced platelet activation. Single domain antibody clones,105 consisting of single heavy and light chain variable domains (11–13 kDa) and single chain antibodies,106 are highly stable and protease-resistant reagents that can be humanized and readily expressed in phage display libraries. As monomers, these reagents also have the advantage of not clustering or crosslinking GPVI, causing unwanted platelet activation.105,107
The signaling pathways utilized by GPVI to transmit activation signals (Figure 2) may also be targeted therapeutically. Existing reagents targeting Syk108 or Btk109 – which are already approved as antitumor reagents for use in patients with lymphoma – have shown strong efficacy in blocking platelet-collagen responses via GPVI.
Interestingly, the anticancer histone deacetylase inhibitors have additional effects on platelet GPVI. They reduce the surface and total expression of GPVI and impair GPVI signaling, which results in the inhibition of the final common pathways of platelet activation.110 New approaches that target GPVI-mediated reactive oxygen species58 or limit GPVI signaling via the activation of nuclear receptors (liver X receptors) on platelets that dampen platelet-collagen responses111 may represent novel, more subtle approaches to diminish but not block GPVI-signaling function. The fact that many of these agents have been used therapeutically in patients and do not seem to cause clinically significant bleeding provides encouragement and possibly underscores the relatively minor role for GPVI in normal hemostasis.
Final remarks
For a number of reasons, GPVI presents as an attractive and feasible target to modulate platelet function. Peripherally, it is found exclusively on platelets, meaning that its modulation is less likely to lead to off-target effects. GPVI is accessible on the surface of platelets and tools and reagents exist to: 1) rapidly block the function or downregulate surface levels of the molecule; and 2) quantify changes in the surface levels of GPVI (by fluorescence-activated cell sorting [FACS]) and/or shed soluble GPVI in plasma (by enzyme-linked immunosorbent assay [ELISA] or bead-based immunoassay).
While the loss of GPVI does not appear to seriously disrupt normal hemostasis, pathological thrombus formation is significantly attenuated by targeting GPVI. Despite these positive milestones, it is valuable to remember that many proteins and gene products contribute to a given platelet phenotype in the complex human system. In the case of mouse GPVI, at least one or more unknown modifier genes in a modifier of hemostasis locus were shown to control the extent to which platelet thrombus formation in vivo was disrupted by the absence of platelet GPVI. Conceivably this could occur by altering the composition and thrombotic nature of the extracellular matrix through the regulation of gene expression in endothelial cells, smooth muscle cells, and/or fibroblasts.112
Further, GPVI-based inhibitors need to be carefully evaluated for safety, efficacy, and potency in the different patient groups and – as a monotherapy – suitability for acute conditions, or in combination with the existing antiplatelet and anticoagulant therapies as part of an approach to chronic treatment. For these reasons, data from human trials are eagerly awaited. However, together with new tools to specifically examine the antithrombotic (and other) effects of new and existing anti-GPVI reagents, such as a mouse expressing human GPVI,113 a reagent that controls GPVI expression and function is practical and a reasonable proposition.
Acknowledgments
This work was supported by the National Health and Medical Research Council of Australia.
Footnotes
Disclosure
The authors report no conflicts of interest in this work.
References
- 1.Rondina MT, Weyrich AS, Zimmerman GA. Platelets as cellular effectors of inflammation in vascular diseases. Circ Res. 2013;112(11):1506–1519. doi: 10.1161/CIRCRESAHA.113.300512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Furie B, Furie BC. Mechanisms of thrombus formation. New Engl J Med. 2008;359(9):938–949. doi: 10.1056/NEJMra0801082. [DOI] [PubMed] [Google Scholar]
- 3.Mackman N. Triggers, targets and treatments for thrombosis. Nature. 2008;451(7181):914–918. doi: 10.1038/nature06797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Jackson SP. Arterial thrombosis – insidious, unpredictable and deadly. Nat Med. 2011;17(11):1423–1436. doi: 10.1038/nm.2515. [DOI] [PubMed] [Google Scholar]
- 5.Ungerer M, Münch G. Novel antiplatelet drugs in clinical development. Thromb Haemost. 2013;110(5):868–875. doi: 10.1160/TH13-02-0084. [DOI] [PubMed] [Google Scholar]
- 6.Antithrombotic Trialists’ Collaboration Collaborative meta-analysis of randomised trials of antiplatelet therapy for prevention of death, myocardial infarction, and stroke in high risk patients. BMJ. 2002;324(7329):71–86. doi: 10.1136/bmj.324.7329.71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hankey GJ. Antiplatelet therapy for the prevention of recurrent stroke and other serious vascular events: a review of the clinical trial data and guidelines. Curr Med Res Opin. 2007;23(6):1453–1462. doi: 10.1185/030079907X199727. [DOI] [PubMed] [Google Scholar]
- 8.Cheng JW. Updates in antiplatelet agents used in cardiovascular diseases. J Cardiovas Pharmacol Ther. 2013;18(6):514–524. doi: 10.1177/1074248413499971. [DOI] [PubMed] [Google Scholar]
- 9.Berger PB, Bhatt DL, Fuster V, et al. CHARISMA Investigators Bleeding complications with dual antiplatelet therapy among patients with stable vascular disease or risk factors for vascular disease: Results from the clopidogrel for high atherothrombotic risk and ischemic stabilization, management, and avoidance (CHARISMA) trial. Circulation. 2010;121(23):2575–2583. doi: 10.1161/CIRCULATIONAHA.109.895342. [DOI] [PubMed] [Google Scholar]
- 10.Bigalke B, Stellos K, Weig HJ, et al. Regulation of platelet glycoprotein VI (GPVI) surface expression and of soluble GPVI in patients with atrial fibrillation (AF) and acute coronary syndrome (ACS) Basic Res Cardiol. 2009;104(3):352–357. doi: 10.1007/s00395-009-0779-7. [DOI] [PubMed] [Google Scholar]
- 11.Bigalke B, Stellos K, Geisler T, et al. Glycoprotein VI for diagnosis of acute coronary syndrome when ECG is ambiguous. Int J Cardiol. 2011;149(2):164–168. doi: 10.1016/j.ijcard.2009.12.026. [DOI] [PubMed] [Google Scholar]
- 12.Bigalke B, Stellos K, Geisler T, Lindemann S, May AE, Gawaz M. Glycoprotein VI as a prognostic biomarker for cardiovascular death in patients with symptomatic coronary artery disease. Clin Res Cardiol. 2010;99(4):227–233. doi: 10.1007/s00392-009-0109-y. [DOI] [PubMed] [Google Scholar]
- 13.Dütting S, Bender M, Nieswandt B. Platelet GPVI: a target for antithrombotic therapy. Trends Pharmacol Sci. 2012;33(11):583–590. doi: 10.1016/j.tips.2012.07.004. [DOI] [PubMed] [Google Scholar]
- 14.Zahid M, Mangin P, Loyau S, et al. The future of glycoprotein VI as an antithrombotic target. J Thromb Haemost. 2012;10(12):2418–2427. doi: 10.1111/jth.12009. [DOI] [PubMed] [Google Scholar]
- 15.Bigalke B, Krämer BF, Seizer P, Fateh-Moghadam S, Gawaz M, Lindemann S. Diagnostic and therapeutic potentials of platelet glycoprotein VI. Semin Thromb Hemost. 2010;36(2):203–211. doi: 10.1055/s-0030-1251505. [DOI] [PubMed] [Google Scholar]
- 16.Gawaz M, Vogel S, Pfannenberg C, Pichler B, Langer H, Bigalke B. Implications of glycoprotein VI for theranostics. Thromb Haemost. 2014;112(1):1–6. doi: 10.1160/TH13-09-0756. [DOI] [PubMed] [Google Scholar]
- 17.Nieswandt B, Watson SP. Platelet-collagen interaction: is GPVI the central receptor. Blood. 2003;102(2):449–461. doi: 10.1182/blood-2002-12-3882. [DOI] [PubMed] [Google Scholar]
- 18.Inoue O, Suzuki-Inoue K, McCarty OJ, et al. Laminin stimulates spreading of platelets through integrin α6β1-dependent activation of GPVI. Blood. 2006;107(4):1405–1412. doi: 10.1182/blood-2005-06-2406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Clemetson JM, Polgar J, Magnenat E, Wells TN, Clemetson KJ. The platelet collagen receptor glycoprotein VI is a member of the immunoglobulin superfamily closely related to FcαR and the natural killer receptors. J Biol Chem. 1999;274(41):29019–29024. doi: 10.1074/jbc.274.41.29019. [DOI] [PubMed] [Google Scholar]
- 20.Burkhart JM, Vaudel M, Gambaryan S, et al. The first comprehensive and quantitative analysis of human platelet protein composition allows the comparative analysis of structural and functional pathways. Blood. 2012;120(15):e73–e82. doi: 10.1182/blood-2012-04-416594. [DOI] [PubMed] [Google Scholar]
- 21.Tsuji M, Ezumi Y, Arai M, Takayama H. A novel association of Fc receptor γ-chain with glycoprotein VI and their co-expression as a collagen receptor in human platelets. J Biol Chem. 1997;272(38):23528–23531. doi: 10.1074/jbc.272.38.23528. [DOI] [PubMed] [Google Scholar]
- 22.Bergmeier W, Stefanini L. Platelet ITAM signaling. Curr Opin Hematol. 2013;20(5):445–450. doi: 10.1097/MOH.0b013e3283642267. [DOI] [PubMed] [Google Scholar]
- 23.Gibbins JM, Okuma M, Farndale R, Barnes M, Watson SP. Glycoprotein VI is the collagen receptor in platelets which underlies tyrosine phosphorylation of the Fc receptor γ-chain. FEBS Lett. 1997;413(2):255–259. doi: 10.1016/s0014-5793(97)00926-5. [DOI] [PubMed] [Google Scholar]
- 24.Andrews RK, Suzuki-Inoue K, Shen Y, Tulasne D, Watson SP, Berndt MC. Interaction of calmodulin with the cytoplasmic domain of platelet glycoprotein VI. Blood. 2002;99(11):4219–4221. doi: 10.1182/blood-2001-11-0008. [DOI] [PubMed] [Google Scholar]
- 25.Lecut C, Arocas V, Ulrichts H, et al. Identification of residues within human glycoprotein VI involved in the binding to collagen: evidence for the existence of distinct binding sites. J Biol Chem. 2004;279(50):52293–52299. doi: 10.1074/jbc.M406342200. [DOI] [PubMed] [Google Scholar]
- 26.Knight CG, Morton LF, Onley DJ, et al. Collagen-platelet interaction: Gly-Pro-Hyp is uniquely specific for platelet Gp VI and mediates platelet activation by collagen. Cardiovasc Res. 1999;41(2):450–457. doi: 10.1016/s0008-6363(98)00306-x. [DOI] [PubMed] [Google Scholar]
- 27.Casasnovas JM, Stehle T, Liu JH, Wang JH, Springer TA. A dimeric crystal structure for the N-terminal two domains of intercellular adhesion molecule-1. Proc Natl Acad Sci USA. 1998;95(8):4134–4139. doi: 10.1073/pnas.95.8.4134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Cornish AL, Freeman S, Forbes G, et al. Characterization of siglec-5, a novel glycoprotein expressed on myeloid cells related to CD33. Blood. 1998;92(6):2123–2132. [PubMed] [Google Scholar]
- 29.Ramsland PA, Farrugia W, Bradford TM, et al. Structural basis for Fc γRIIa recognition of human IgG and formation of inflammatory signaling complexes. J Immunol. 2011;187(6):3208–3217. doi: 10.4049/jimmunol.1101467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Jung SM, Moroi M, Soejima K, et al. Constitutive dimerization of glycoprotein VI (GPVI) in resting platelets is essential for binding to collagen and activation in flowing blood. J Biol Chem. 2012;287(35):30000–30013. doi: 10.1074/jbc.M112.359125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Arthur JF, Shen Y, Kahn ML, Berndt MC, Andrews RK, Gardiner EE. Ligand binding rapidly induces disulfide-dependent dimerization of glycoprotein VI on the platelet plasma membrane. J Biol Chem. 2007;282(42):30434–30441. doi: 10.1074/jbc.M701330200. [DOI] [PubMed] [Google Scholar]
- 32.Loyau S, Dumont B, Ollivier V, et al. Platelet glycoprotein VI dimerization, an active process inducing receptor competence, is an indicator of platelet reactivity. Arterioscler Thromb Vasc Biol. 2012;32(3):778–785. doi: 10.1161/ATVBAHA.111.241067. [DOI] [PubMed] [Google Scholar]
- 33.Horii K, Kahn ML, Herr AB. Structural basis for platelet collagen responses by the immune-type receptor glycoprotein VI. Blood. 2006;108(3):936–942. doi: 10.1182/blood-2006-01-010215. [DOI] [PubMed] [Google Scholar]
- 34.Miura Y, Takahashi T, Jung SM, Moroi M. Analysis of the interaction of platelet collagen receptor glycoprotein VI (GPVI) with collagen. A dimeric form of GPVI, but not the monomeric form, shows affinity to fibrous collagen. J Biol Chem. 2002;277(48):46197–46204. doi: 10.1074/jbc.M204029200. [DOI] [PubMed] [Google Scholar]
- 35.Al-Tamimi M, Arthur JF, Gardiner EE, Andrews RK. Focusing on plasma glycoprotein VI. Thromb Haemost. 2012;107(4):648–655. doi: 10.1160/TH11-10-0745. [DOI] [PubMed] [Google Scholar]
- 36.Gardiner EE, Arthur JF, Kahn ML, Berndt MC, Andrews RK. Regulation of platelet membrane levels of glycoprotein VI by a platelet-derived metalloproteinase. Blood. 2004;104(12):3611–3617. doi: 10.1182/blood-2004-04-1549. [DOI] [PubMed] [Google Scholar]
- 37.Gardiner EE, Karunakaran D, Arthur JF, et al. Dual ITAM-mediated proteolytic pathways for irreversible inactivation of platelet receptors: de-ITAM-izing FcγRIIa. Blood. 2008;111(1):165–174. doi: 10.1182/blood-2007-04-086983. [DOI] [PubMed] [Google Scholar]
- 38.Al-Tamimi M, Grigoriadis G, Tran H, et al. Coagulation-induced shedding of platelet glycoprotein VI mediated by factor Xa. Blood. 2011;117(14):3912–3920. doi: 10.1182/blood-2010-08-301523. [DOI] [PubMed] [Google Scholar]
- 39.Al-Tamimi M, Tan CW, Qiao J, et al. Pathological shear triggers shedding of vascular receptors: a novel mechanism for down-regulation of platelet glycoprotein VI in stenosed coronary vessels. Blood. 2012;119(18):4311–4320. doi: 10.1182/blood-2011-10-386607. [DOI] [PubMed] [Google Scholar]
- 40.Rabie T, Varga-Szabo D, Bender M, et al. Diverging signaling events control the pathway of GPVI down-regulation in vivo. Blood. 2007;110(2):529–535. doi: 10.1182/blood-2006-11-058107. [DOI] [PubMed] [Google Scholar]
- 41.Takayama H, Hosaka Y, Nakayama K, et al. A novel antiplatelet antibody therapy that induces cAMP-dependent endocytosis of the GPVI/Fc receptor γ-chain complex. J Clin Invest. 2008;118(5):1785–1795. doi: 10.1172/JCI32513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Murphy G. Regulation of the proteolytic disintegrin metalloproteinases, the ‘Sheddases’. Semin Cell Dev Biol. 2009;20(2):138–145. doi: 10.1016/j.semcdb.2008.09.004. [DOI] [PubMed] [Google Scholar]
- 43.van der Vorst EP, Keijbeck AA, de Winther MP, Donners MM. A disintegrin and metalloproteases: molecular scissors in angiogenesis, inflammation and atherosclerosis. Atherosclerosis. 2012;224(2):302–308. doi: 10.1016/j.atherosclerosis.2012.04.023. [DOI] [PubMed] [Google Scholar]
- 44.Hartmann M, Herrlich A, Herrlich P. Who decides when to cleave an ectodomain. Trends Biochem Sci. 2013;38(3):111–120. doi: 10.1016/j.tibs.2012.12.002. [DOI] [PubMed] [Google Scholar]
- 45.Gardiner EE, Karunakaran D, Shen Y, Arthur JF, Andrews RK, Berndt MC. Controlled shedding of platelet glycoprotein (GP)VI and GPIb-IX-V by ADAM family metalloproteinases. J Thromb Haemost. 2007;5(7):1530–1537. doi: 10.1111/j.1538-7836.2007.02590.x. [DOI] [PubMed] [Google Scholar]
- 46.Ludwig A, Hundhausen C, Lambert MH, et al. Metalloproteinase inhibitors for the disintegrin-like metalloproteinases ADAM10 and ADAM17 that differentially block constitutive and phorbol ester-inducible shedding of cell surface molecules. Comb Chem High Throughput Screen. 2005;8(2):161–171. doi: 10.2174/1386207053258488. [DOI] [PubMed] [Google Scholar]
- 47.Andrews RK, Berndt MC. Bernard–Soulier syndrome: an update. Semin Thromb Hemost. 2013;39(6):656–662. doi: 10.1055/s-0033-1353390. [DOI] [PubMed] [Google Scholar]
- 48.Kroll MH, Hellums JD, McIntire LV, Schafer AI, Moake JL. Platelets and shear stress. Blood. 1996;88(5):1525–1541. [PubMed] [Google Scholar]
- 49.Andrews RK, Gardiner EE, Shen Y, Berndt MC. Platelet interactions in thrombosis. IUBMB Life. 2004;56(1):13–18. doi: 10.1080/15216540310001649831. [DOI] [PubMed] [Google Scholar]
- 50.McFadyen JD, Jackson SP. Differentiating haemostasis from thrombosis for therapeutic benefit. Thromb Haemost. 2013;110(5):859–867. doi: 10.1160/TH13-05-0379. [DOI] [PubMed] [Google Scholar]
- 51.Rabie T, Strehl A, Ludwig A, Nieswandt B. Evidence for a role of ADAM17 (TACE) in the regulation of platelet glycoprotein V. J Biol Chem. 2005;280(15):14462–14468. doi: 10.1074/jbc.M500041200. [DOI] [PubMed] [Google Scholar]
- 52.Arthur JF, Gardiner EE, Matzaris M, et al. Glycoprotein VI is associated with GPIb-IX-V on the membrane of resting and activated platelets. Thromb Haemost. 2005;93(4):716–723. doi: 10.1160/TH04-09-0584. [DOI] [PubMed] [Google Scholar]
- 53.Ozaki Y, Asazuma N, Suzuki-Inoue K, Berndt MC. Platelet GPIb-IX-V-dependent signaling. J Thromb Haemost. 2005;3(8):1745–1751. doi: 10.1111/j.1538-7836.2005.01379.x. [DOI] [PubMed] [Google Scholar]
- 54.Watson SP, Herbert JM, Pollitt AY. GPVI and CLEC-2 in hemostasis and vascular integrity. J Thromb Haemost. 2010;8(7):1456–1467. doi: 10.1111/j.1538-7836.2010.03875.x. [DOI] [PubMed] [Google Scholar]
- 55.Suzuki-Inoue K, Wilde JI, Andrews RK, et al. Glycoproteins VI and Ib-IX-V stimulate tyrosine phosphorylation of tyrosine kinase Syk and phospholipase Cγ2 at distinct sites. Biochem J. 2004;378(Pt 3):1023–1029. doi: 10.1042/BJ20031430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Gardiner EE, Arthur JF, Shen Y, et al. GPIbα-selective activation of platelets induces platelet signaling events comparable to GPVI activation events. Platelets. 2010;21(4):244–252. doi: 10.3109/09537101003695339. [DOI] [PubMed] [Google Scholar]
- 57.Arthur JF, Qiao J, Shen Y, et al. ITAM receptor-mediated generation of reactive oxygen species in human platelets occurs via Syk-dependent and Syk-independent pathways. J Thromb Haemost. 2012;10(6):1133–1141. doi: 10.1111/j.1538-7836.2012.04734.x. [DOI] [PubMed] [Google Scholar]
- 58.Vara D, Campanella M, Pula G. The novel NOX inhibitor 2- acetylphenothiazine impairs collagen-dependent thrombus formation in a GPVI-dependent manner. Br J Pharmacol. 2013;168(1):212–224. doi: 10.1111/j.1476-5381.2012.02130.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Walsh TG, Berndt MC, Carrim N, Cowman J, Kenny D, Metharom P. The role of Nox1 and Nox2 in GPVI-dependent platelet activation and thrombus formation. Redox Biol. 2014;2:178–186. doi: 10.1016/j.redox.2013.12.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Arthur JF, Shen Y, Gardiner EE, et al. TNF receptor-associated factor 4 (TRAF4) is a novel binding partner of glycoprotein Ib and glycoprotein VI in human platelets. J Thromb Haemost. 2011;9(1):163–172. doi: 10.1111/j.1538-7836.2010.04091.x. [DOI] [PubMed] [Google Scholar]
- 61.Miller MW, Basra S, Kulp DW, et al. Small-molecule inhibitors of integrin α2β1 that prevent pathological thrombus formation via an allosteric mechanism. Proc Natl Acad Sci USA. 2009;106(3):719–724. doi: 10.1073/pnas.0811622106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Nissinen L, Pentikäinen OT, Jouppila A, et al. A small-molecule inhibitor of integrin α2 β1 introduces a new strategy for antithrombotic therapy. Thromb Haemost. 2010;103(2):387–397. doi: 10.1160/TH09-06-0358. [DOI] [PubMed] [Google Scholar]
- 63.Schaff M, Tang C, Maurer E, et al. Integrin α6β1 is the main receptor for vascular laminins and plays a role in platelet adhesion, activation, and arterial thrombosis. Circulation. 2013;128(5):541–552. doi: 10.1161/CIRCULATIONAHA.112.000799. [DOI] [PubMed] [Google Scholar]
- 64.Müller F, Mutch NJ, Schenk WA, et al. Platelet polyphosphates are proinflammatory and procoagulant mediators in vivo. Cell. 2009;139(6):1143–1156. doi: 10.1016/j.cell.2009.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Schulz C, Leuschen NV, Fröhlich T, et al. Identification of novel downstream targets of platelet glycoprotein VI activation by differential proteome analysis: implications for thrombus formation. Blood. 2010;115(20):4102–4110. doi: 10.1182/blood-2009-07-230268. [DOI] [PubMed] [Google Scholar]
- 66.Arachiche A, Kerbiriou-Nabias D, Garcin I, Letellier T, Dachary-Prigent J. Rapid procoagulant phosphatidylserine exposure relies on high cytosolic calcium rather than on mitochondrial depolarization. Arterioscler Thromb Vasc Biol. 2009;29(11):1883–1889. doi: 10.1161/ATVBAHA.109.190926. [DOI] [PubMed] [Google Scholar]
- 67.Choo HJ, Saafir TB, Mkumba L, Wagner MB, Jobe SM. Mitochondrial calcium and reactive oxygen species regulate agonist-initiated platelet phosphatidylserine exposure. Arterioscler Thromb Vas Biol. 2012;32(12):2946–2955. doi: 10.1161/ATVBAHA.112.300433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Moroi M, Jung SM, Okuma M, Shinmyozu K. A patient with platelets deficient in glycoprotein VI that lack both collagen-induced aggregation and adhesion. J Clin Invest. 1989;84(5):1440–1445. doi: 10.1172/JCI114318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Gardiner EE, Al-Tamimi M, Mu FT, et al. Compromised ITAM-based platelet receptor function in a patient with immune thrombocytopenic purpura. J Thromb Haemost. 2008;6(7):1175–1182. doi: 10.1111/j.1538-7836.2008.03016.x. [DOI] [PubMed] [Google Scholar]
- 70.Dumont B, Lasne D, Rothschild C, et al. Absence of collagen-induced platelet activation caused by compound heterozygous GPVI mutations. Blood. 2009;114(9):1900–1903. doi: 10.1182/blood-2009-03-213504. [DOI] [PubMed] [Google Scholar]
- 71.Hermans C, Wittevrongel C, Thys C, Smethurst PA, Van Geet C, Freson K. A compound heterozygous mutation in glycoprotein VI in a patient with a bleeding disorder. J Thromb Haemost. 2009;7(8):1356–1363. doi: 10.1111/j.1538-7836.2009.03520.x. [DOI] [PubMed] [Google Scholar]
- 72.Arthur JF, Dunkley S, Andrews RK. Platelet glycoprotein VI-related clinical defects. Brit J Haematol. 2007;139(3):363–372. doi: 10.1111/j.1365-2141.2007.06799.x. [DOI] [PubMed] [Google Scholar]
- 73.Qiao J, Arthur JF, Collecutt M, et al. An acquired defect associated with abnormal signaling of the platelet collagen receptor glycoprotein VI. Acta Haematol. 2012;128(4):233–241. doi: 10.1159/000340048. [DOI] [PubMed] [Google Scholar]
- 74.Bellucci S, Huisse MG, Boval B, et al. Defective collagen-induced platelet activation in two patients with malignant haemopathies is related to a defect in the GPVI-coupled signaling pathway. Thromb Haemost. 2005;93(1):130–138. doi: 10.1160/TH04-05-0312. [DOI] [PubMed] [Google Scholar]
- 75.Lockyer S, Okuyama K, Begum S, et al. GPVI-deficient mice lack collagen responses and are protected against experimentally induced pulmonary thromboembolism. Thromb Res. 2006;118(3):371–380. doi: 10.1016/j.thromres.2005.08.001. [DOI] [PubMed] [Google Scholar]
- 76.Nieswandt B, Bergmeier W, Schulte V, Rackebrandt K, Gessner JE, Zirngibl H. Expression and function of the mouse collagen receptor glycoprotein VI is strictly dependent on its association with the FcRγ chain. J Biol Chem. 2000;275(31):23998–24002. doi: 10.1074/jbc.M003803200. [DOI] [PubMed] [Google Scholar]
- 77.Massberg S, Gawaz M, Grüner S, et al. A crucial role of glycoprotein VI for platelet recruitment to the injured arterial wall in vivo. J Exp Med. 2003;197(1):41–49. doi: 10.1084/jem.20020945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Dubois C, Panicot-Dubois L, Gainor JF, Furie BC, Furie B. Thrombin-initiated platelet activation in vivo is vWF independent during thrombus formation in a laser injury model. J Clin Invest. 2007;117(4):953–960. doi: 10.1172/JCI30537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Mangin P, Yap CL, Nonne C, et al. Thrombin overcomes the thrombosis defect associated with platelet GPVI/FcRγ deficiency. Blood. 2006;107(11):4346–4353. doi: 10.1182/blood-2005-10-4244. [DOI] [PubMed] [Google Scholar]
- 80.Kato K, Kanaji T, Russell S, et al. The contribution of glycoprotein VI to stable platelet adhesion and thrombus formation illustrated by targeted gene deletion. Blood. 2003;102(5):1701–1707. doi: 10.1182/blood-2003-03-0717. [DOI] [PubMed] [Google Scholar]
- 81.Bender M, Hagedorn I, Nieswandt B. Genetic and antibody-induced glycoprotein VI deficiency equally protects mice from mechanically and FeCl(3)-induced thrombosis. J Thromb Haemost. 2011;9(7):1423–1426. doi: 10.1111/j.1538-7836.2011.04328.x. [DOI] [PubMed] [Google Scholar]
- 82.Grüner S, Prostredna M, Aktas B, et al. Anti-glycoprotein VI treatment severely compromises hemostasis in mice with reduced α2β1 levels or concomitant aspirin therapy. Circulation. 2004;110(18):2946–2951. doi: 10.1161/01.CIR.0000146341.63677.3C. [DOI] [PubMed] [Google Scholar]
- 83.Dubois C, Panicot-Dubois L, Merrill-Skoloff G, Furie B, Furie BC. Glycoprotein VI-dependent and -independent pathways of thrombus formation in vivo. Blood. 2006;107(10):3902–3906. doi: 10.1182/blood-2005-09-3687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Yousuf O, Bhatt DL. The evolution of antiplatelet therapy in cardiovascular disease. Nat Rev Cardiol. 2011;8(10):547–559. doi: 10.1038/nrcardio.2011.96. [DOI] [PubMed] [Google Scholar]
- 85.Moser M, Olivier CB, Bode C. Triple antithrombotic therapy in cardiac patients: more questions than answers. Eur Heart J. 2014;35(4):216–223. doi: 10.1093/eurheartj/eht461. [DOI] [PubMed] [Google Scholar]
- 86.van der Hulle T, Kooiman J, den Exter PL, Dekkers OM, Klok FA, Huisman MV. Effectiveness and safety of novel oral anticoagulants compared with vitamin K antagonists in the treatment of acute symptomatic venous thromboembolism: a systematic review and meta-analysis. J Thromb Haemost. 2014;12(3):320–328. doi: 10.1111/jth.12485. [DOI] [PubMed] [Google Scholar]
- 87.Ahrens I, Lip GY, Peter K. New oral anticoagulant drugs in cardiovascular disease. Thromb Haemost. 2010;104(1):49–60. doi: 10.1160/TH09-05-0327. [DOI] [PubMed] [Google Scholar]
- 88.Aradi D, Komócsi A, Vorobcsuk A, Serebruany VL. Impact of clopidogrel and potent P2Y 12 -inhibitors on mortality and stroke in patients with acute coronary syndrome or undergoing percutaneous coronary intervention: a systematic review and meta-analysis. Thromb Haemost. 2013;109(1):93–101. doi: 10.1160/TH12-06-0377. [DOI] [PubMed] [Google Scholar]
- 89.Muñiz-Lozano A, Rollini F, Franchi F, Angiolillo DJ. Update on platelet glycoprotein IIb/IIIa inhibitors: recommendations for clinical practice. Ther Adv Cardiovasc Dis. 2013;7(4):197–213. doi: 10.1177/1753944713487781. [DOI] [PubMed] [Google Scholar]
- 90.Swieringa F, Kuijpers MJ, Heemskerk JW, van der Meijden PE. Targeting platelet receptor function in thrombus formation: the risk of bleeding. Blood Rev. 2014;28(1):9–21. doi: 10.1016/j.blre.2013.12.001. [DOI] [PubMed] [Google Scholar]
- 91.Dahlen JR, Price MJ, Parise H, Gurbel PA. Evaluating the clinical usefulness of platelet function testing: considerations for the proper application and interpretation of performance measures. Thromb Haemost. 2013;109(5):808–816. doi: 10.1160/TH12-08-0608. [DOI] [PubMed] [Google Scholar]
- 92.Harrison P, Lordkipanidzé M. Testing platelet function. Hematol Oncol Clin North Am. 2013;27(3):411–441. doi: 10.1016/j.hoc.2013.03.003. [DOI] [PubMed] [Google Scholar]
- 93.Berlanga O, Bobe R, Becker M, et al. Expression of the collagen receptor glycoprotein VI during megakaryocyte differentiation. Blood. 2000;96(8):2740–2745. [PubMed] [Google Scholar]
- 94.Nieswandt B, Schulte V, Bergmeier W, et al. Long-term antithrombotic protection by in vivo depletion of platelet glycoprotein VI in mice. J Exp Med. 2001;193(4):459–469. doi: 10.1084/jem.193.4.459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Boilard E, Nigrovic PA, Larabee K, et al. Platelets amplify inflammation in arthritis via collagen-dependent microparticle production. Science. 2010;327(5965):580–583. doi: 10.1126/science.1181928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Kleinschnitz C, Pozgajova M, Pham M, Bendszus M, Nieswandt B, Stoll G. Targeting platelets in acute experimental stroke: impact of glycoprotein Ib, VI, and IIb/IIIa blockade on infarct size, functional outcome, and intracranial bleeding. Circulation. 2007;115(17):2323–2330. doi: 10.1161/CIRCULATIONAHA.107.691279. [DOI] [PubMed] [Google Scholar]
- 97.Devi S, Kuligowski MP, Kwan RY, et al. Platelet recruitment to the inflamed glomerulus occurs via an αIIbβ3/GPVI-dependent pathway. Am J Pathol. 2010;177(3):1131–1142. doi: 10.2353/ajpath.2010.091143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Massberg S, Konrad I, Bültmann A, et al. Soluble glycoprotein VI dimer inhibits platelet adhesion and aggregation to the injured vessel wall in vivo. FASEB J. 2004;18(2):397–399. doi: 10.1096/fj.03-0464fje. [DOI] [PubMed] [Google Scholar]
- 99.Bültmann A, Herdeg C, Li Z, et al. Local delivery of soluble platelet collagen receptor glycoprotein VI inhibits thrombus formation in vivo. Thromb Haemost. 2006;95(5):763–766. [PubMed] [Google Scholar]
- 100.Ungerer M, Li Z, Baumgartner C, et al. The GPVI-Fc fusion protein Revacept reduces thrombus formation and improves vascular dysfunction in atherosclerosis without any impact on bleeding times. PLoS One. 2013;8(8):e71193. doi: 10.1371/journal.pone.0071193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Bigalke B, Phinikaridou A, Andia ME, et al. Positron emission tomography/computed tomographic and magnetic resonance imaging in a murine model of progressive atherosclerosis using (64)Cu-labeled glycoprotein VI-Fc. Circ Cardiovasc Imaging. 2013;6(6):957–964. doi: 10.1161/CIRCIMAGING.113.000488. [DOI] [PubMed] [Google Scholar]
- 102.Goebel S, Li Z, Vogelmann J, et al. The GPVI-Fc fusion protein Revacept improves cerebral infarct volume and functional outcome in stroke. PLoS One. 2013;8(7):e66960. doi: 10.1371/journal.pone.0066960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Ungerer M, Rosport K, Bültmann A, et al. Novel antiplatelet drug revacept (Dimeric Glycoprotein VI-Fc) specifically and efficiently inhibited collagen-induced platelet aggregation without affecting general hemostasis in humans. Circulation. 2011;123(17):1891–1899. doi: 10.1161/CIRCULATIONAHA.110.980623. [DOI] [PubMed] [Google Scholar]
- 104.Grüner S, Prostredna M, Koch M, et al. Relative antithrombotic effect of soluble GPVI dimer compared with anti-GPVI antibodies in mice. Blood. 2005;105(4):1492–1499. doi: 10.1182/blood-2004-06-2391. [DOI] [PubMed] [Google Scholar]
- 105.Walker A, Pugh N, Garner SF, et al. Bloodomics Consortium Single domain antibodies against the collagen signaling receptor glycoprotein VI are inhibitors of collagen induced thrombus formation. Platelets. 2009;20(4):268–276. doi: 10.1080/09537100902893792. [DOI] [PubMed] [Google Scholar]
- 106.Muzard J, Bouabdelli M, Zahid M, et al. Design and humanization of a murine scFv that blocks human platelet glycoprotein VI in vitro. FEBS J. 2009;276(15):4207–4222. doi: 10.1111/j.1742-4658.2009.07129.x. [DOI] [PubMed] [Google Scholar]
- 107.Zahid M, Loyau S, Bouabdelli M, Aubrey N, Jandrot-Perrus M, Billiald P. Design and reshaping of an scFv directed against human platelet glycoprotein VI with diagnostic potential. Anal Biochem. 2011;417(2):274–282. doi: 10.1016/j.ab.2011.06.036. [DOI] [PubMed] [Google Scholar]
- 108.Spalton JC, Mori J, Pollitt AY, Hughes CE, Eble JA, Watson SP. The novel Syk inhibitor R406 reveals mechanistic differences in the initiation of GPVI and CLEC-2 signaling in platelets. J Thromb Haemost. 2009;7(7):1192–1199. doi: 10.1111/j.1538-7836.2009.03451.x. [DOI] [PubMed] [Google Scholar]
- 109.Uckun FM, Vassilev A, Bartell S, Zheng Y, Mahajan S, Tibbles HE. The anti-leukemic Bruton’s tyrosine kinase inhibitor alpha-cyano-beta-hydroxy-beta-methyl-N-(2,5-dibromophenyl) propenamide (LFM-A13) prevents fatal thromboembolism. Leuk Lymphoma. 2003;44(9):1569–1577. doi: 10.3109/10428190309178781. [DOI] [PubMed] [Google Scholar]
- 110.Bishton MJ, Gardiner EE, Harrison SJ, Prince HM, Johnstone RW. Histone deacetylase inhibitors reduce glycoprotein VI expression and platelet responses to collagen related peptide. Thromb Res. 2013;131(6):514–520. doi: 10.1016/j.thromres.2013.02.013. [DOI] [PubMed] [Google Scholar]
- 111.Spyridon M, Moraes LA, Jones CI, et al. LXR as a novel antithrombotic target. Blood. 2011;117(21):5751–5761. doi: 10.1182/blood-2010-09-306142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Cheli Y, Jensen D, Marchese P, et al. The Modifier of hemostasis (Mh) locus on chromosome 4 controls in vivo hemostasis of Gp6−/− mice. Blood. 2008;111(3):1266–1273. doi: 10.1182/blood-2007-09-111369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Mangin PH, Tang C, Bourdon C, et al. A humanized glycoprotein VI (GPVI) mouse model to assess the antithrombotic efficacies of anti-GPVI agents. J Pharmacol Exp Ther. 2012;341(1):156–163. doi: 10.1124/jpet.111.189050. [DOI] [PubMed] [Google Scholar]
- 114.Cosemans JM, Kuijpers MJ, Lecut C, et al. Contribution of platelet glycoprotein VI to the thrombogenic effect of collagens in fibrous atherosclerotic lesions. Atherosclerosis. 2005;181(1):19–27. doi: 10.1016/j.atherosclerosis.2004.12.037. [DOI] [PubMed] [Google Scholar]
- 115.Kuijpers MJ, Gilio K, Reitsma S, et al. Complementary roles of platelets and coagulation in thrombus formation on plaques acutely ruptured by targeted ultrasound treatment: a novel intravital model. J Thromb Haemost. 2009;7(1):152–161. doi: 10.1111/j.1538-7836.2008.03186.x. [DOI] [PubMed] [Google Scholar]
- 116.Al-Tamimi M, Gardiner EE, Thom JY, et al. Soluble glycoprotein VI is raised in the plasma of patients with acute ischemic stroke. Stroke. 2011;42(2):498–500. doi: 10.1161/STROKEAHA.110.602532. [DOI] [PubMed] [Google Scholar]
- 117.Bigalke B, Stellos K, Geisler T, et al. Expression of platelet glycoprotein VI is associated with transient ischemic attack and stroke. Eur J Neurol. 2010;17(1):111–117. doi: 10.1111/j.1468-1331.2009.02754.x. [DOI] [PubMed] [Google Scholar]
- 118.Boulaftali Y, Hess PR, Getz TM, et al. Platelet ITAM signaling is critical for vascular integrity in inflammation. J Clin Invest. 2013;123(2):908–916. doi: 10.1172/JCI65154. [DOI] [PMC free article] [PubMed] [Google Scholar]