

Summary
Small cell lung carcinoma (SCLC) is a highly lethal, smoking-associated cancer with few known targetable genetic alterations. Using genome sequencing, we characterized the somatic evolution of a genetically engineered mouse model (GEMM) of SCLC initiated by loss of Trp53 and Rb1. We identified alterations in DNA copy number and complex genomic rearrangements and demonstrated a low somatic point mutation frequency in the absence of tobacco mutagens. Alterations targeting the tumor suppressor Pten occurred in the majority of murine SCLC studied, and engineered Pten deletion accelerated murine SCLC and abrogated loss of Chr19 in Trp53; Rb1; Pten compound mutant tumors. Finally, we found evidence for polyclonal and sequential metastatic spread of murine SCLC by comparative sequencing of families of related primary tumors and metastases. We propose a temporal model of SCLC tumorigenesis with implications for human SCLC therapeutics and the nature of cancer-genome evolution in GEMMs.
Introduction
Human tumors are believed to arise through reiterated Darwinian cycles of spontaneous mutation and selection (Nowell, 1976). By the time a tumor is clinically detected, individual tumor cells harbor numerous acquired mutational events under selection (“drivers”) and an even greater number of events offering no selective advantage (“passengers”). The identification of driver mutations in human cancers remains a major obstacle for cancer genome sequencing efforts. Although several approaches have been recently described, the statistical power of these approaches is critically dependent on large sample numbers in part due to high-observed mutation frequencies, particularly in mutagen-associated cancers (Lawrence et al., 2013).
As an example of these challenges, two recent small-cell lung carcinoma (SCLC)-sequencing studies identified distinct novel putative driver alterations (Peifer et al., 2012; Rudin et al., 2012). Whether this reflects true biological differences in the tumor cohorts or differences in analytical methods is unknown. A limitation of these and many human cancer-genome characterization studies to date is the lack of rigorous in vivo functional validation. Studies that include functional data largely rely upon cultured cells, which lack many hallmark features of naturally arising tumors (Hanahan and Weinberg, 2011).
SCLC is nearly always associated with extended tobacco use and has lagged behind other solid tumors with respect to identification of targetable driver mutations (Califano et al., 2012; Jackman and Johnson, 2005). In addition, patients usually present with highly advanced, metastatic disease. Although there is often a significant response to systemic chemotherapy, the disease invariably relapses and the median 5-year overall survival is less than 5%. Moreover, surgical resection is rarely performed, and the consequent lack of available SCLC tissue is a significant barrier to molecular studies. Human SCLC harbor mutations in TP53 and RB1 at very high frequency; therefore, a mouse model of SCLC was generated by lung-specific compound deletion of Trp53 and Rb1 (hereafter referred to as “PR mSCLC” for p53, Rb1 mutant murine SCLC; Meuwissen et al., 2003). In the absence of tobacco-associated mutagens, these animals develop SCLC that progresses from small neuroendocrine bodies (NEBs) and recapitulates many features of the human disease, including frequent distant metastases to sites commonly seeded by human SCLC.
We hypothesized that several features of the PR mSCLC model were well-suited for a comparative cancer genome sequencing study. First, unlike many models, mSCLC is initiated by engineered deletion of two tumor suppressors (Rb and p53). In addition, somatic tumor-genome evolution, including the conserved oncogenic DNA copy number amplifications of Mycl1 and Nfib, has been demonstrated during tumor progression in mSCLC (Calbó et al., 2005; Dooley et al., 2011). The long latency (12–14 months) and high frequency of metastases in this model also mirrors the progression of most adult-onset human cancers that are believed to evolve over many years (Jones et al., 2008). We also predicted that the absence of tobacco mutagens would reduce the background mutation frequency and improve statistical power to detect recurrent driver mutations. Here, we characterize a panel of PR mSCLC primary and metastatic tumors at single-nucleotide resolution using exome and genome sequencing in order to elucidate mechanisms of tumor progression and identify conserved acquired somatic drivers of SCLC.
Results
Murine SCLCs Harbor Complex Genomes
We generated a PR mSCLC tissue bank consisting of primary tumors and metastases as previously described (Dooley et al., 2011; Table S1A available online). Exome sequencing was performed on 27 SCLC primary tumors and metastases isolated from six individual animals with matched control DNA isolated from tail clippings (total of 33 exomes). Fourteen tumors and paired control DNA also underwent whole-genome sequencing (WGS; total 18). Raw data were processed through a mouse-specific analysis pipeline (Extended Experimental Procedures).
Considering the role of previously characterized acquired DNA copy number alterations in this model, we first identified recurrent somatic DNA copy number alterations in primary mSCLC tumors (Extended Experimental Procedures; Tables S1B-S1D; Beroukhim et al., 2007; Mermel et al., 2011). As shown in Figures 1A–1C the most frequent alterations in somatic DNA copy number were whole chromosomal alterations, including losses of Chr19 (9 of 17 primary tumors; GISTIC q value: <1 × 10−15), followed by gain of Chr4 (6/17 primary tumors; GISTIC q value: <1 × 10−15). mSCLC tumors also harbored recurrent loss of Chr8, Chr12, Chr14, and Chr16 (GISTIC q value: 8.34 × 10−7; <1 × 10−15, <1 × 10−15, and 8.34 × 10−7, respectively) and gain of Chr16 (q value 1.6 × 10−6). In all cases, whole chromosome losses were hemizygous.
Figure 1. Genome Remodeling during mSCLC Progression.
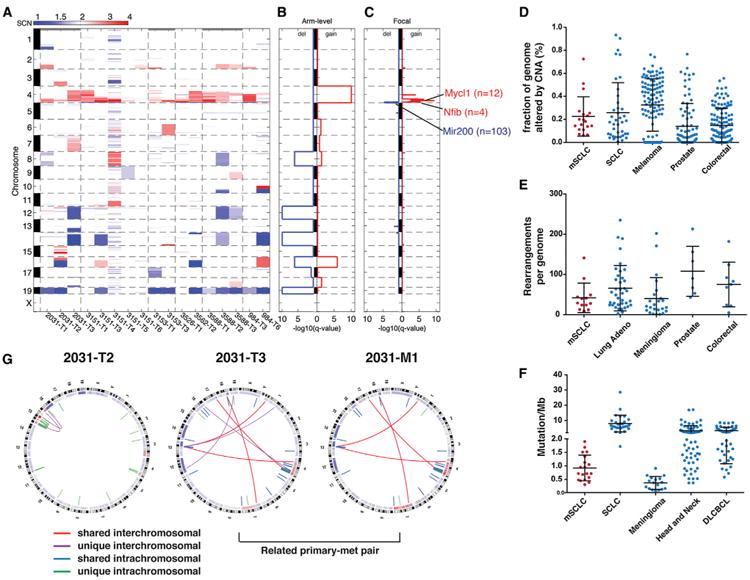
(A) Global DNA copy number alterations (CNAs) shown for mSCLC primary tumors. Blue represents copy loss; red represents copy gain. Individual tumors shown horizontally, chromosomes depicted on vertical axis.
(B) GISTIC analysis of recurrent whole-chromosome CNAs in mSCLC identifies recurrent loss of Chr19, Chr12, Chr14, and Chr16 and recurrent gain of Chr4.
(C) GISTIC analysis of recurrent focal CNAs identifies six recurrent amplification peaks on Chr4 and a single area of recurrent copy number loss on Chr4. Genes of interest are listed next to focal gains and losses, and the number of genes in the peak of the CNA is shown in parentheses.
(D) Comparison between mSCLC and human cancers with respect to the fraction of the genome altered by CNAs.
(E) Comparison of mSCLC and human cancers with respect to the number of rearrangements per tumor.
(F) mSCLCs exhibit a lower point mutation frequency compared to human cancers.
(G) Circos diagrams depicting whole-genome sequencing of mSCLC. Shared rearrangements between primary tumors and metastases identify lineage relationships. Outer band represents metaphase banding pattern; inner track shows DNA copy number alterations. Intrachromosomal rearrangements are shown as blue (shared between multiple tumors) or green (unique); interchromosomal events are shown as red lines (shared between multiple tumors) or purple (unique). See also Figure S1 and Table S1. Error bars (D–F) represent mean and SD.
We also detected focal Chr4 amplifications that harbored well-established oncogenes previously described in PR mSCLC, including Mycl1 (8/17 tumors; GISTIC q value: 3.7 × 10−7) and Nfib (4/17 tumors; GISTIC q value 1.0 × 10−4; Figure 1C; Calbó et al., 2005; Dooley et al., 2011). We identified a recurrent focal deletion encompassing Mir200a/Mir200b and the hairy enhancer of split (Hes2, Hes3, and Hes5) family of transcriptional effectors of the Notch signaling pathway (q value 2.39 × 10−5). Whether these amplified and deleted regions drive progression of mSCLC remains to be tested, but analysis with GISTIC demonstrates that these alterations occurred more frequently than expected by chance, suggesting these events provide selective advantage during tumor progression.
We identified genomic rearrangements in 14 mSCLC tumors by WGS (Table S1E; Extended Experimental Procedures). No specific classes of rearrangements were enriched in these samples, nor were recurrent rearrangements identified in mSCLC (Figure S1A). However, the vast majority of events occurred on Chr4 (Figure S1B). We investigated the process underlying the complex amplifications on Chr4 by reconstruction of individual Chr4 maps (Figure S1C). This suggested that serial rearrangement cycles, rather than a single event (i.e., chromothripsis), led to generation of the Chr4 oncogenic amplifications in a punctuated manner (Baca et al., 2013; Korbel and Campbell, 2013). Interestingly, the tumor-suppressor loci Cdkn2a and Cdkn2b are located between Nfib and Mycl1 on Chr4 and therefore may pose a selective pressure against whole chromosomal gain or chromothripsis of Chr4 as a mechanism to amplify Mycl1 and Nfib function in this setting. PCR-based validation of a subset of putative rearrangements suggested a high degree of confidence in rearrangement calls (Figure S1D; Experimental Procedures). We detected rearrangements with putative function, including a focal deletion of Magi2, an upstream component of the phosphatase and tensin homolog (PTEN)/phosphatidylinositol 3-kinase (PI3K) signaling network (Wu et al., 2000; Zmajkovicova et al., 2013). We also noted intrachromosomal rearrangements generating in-frame fusions of Abl1 kinase (Abl1-Nup214) and transcriptional regulators Cited4 (Cited4-Asap3 and 9030409GRik-Cited4; Figure S1E).
We next identified point mutations in the exonic sequences of 27 primary tumor and metastasis using muTect (Cibulskis et al., 2013). Exome sequencing revealed a mean of 0.91 protein-altering point mutations per Mb sequenced, or 27.9 protein-altering mutations per tumor (Figure S2A; Table S1F). Given the general lack of experience with these methods in model organisms, we performed two independent validation exercises that suggested these methods were accurate and estimates of mutation frequencies were valid (see Extended Experimental Procedures; Table S1G; Figure S2B).
To begin cross-species analysis of human cancer and murine SCLC genomes, we first compared the overall frequencies of DNA copy number alterations, point mutations, and rearrangements observed in mSCLC to several human tumor types (Extended Experimental Procedures). Interestingly, mSCLCs exhibited a comparable frequency of genomic rearrangements and copy number alterations compared to human cancers but harbored a significantly lower number of point mutations (Figures 1D–1F). Although the mechanisms underlying these observations remain unclear, the low point mutation frequency is likely in part a result of the absence of tobacco-associated mutagens. The high frequency of rearrangements, including complex punctuated rearrangements of Chr4, may be in part a reflection of p53 loss (Rausch et al., 2012). In order to specifically search for shared driver genes in SCLC, we also compared recurrently amplified or deleted regions within mSCLC genomes to available data from human SCLC genome-sequencing studies (Table S1H; Extended Experimental Procedures; Peifer et al., 2012; Rudin et al., 2012). Regions within Chr4 amplification and Chr14 loss in mSCLC were conserved in human SCLC. The region of recurrent amplification on Chr4 encompassing Mycl1 corresponded to a region of amplification on Chr1 in human SCLC harboring MYCL1. In addition, a set of genes frequently deleted within the Chr3p region in human SCLC, including the putative tumor suppressor FHIT, was syntenic to murine Chr14, which was subject to recurrent hemizygous loss (Wistuba et al., 2001).
The mechanisms that give rise to point mutations in this model are not the focus of this work; however, we observed a noteworthy mutation signature in mSCLC that would bear further investigation (Figures S2C and S2D). This signature was also dominant in the set of validated mutations (Extended Experimental Procedures). Interestingly, the signature was not detected in a KrasG12D; p53-null driven mouse model of lung adenocarcinoma (data not shown; D.G.M, G.G., and T.J., unpublished data). This suggested the mSCLC signature is unique to this model and not a universal feature of genetically engineered models or p53-null murine cancer cells. These mutations did not occur preferentially in regions of DNA copy number alteration or near rearrangement breakpoints (data not shown), and therefore, we suggest that this mutational signature is the product of an as yet unknown mechanism.
mSCLCs Acquire Recurrent Alterations in the Pten Tumor Suppressor Pathway
We identified two statistically significant recurrently mutated genes using MutSig, Pten and Olfr811 (Figure 2A; Table S1I; Extended Experimental Procedures). Three independent point mutations in Pten were identified and validated (Figures S2E and S2F). Review of individual Pten mutations provided strong evidence for functional impact of these mutations: the T131P mutation corresponds to a hot spot mutation site within the catalytic core of the phosphatase domain, and the T26P and R267-splice mutations are documented germline events in Cowden's syndrome (Forbes et al., 2011; Pilarski et al., 2011; Tsou et al., 1998). We also identified recurrent nonsynonymous mutations in genes encoding other Pten signaling components: Magi1, a scaffolding protein involved in shuttling Pten to the plasma membrane; Eef2k, a downstream PI3K effector involved in the regulation of translation elongation; and Ikbkb, a gene product linking nuclear factor kB to PI3K signaling (Häcker and Karin, 2006; Kotelevets et al., 2005; Zmajkovicova et al., 2013). Each of these genes was mutated in two clonally independent tumors. This suggests that disruption of Pten signaling at multiple points in the pathway may promote tumor progression in mSCLC. Indeed, ingenuity pathway analysis of all protein-altering point mutations identified in mSCLC revealed enrichment of the PI3K/PTEN networks (Table S1J). In addition, PTEN alterations have been reported in human SCLC, further supporting a conserved tumor-suppressive role (Dacic et al., 2002; Forgacs et al., 1998; Yokomizo et al., 1998).
Figure 2. mSCLCs Acquire Recurrent Pten Alterations.
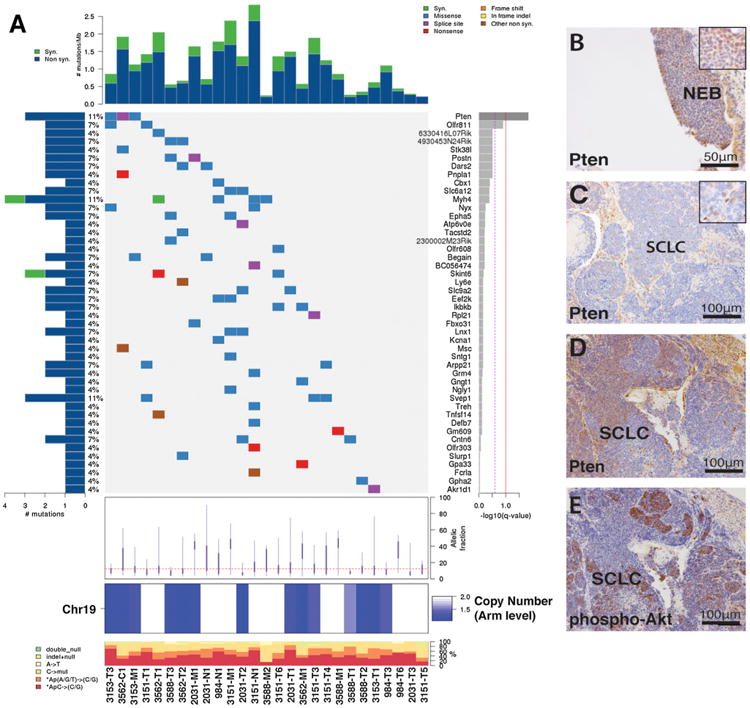
(A) CoMut plot showing the individual mSCLC tumors on the x axis and mutation information on the y axes. Two genes are considered statistically significantly mutated: Pten and Olfr811. Left-sided histogram shows percent of samples with a mutation in the given gene in the right side of plot. Average allelic fraction of mutations in individual tumors shown below mutation plot, and Chr19 copy status shown at bottom of diagram. Number of mutations/sample shown on top histogram.
(B) Pten immunohistochemistry (IHC) showing positive Pten staining in early premalignant neuroendocrine bodies (NEBs).
(C) Pten IHC depicting a Pten-negative tumor.
(D and E) Pten (D) and phospho-AKT(S473) (E) mutually exclusive IHC staining in the same mSCLC tumor.
See also Figure S2 and Table S1.
In addition to alterations in Pten signaling, we detected recurrent mutations in the semaphorin (Sema5b, Sema3c, and Sema4f) and ephrin (Epha5 and Epha7) gene families, which regulate cellular migration during embryonic development. Interestingly, axonal guidance pathways have been implicated as drivers in pancreatic cancer by recent cross-species sequencing and transposon-based screens in pancreatic cancer (Biankin et al., 2012).
We also compared genes mutated in mSCLC to two recent human-SCLC-sequencing reports (Peifer et al., 2012; Rudin et al., 2012). Because the mutational burden in human SCLC was high, we analyzed genes mutated in mSCLC relative to the published significantly mutated, hot spot, and clustered genes from both studies (Table S1K). Several genes were mutated in both mSCLC and human SCLC, including PTEN and EPHA7. However, whether this is the product of the high mutation frequency observed in human SCLC or reflects a conserved functional role for shared events remains to be determined. Of note, only one gene besides TP53 and RB1, TMEM132D, was reported as significantly mutated by both human studies. This highlights the challenges associated with genomic analysis of highly mutated human cancer genomes.
The genomic location of Pten, Chr19 28.14cM, also raised the possibility that Chr19 loss, the most frequent observed alteration in DNA copy number in mSCLC, may be driven primarily by impairment of Pten function. All tumors harboring Pten point mutations also exhibited evidence for loss of Chr19, suggesting that complete loss of Pten function may be advantageous and occur via multiple mechanisms in mSCLC.
To further define the frequency and timing of Pten loss in mSCLC, we examined Pten expression by immunohistochemistry (IHC) in early neuroendocrine bodies and in advanced tumors (Figures 2B–2D). Qualitative analysis of Pten staining revealed that the majority of high-grade mSCLCs exhibit complete loss of Pten as compared to precursor neuroendocrine bodies (Figure S2G), supporting the notion that Pten loss may facilitate tumor outgrowth. We also observed several mSCLC tumors with a mixed Pten status (Figure 2D). Although PI3K independent functions of Pten have been demonstrated, a hallmark of Pten loss is activation of the canonical PI3K/AKT signaling pathway. Therefore, we assessed Pten status and PI3K signaling by IHC for Pten and phospho-Akt (pAktS473). Pten-negative areas showed a reciprocal increase in pAkt staining by IHC (Figures 2D and 2E), implicating canonical PI3K signaling as a major output of Pten loss during mSCLC progression. In addition, a tumor harboring an inactivating Pten point mutation (T131P), and another sample with a Magi2 deletion, did not show loss of Pten IHC staining but exhibited increased pAkt staining (data not shown).
To test whether mutations in upstream signaling components impact canonical PI3K signaling in SCLC, we depleted Magi1 using small hairpin RNAs (shRNAs) in mSCLC cell lines. Consistent with this model, Magi1 knockdown resulted in increased pAkt signaling uniquely in cells with wild-type Pten status (Figure S3A). We detected modestly diminished cellular growth in response to Magi knockdown in mSCLC cells, independent of Pten status, suggesting that Magi1 was required for PI3K-independent functions in mSCLC cells, consistent with prior studies (Wegmann et al., 2004).
Pten Deletion in mSCLC Accelerates Tumorigenesis
In order to define the role of Pten loss in mSCLC, we crossed a cre-regulated, conditional Pten allele into the PR mSCLC model (hereafter referred to as PRPt for p53, Rb1 and Pten; Lesche et al., 2002). To assess tumor growth kinetics in living animals, a cohort of PRPt animals also harbored a cre-activated luciferase reporter allele. We initiated tumors in PRPt (Trp53FL/FL; Rb1FL/FL; PtenFL/FL), PRPt/+ (Trp53FL/FL; Rb1FL/FL; PtenFL/+), and PR (Trp53FL/FL; Rb1FL/FL; Pten+/+) animals by intratracheal administration of adenovirus expressing cre recombinase under the control of the neuroendocrine-specific CGRP promoter in order to selectively delete Rb1, Trp53, and Pten in pulmonary neuroendocrine cells (Sutherland et al., 2011). Measurement of in vivo luciferase activity at 4, 5, and 6 months posttumor induction revealed acceleration of tumor growth in PRPt versus PR animals (Figures 3A and 3B).
Figure 3. Pten Deletion Accelerates mSCLC Progression.
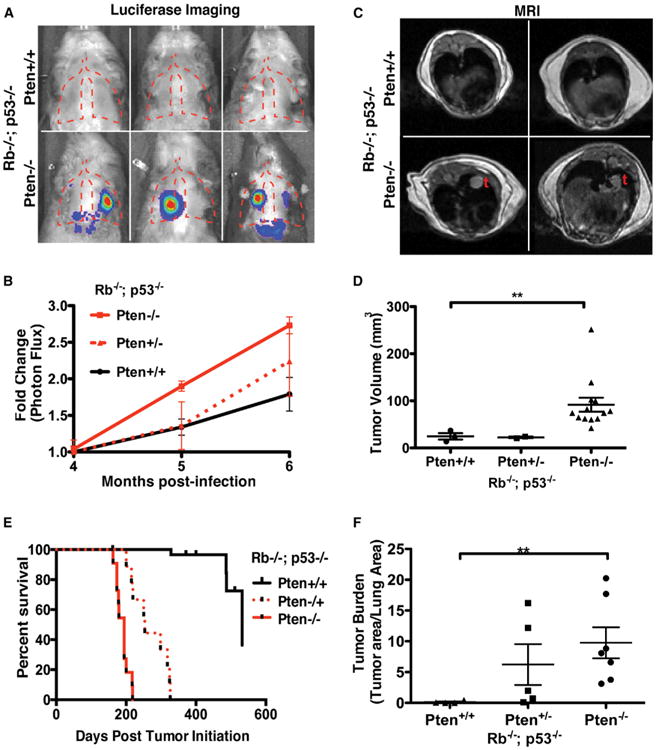
(A) Representative bright field/luminescence images.
(B) Quantitation of luminescence (photon flux) of PR and PRPt animals. Relative photon flux calculated by normalizing all time points per animal to initial measurements at 4 months postinfection.
(C) Representative axial MRI sections of PR and PRPt animals at 5 months postinfection.
(D) MRI tumor volume measurements (mm3).
(E) Overall survival of SCLC cohorts.
(F) Relative tumor burden determined by quantitative hematoxylin and eosin staining microscopy. **p < 0.01.
(B, D, and F) Mean and SEM shown by error bars. See also Figure S3 and Table S1.
We also assessed tumor burden in cohorts of PR, PRPt/+, and PRPt animals 5 months following tumor induction using small-animal MRI. We detected significantly greater tumor volume in PRPt animals (Figures 3C and 3D). Furthermore, we observed a significant reduction in mSCLC tumor latency and significantly reduced overall survival in PRPt (6.3 months) and PRPt/+ (8.3 months) compared to PR animals (17.5 months; Figure 3E). We confirmed the in vivo imaging results in cohorts of PR, PRPt/+, and PRPt animals at 5 months postinduction using quantitative histology, which showed a striking difference in tumor burden (Figure 3F). To ensure a change in tumor spectrum did not underlie the acceleration of tumor progression in PRPt animals, we performed histological analysis of tumor-bearing lung sections. The majority of the tumors identified displayed neuroendocrine features and stained positive for the neuroendocrine marker CGRP, consistent with a role for Pten loss as a driver of SCLC progression (Figures S3B, S3E, and S3H).
The state of PI3K signaling was assessed using phosphospecific IHC for Akt. High levels of pAkt were present in PRPt tumors at 6 months posttumor induction (Figures S3D and S3G). Pten loss and activated Akt signaling are known to promote proliferation and oppose apoptosis. Therefore, we assayed apoptosis and proliferation by IHC. We detected a significant increase in phospho-histone H3, a mitotic marker, in the PRPt as compared to PR tumors 6 months after tumor initiation (Figure S3I). Conversely, we observed a decrease in the levels of cleaved caspase-3, a marker of apoptosis, in Pten-null tumors (Figure S3J). These data support the tumor-suppressive role of Pten in mSCLC and provide evidence that loss of Pten can drive tumor progression, in part through increased proliferation and diminished apoptosis.
Combinations of Engineered Events Alter the Path of mSCLC Genomic Evolution
Detection of recurrent Pten point mutations and the in vivo functional studies provide evidence that Pten loss is a crucial event during mSCLC tumor progression. It is therefore reasonable to hypothesize that Pten mutation is the driving force behind Chr19 loss in PR mSCLC. To test this notion, we utilized PRPt triple-mutant mSCLC to characterize tumor-genome evolution in the context of Pten loss at the time of tumor initiation. We performed low-read-depth whole-genome sequencing of 11 PRPt (nine primary tumors and two metastases) and two PRPt/+ mSCLC tumors to characterize acquired DNA copy number alterations. These tumors were of similar size and histologic grade to PR tumors used in our initial analyses. Consistent with the hypothesis that selective advantage of Pten impairment underlies loss of Chr19 in this model, all PRPt mSCLC exhibited a DNA copy number of two for Chr19 (Figures 4A and 4B; Table S1L). Therefore, deletion of a single exon of Pten provided the selective advantage for loss of an entire chromosome in this model. Interestingly, two PRPt/+ tumors exhibited hemizygous loss of Chr19, and review of the sequencing reads demonstrated that the mutant chromosome 19 was retained, suggesting additional selective advantage of complete Pten loss (Figure 4C).
Figure 4. Pten Deletion Alters Genome Evolution in mSCLC.
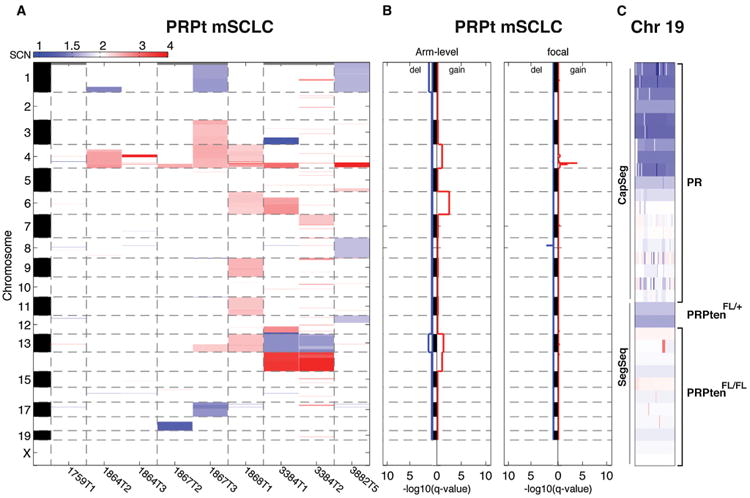
(A) CNA heatmap for PRPt mSCLC tumors analyzed by SegSeq.
(B) GISTIC results for recurrent CNAs. Note the absence of Chr19 deletion and presence of Chr4 focal amplification.
(C) Chr19 copy number map for all primary tumors analyzed. Genotype is listed to the right. Analysis method (CapSeg or SegSeq) listed to the left.
See also Figure S4 and Table S1.
Despite the decreased tumor latency and absence of Chr19 loss in the PRPt mSCLC tumors, we detected recurrent focal amplifications on Chr4 in these samples (Figure 4B; Table S1M). Although GISTIC analysis did not show Mycl1 within the predicted amplification peak, our sequencing read depth was designed to detect whole-chromosome gains and losses rather than focal alterations. Therefore, we used quantitative PCR to assess Mycl1 amplification in these samples. Mycl1 copy number was amplified in these tumors, suggesting an important role for Mycl1 amplification during the initial outgrowth of mSCLC (Figure S4).
Intratumoral Clonal Heterogeneity in mSCLC
We established tumor lineage relationships between primary tumors and metastases from individual mice by identification of shared DNA rearrangements and point mutations (example, Figure 1G). Using this approach, we identified related primary-metastasis pairs from six animals, including three tumor families from which we sequenced multiple metastatic lesions (Table S1N). Although shared somatic mutations can identify basic lineage relationships between tumors, more sophisticated methods are required to reconstruct the clonal architecture of tumor progression. We first examined the allelic fraction of somatic mutations in mSCLC tumors. The observed distribution of allelic fractions for all somatic mutations observed in primary tumors revealed an abundance of mutations with low allelic fraction. However, metastases exhibited a greater proportion of mutations with high allelic fraction, consistent with a clonal bottleneck during metastatic seeding (Figures S5A and S5B).
The allelic fraction of a mutation depends on tumor purity, local DNA copy number, and the fraction of cells in a tumor that harbor the mutation. Therefore, in order to systematically characterize the clonal structure of PR mSCLC tumors, we used ABSOLUTE, a method that utilizes the copy number of DNA in the vicinity of a mutation and tumor purity to estimate the fraction of cancer cells harboring a given mutation, called the cancer cell fraction (CCF) (Carter et al., 2012; Landau et al., 2013). ABSOLUTE analysis confirmed the presence of clonal heterogeneity in all mSCLC tumors, with a range of two to five subclones within individual tumors (Tables S1O–S1Q). We compared the CCF for individual mutations between primary tumors and metastases using two-dimensional plots in order to define the clonal structure of mSCLC (Figure 5). Clonal lineages were identified by clustering mutations exhibiting shared CCF (Landau et al., 2013). As a tumor subclone colonizes metastatic sites, it passes through population bottlenecks in which the preexisting mutations are enriched and additional mutations are subsequently acquired. We denote clonal lineages by a common nomenclature (for example, clone 1a is a descendent of clone 1 and clone 1b is a descendent of clone 1a, whereas clone 2 is a related, but independent, sibling of clone 1). The number of mutations delineating each clonal transition is shown in Figure 5, and all mutations in the tumor families detailed below are annotated with the CCF and subclone assignment in Tables S1O–S1Q.
Figure 5. Clonal Evolution of mSCLC during Metastatic Spread.
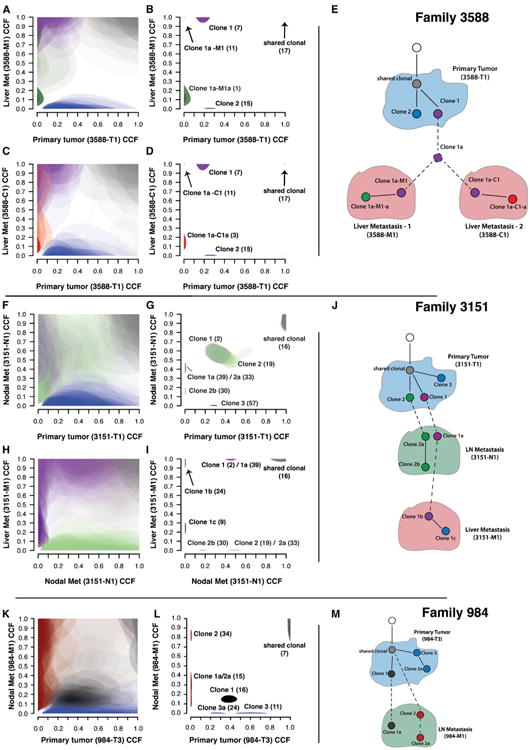
Each tier (A–E, F–J, and K–M) shows ABSOLUTE analysis of related primary and metastatic tumors in an individual animal. Left column of plots (A), (C), (F), (H), and (K) shows unclustered CCF results for individual mutations in tumor pairs. Each cloud represents the 95% confidence interval of the predicted CCF for each mutation. Middle plots (B, D, G, I, and L) show the 95% confidence intervals following a Bayesian clustering procedure that groups mutations into predicted subclones. Clonal models based on ABSOLUTE results are shown in (E), (J), and (M). Dashed lines represent assumed transitions that are not directly observed in the data. In all panels, cloud color denotes membership in corresponding node (clone) in the model diagram. (A–D) CCF results demonstrating a single subclone (purple) from the primary tumor seeded two independent liver metastases. (F and G) shows multiple subclones from the primary tumor (clone 1 and clone 2) seeding a lymph node metastasis. Clone 1b (purple), a descendent of clone 1a, seeded a tertiary liver metastasis from the lymph node (H and I). (K and L) CCF results showing polyclonal seeding of a lymph node met (gray, clone 1a, and red, clone 2).
See also Figure S5 and Table S1.
Parallel Seeding of Multiple Liver Metastases in mSCLC
From animal 3588, we sequenced three primary tumors and two independent liver metastases (Figures 5A–5E; Table S1O). Both liver metastases (3588-C1 and 3588-M1) shared 17 clonal mutations with one primary tumor, 3588-T1, demonstrating that these arose from a single primary tumor. 3588-T1 harbored two independent subclones existing at approximately 0.2 CCF (clone 1, purple) and 0.25 CCF (clone 2, blue; see Figures 5B–5D). Interestingly, both liver metastases originated from closely related subclones (clone 1a-M1 and clone 1a-C1) that were descended from a common ancestor (clone 1a). Clone 1a was a descendent of clone 1, as demonstrated by the enrichment of the CCF of clone 1 mutations in both descendent metastases (Figures 5B and 5D, purple clusters). We could not discriminate if clone 1a was present in the primary tumor outside of the sampled region or below the level of detection or whether it first seeded another site prior to founding the liver metastases. Each metastatic lesion also continued to evolve during outgrowth as evidenced by acquisition of subclonal mutations (clone 1a-M1-a, green, and clone 1a-C1-a, red; Figures 5B, 5D, and 5E).
Polyclonal Seeding of Local Lymph Node Metastases in mSCLC
We sequenced five primary tumors, one thoracic lymph node metastasis, and one liver metastasis from animal 3151. Again, in this case, both metastases shared acquired clonal mutations with one primary tumor, confirming their shared origin (light gray clusters, Figures 5G and 5I; Table S1P). Comparison of the primary tumor (3151-T1) and lymph node metastasis (3151-N1) revealed a large cluster of mutations (green-light purple central cluster, Figure 5G) present at subclonal CCF in both the primary tumor and metastasis. A single-cell-origin model would require mutations to become clonal in a descendent metastasis. Therefore, these data were inconsistent with monoclonal origin of the lymph node metastasis (Figures 5F–5J). Comparison of the lymph node and liver metastases (3151-M1) revealed that only a subset of mutations in this cluster was shared with the liver metastasis (Figures 5H and 5I; clone 1 mutations). In addition, mutations in clone 1a from the nodal metastasis (3151-N1) were enriched to a clonal CCF (1.0) in the distant liver metastasis (Figure 5I). We therefore concluded that two related primary tumor subclones (clone 1, light purple, and clone 2, green; Figure 5G) seeded the lymph node metastasis and gave rise to clones 1a and 2a (Figure 5G). However, only one of these clones spread to the liver (clone 1b, a direct descendent of clone 1a from the lymph node metastasis; Figure 5J). Clone 1b was the founding clone of the liver metastasis and was a recent descendent of clone 1a, as shown by the enrichment of clone 1a CCF (∼0.5 to 1.0) in the distant liver metastasis (3151-M1; Figure 5I). We cannot formally exclude the possibility that two highly related clone 1 cells spread directly from the primary tumor to both the lymph node and liver metastasis. However, in this scenario, we would not have expected all clone 1a mutations to be shared with the liver metastasis, as we observed. Therefore, the most parsimonious explanation for the observed data is that two primary tumor subclones first seeded the nodal metastasis and that clone 1a further evolved into clone 1b and founded a tertiary liver metastasis.
Two primary tumors and one lymph node metastasis were sequenced from animal 984. Shared somatic mutations demonstrated that primary tumor 984-T3 founded the lymph node metastasis 984-M1 (Table S1Q). Analysis of this relationship using ABSOLUTE revealed that clone 1 (dark gray, Figures 5K and 5L) was present at subclonal CCF in both the primary tumor and metastasis. This pattern is also inconsistent with monoclonal origin of the nodal metastasis and indicated that multiple primary tumor (984-T3) subclones (clone 1, dark gray, and clone 2, red) spread to the local draining lymph node. Although clone 2 mutations were not detected in the primary tumor, the presence of shared clonal mutations between 984-T3 and 984-M1 (light gray cluster, Figure 5L) confirmed that both subclones within the local lymph node arose from a single primary tumor (984-T3; Figure 5M). In each of these analyses, although we identified specific mutations present within individual subclones, our data did not address whether individual mutations acted as drivers or passengers.
Discussion
Comparative Genomics to Identify Driver Genes in Cancer
A longstanding goal of cancer genetics has been to exploit the evolutionary conservation of the major tumor-suppressive and proto-oncogenic cellular pathways between humans and mice for biological discovery and preclinical modeling. Recently, this has included analysis of acquired mutations in the genomes of mouse cancer models. For example, focused DNA sequencing of known proto-oncogenes in genetically engineered mouse models initiated by “weak” cancer initiators has identified somatically acquired activating mutations in Hras and Notch1 (O'Neil et al., 2006; Podsypanina et al., 2004). In addition, small-scale exome and genome sequencing of murine models of leukemia and breast cancers has identified conserved somatic Jak1 and Trp53 mutations (Wartman et al., 2011; Yuan et al., 2012).
The present study is to our knowledge the most complete description of the somatic genome of a genetically engineered mouse cancer model to date and also quantitatively assesses intratumoral clonal heterogeneity and clonal evolution during metastatic colonization. In the absence of tobacco mutagens, we detected relatively few somatic point mutations compared to human SCLC and mutagen-induced murine tumors (Matsushita et al., 2012). The reduced mutational “noise” facilitated identification of Pten loss as an important driver of tumor progression in mSCLC. We detected recurrent somatic alterations targeting Pten, including point mutations previously described in human cancer and Cowden syndrome. In addition, we detected acquired point mutations in other components of the PTEN/PI3K pathway, including Magi1, Eef2k, Ikbkb, Insr, and Bcar1. The majority of PR mSCLC tumors harbor a mutation in this pathway and/or Chr19 loss. We also provide a functional validation of the role of Pten by compound deletion of Pten in the autochthonous mSCLC model. These results are consistent with a prior publication, suggesting engineered Pten loss accelerates early tumorigenesis in a similar SCLC model (Song et al., 2012), and we additionally characterize the effect of Pten deletion at all stages of SCLC progression, including a dramatic decrease in survival of PRPt animals.
Recent sequencing of human SCLC identified multiple mutations in the PTEN/PI3K pathway, including PTEN, PIK3CB, PIK3R3, MAGI1, and MAGI2 mutations. Although PTEN was not identified as a recurrent target of deletion in SCLC, Chr10 copy number loss encompassing PTEN was evident in one study (Peifer et al., 2012). In addition, prior studies have demonstrated PTEN alterations in approximately 20% of SCLC (Dacic et al., 2002; Forgacs et al., 1998; Yokomizo et al., 1998). Therefore, we suggest that PR mSCLC models a subclass of human SCLC harboring alterations in the PTEN pathway.
Our data support a model in which Pten acts primarily as a classical, rather than haploinsufficient, tumor suppressor in mSCLC. First, Pten protein expression was lost completely in the majority of high-grade SCLC. In addition, all Pten point mutations occurred in tumors with copy number loss at Chr19, suggesting that Pten function was completely abrogated in these cells. Finally, PRPt/+ animals showed evidence for loss of the wild-type (WT) Pten allele, suggesting additional selective advantage of complete Pten loss.
Single Locus Control of Chromosomal Alterations in Cancer
Whole-chromosome gains and losses are frequently observed in human cancers, yet it remains unclear whether the selective advantage for these events results from one or multiple loci. Several studies in mouse models have suggested that multiple loci confer selective advantage for whole chromosome gain and loss. For example, Kras-LA2 animals developed spontaneous lung adenomas, and these tumors frequently exhibited whole-chromosome gains of the LA2-bearing mutant Chr6, whereas focal Kras-LA2 amplifications were uncommon (Sweet-Cordero et al., 2006; To et al., 2011). This led to the speculation that additional oncogenic Chr6 loci are under selection, including other components of Mapk signaling (To et al., 2011). In addition, prior studies of radiation-induced lymphomas from Trp53+/− animals identified frequent loss of heterozygosity (LOH) at Chr19 and focal deletions involving Pten (Mao et al., 2003). Interestingly, whole-chromosome loss of Chr19 was also noted in Pten+/− and Pten+/−; Trp53+/− lymphomas and was interpreted as evidence for the existence of additional tumor-suppressor loci.
The data presented in this study implicate a single gene, Pten, at the driving force behind loss of Chr19 in mSCLC. Of note, Mao et al. (2004) showed frequent whole-chromosome loss of Chr3 in radiation-induced lymphomas in Trp53+/− mice and identified Fbxw7 as a candidate tumor suppressor in a small focal deletion. Heterozygous loss of Fbxw7 accelerated tumorigenesis and abrogated Fbxw7 LOH, which was inferred to reflect maintenance of the entire chromosome. However, these studies restricted LOH analysis to the Fbxw7 locus. The data presented here extend the concept that a selective advantage for impairment of a single locus can drive loss of an entire chromosome and definitively demonstrate retention of Chr19 in PRPt mSCLC using whole-genome methods. The fact that no PRPt tumor analyzed exhibited DNA copy number loss of Chr19 is a powerful demonstration that loss of the Pten tumor suppressor is the driving selective mechanism behind loss of Chr19 in PR mSCLC.
Patterns of Clonal Progression in Msclc
The description of clonal evolution between primary tumors and metastases in solid human malignancies has been achieved primarily by sequencing several distinct tumor regions (Campbell et al., 2010; Gerlinger et al., 2012; Yachida et al., 2010). However, in contrast to most human biopsies, which sample only a small part of a tumor, we utilized approximately 50% of the individual mSCLC tumor mass for nucleic acid preparation. Thus, a large fraction of tumor sampling coupled with a deep sequencing coverage (approximately 100×) may have helped elucidate the patterns of clonal evolution between primary tumors and metastases in the mSCLC model. We also capitalized on the high frequency of macroscopic metastases in the mSCLC model and our ability to harvest these diverse sites of disease at animal necropsy. We believe our finding of spread of multiple primary tumor subclones to a local lymph node metastasis is unprecedented and the first demonstration of polyclonal seeding of metastases using deep sequencing. Considering the role of the metastatic niche in mediating treatment resistance (Gilbert and Hemann, 2010), the fact that multiple tumor subclones establish themselves in environments such as the lymph node may have therapeutic implications.
We provide evidence of spread of a tumor subclone from a lymph node metastasis to a distant site (Figure 5J). Sequential spread of metastasis from one site to another has also been demonstrated in human pancreatic cancer (Campbell et al., 2010; Yachida et al., 2010). These and our data raise the possibility that lymph nodes can act as the site of collection of multiple tumor subclones and may serve as a gateway for distant metastases. These data are consistent with the prognostic implication of sentinel lymph node biopsy in several cancer types (Chen et al., 2006). However, whether seeding of lymph node metastases prior to systemic dissemination is a more uniform property of cancer metastasis will require additional studies. In addition, it will be important to determine if additional genetic and or epigenetic alterations acquired in the local lymph node microenvironment contribute to systemic dissemination. The ability to illuminate the dynamic clonal architecture within metastatic mSCLC tumors highlights the opportunity to more completely dissect clonal metastatic progression in this experimentally tractable system.
A Model for Tumorigenesis in SCLC
The genomic and functional data presented here begin to establish a framework for tumorigenesis in SCLC (Figure 6). The near-ubiquitous presence of TP53 and RB1 mutations in human SCLC suggests that these are among the earliest events in these tumors. In this genetic context, we believe Mycl1 amplification events provide a strong proliferative advantage. Although the clonality analysis described in this study did not allow for characterization of high-level amplifications, the ease of detection of the Chr4 amplifications suggests that these amplifications arise during early outgrowth of the primary tumor. In addition, the occurrence of focal Chr4 amplifications in the setting of engineered Pten deletion is a strong demonstration of the requisite nature of Mycl1 amplification in early mSCLC outgrowth. These data also suggest that Pten mutations are selected for at later stages of tumor progression, a notion supported by our Pten IHC analysis showing tumors with areas both positive and negative for Pten expression.
Figure 6. Model of SCLC Tumorigenesis.
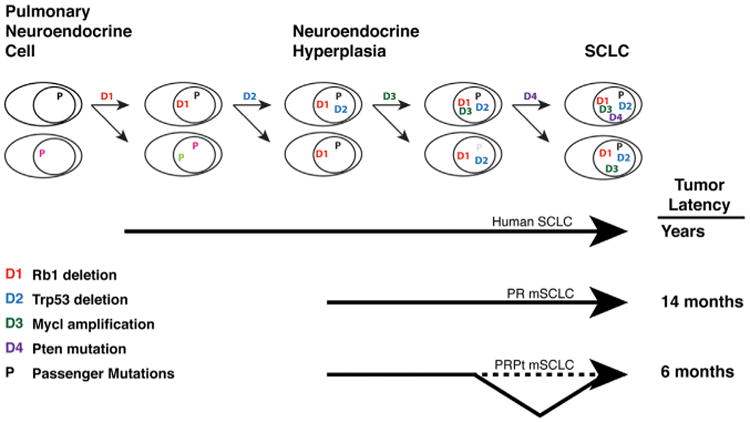
Stepwise acquisition of recurrent driver mutations identified by comparative SCLC sequencing (D1–D4) promotes murine SCLC progression from the pulmonary neuroendocrine cell to bona fide SCLC. As additional drivers are engineered into the mouse model, tumor latency is reduced and progression is accelerated. P, passenger mutations fixed in the tumor population during progression through the series of bottlenecks.
Therapeutic targeting of Pten signaling with available high-potency inhibitors of PI3K might therefore be warranted for human SCLC. However, although we detect increased phospho-Akt in advanced mSCLC, it is not certain that the effects of Pten loss are entirely mediated via increased PI3K activity. In addition, increased MYC DNA copy number or gene expression has been shown to mediate primary resistance to PI3K inhibition (Ilic et al., 2011; Shepherd et al., 2013). Considering the frequent occurrence of Chr4 amplifications involving Mycl1, combination therapy targeting L-myc or biological effectors of L-myc may be necessary to achieve effective responses.
Implications for Tumor Evolution in Mouse Cancer Models
Characterization of PR tumors by exome and genome sequencing revealed a highly molded somatic genome with a low somatic point mutation frequency relative to human cancers. Interestingly, PRPt tumors exhibited a less-complex tumor genome by DNA copy number analysis. This may reflect the addition of a third driver event (Pten loss) at tumor initiation that bypasses a critical bottleneck during tumor outgrowth. This notion is supported by previous studies showing recurrent acquisition of activating oncogene mutations in models initiated by transgenes with relatively modest impact on proliferation (Podsypanina et al., 2004). Therefore, although the initiating engineered mutations are often credited with the entire tumor phenotype, somatically acquired events should be considered when interpreting results of both discovery efforts and preclinical studies using mouse cancer models. The work described here demonstrates the potential of harnessing the shared evolution of GEMMs and human cancers using comparative genome sequencing to identify acquired drivers of cancer progression and dissect stepwise tumorigenesis.
Experimental Procedures
Mouse Model
p53fl/fl;Rbfl/fl and p53fl/fl;Rbfl/fl;Rosa26LSL-Luc/LSL-Luc mice have been previously described (Dooley et al., 2011; Rosa26LSL-Luc/LSL-Luc mice are from E. Jackson and T.J., unpublished data). To obtain p53fl/fl;Rbfl/fl;Rosa26LSL-Luc/LSL-Luc; Ptenfl/fl mice, we obtained Ptenfl/fl animals from Jackson Laboratories (Ptentm1Hwu/j; Lesche et al., 2002). Tumors were initiated by adenoviral delivery of Cre, as described previously (DuPage et al., 2009). The Massachusetts Institute of Technology (MIT) Institutional Animal Care and Use Committee approved all animal studies and procedures. Primary tumors and metastases from the mouse model were dissected and snap-frozen for DNA and/or RNA isolation as previously described (Dooley et al., 2011). In parallel, part of each tumor was kept for histology to verify SCLC features. Live-animal imaging was performed using an IVIS Spectrum (PerkinElmer) and Varian 7T/310/ASR MRI system (Varian/Agilent Technologies).
Genome, Exome Sequencing
Whole-exome sequencing was performed as previously described (Gnirke et al., 2009). Whole-genome sequencing was performed using a protocol developed for human whole-genome studies (Berger et al., 2011). Low-coverage whole-genome sequencing and DNA copy number analysis was performed as previously described (Dooley et al., 2011). Mutations were identified using MuTect (Cibulskis et al., 2013). Recurrence was assessed using MutSig 1.0 with minor modifications for mouse data (Berger et al., 2011; Getz et al., 2007). All somatic mutations were reviewed manually, and validation of selected mutations was performed by targeted resequencing using microfluidic PCR (Access array system; Fluidigm) and the MiSeq sequencing system (Illumina). Genomic rearrangements were identified by dRanger (Berger et al., 2011). DNA copy number alterations were identified from exome data using CapSeg (A.M., B. Hernandez, M. Meyerson, G.G., and S.L.C., unpublished data), and GISTIC 2.0 was used to identify recurrent alterations (Beroukhim et al., 2010; Mermel et al., 2011). SegSeq was used to identify copy number alterations (CNAs) from WGS data (Chiang et al., 2009). See Extended Experimental Procedures for additional details.
Immunohistochemical Analysis
IHC was performed on formalin-fixed paraffin-embedded 4 μm sections using the ImmPRESS Peroxidase Polymer Detection Kit (Vector Laboratories) with recommended dilutions of primary antibodies (see Extended Experimental Procedures).
Murine SCLC Cell Line Functional Experiments
Murine SCLC cell lines were maintained as previously described (Dooley et al., 2011). Broad TRC shRNAs (shMagi1_83: TRCN0000079083, shMagi1_83: TRCN0000079084) were used for Magi1 knockdown. Following infection with Magi1 or control shRNA-expressing lentiviruses, cells were selected using puromycin (Sigma). After puromycin selection, cells viability was assessed by Cell Titer Glo (Promega).
ABSOLUTE Analysis and Deductive Logic of Clonal Evolution Mapping
ABSOLUTE was performed as previously described (Carter et al., 2012; Landau et al., 2013). All mutations that define clonal lineages underwent extensive manual review using IGV (Robinson et al., 2011) to ensure models were generated using high-confidence mutations.
Supplementary Material
Acknowledgments
We acknowledge Denise Crowley and the Swanson Biotechnology Core Facility for histology preparation and IHC; Harold Varmus, Nadya Dimitrova, and Eric L. Snyder for critical review of the manuscript; and Peter Campbell for a fruitful discussion. This work was supported by the Ludwig Center for Molecular Oncology at MIT (to T.J.), the Howard Hughes Medical Institute (to T.J.), the National Human Genome Research Institute (to S.G. and G.G.), NIH-NCI Career Development award K08CA160658 (to D.G.M.), and a Hope Funds for Cancer Research Fellowship (to T.P.). G.G. is the Paul C. Zamecnik, MD, Chair in Oncology at Massachusetts General Hospital. T.J. is the David H. Koch Professor of Biology and a Daniel K. Ludwig Scholar at MIT. The authors wish to dedicate this paper to the memory of Officer Sean Collier, for his caring service to the MIT community and for his sacrifice.
Footnotes
Accession Numbers: The NCBI BioProject accession number for the sequences reported in this paper is PRJNA223640.
Supplemental Information: Supplemental Information includes Extended Experimental Procedures, five figures, and one table and can be found with this article online at http://dx.doi.org/10.1016/j.cell.2014.02.031.
Author Contributions: D.G.M., T.J., and G.G. designed the study, and D.G.M. wrote the manuscript. D.G.M. and T.P. equally contributed to biological interpretation of sequencing results and performed experiments. A.T.-W. and C.S. contributed equally to analysis of sequencing data. S.C. performed ABSOLUTE analysis of sequencing data. Inference of clonal evolution was performed by S.C., D.G.M., T.P., and A.T.-W. G.G. and T.J. supervised data analysis and experiments.
References
- Baca SC, Prandi D, Lawrence MS, Mosquera JM, Romanel A, Drier Y, Park K, Kitabayashi N, MacDonald TY, Ghandi M, et al. Punctuated evolution of prostate cancer genomes. Cell. 2013;153:666–677. doi: 10.1016/j.cell.2013.03.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berger MF, Lawrence MS, Demichelis F, Drier Y, Cibulskis K, Siva-chenko AY, Sboner A, Esgueva R, Pflueger D, Sougnez C, et al. The genomic complexity of primary human prostate cancer. Nature. 2011;470:214–220. doi: 10.1038/nature09744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beroukhim R, Getz G, Nghiemphu L, Barretina J, Hsueh T, Linhart D, Vivanco I, Lee JC, Huang JH, Alexander S, et al. Assessing the significance of chromosomal aberrations in cancer: methodology and application to glioma. Proc Natl Acad Sci USA. 2007;104:20007–20012. doi: 10.1073/pnas.0710052104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beroukhim R, Mermel CH, Porter D, Wei G, Raychaudhuri S, Donovan J, Barretina J, Boehm JS, Dobson J, Urashima M, et al. The landscape of somatic copy-number alteration across human cancers. Nature. 2010;463:899–905. doi: 10.1038/nature08822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biankin AV, Waddell N, Kassahn KS, Gingras MC, Muthuswamy LB, Johns AL, Miller DK, Wilson PJ, Patch AM, Wu J, et al. Pancreatic cancer genomes reveal aberrations in axon guidance pathway genes. Nature. 2012;491:399–405. doi: 10.1038/nature11547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calbó J, Meuwissen R, van Montfort E, van Tellingen O, Berns A. Genotype-phenotype relationships in a mouse model for human small-cell lung cancer. Cold Spring Harb Symp Quant Biol. 2005;70:225–232. doi: 10.1101/sqb.2005.70.026. [DOI] [PubMed] [Google Scholar]
- Califano R, Abidin AZ, Peck R, Faivre-Finn C, Lorigan P. Management of small cell lung cancer: recent developments for optimal care. Drugs. 2012;72:471–490. doi: 10.2165/11597640-000000000-00000. [DOI] [PubMed] [Google Scholar]
- Campbell PJ, Yachida S, Mudie LJ, Stephens PJ, Pleasance ED, Stebbings LA, Morsberger LA, Latimer C, McLaren S, Lin ML, et al. The patterns and dynamics of genomic instability in metastatic pancreatic cancer. Nature. 2010;467:1109–1113. doi: 10.1038/nature09460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carter SL, Cibulskis K, Helman E, McKenna A, Shen H, Zack T, Laird PW, Onofrio RC, Winckler W, Weir BA, et al. Absolute quantification of somatic DNA alterations in human cancer. Nat Biotechnol. 2012;30:413–421. doi: 10.1038/nbt.2203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen SL, Iddings DM, Scheri RP, Bilchik AJ. Lymphatic mapping and sentinel node analysis: current concepts and applications. CA Cancer J Clin. 2006;56:292–309. doi: 10.3322/canjclin.56.5.292. [DOI] [PubMed] [Google Scholar]
- Chiang DY, Getz G, Jaffe DB, O’Kelly MJ, Zhao X, Carter SL, Russ C, Nusbaum C, Meyerson M, Lander ES. High-resolution mapping of copy-number alterations with massively parallel sequencing. Nat Methods. 2009;6:99–103. doi: 10.1038/nmeth.1276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cibulskis K, Lawrence MS, Carter SL, Sivachenko A, Jaffe D, Sougnez C, Gabriel S, Meyerson M, Lander ES, Getz G. Sensitive detection of somatic point mutations in impure and heterogeneous cancer samples. Nat Biotechnol. 2013;31:213–219. doi: 10.1038/nbt.2514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dacic S, Finkelstein SD, Baksh FK, Swalsky PA, Barnes LE, Yousem SA. Small-cell neuroendocrine carcinoma displays unique profiles of tumor-suppressor gene loss in relationship to the primary site of formation. Hum Pathol. 2002;33:927–932. doi: 10.1053/hupa.2002.126875. [DOI] [PubMed] [Google Scholar]
- Dooley AL, Winslow MM, Chiang DY, Banerji S, Stransky N, Dayton TL, Snyder EL, Senna S, Whittaker CA, Bronson RT, et al. Nuclear factor I/B is an oncogene in small cell lung cancer. Genes Dev. 2011;25:1470–1475. doi: 10.1101/gad.2046711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DuPage M, Dooley AL, Jacks T. Conditional mouse lung cancer models using adenoviral or lentiviral delivery of Cre recombinase. Nat Protoc. 2009;4:1064–1072. doi: 10.1038/nprot.2009.95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forbes SA, Bindal N, Bamford S, Cole C, Kok CY, Beare D, Jia M, Shepherd R, Leung K, Menzies A, et al. COSMIC: mining complete cancer genomes in the Catalogue of Somatic Mutations in Cancer. Nucleic Acids Res. 2011;39(Database issue):D945–D950. doi: 10.1093/nar/gkq929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forgacs E, Biesterveld EJ, Sekido Y, Fong K, Muneer S, Wistuba II, Milchgrub S, Brezinschek R, Virmani A, Gazdar AF, Minna JD. Mutation analysis of the PTEN/MMAC1 gene in lung cancer. Oncogene. 1998;17:1557–1565. doi: 10.1038/sj.onc.1202070. [DOI] [PubMed] [Google Scholar]
- Gerlinger M, Rowan AJ, Horswell S, Larkin J, Endesfelder D, Gronroos E, Martinez P, Matthews N, Stewart A, Tarpey P, et al. Intratumor heterogeneity and branched evolution revealed by multiregion sequencing. N Engl J Med. 2012;366:883–892. doi: 10.1056/NEJMoa1113205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Getz G, Höfling H, Mesirov JP, Golub TR, Meyerson M, Tibshirani R, Lander ES. Comment on “The consensus coding sequences of human breast and colorectal cancers”. Science. 2007;317:1500. doi: 10.1126/science.1138764. [DOI] [PubMed] [Google Scholar]
- Gilbert LA, Hemann MT. DNA damage-mediated induction of a chemoresistant niche. Cell. 2010;143:355–366. doi: 10.1016/j.cell.2010.09.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gnirke A, Melnikov A, Maguire J, Rogov P, LeProust EM, Brockman W, Fennell T, Giannoukos G, Fisher S, Russ C, et al. Solution hybrid selection with ultra-long oligonucleotides for massively parallel targeted sequencing. Nat Biotechnol. 2009;27:182–189. doi: 10.1038/nbt.1523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Häcker H, Karin M. Regulation and function of IKK and IKK- related kinases. Sci STKE. 2006;2006:re13. doi: 10.1126/stke.3572006re13. [DOI] [PubMed] [Google Scholar]
- Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144:646–674. doi: 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- Ilic N, Utermark T, Widlund HR, Roberts TM. PI3K-targeted therapy can be evaded by gene amplification along the MYC-eukaryotic translation initiation factor 4E (eIF4E) axis. Proc Natl Acad Sci USA. 2011;108:E699–E708. doi: 10.1073/pnas.1108237108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackman DM, Johnson BE. Small-cell lung cancer. Lancet. 2005;366:1385–1396. doi: 10.1016/S0140-6736(05)67569-1. [DOI] [PubMed] [Google Scholar]
- Jones S, Chen WD, Parmigiani G, Diehl F, Beerenwinkel N, Antal T, Traulsen A, Nowak MA, Siegel C, Velculescu VE, et al. Comparative lesion sequencing provides insights into tumor evolution. Proc Natl Acad Sci USA. 2008;105:4283–4288. doi: 10.1073/pnas.0712345105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korbel JO, Campbell PJ. Criteria for inference of chromothripsis in cancer genomes. Cell. 2013;152:1226–1236. doi: 10.1016/j.cell.2013.02.023. [DOI] [PubMed] [Google Scholar]
- Kotelevets L, van Hengel J, Bruyneel E, Mareel M, van Roy F, Chastre E. Implication of the MAGI-1b/PTEN signalosome in stabilization of adherens junctions and suppression of invasiveness. FASEB J. 2005;19:115–117. doi: 10.1096/fj.04-1942fje. [DOI] [PubMed] [Google Scholar]
- Landau DA, Carter SL, Stojanov P, McKenna A, Stevenson K, Lawrence MS, Sougnez C, Stewart C, Sivachenko A, Wang L, et al. Evolution and impact of subclonal mutations in chronic lymphocytic leukemia. Cell. 2013;152:714–726. doi: 10.1016/j.cell.2013.01.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lawrence MS, Stojanov P, Polak P, Kryukov GV, Cibulskis K, Sivachenko A, Carter SL, Stewart C, Mermel CH, Roberts SA, et al. Mutational heterogeneity in cancer and the search for new cancer-associated genes. Nature. 2013;499:214–218. doi: 10.1038/nature12213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lesche R, Groszer M, Gao J, Wang Y, Messing A, Sun H, Liu X, Wu H. Cre/loxP-mediated inactivation of the murine Pten tumor suppressor gene. Genesis. 2002;32:148–149. doi: 10.1002/gene.10036. [DOI] [PubMed] [Google Scholar]
- Mao JH, Wu D, Perez-Losada J, Nagase H, DelRosario R, Balmain A. Genetic interactions between Pten and p53 in radiation-induced lymphoma development. Oncogene. 2003;22:8379–8385. doi: 10.1038/sj.onc.1207083. [DOI] [PubMed] [Google Scholar]
- Mao JH, Perez-Losada J, Wu D, Delrosario R, Tsunematsu R, Nakayama KI, Brown K, Bryson S, Balmain A. Fbxw7/Cdc4 is a p53-dependent, haploinsufficient tumour suppressor gene. Nature. 2004;432:775–779. doi: 10.1038/nature03155. [DOI] [PubMed] [Google Scholar]
- Matsushita H, Vesely MD, Koboldt DC, Rickert CG, Uppaluri R, Magrini VJ, Arthur CD, White JM, Chen YS, Shea LK, et al. Cancer exome analysis reveals a T-cell-dependent mechanism of cancer immunoediting. Nature. 2012;482:400–404. doi: 10.1038/nature10755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mermel CH, Schumacher SE, Hill B, Meyerson ML, Beroukhim R, Getz G. GISTIC2.0 facilitates sensitive and confident localization of the targets of focal somatic copy-number alteration in human cancers. Genome Biol. 2011;12:R41. doi: 10.1186/gb-2011-12-4-r41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meuwissen R, Linn SC, Linnoila RI, Zevenhoven J, Mooi WJ, Berns A. Induction of small cell lung cancer by somatic inactivation of both Trp53 and Rb1 in a conditional mouse model. Cancer Cell. 2003;4:181–189. doi: 10.1016/s1535-6108(03)00220-4. [DOI] [PubMed] [Google Scholar]
- Nowell PC. The clonal evolution of tumor cell populations. Science. 1976;194:23–28. doi: 10.1126/science.959840. [DOI] [PubMed] [Google Scholar]
- O'Neil J, Calvo J, McKenna K, Krishnamoorthy V, Aster JC, Bassing CH, Alt FW, Kelliher M, Look AT. Activating Notch1 mutations in mouse models of T-ALL. Blood. 2006;107:781–785. doi: 10.1182/blood-2005-06-2553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peifer M, Fernández-Cuesta L, Sos ML, George J, Seidel D, Kasper LH, Plenker D, Leenders F, Sun R, Zander T, et al. Integrative genome analyses identify key somatic driver mutations of small-cell lung cancer. Nat Genet. 2012;44:1104–1110. doi: 10.1038/ng.2396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pilarski R, Stephens JA, Noss R, Fisher JL, Prior TW. Predicting PTEN mutations: an evaluation of Cowden syndrome and Bannayan-Riley-Ruvalcaba syndrome clinical features. J Med Genet. 2011;48:505–512. doi: 10.1136/jmg.2011.088807. [DOI] [PubMed] [Google Scholar]
- Podsypanina K, Li Y, Varmus HE. Evolution of somatic mutations in mammary tumors in transgenic mice is influenced by the inherited genotype. BMC Med. 2004;2:24. doi: 10.1186/1741-7015-2-24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rausch T, Jones DT, Zapatka M, Stütz AM, Zichner T, Weischenfeldt J, Jäger N, Remke M, Shih D, Northcott PA, et al. Genome sequencing of pediatric medulloblastoma links catastrophic DNA rearrangements with TP53 mutations. Cell. 2012;148:59–71. doi: 10.1016/j.cell.2011.12.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson JT, Thorvaldsdottir H, Winckler W, Guttman M, Lander ES, Getz G, Mesirov JP. Integrative genomics viewer. Nat Biotechnol. 2011;29:24–26. doi: 10.1038/nbt.1754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rudin CM, Durinck S, Stawiski EW, Poirier JT, Modrusan Z, Shames DS, Bergbower EA, Guan Y, Shin J, Guillory J, et al. Comprehensive genomic analysis identifies SOX2 as a frequently amplified gene in small-cell lung cancer. Nat Genet. 2012;44:1111–1116. doi: 10.1038/ng.2405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shepherd C, Banerjee L, Cheung CW, Mansour MR, Jenkinson S, Gale RE, Khwaja A. PI3K/mTOR inhibition upregulates NOTCH-MYC signalling leading to an impaired cytotoxic response. Leukemia. 2013;27:650–660. doi: 10.1038/leu.2012.285. [DOI] [PubMed] [Google Scholar]
- Song H, Yao E, Lin C, Gacayan R, Chen MH, Chuang PT. Functional characterization of pulmonary neuroendocrine cells in lung development, injury, and tumorigenesis. Proc Natl Acad Sci USA. 2012;109:17531–17536. doi: 10.1073/pnas.1207238109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sutherland KD, Proost N, Brouns I, Adriaensen D, Song JY, Berns A. Cell of origin of small cell lung cancer: inactivation of Trp53 and Rb1 in distinct cell types of adult mouse lung. Cancer Cell. 2011;19:754–764. doi: 10.1016/j.ccr.2011.04.019. [DOI] [PubMed] [Google Scholar]
- Sweet-Cordero A, Tseng GC, You H, Douglass M, Huey B, Albertson D, Jacks T. Comparison of gene expression and DNA copy number changes in a murine model of lung cancer. Genes Chromosomes Cancer. 2006;45:338–348. doi: 10.1002/gcc.20296. [DOI] [PubMed] [Google Scholar]
- To MD, Quigley DA, Mao JH, Del Rosario R, Hsu J, Hodgson G, Jacks T, Balmain A. Progressive genomic instability in the FVB/Kras(LA2) mouse model of lung cancer. Mol Cancer Res. 2011;9:1339–1345. doi: 10.1158/1541-7786.MCR-11-0219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsou HC, Ping XL, Xie XX, Gruener AC, Zhang H, Nini R, Swisshelm K, Sybert V, Diamond TM, Sutphen R, Peacocke M. The genetic basis of Cowden's syndrome: three novel mutations in PTEN/MMAC1/TEP1. Hum Genet. 1998;102:467–473. doi: 10.1007/s004390050723. [DOI] [PubMed] [Google Scholar]
- Wartman LD, Larson DE, Xiang Z, Ding L, Chen K, Lin L, Cahan P, Klco JM, Welch JS, Li C, et al. Sequencing a mouse acute promyelocytic leukemia genome reveals genetic events relevant for disease progression. J Clin Invest. 2011;121:1445–1455. doi: 10.1172/JCI45284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wegmann F, Ebnet K, Du Pasquier L, Vestweber D, Butz S. Endothelial adhesion molecule ESAM binds directly to the multidomain adaptor MAGI-1 and recruits it to cell contacts. Exp Cell Res. 2004;300:121–133. doi: 10.1016/j.yexcr.2004.07.010. [DOI] [PubMed] [Google Scholar]
- Wistuba II, Gazdar AF, Minna JD. Molecular genetics of small cell lung carcinoma. Semin Oncol. 2001;28(2, Suppl 4):3–13. [PubMed] [Google Scholar]
- Wu X, Hepner K, Castelino-Prabhu S, Do D, Kaye MB, Yuan XJ, Wood J, Ross C, Sawyers CL, Whang YE. Evidence for regulation of the PTEN tumor suppressor by a membrane-localized multi-PDZ domain containing scaffold protein MAGI-2. Proc Natl Acad Sci USA. 2000;97:4233–4238. doi: 10.1073/pnas.97.8.4233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yachida S, Jones S, Bozic I, Antal T, Leary R, Fu B, Kamiyama M, Hruban RH, Eshleman JR, Nowak MA, et al. Distant metastasis occurs late during the genetic evolution of pancreatic cancer. Nature. 2010;467:1114–1117. doi: 10.1038/nature09515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yokomizo A, Tindall DJ, Drabkin H, Gemmill R, Franklin W, Yang P, Sugio K, Smith DI, Liu W. PTEN/MMAC1 mutations identified in small cell, but not in non-small cell lung cancers. Oncogene. 1998;17:475–479. doi: 10.1038/sj.onc.1201956. [DOI] [PubMed] [Google Scholar]
- Yuan W, Stawiski E, Janakiraman V, Chan E, Durinck S, Edgar KA, Kljavin NM, Rivers CS, Gnad F, Roose-Girma M, et al. Conditional activation of Pik3ca(H1047R) in a knock-in mouse model promotes mammary tumorigenesis and emergence of mutations. Oncogene. 2012;32:318–326. doi: 10.1038/onc.2012.53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zmajkovicova K, Jesenberger V, Catalanotti F, Baumgartner C, Reyes G, Baccarini M. MEK1 is required for PTEN membrane recruitment, AKT regulation, and the maintenance of peripheral tolerance. Mol Cell. 2013;50:43–55. doi: 10.1016/j.molcel.2013.01.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.